
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkali-induced catalytic tuning at metal and metal oxide interfaces
Wenjie
Liao
 *a,
An
Nguyen
b and
Ping
Liu
*ab
*a,
An
Nguyen
b and
Ping
Liu
*ab
aChemistry Division, Brookhaven National Laboratory, Upton, NY 11973, USA. E-mail: wliao@bnl.gov; pingliu3@bnl.gov
bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, USA
First published on 1st April 2025
Abstract
Alkali metals have been recognized as effective promoters in heterogeneous catalysis, capable of enhancing catalytic activity and tuning product distributions. Over the past few decades, significant efforts have been made aiming to reveal the mechanisms underlying the promoting effect of alkalis. However, the roles that alkali metals play in the catalytic process remain elusive due to challenges in capturing their catalytic behaviours upon exposure to reactive environments. This review summarizes recent surface science and theoretical studies of alkali (potassium, cesium)-decorated metal and metal oxide model catalysts, revealing the crucial tuning by alkalis of activity and selectivity for CO2 hydrogenation. The analysis of electronic structures identifies the selective binding mechanism of the positively charged alkali ions on the surface, being able to reduce the surface work function and lead to strong electron polarization on the surfaces. Depending on the alkali–support interaction, the deposition of alkalis can selectively modify the bindings of reaction intermediates involved in CO2 hydrogenation via the interplay among the ionic, covalent and electrostatic tunings. As a result, CO2 can be effectively activated and converted into diverse products at the alkali–support interface, ranging from formic acid to methanol and ethanol. The identified selective bond-tuning advances the application of alkalis in promoting catalytic activity and controlling catalytic selectivity at alkali–support interfaces.
 Wenjie Liao | Wenjie Liao received his BS in Chemical Engineering from Tianjin University in 2017 and earned his PhD in Chemistry from Stony Brook University in 2023 under the supervision of Dr Ping Liu. He is currently a postdoctoral researcher in Dr Ping Liu's group at Brookhaven National Laboratory. His research focuses on employing computational chemistry tools and machine learning models to understand the nature of active sites, identify the operational reaction pathways, and promote catalytic performance in the conversion of C1 molecules. |
 An Nguyen | Phuc-An Nguyen earned a BS in Integrated Science from Fulbright University Vietnam in Saigon in 2023 as part of the institution's inaugural graduating class. Currently, Nguyen is a second-year PhD candidate in Chemistry at Stony Brook University, working under the guidance of Dr Ping Liu. As a researcher in Dr Liu's group at Brookhaven National Laboratory, Nguyen leverages computational chemistry techniques and machine learning methodologies to explore the nature of catalytic active sites, elucidate key reaction mechanisms, and enhance catalytic efficiency. |
 Ping Liu | Ping Liu is a senior chemist in the Chemistry Division, Brookhaven National Laboratory and an adjunct faculty member of the Chemistry Department, Stony Brook University. Her research focuses on theoretical description of multifunctional catalysts and their catalytic applications in heterogeneous catalysis and electrocatalysis, going from a fundamental understanding of catalytic processes under reaction conditions to providing guidance for catalyst design. Dr Liu has been recognized as a Highly Cited Researcher in Cross-Field research by Clarivate Analytics. |
1. Introduction
Alkali metals, especially potassium (K) and cesium (Cs), have been widely used as promoters to tune the activity and selectivity of heterogenous catalysts for many crucial chemical processes such as Fischer–Tropsch synthesis (FTS), water–gas shift reactions, methanol (CH3OH) synthesis from carbon monoxide (CO)/carbon dioxide (CO2) hydrogenation, and ammonia synthesis.1–9 It has been reported previously through both experimental10–14 and theoretical studies15–25 that doping with alkali metals enables tuning of the adsorption and dissociation of molecules, such as CO,10,26 CO2,11,27 oxygen (O2),16,28,29 nitrogen (N2),7,30 and water (H2O),22,31,32 at the transition metal sites. So far, there has been no generally accepted mechanism to pinpoint the origin of the catalytic promoting effects of alkalis. Alternatively, several mechanisms have been proposed for the alkali-induced bond-tuning, including (1) electronic effects that modify the electronic structure of neighbouring transition metal sites via electron transfer from the alkali;12,33–35 (2) reduction of the work function on transition metal surfaces,36–39 thus affecting the ionization potential and Lewis basicity;40,41 (3) electrostatic effects via dipole–dipole interactions between positively charged alkali ions and polarized adsorbates;10,15–17,42 (4) steric effects including active site blocking20,43,44 that inhibit adsorption of intermediates,17 and surface reconstruction;45 (5) synergistic or ensemble effects between alkalis and neighboring active sites which are necessary to provide effective binding to the reaction intermediates.33The understanding of how alkalis function during catalytic processes is hindered by limited understanding of configurations of alkali deposits and their binding conformations with reaction intermediates under reaction conditions. This is extremely difficult to achieve at an atomic level using the current experimental tools. Instead, diverse structures have been proposed theoretically, which may not be directly relevant to the catalysis. Alkali promoters typically present in the form of oxides or salts with various morphologies rather than metallic phases upon interaction with reactive environments, which is often accompanied by structural changes. Accurate description of such complex transitions is crucial to elucidate the alkali-induced variation in reaction mechanisms and catalytic performances, which can greatly promote rational catalyst design using alkalis.
The present review discusses the recent surface science and theoretical studies of alkali-induced tuning effects in heterogenous catalysis, providing a bottom-up understanding going from surface morphology, electronic and atomic structures, and binding properties to catalytic activity and selectivity. CO2 hydrogenation on metal and metal oxide catalysts was focused, which is of catalytic interest due to the importance in converting greenhouse gases into value-added chemicals. In particular, the production of methanol (CO2 + 3H2 → CH3OH + H2O, ΔH = −49.5 kJ mol−1) has attracted considerable attention. The current industrial catalyst for CH3OH synthesis from CO2 hydrogenation, Cu–Zn/Al2O3, suffers from a low CO2 conversion rate and CH3OH selectivity due to the side reaction, reverse water–gas-shift reaction (RWGS: CO2 + H2 → CO + H2O, ΔH = 41.2 kJ mol−1).46–48 According to previous studies on metallic Cu surfaces and nanoparticles,49–51 the CH3OH yield could be improved by stabilizing the key intermediates, e.g. *CO2, formic acid (*HCOOH), carbon monoxide (*CO), or formyl (*CHO). Extensive studies have focused on optimizing the binding energy of these key intermediates and thus the activity of Cu using metal dopants50,51 or oxide promoters,48,52–56 while the promoting effect on alcohol selectivity is rather small and CO remains the major product. Cs has been reported to be an efficient promoter for CO2 hydrogenation particularly on Cu-based catalysts,57 not only promoting the adsorption and activation of CO2,26,58–60 but also enhancing the selectivity of CH3OH,61–63 ethanol (C2H5OH) and other higher alcohols.64–66 In addition, well distributed K over Cu/SiO2 was also found to facilitate CO2 conversion,67 while the CH3OH selectivity was enhanced by the addition of K during CO hydrogenation over unsupported Cu catalysts.68,69 Yet, the roles that Cs and K play during the CO2 hydrogenation is less understood.
This review rationalizes how the multiple active sites at the alkali–support interfaces behave to tune the reaction mechanism, and explores the general design principle, challenges and opportunities in optimizing alkali–support interfaces towards selective CO2 conversion to alcohols. CO2 hydrogenation is rather complex, and typically involves multiple pathways, including formate, RWGS, CO hydrogenation and methanation pathways, as well as products, including methane (CH4), CO, HCOOH, formaldehyde (CH2O) and alcohols (i.e., CH3OH and C2H5OH, Fig. 1). Product separation can be cost effective in practical applications. Thus, the selectivity was focused on here, that is, tuning the alkali–support interface to improve CO2 hydrogenation toward CH3OH and higher alcohols, which are chemically more valuable than CO and CH4. This review first summarizes the morphologies of alkali-decorated surfaces upon exposure to reactive environments. This summary is followed by identification of the tunable binding nature of alkali sites via alkali–support interactions and the corresponding impact on catalytic selectivity during CO2 hydrogenation. Finally, the challenges and opportunities for optimizing alkali–support interfaces to advance selective CO2 hydrogenation are discussed.
 | ||
| Fig. 1 Schematics of the reaction network for CO2 hydrogenation to C1 products. “*X” represents an adsorbed species on the surface. | ||
2. Morphology of alkali-decorated metal surfaces
2.1. K on Cu(111)
Decoration with K has played essential roles in promoting catalytic reactions in many established industrial catalytic processes including WGS, Haber–Bosch and FTS processes.70–72 A combined density functional theory (DFT) and scanning tunneling microscopy (STM) study showed that both K and Cu(111) undergo oxidation, and deposition of K leads to the further oxidation of CuxO film supported on Cu(111) or K/CuxO/Cu(111) (x ≤ 2) by sequential annealing under an oxygen atmosphere (Fig. 2a).73 The DFT calculations identified a pseudomorphic growth mode, which is mainly controlled by strong interaction with chemisorbed oxygen at the center of the Oδ−–Cuδ+–Oδ− hexagonal ring. The coverage can reach up to 0.19 monolayer (ML), which is consistent with the STM observations (Fig. 2b). | ||
| Fig. 2 (a) STM image of the K/CuxO/Cu(111) surface scanned at room temperature, which is followed by sequential annealing in 5 × 10−7 Torr O2 at 500 K for 10 min: 0.87 nA, 1.68 V. Inset: A 20 × 20 nm2 STM image of the surface prepared by room temperature deposition: 0.52 nA, 0.49 V. (b) DFT-optimized structure for K/CuxO/Cu(111) and the corresponding simulated STM image in constant current mode at −1.2 V sample bias. (c) Calculated electron localization function (ELF) of the CuxO/Cu(111) before (left) and after (right) the deposition of K, in which the projected 2D slices over and normal to the surface are displayed. The iso-surface level was chosen as 0.3e/a03 (a0 represents Bohr radius). Side views of DFT optimized structures for (d) *CO and (e) *CO2 on K/CuxO/Cu(111) with the calculated charge density difference. Color code: red – O; brown – Cu; purple – K; grey – C; yellow iso-surfaces – charge accumulation; cyan iso-surfaces – charge depletion at 0.001e/a03 (a0: Bohr radius). Copyright 2015, Wiley.73 | ||
The deposition of K has a significant impact on the surface electronic structure of CuxO/Cu(111).73 A reduction in work function from 5.3 eV to 2.0 eV was observed. This leads to strong electron polarization on the surfaces in a selective way, that is, enhancing the Lewis basicity of the surface, but hardly altering the weak Lewis acidity (Fig. 2c). As a result, the tuning of K on the chemical activities of CuxO/Cu(111) is selective. For bindings of CO and CO2 involved in CO oxidation, the K-induced effect destabilizes *CO (ΔBE = 0.25 eV), but stabilizes *CO2 binding in a bent O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO motif (ΔBE = −0.22 eV, Table 1) compared with CuxO/Cu(111). In both cases, the deposited K donates electrons to CuxO/Cu(111) (electronic effect) and there is a synergy between K sites and Cu sites to accommodate the O anchor via electrostatic Oδ−–Kδ+ interaction and C anchor via Cu–C interaction (ensemble effect, Fig. 2d and e). While the opposite bond-tuning is likely dominated by the K-induced reduction in work function and accumulation of electron densities over the surface of CuxO/Cu(111), which facilitates charge transfer to *CO2 and formation of stable carboxylic-like species, but destabilizes *CO likely due to the nature of weak electron-acceptor and weak C–Cuδ+ interactions (Fig. 2d).
CO motif (ΔBE = −0.22 eV, Table 1) compared with CuxO/Cu(111). In both cases, the deposited K donates electrons to CuxO/Cu(111) (electronic effect) and there is a synergy between K sites and Cu sites to accommodate the O anchor via electrostatic Oδ−–Kδ+ interaction and C anchor via Cu–C interaction (ensemble effect, Fig. 2d and e). While the opposite bond-tuning is likely dominated by the K-induced reduction in work function and accumulation of electron densities over the surface of CuxO/Cu(111), which facilitates charge transfer to *CO2 and formation of stable carboxylic-like species, but destabilizes *CO likely due to the nature of weak electron-acceptor and weak C–Cuδ+ interactions (Fig. 2d).
| Species | BE (eV) | ||||
|---|---|---|---|---|---|
| Cu(111)74 | Cs3O4H3/Cu(111)66 | K/CuxO/Cu(111)74 | K2O/Au(111)75 | Cs3O4H3/Au(111)76 | |
| *CO2 | −0.06 | −0.86 | −0.32 | −0.40 | −0.03 |
| *HCOO | −2.84 | −3.50 | −3.75 | — | −2.98 |
| *COOH | −1.66 | −1.53 | −2.39 | — | −0.84 |
| *H2COO | −4.03 | −3.55 | −3.66 | — | −2.40 |
| *HCOOH | −0.24 | −0.41 | −0.84 | — | −0.36 |
| *H2COOH | −2.19 | −2.84 | −2.66 | — | −2.17 |
| *H2C(OH)2 | −0.25 | −0.50 | −0.92 | — | −0.42 |
| *CO | −0.89 | −0.17 | −0.41 | −0.13 | −0.04 |
| *CHO | −1.29 | −0.73 | — | — | −0.21 |
| *CH2O | −0.11 | −0.27 | −0.51 | — | −0.15 |
| *CH3O | −2.44 | −2.33 | −2.42 | — | −1.81 |
| *CH3OH | −0.29 | −0.40 | −0.85 | — | −0.34 |
| *OH | −2.92 | −3.55 | −3.41 | −2.57 | −2.60 |
| *H2O | −0.24 | −0.48 | −0.94 | −0.88 | −0.49 |
2.2. K on Au(111)
The situation for K deposition on Au(111) is different from that of Cu(111). Upon deposition of K under an oxygen atmosphere on Au(111), small KOx clusters at low K loading were observed at the undercoordinated elbow sites of the herringbone reconstruction by scanning tunneling microscopy (STM) (Fig. 3a).75 At low temperature (<305 Kelvin), the formation of K2O2 on Au(111) is preferred by dissociative O2 adsorption at a K coverage of 0.125 ML (2*K+ O2(g) → *K2O2 + *, Fig. 3b) according to the phase diagram that was calculated using DFT. Herein, the O–O bond distance in *K2O2 (1.46 Å) is closer to a peroxo (1.49 Å) instead of a superoxo (1.28 Å) which well describes the STM data collected at ∼0.1 ML coverage.15 | ||
| Fig. 3 (a) The multiple growth modes of potassium oxide modelled by DFT calculations (upper row) and observed by STM (lower row). Color code: purple – K; gold – Au; red – O. (b) Calculated phase diagram of potassium oxide species on Au(111) under oxidation conditions. Copyright 2022, Wiley.75 | ||
As temperature increases (300–465 Kelvin), the O–O bond dissociation of K2O2 and subsequent reduction by metallic K becomes favorable (1/2*K2O2 + *K → *K2O, Fig. 3b), leading to the formation of K2O/Au(111) (Fig. 3a, left column). At higher temperatures (>465 Kelvin), the entropic contribution of O2 dominates the thermodynamics, which drives the decomposition of *K2O2 to *K2O (K2O2 → *K2O + 1/2O2(g), Fig. 3b). Meanwhile, the formation of K2O2 is no longer a thermodynamic spontaneous process, so K2O2 is not a stable phase at this elevated temperature. At medium to high K coverage (∼0.2 ML, Fig. 3a), small KOx clusters can aggregate to form large islands as observed in STM, which likely adopts K3O3 overlayer on Au(111) according to the DFT calculations.75 Notably, the K+ ions are arranged in a hexagonal overlayer corresponding to a periodicity of 8.8 Å. Such hexagonal conformation cannot be maintained in other K3Oy/Au(111) (y = 1, 2, 4, 5, 6) overlayer systems, which often require the rearrangement of K and interacted Au to maximize their oxygen coordination.
Using CO oxidation as a probing reaction, it was observed experimentally that the rate of CO2 production on exposure to 4 Torr of CO and 2 Torr of O2 at 473 Kelvin is zero on Au(111) and reaches a maximum around 0.1 ML of KOx on Au(111).75 When the coverage of KOx goes above 0.15 ML, the activity starts to drop. Under this condition, K2O/Au(111) is likely formed according to the DFT-calculated phase diagram (Fig. 3b), which displays an optimal activity and resistance to deactivation by carbonate (*CO3), a potential poison, as compared to K2O2/Au(111) and K3O3 overlayer/Au(111).
Due to accumulated electron density on the top of Oδ− (Fig. 4a), the oxygen site acts as a strong anchor for CO to facilitate the production of *CO2 (BE = −0.40 eV, Fig. 4b and Table 1) via the Mars–van-Krevelen (MvK) mechanism. In addition, due to the limited oxygen available for *K2O, the formation of *CO3 is hindered (BE = −0.85 eV, Fig. 4b). While with the decreased K![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio, *K2O2 and *K3O3 can provide more oxygen, tuning the selectivity gradually from CO2 toward *CO3 (BE = −2.34 eV, Fig. 4c and d). This is well demonstrated by the experimentally observed decrease in activity at high K coverage. Herein, both Au and KOx participate in binding, as seen for K/CuxO/Cu(111) (Fig. 2). The high CO oxidation activity of K2O/Au(111) depends on the combined contribution from the K-induced reduction in work function and surface accumulation of electron density to activate the oxygen sites, ensemble effect of surface Au sites and electrostatic effect provided by K, which provides active sites to stabilize the C-anchor and O-anchor of CO2 respectively.75
:O ratio, *K2O2 and *K3O3 can provide more oxygen, tuning the selectivity gradually from CO2 toward *CO3 (BE = −2.34 eV, Fig. 4c and d). This is well demonstrated by the experimentally observed decrease in activity at high K coverage. Herein, both Au and KOx participate in binding, as seen for K/CuxO/Cu(111) (Fig. 2). The high CO oxidation activity of K2O/Au(111) depends on the combined contribution from the K-induced reduction in work function and surface accumulation of electron density to activate the oxygen sites, ensemble effect of surface Au sites and electrostatic effect provided by K, which provides active sites to stabilize the C-anchor and O-anchor of CO2 respectively.75
 | ||
| Fig. 4 (a) Contour plot of charge density difference for K2O/Au(111). The blue region and red region indicate the accumulation and depletion of electron density, respectively. Side views of the DFT optimized structures for (b) *CO2 on K2O/Au(111), (c) *CO3 on K2O/Au(111), and (d) *CO3 on K3O3/Au(111). Color code: purple – K; gold – Au; red – O; grey – C. Copyright 2022, Wiley.77 | ||
2.3. Cs on Au(111)
The deposition of Cs on Au(111) behaves in a similar way to that of K, demonstrating a coverage-dependent growth mode akin to that observed with KOx/Au(111) (Fig. 3a).78 Again, STM captured CsOx clusters anchored at the herringbone elbows after depositing 0.05 ML on Au(111). The deposited cluster features a Cs:O ratio of ∼1:1 and 1.5 Å in height and 2.5 nm in width (Fig. 5a).78 When the Cs coverage is increased to 0.2 ML (Fig. 5b), 2D islands that contain Cs2O2 and a CsyO (y ≥ 2) suboxide emerge. When the coverage of CsOx is increased to 0.6 ML, the CsOx islands grow into a continuous film with a rectangular growth pattern (zone A, Fig. 5c) or hexagonal pattern (zone B, Fig. 5c), covering most of Au(111).78
 | ||
| Fig. 5 STM images of CsOx with different coverages on Au(111): (a) 0.05 ML; (b) 0.2 ML; (c) 0.6 ML. Copyright 2024, American Chemical Society.78 Side views of DFT-optimized structures for (d) the Cs3O4H3/Au(111) model surface with (e) *OH and (f) *H2O adsorption. Color code: dark purple – Cs; gold – Au; red – O; white – H. Copyright 2023, American Chemical Society.76 | ||
The WGS reaction was selected to probe the intrinsic activity of CsOx/Au(111), where Au(111) alone exhibits negligible reactivity.79,80 While with coverage less than 0.1 ML, CsOx-deposited Au(111) effectively dissociates H2O via formation of a Cs–OH bond, which was previously identified as the rate-determining step in the WGS reaction on metal surfaces,31 hereby catalyzing the overall reaction as observed experimentally.78 The catalytic activity of CsOx/Au(111) (apparent activation energy: 0.48 eV) is even lower than that on Cu(111) (apparent activation energy: 0.78 eV). To rationalize the Cs promoting effect, the CsOx clusters at low coverage were described using the Cs3O4H3/Au(111) model in DFT calculations, where a Cs:O ratio of 0.75 and coverage of 0.13 ML closely resembled the experimental observations (Fig. 5d). DFT calculations indicated that the deposition of CsOx destabilizes *OH (Fig. 5e; BE = −2.60 eV vs. −2.92 eV, Table 1) but stabilizes H2O (Fig. 5f; BE = −0.49 eV vs. −0.24 eV) with respect to Cu(111). Consequently, *H2O desorption is effectively hindered, but still enables an exothermic dissociation (reaction energy, ΔE = −0.53 eV) with a low activation barrier (barrier: Ea = 0.13 eV).76 The preferential hydroxylation of CsOx clusters weakens the bindings of both *CO and *CO2 on Cs3O4H3/Au(111) (Table 1) and effectively hinders *CO3 poisoning as seen for KO/Au(111), while it effectively facilitates the oxidation of CO by *OH and the removal of *CO2. Again, the promoted WGS reaction by Cs decoration is likely associated with the interplay of reduction in work function with the electrostatic effect, which provides selective bond-tuning of reaction intermediates via Cs-facilitated and mediated charge transfer from Au(111). Note that Au sites are not involved in the binding directly and the ensemble effect is inhibited in this case.
2.4. Cs on Cu(111)
Different from K/CuxO/Cu(111), Cs/CuxO/Cu(111) was observed to be unstable and undergo reduction to CsOx/Cu(111) by annealing at 650 Kelvin as observed by STM (Fig. 6a) and X-ray photoemission spectroscopy (XPS) measurements.34 According to STM observations, CsOx nanoclusters are randomly dispersed over Cu(111), which adopts an average of 2 nm in width and ∼1 Å in height. This structural motif is well captured by a triangle Cs3O4H3 cluster over Cu(111) (Fig. 6b), where the hydroxylation was also considered to account for the reducing condition of CO2 hydrogenation. | ||
| Fig. 6 (a) STM image of CsOx nanostructures deposited on Cu(111). The CsOx/Cu(111) surface was prepared by annealing a CsOx/Cu2O/Cu(111) system at 650 K. Scanning conditions: 30 nm2, Vt = 1.25 V, It = 0.14 nA. Copyright 2020, AIP Publishing.34 DFT-optimized structures of (b) Cs3O4H3/Cu(111) model surface and (c) *CO2 on Cs3O4H3/Cu(111). Colour code: brown – Cu; dark purple – Cs; red – O; grey – C; white – H. Copyright 2021, American Chemical Society.66 | ||
The introduced Cs3O4H3 was found to distinctly stabilize the probing CO2 molecule (BE = −0.86 eV vs. −0.06 eV for Cu), which is more prominent than that of Cs on Au(111) and K on Cu(111) (Table 1). Compared to Cs3O4H3/Au(111) and K/CuxO/Cu(111), Cs3O4H3/Cu(111) has stronger alkali–support interactions and lower-coordinated sites of small clusters with higher fluxionality, respectively. Both factors enable a significant reduction in work function from 5.3 eV to 3.8 eV, Cs-mediated transfer of ∼0.8 electrons and strengthened *CO2 binding (Table 1). Upon exposure to CO2, the *OH group of the Cu(111)-supported Cs3O4H3 cluster is highly flexible and chemically active forming C–O bonds and *HCO3 species at the Cs sites (Fig. 6c). In addition, the negatively charged *CO2 corresponds to a charge accumulation over the O anchor, which provides additional stabilization to the produced *HCO3 by strengthening the electrostatic Oδ−–Csδ+ interaction (Fig. 6d). Indeed, XPS showed that on exposure to CO2 the O 1s region of Cs3O4H3/Cu(111) was drastically modified and produced a clear feature at ∼531.5 eV corresponding to adsorbed carbonate species.34
Overall, the K and Cs metals supported on metal surfaces can be highly sensitive to the exposed environment. Depending on the alkali–support interaction, the coverage and reactive gases exposed, distinct morphologies can be formed, ranging from well-dispersed clusters to aggregated islands. Wherein, the presence of alkali always promotes surface oxidation. Such structural variation directly affects the binding properties of the support, where the previously proposed reduction in work function, electronic effect, electrostatic effect and ensemble effect can all contribute. However, how these alkali-induced effects interlay to stabilize CO2 and likely promote the hydrogenation reaction on Au and Cu surfaces remain elusive. In addition, the alkali-tuned binding can be more complex on moving from CO2 to other reaction intermediates involved in CO2 hydrogenation (Fig. 1). To address this complexity and thus the corresponding effect on the overall reaction, an in-depth understanding of the binding nature of alkali-sites is crucial.
3. Selective binding nature of alkalis
In order to understand the nature of K and Cs-introduced bond-tuning, the binding energies of reaction intermediates involved in CO2 hydrogenation on various surfaces were summarized, including on K/CuxO/Cu(111), CsOx/Cu(111), and CsOx/Au(111) surfaces (Table 1). Cu(111) was also included for comparison. Note that CuxO/Cu(111) was not included, which was reported to undergo reduction to Cu(111) under the reducing condition of CO2 hydrogenation,52 while the K deposition promoted the stability via strong K–O interaction.733.1 K/CuxO/Cu(111)
On K/CuxO/Cu(111), DFT calculations indicated that the K sites are catalytically active and directly interact with the reaction intermediates.74 Importantly, these interactions are selective, depending on the nature of adsorbate, as clearly demonstrated for *CO2 and *CO (Fig. 2d and e). Generally, K-induced stabilization is more pronounced for open-shell intermediates, e.g., *HCOO and methoxy (*CH3O), than closed-shell intermediates, e.g., *HCOOH and *CH3OH. While the corresponding magnitude varies based on the binding conformation: the adsorbates anchored merely via O or O-anchored, such as *HCOO, *H2COO, *HCOOH, methoxyhydroxy (*H2COOH), methanediol (*H2C(OH)2), *CH2O, *CH3O, *CH3OH, *OH and *H2O along the formate pathway; the adsorbates anchored via both O and C or O-/C-co-anchored, such as *CO, *CO2, *CH2OH, carboxyl (*COOH) and formyl (*CHO), primarily involved in the RWGS and CO hydrogenation pathways (Fig. 1 and 7). | ||
| Fig. 7 Side views of the DFT optimized adsorption configurations for selected intermediates on the K/CuxO/Cu(111) model surface. (a) *CO2, (b) *HCOO, (c) *COOH, (d) *H2COO, (e) *HCOOH, (f) *H2COOH, (g) *H2C(OH)2, (h) *CHO, (i) *CH2O, (j) *CH3O, (k) *CH3OH, (l) *CO, (m) *H2O, (n) *H2COH, (o) *H, and (p) *OH. Color code: brown – Cu; purple – K; red – O; grey – C; white – H. Copyright 2020, American Chemical Society.74 | ||
 | ||
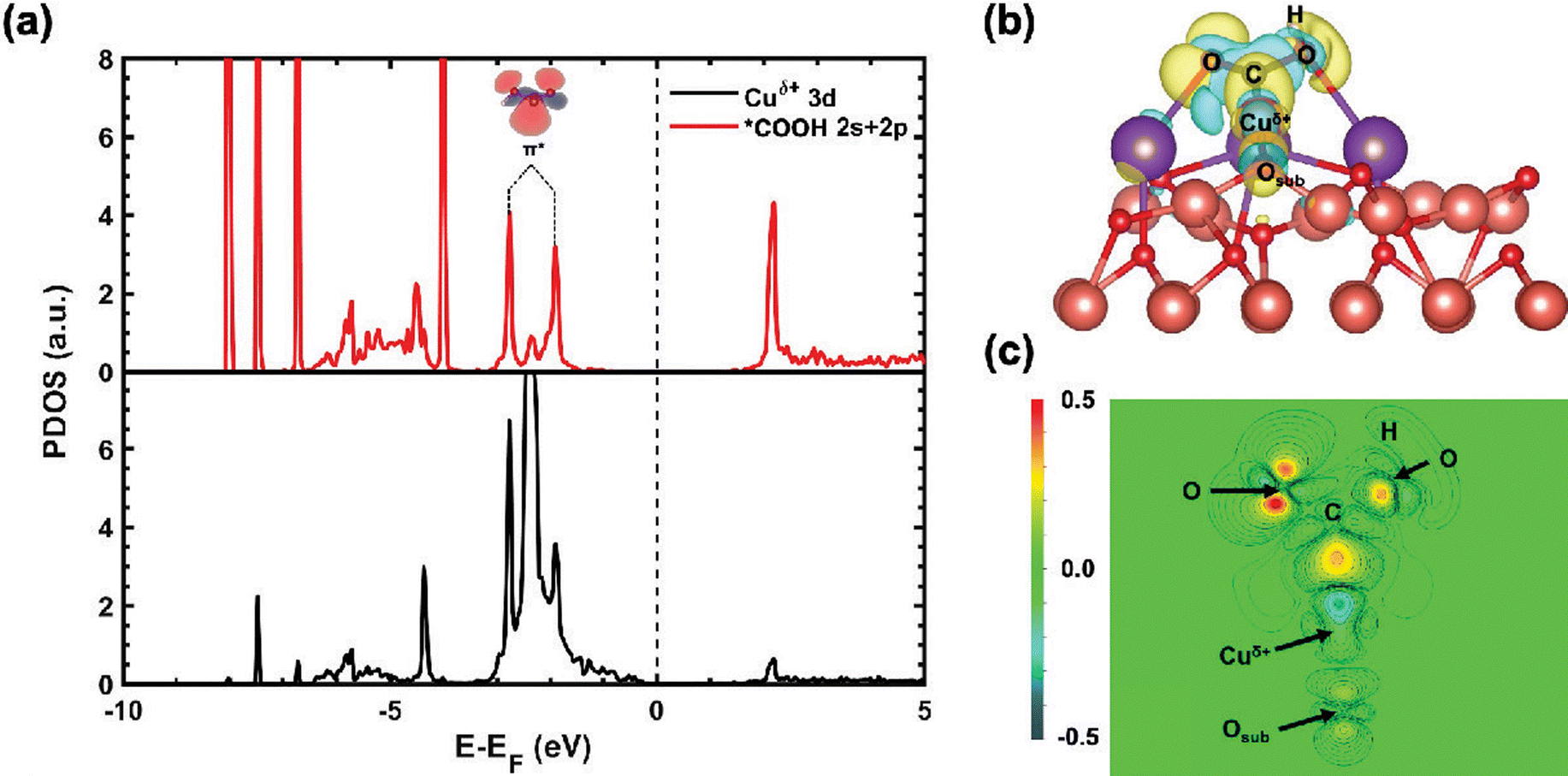
| Fig. 8 Electronic structure of *HCOO on K/CuxO/Cu(111). (a) Calculated projected density of states (PDOS) for the 3d states of Cu0 on Cu(111) (left) and Cuδ+ supported CuxO layer (middle) before (dashed blue line) and after (solid black line) the adsorption of *HCOO, together with the calculated total DOS of *HCOO (right). (b) Side view of the calculated electron density difference of *HCOO on K/CuxO/Cu(111); (c) contour plot of (b), where the cross-section sits on the plane of Cuδ+, K and O. Color code for atoms: brown – Cu; purple – K; red – O; grey – C; white – H. Color code for iso-surfaces: yellow iso-surfaces – charge accumulation; cyan iso-surfaces – charge depletion at 0.001e/a03 (a0: Bohr radius). Copyright 2020, American Chemical Society.74 | ||
Other open-shell O-anchored adsorbates, i.e., *OH, *CH3O, *H2COO and *H2COOH, share the same binding mechanism as *HCOO (Fig. 7). Their interactions with K/CuxO/Cu(111) are also strong (BE < −2 eV) and feature an ionic binding nature via an electron transfer from the surface to the adsorbate (∼−0.8e). On the other hand, the tuning magnitude by K (ΔBE > −0.5 eV) is not as significant as that of *HCOO (ΔBE = −0.91 eV, Table 1). This reduced impact can be related to factors including the elevated energy of the lowest unoccupied state, elongated K–O bond length, reduced number of oxygen anchors and local structural distortions on interaction.
 | ||
| Fig. 9 Structure of *HCOOH on K/CuxO/Cu(111). (a) Side view of the calculated electron density difference: yellow and cyan area depict the iso-surface for the accumulation and the depletion of charge density at 0.001e/a03 (a0: Bohr radius); (b) contour plot of (a), the cross-section sits on the molecular plane of *HCOOH. Copyright 2020, American Chemical Society.74 | ||
Similar situations were observed for *H2O and *CH3OH with the binding energies strengthened from −0.24 eV and −0.29 eV on Cu(111) to −0.94 eV and −0.85 eV on K/CuxO/Cu(111), respectively (Table 1). Again, the electrostatic interactions together with the contribution from hydrogen bonds play a major role, while the contribution due to charge transfer is rather limited (−0.15e and −0.12e, respectively). For *H2C(OH)2 (BE = −0.92 eV vs. −0.25 eV for Cu(111), Table 1) and *CH2O (BE = −0.51 eV vs. −0.11 eV for Cu(111), Table 1), although there is a lack of hydrogen bonds, the K-enhanced binding is still maintained, indicating the dominant contribution from the electrostatic stabilization to the oxygen anchors.
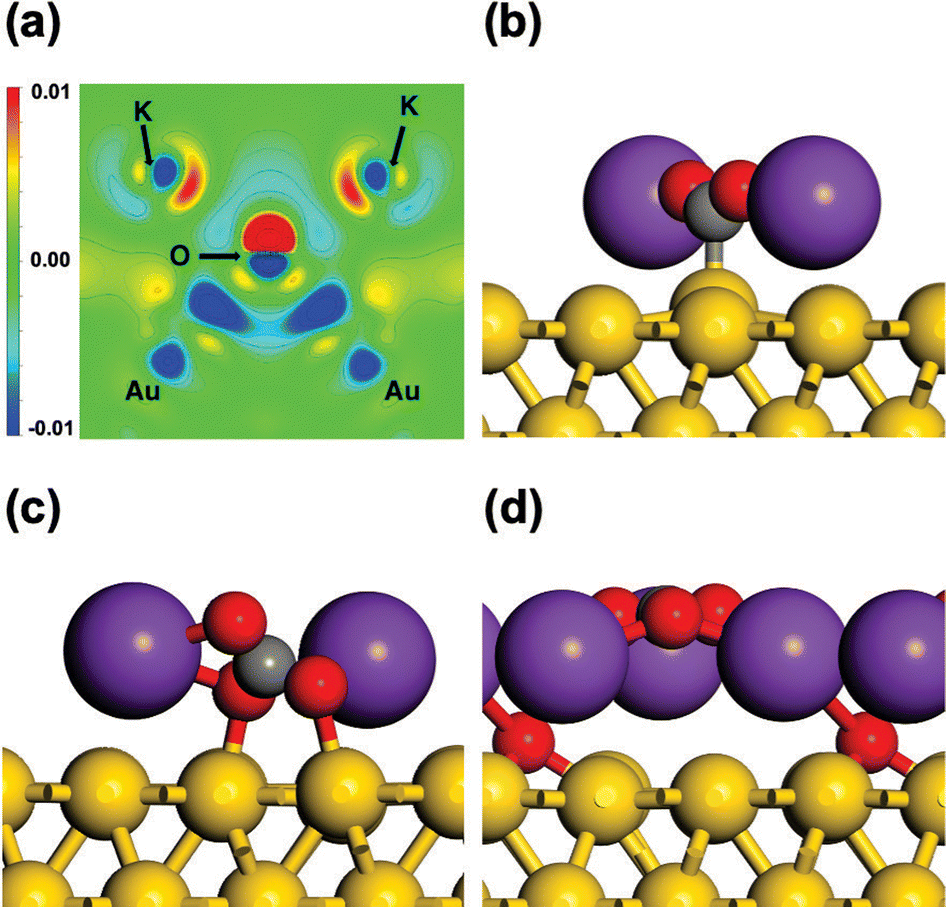
 | ||
| Fig. 10 Structure of *COOH on K/CuxO/Cu(111). (a) Calculated projected density of states (PDOS) for the 3d states of Cuδ+ (black line) and the interacting *COOH (red line). Inset depicts the calculated wave function of π* states in –COOH(g). (b) Side view of the calculated electron density difference: yellow and cyan areas depict the iso-surface for the accumulation and the depletion of charge density at 0.001e/a03 (a0: Bohr radius); (c) contour plot of (b), the cross-section sits on the molecular plane of *COOH. Copyright 2020, American Chemical Society.74 | ||
In general, the deposition of K on Cu(111) and thus the formation of K/CuxO/Cu(111) effectively tune the binding of intermediates involved in CO2 hydrogenation (Table 1). K participates in the binding directly. Depending on the binding mechanism, the bond-tuning can be divided into three groups. One is the ionic tuning that takes advantage of the K-induced reduction in work function and surface accumulation of electron density. It typically results in significant charge transfer and bond-strengthening as seen for O-anchored open-shell intermediates at K sites. Another is the covalent tuning, which depends on the ensemble of K and Cu sites and is well demonstrated for adsorptions of the C-/O-co-anchored intermediates. Specifically, the contribution from the surface Cu site is essential to stabilize the C-anchor of adsorbates via covalent C–Cuδ+ bonds. Meanwhile, the K site also helps by accommodating the oxygen anchor via electrostatic Oδ−–Kδ+ interaction. Finally, it is the electrostatic tuning which benefits the bindings of all intermediates considered via Oδ−–Kδ+ interaction. The intermediates with more O-anchors may form additional K–O bonds to further promote binding. For the intermediates that possess hydroxyl groups, the formation of hydrogen bonding with surface oxygen sites stabilized by K can gain additional stability. Depending on the nature of the intermediates, the contributions from the three types of tunings may vary, thus enabling selective bond-tuning.
3.2. CsOx/Cu(111)
A similar tuning effect to that for K was also observed for Cs deposited on Cu(111) in the form of CsOx/Cu(111) (Table 1).66 Again, for O-anchored intermediates, the Cs-induced stabilization effect is more prominent for open-shell intermediates via the ionic tuning than that on closed-shell intermediates via the electrostatic bonding. For example, the bond-strengthening introduced by Cs3O4 deposition for open-shell O-anchored *HCOO (ΔBE = −0.66 eV) is more significant than that of closed-shell *HCOOH (ΔBE = −0.17 eV, Fig. 11 and Table 1). | ||
| Fig. 11 Side views for DFT optimized structures for selected intermediates in CO2 hydrogenation on Cs3O4H3/Cu(111). (a) *CO2, (b) *HCOO, (c) *COOH, (d) *HCOOH, (e) *CO, (f) *CHO, and (g) *CH3O. Color code: brown – Cu; dark purple – Cs; red – O; grey – C; white – H. Copyright 2021, American Chemical Society.66 | ||
C-/O-co-Anchored intermediates are destabilized on Cs deposition. The origin of Cs-induced destabilization is likely associated with the absence of an ensemble effect at the CsOx–Cu(111) interface. As demonstrated in the case of K/CuxO/Cu(111), the stabilization of C-/O-co-anchored intermediates requires the participation of Cu sites to form covalent Cu–C bonds. However, the formation of CsOx clusters on Cu(111) completely blocks the access of adsorbed species at the Cs sites to the surface Cu sites (Fig. 11). The binding of *COOH at the Cs hollow site, for instance, merely depends on the two oxygen anchors via electrostatic Oδ−–Csδ+ interactions (Fig. 11). In addition, the charge density in *COOH is accumulated at the dangling C-anchor, likely weakening the electrostatic attraction. As a result, the Cs-decoration does not strengthen the binding of *COOH as seen for K on Cu(111) (ΔBE = −0.73 eV), but results in bond-weakening in this case (ΔBE = 0.13 eV, Table 1).
3.4. CsOx/Au(111)
Upon substituting Cu(111) for Au(111), the local structure of CsOx remains unchanged (Fig. 5), and the impact on the binding of O-anchored closed-shell intermediates is small.76 As seen for *HCOOH, the variation in binding energy is only 0.05 eV (Table 1). The stabilization of *HCOOH requires the electrostatic tuning via the Oδ−–Csδ+ interaction to be effectively enhanced. However, the DFT results showed the identical charge of Cs and O as well as the same Cs–O distance on Au(111) and Cu(111).66,76 These results suggest that such electrostatic binding is more sensitive to the local structure of the alkali oxide cluster than the type of substrate.The O-anchored open-shell intermediates behave differently, where the strong binding depends on the Cs-induced ionic tuning. Upon interaction with *HCOO, for example, both Au(111) and Cu(111) transfer approximately one electron to *HCOO, which is mediated by Cs. However, the surface metal sites that interact directly with the CsOx cluster behave differently. In the case of CsOx/Cu(111), a drastic downshift in Cu 3d orbital is observed. That is, the adsorption of *HCOO promotes the oxidation of surface Cu, which also helps to lower the corresponding binding energy. By comparison, such downshift is far less profound in Au 5d states. Consequently, *HCOO is more stabilized on the Cs sites when using Cu(111) as the substrate (BE = −3.50 eV) instead of Au(111) (−2.98 eV, Table 1).
The alkali-induced selective binding-tuning to the key reaction intermediates involved in CO2 hydrogenation can offer a great opportunity to control the operating reaction pathways and the catalytic activity and selectivity. In particular, the preferentially stabilized O-anchored over the C-/O-co-anchored intermediates likely leads to a suppressed RWGS pathway toward CO production but promoting CO2 activation via the formate pathway toward HCOOH or CH3OH production (Fig. 1 and Table 1). To validate the predictions based on the binding energies of reaction intermediates, detailed studies to map the reaction network and evaluate the catalytic activity and selectivity are necessary.
4. CO2 hydrogenation at alkali–support interfaces
4.1. K–CuxO/Cu(111) interface
Upon depositing K on CuxO/Cu(111), Cuδ+ ions were found to remain intact under the reducing condition of CO2 hydrogenation due to the stabilization of K on interfacial O species, while on CuxO/Cu(111) a reduction to Cu0 was observed.73,74 As demonstrated below, similar surface oxidation promoted by alkali was also observed for other alkali–support systems. The generated interfaces offer multifunctional centers, including alkali cations, metal cations, and metal and oxygen sites, which can work synergistically to allow selective binding of reaction intermediates and effectively tune the selectivity of CO2 hydrogenation.The significantly strengthened K–OOCH bond on K/CuxO/Cu(111) promotes the initial CO2 hydrogenation to *HCOO along the formate pathway according to the DFT-calculated binding energies (Table 1). However, *HCOO may also be over-stabilized to suppress the overall conversion. Although the presence of K also helps to stabilize *HCOOH, the effect is not as significant as that for *HCOO (Table 1). In term of selectivity, the K-populated formate pathway can lead to the production of HCOOH and CH3OH (Fig. 1), which is difficult to predict merely based on the binding energies. A theoretical study was carried out to obtain atomic-level insight into the reaction pathways and kinetics, where the intermediates and transition states along both the formate pathway and the RWGS + CO hydrogenation pathway were taken into consideration.74
According to DFT calculations and kinetic Monte Carlo (kMC) simulations under typical experimental conditions (temperature: 400–600 Kelvin; pressure ratio of CO2:H2 = 1:9),48,50,51,74 the formate pathway was indeed found to dominate the overall CO2 hydrogenation over K/CuxO/Cu(111). The reaction starts with CO2 adsorption at the K bridge site and is followed by sequential hydrogenations to *HCOO, *HCOOH, *H2COOH, H2C(OH)2 and eventually *CH3OH (black line, Fig. 12a). By comparison, the formate pathway via *H2CO (blue line, Fig. 12a) and the RWGS pathway via the *COOH intermediate are less competitive. While on the Cu surfaces, it was reported previously that the *H2CO-mediated formate pathway produces CH3OH, which is ∼2–3 orders of magnitude lower in rate as compared to the CO production via the RWGS pathway.50 In addition, along the RWGS pathway *COOH does not undergo typical C–O bond dissociation,49,50 but favors hydrogenation to produce *HCOOH, which leads to a merging of the RWGS pathway into the formate pathway. Note that the methanation pathway via direct CO2 dissociation is not preferred on both K/CuxO/Cu(111) and Cu surfaces.74
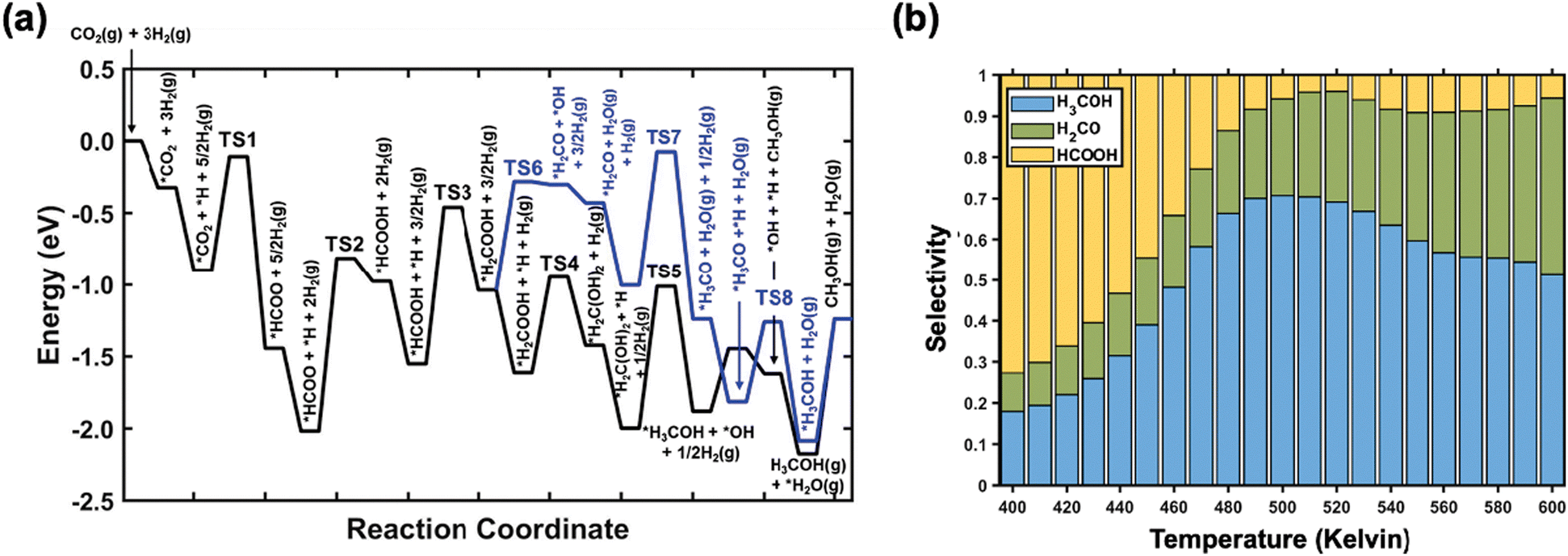 | ||
| Fig. 12 (a) Potential energy diagrams for the hydrogenation of CO2 to CH3OH on the K/CuxO/Cu(111) surface including energetics for reaction intermediates and transition states (TS). The blue line indicates the CH2O-mediated formate pathway, and the black line indicates the H2C(OH)2-mediated formate pathway. (b) Variation in kMC simulated selectivity for carbon-based products, including CH3OH (blue), CH2O (green), and HCOOH (yellow), as a function of temperature during CO2 hydrogenation over K/CuxO/Cu(111). Copyright 2020, American Chemical Society.74 | ||
Upon exposure of a CO2/H2 mixture to K/CuxO/Cu(111) ranging from 400 to 600 Kelvin, the kMC simulation observed a variation in selectivity of carbon-based products (Fig. 12b).74 At 400 Kelvin, the hydrogenation of *HCOOH to H2COOH is still limited by the high activation barrier (Fig. 12a). Thus, *HCOOH prefers to desorb and HCOOH is the major product (72.6%, Fig. 12b). By comparison, the selectivity to CH3OH (17.9%) and CH2O (9.3%) is much lower. Upon going from 400 to 500 Kelvin, the hydrogenation of *HCOOH to H2COOH and *H2C(OH)2 is greatly facilitated (black line, Fig. 12a), which lowers the production of HCOOH and maximizes the CH3OH selectivity (∼70.7%). During this process, the CH2O selectivity also increases from 9.3% to 23.4%. In contrast, the effect on the dissociation of *H2COOH to *CH2O (blue line, Fig. 12a), which is a more difficult step, is rather small. With the temperature elevated to 600 Kelvin, the CH2O selectivity is enhanced the most significantly to 43.0%, which is driven by the increased entropic contribution to *CH2O desorption. Nevertheless, CH3OH remains the most selective carbon-based product at 600 Kelvin (49.3%). Notably, the selectivity to CO, which is the major product on Cu,50 remains as low as 0.3% at the temperatures considered.
The deposited K on CuxO/Cu(111) clearly shows the promoting effect on the CO2 conversion and more importantly tunes the major product from CO as seen for Cu(111) to CH3OH. During this process, K is not a building block, a simple electron donor or a secondary active site.10,15,16,42,93 Instead, it is the major active center. According to the sensitivity analysis,74 reducing the binding strength of O-anchored open-shell *HCOO can effectively promote the CO2 conversion rate, while stabilizing O-anchored closed-shell *HCOOH is critical to tuning the selectivity toward CH3OH. The K-induced selective stabilization on *HCOOH via the electrostatic Oδ−–Kδ+ interaction slows down the desorption, so that *HCOOH has a lifetime long enough to be hydrogenated and thus produce CH3OH (Fig. 9 and 12). However, the ionic tuning by K decoration makes *HCOO too stable to enable facile CO2 conversion. The fine balance between alkali-induced electrostatic tuning and ionic tuning can be crucial to maximize the selective CO2 to CH3OH conversion.
Similar K-promoting effects were also observed for the RWGS reaction over Cu(110), while they were not as prominent as those on Cu(111).22 Due to the higher-lying Cu d-band center, Cu(110) stabilizes K more significantly than Cu(111), likely hindering the structural fluxionality upon interaction with reaction intermediates. In addition, the work function reduction induced by K adsorption on Cu(110) was also found to be less.94 As a result, the K-induced bond strengthening of O-containing intermediates was reported to be more significant on Cu(111) than on Cu(110).22,94
4.2. CsOx–Cu(111) interface
Replacing K for Cs on Cu(111) transforms the interface from KOx–CuxO to CsOx–Cu (Fig. 7 and 11) and the structure of alkali oxide from an overlayer to small clusters, which lead to bond-weakening for most of the reaction intermediates involved in the formate and RWGS pathways (Table 1).66,74 The CsOx–Cu(111) interface helps to stabilize *HCOOH and *HCOO at the Cs sites as compared to the Cu sites on Cu(111), although the effect is less pronounced than that observed on K/CuxO/Cu(111) (Table 1). Therefore, not only CO2 conversion, but also methanol selectivity is likely to be improved. This was confirmed by an experimental study, where CsOx/Cu(111) was identified as the active phase of CsOx/CuxO/Cu(111) under 0.5 atm CO2 and 4.5 atm H2, able to produce CH3OH 10 times faster than that on Cu(111) (Fig. 13).66 The difference in CO2 hydrogenation between K and Cs modification on Cu remains elusive. According to the DFT calculations (Table 1), instead, compared to K/CuxO/Cu(111), the destabilized *HCOO by CsOx/Cu(111) may facilitate the CO2 conversion, but the weakened *HCOOH binding may reduce the selectivity of CH3OH. However, this prediction still needs to be validated by performing a direct comparison of catalytic performance based on theoretical calculations and experimental studies.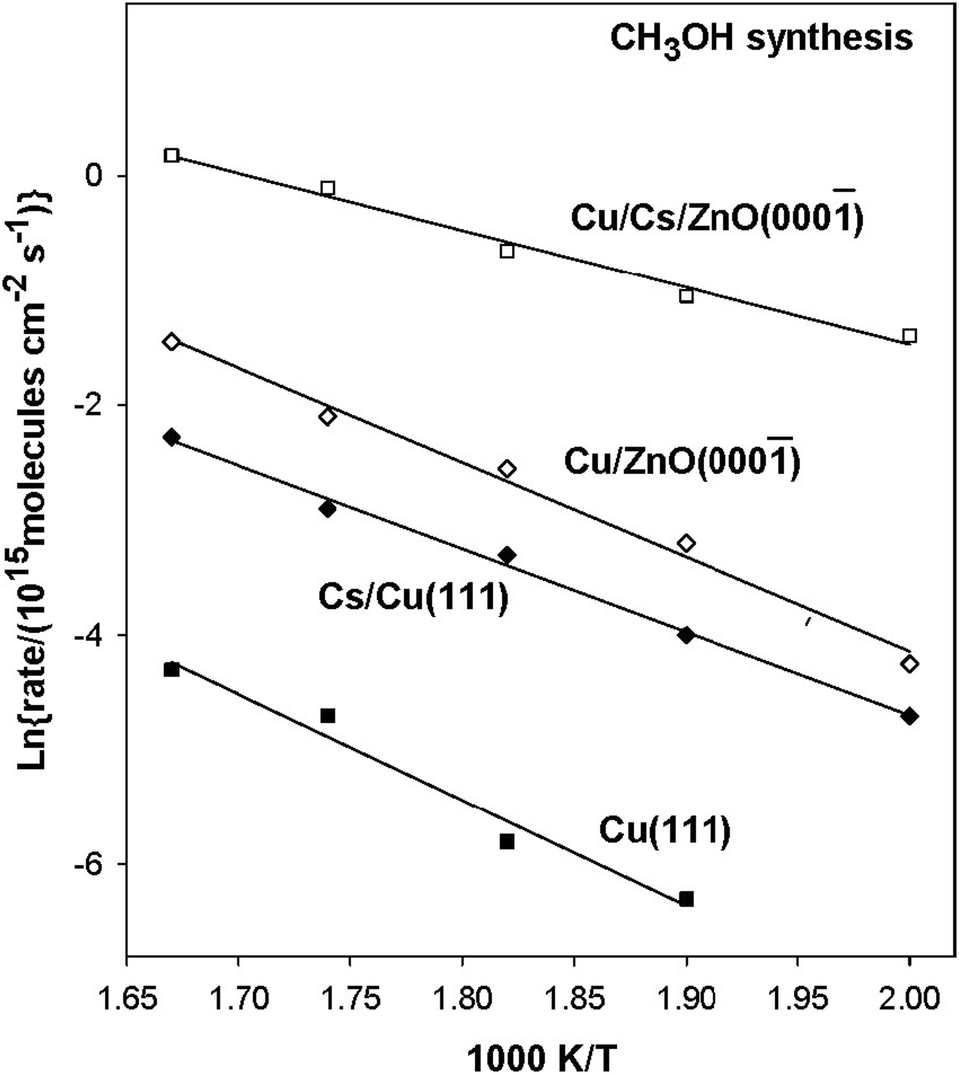 | ||
Fig. 13 Arrhenius plot for methanol synthesis on Cu(111), 0.1 ML of Cs on Cu(111), 0.4 ML of Cu on ZnO(000![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), and 0.1 ML Cs on ZnO(000) surfaces pre-covered by 0.4 ML of Cu. The catalytic tests were done with 0.049 MPa (0.5 atm) of CO2 plus 0.441 MPa (4.5 atm) of H2. Copyright 2021, American Chemical Society.66 ), and 0.1 ML Cs on ZnO(000) surfaces pre-covered by 0.4 ML of Cu. The catalytic tests were done with 0.049 MPa (0.5 atm) of CO2 plus 0.441 MPa (4.5 atm) of H2. Copyright 2021, American Chemical Society.66 | ||
4.3. CsOx–Au(111) interface
Moving from Cu(111) to Au(111) destabilizes all reaction intermediates in the Cs sites at the interface (Table 1). Yet, the bindings of *HCOO and *HCOOH are still more stable than on Cu(111). Thus, the enhanced CO2 conversion via the formate pathway and selective production of CH3OH are expected, which was validated experimentally. Herein, the activation of CO2 over CsOx/Au(111) was observed even at room temperature, which is not feasible on Cu(111).49,50,76 However, only HCOOH is produced.According to the DFT calculations, indeed the RWGS pathway via the *COOH intermediate cannot compete with the formate pathway as observed for K-modified CuxO/Cu(111) (Fig. 14a).74,76 With the suppressed contribution of charge transfer into the bindings of the open-shell intermediates, the initial hydrogenation of *CO2 into *COOH is even more endothermic on CsOx/Au(111) than that on CsOx/Cu(111), by 0.28 eV, though the same adsorption conformation is adopted (Fig. 14b). Instead, the formate pathway is more preferred. Due to the selective destabilization of *HCOO over *HCOOH on going from Cu(111) to Au(111), the hydrogenation from *HCOO to *HCOOH is greatly facilitated on CsOx/Au(111), corresponding to a reduced reaction energy from 1.10 eV to 0.14 eV along with a low activation barrier of 0.42 eV (Fig. 14a). In this case, *HCOOH is stable enough to allow *HCOO hydrogenation. However, it is not ready for the further hydrogenation and decomposition to produce CH3OH, instead overcoming a 0.36 eV barrier for facile desorption.
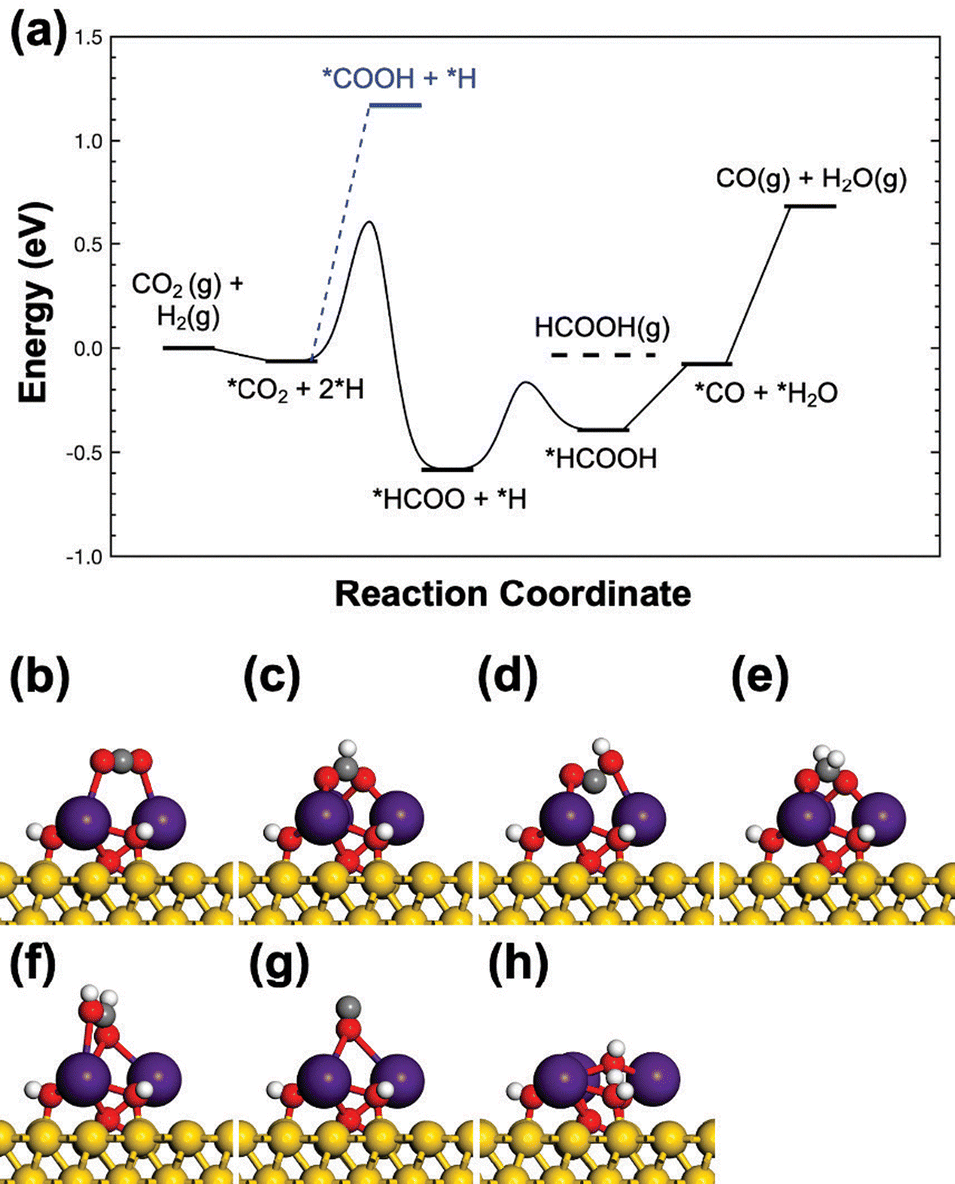 | ||
| Fig. 14 (a) Potential energy diagram for CO2 hydrogenation on the Cs3O4H3/Au(111) surface. Front views of DFT optimized adsorption configurations for the selected intermediates: (b) *CO2, (c) *HCOO, (d) *COOH, (e) *H2COO, (f) *HCOOH, (g) *CO, and (h) *H2O. Color code: gold – Au; dark purple – Cs; red – O; grey – C; white – H. Copyright 2023, American Chemical Society.76 | ||
4.4. Cs–Cu–ZnO(000![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) ) interface
) interface
The tri-phase interface of Cs–Cu–ZnO(000) offers a distinct opportunity to take advantage of the ensemble effect among alkali, metal and metal oxide sites as compared to the dual-phase interfaces, alkali–metal and alkali–oxide, as demonstrated above. A gradual increase in the production of CH3OH from CO2 hydrogenation when going from Cu(111), CsOx/Cu(111), Cu/ZnO(000) to Cs/Cu/ZnO(000) was observed experimentally under 0.5 atm of CO2 and 4.5 atm of H2 (Fig. 13).66 This promoting effect was well captured by the DFT-based kMC simulations. The results showed that under the same condition the average rate for CH3OH synthesis on Cu/ZnO(000) was increased by about four orders of magnitude by the Cs decoration, in reasonable agreement with the experimentally measured increase of about two orders of magnitude (Fig. 13).66
According to the kMC analysis, under these reaction conditions the CO2 hydrogenation undergoes via both the formate pathway and RWGS + CO hydrogenation pathway, which leads to *CHO and eventually *CH3OH formations on Cs/Cu/ZnO(000) (Fig. 15a and b).66 Herein, the Cs–Cu–ZnO(000) interface was identified as the active sites (Fig. 15c–j). The introduction of Cu nanoparticles and thus the low-coordinated Cu sites can greatly enhance the binding properties compared with the Cu sites on the CuxO layer and Cu(111) as reported previously.50,51,74 Given this, the ensemble effect to stabilize the C-anchors of reaction intermediates can be advanced. Meanwhile, the nearby Cs sites also help to interact with the O-anchor as seen for other alkali-decorated systems.66 The ensemble effect via the synergy between Cs and Cu at the Cs–Cu–ZnO(000) interface significantly strengthens the binding of C-/O-co-anchored intermediates, e.g. *CO2, *COOH, *CO and *CHO, which are involved in the RWGS + CO hydrogenation pathway (Fig. 15a and Table 2).66 As a result, CO2 can be activated to form carboxylate-like species, which allows an additional contribution from the ionic tuning to strengthen the binding by 1.19 eV as compared to CsOx/Cu(111) and 1.45 eV as compared to Cu/ZnO(000) (Fig. 15c and Table 2). The kMC results confirmed that the stabilized *CO2 at the interface promotes CO2 conversion to *CHO and to CH3OH. Similar promoting behaviors were also observed for *COOH, *CHO and *CO (Table 2).
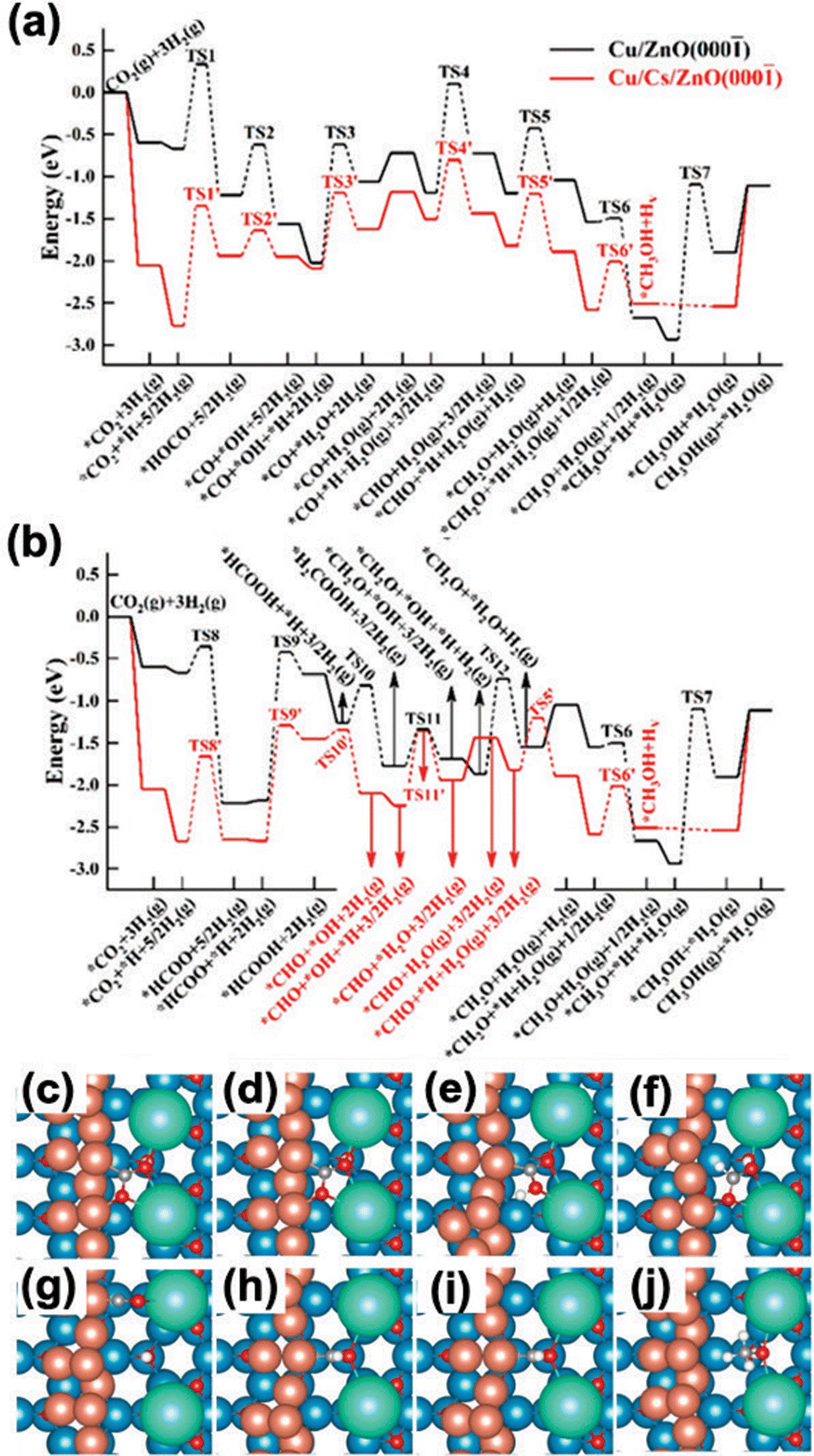 | ||
| Fig. 15 Potential energy diagram for CH3OH synthesis from CO2 hydrogenation on Cu/ZnO(000) (black lines) and Cs/Cu/ZnO(000) (red lines) via the (a) RWGS + CO hydrogenation pathway and (b) formate pathway. Top views of the DFT optimized structures of selected key intermediates: (c) *CO2, (d) *HCOO, (e) *COOH, (f) *HCOOH, (g) *CO, (h) *CHO, (i) *CH2O, and (j) *CH3O. Color code: blue – Zn; brown – Cu; green – Cs; grey – C; white – H. Copyright 2021, American Chemical Society.66 | ||
), CsOx/Cu(111), and Cs/Cu/ZnO(000) surfaces66
| Species | BE (eV) | ||
|---|---|---|---|
| Cu/ZnO(000) |
Cs3O4H3/Cu(111) | Cs/Cu/ZnO(000) |
|
| *CO2 | −0.60 | −0.86 | −2.05 |
| *HCOO | −4.74 | −3.50 | −5.16 |
| *COOH | −3.52 | −1.73 | −4.23 |
| *HCOOH | −0.72 | −0.41 | −1.49 |
| *CO | −1.45 | −0.17 | −1.93 |
| *CHO | −2.57 | −0.73 | −3.28 |
Compared to the C-/O-co-anchored intermediates, stabilization of the O-anchored intermediates at the Cs–Cu–ZnO interface is generally less significant (Table 2). Although *HCOO is anchored at the interface via both O–Cs and O–Cu bonds (Fig. 15d), the missing Cu–C bond limits the contribution from the covalent tuning and the corresponding binding is only stabilized by 0.42 eV compared to Cu/ZnO(000) (Table 2). Yet, an increase in binding of 1.66 eV is gained as compared to CsOx/Cu(111), demonstrating the significant contribution from low-coordinated Cu sites. *HCOOH is adsorbed at the interface in a similar configuration as that of *HCOO, however, in addition to O–Cs and O–Cu bonds, a hydrogen bond is formed with ZnO(000), which results in a stronger binding than that of Cu/ZnO(000) and CsOx/Cu(111) (Fig. 15e and Table 1).
Due to selective bond-strengthening of C-/O-co-anchored intermediates over O-anchored intermediates at the Cs–Cu–ZnO(000) interface, the formate and RWGS + CO hydrogenation pathways are competitive in producing *CHO according to the kMC simulation.66 While the formate pathway is highly preferred for the single phase, Cu, and dual-phase interfaces, K–CuxO, Cs–Cu, K–Au, Cs–Au and Cu–ZnO. In particular, the stability of *CHO is fine-tuned on Cs/Cu/ZnO(000). It is stable enough to drive the C–O bond scission of *HCOOH and prevent the further decomposition to CO, which has been observed for Cu-based systems,50,51,95 but weak enough to be readily available for hydrogenation to produce CH3OH (Fig. 15a and b). More importantly, the appropriate tuning for *CHO at the Cs–Cu–ZnO(000) interface was also found to enable the formation of OHC*–*CHO pairs and facilitate the C–C coupling to produce C2H5OH, though the C2H5OH selectivity is much lower than that of CH3OH.66
5. Summary and outlook
Recent advancements in surface science and theoretical studies of model catalysts have contributed to an in-depth understanding of the morphology of alkali–support interfaces, the selective binding nature of alkali sites, and rational tuning of catalytic activity and selectivity by alkali promoters. This review clearly demonstrates that the variation in combination and local atomic arrangement of alkali (K, Cs) – metal (Cu, Au) or metal oxide (CuxO, ZnO) interfaces under CO2 hydrogenation conditions can effectively facilitate the conversion and selectivity towards various value-added chemicals: formic acid, methanol, and ethanol.The morphology of K, Cs–Cu(111), and Au(111) interfaces strongly depends on alkali–support interactions, coverages, and reactive environments, ranging from dispersed clusters to aggregated islands.34,76,78,96 The role of an alkali is multifunctional, as it promotes surface oxidation, reduces work function, induces polarization of electron densities to the surface, and thus tunes the binding of adsorbates via facilitated charge transfer from the surface, electrostatic interactions and synergy with nearby sites at the interfaces.34,74,76 As a result, the alkali-induced bond-tuning is selective to the reaction intermediates involved in CO2 hydrogenation via three mechanisms: (1) ionic tuning, which leverages alkali-induced reductions in work function and accumulations of surface electron density to particularly stabilize O-anchored open-shell intermediates at alkali sites; (2) covalent tuning, which is reliant on the ensemble effect between alkali and metal sites at the interface to interact with C-/O-co-anchored intermediates; (3) electrostatic tuning, which benefits all intermediates by enhancing Oδ−–K/Csδ+ interactions.66,74,76
Moving from Cs–Cu(111) to K–Cu(111) interface introduces covalent tuning, which interacts with the ionic and electrostatic tunings at the K–CuxO/Cu(111) interface to enhance CO2 activation and methanol production along the alkali-populated formate pathway. However, the destabilized open-shell *HCOO helps alleviate surface poisoning, and the stabilized closed-shell *HCOOH enhances selective CO2 conversion to CH3OH.66,74 Adjusting the substrate from Cu(111) to Au(111) limits ionic tuning and electrostatic tuning and results in destabilized *HCOOH and *HCOO, while covalent tuning remains inhibited. Consequently, CsOx/Au(111) selectively produces HCOOH from CO2 rather than CH3OH, suggesting the critical role of covalent tuning in selective conversion to alcohols.76 Indeed, the Cs–Cu–ZnO(000) interface significantly enhances CH3OH turnover frequency compared to CsOx/Cu(111) by providing the low-coordinated Cu sites from Cu nanoparticles to enhance covalent tuning. In particular, the binding of *CHO can be tuned sufficiently, not only to facilitate the decomposition of *HCOOH and sequential hydrogenation to CH3OH, but also to initiate C–C coupling and ethanol C2H5OH.66
Despite the advances on well-defined model surfaces, insight into the alkali-tuning effect for CO2 hydrogenation over practical powder catalysts remains limited, though the impact is significant. So far, different alkali-induced tuning effects have been proposed. Depositing alkali metals on Co2C/SiO2,97 Ni/CeO2,98 Cu/SiO2,67 Pt/mullite,33 and Rh1/ZrO299 was shown to modify the binding properties of the support and enhance metal dispersion or mitigate sintering, thereby increasing CO2 conversion. In addition, introducing K was reported to reduce the specific surface area and the number of exposed active sites over Cu/SiO2,67 CoCu/TiO2,27 Co/ZrO2,100 Ni/CeO2,98 Ni/Al2O3,101 Co/Al2O3,102 CuO/CeO2,26 and Zr–Co/anatase-TiO2,97 lowering the activity, while the impact on selectivity was less addressed. Significant selectivity-tuning by alkalis was observed for supported Cu catalysts, being able to completely suppress methanol synthesis but produce CO via the RWGS reaction.67 However, for Fischer–Tropsch catalysts such as Co/Al2O3,102 CoCu/TiO2,27 Co2C/SiO2,97 (Co3O4 + Co2C)@Co0,103 Fe/C,104 and Fe3O4@(Fe5C2 + Fe3O4),105 the formation of C2+ products from CO2 hydrogenation, including long-chain hydrocarbons and higher alcohols, is promoted by alkalis. Nevertheless, the underlying mechanisms in all cases remain elusive. Efforts along the following aspects are imperative to enhance the complexity of current model studies, gradually building the atomic understanding of behaviors of alkali-decorated powder catalysts under reaction conditions and advancing the practical application of alkali promoters in CO2 hydrogenation:
(1) Bridging pressure gap to describe interactions between model alkali–support interfaces and reaction conditions. The majority of theoretical studies primarily described the interface by simply depositing alkali on the support, e.g., Ni(111),18 Rh(111),20 Au(111),106 PdPt(111),107 FeO(100),108 MoP(001),109 MoS2(100)110 and Pt/TiO2(110).111 However, these models often lack sufficient fidelity to capture the diversity of alkali morphologies on interaction with the reactive environment. Recently, a computational framework was developed that is capable of accurately predicting activity and selectivity of Pd-based catalysts during CO2 hydrogenation by explicitly accounting for the effect of reaction pressure and temperature on catalyst surface structure and dynamics.112 Herein, the structures of the as-prepared and spent catalysts are different from those during reaction, which are relevant to catalysis. Similar studies will help to map the diverse surface configurations under relevant conditions, which have been demonstrated to effectively impact the alkali-tuned binding to reaction intermediates and thus the catalytic behaviors. Besides, the coordinated in situ characterization on the structures and binding properties of alkali-modified model surfaces is essential to ensure the precision of theoretical prediction on surface structures, which can differ significantly from the ex situ characterization.113–117
(2) Bridging the material gap to capture the structural complexity of powder catalysts. Even though the well-characterized model surfaces can improve the mechanistic understanding of alkali-induced catalytic tuning, in some cases the models are not complex enough to capture the catalytic behaviors of practical power catalysts. For instance, the model study of CO2 hydrogenation over K-modified Cu(111) suggested selective CH3OH synthesis via the promoted formate pathway, with suppression of the RWGS reaction.74 However, the results for K-modified Cu/SiO2 powder catalysts showed selective CO production via a promoted RWGS reaction.67,118–120 Although both systems involve surface oxidation of Cu and the formation of a K–CuxO interface,120 the model surface may not adequately mimic the details such as the local K coverage, the low-coordinated Cu sites of Cu nanoparticles and their preferential motif in interaction with the support and K, thus limiting the capability of model studies to aid in the understanding of power catalysts. Indeed, upon going from Cs/Cu(111) to Cs/Cu/ZnO(000), as demonstrated, not only significant improvement in CO2 conversion to methanol, but also the emerging of ethanol production was observed by including the effect of support and nanoscale Cu catalysts.66 Given that, the complexity of model catalysts needs to be further enhanced to reduce the gap with powder catalysts, which can potentially facilitate the design of alkali-modified catalysts.
(3) Controlling synthesis to ensure coordination of multiple active sites at interfaces. Morphology control of particles is important in heterogeneous catalysis by changing the crystallographic orientation and providing diverse atomic arrangements and active sites.121–124 The stabilized *CHO on Cs/Cu/ZnO(000), for example, enabled the C–C coupling with low C2H5OH production from CO2 on combination of Cs sites and Cu nanoparticles.66 By reducing the size of Cu particles with a MOF structure, Cs/Z12-bpdc-Cu, the combination of Cs and Cu2 centers was found to be highly selective for CO2 hydrogenation to C2H5OH with >99% selectivity.125 Besides the size and shape of metal or metal oxide particles, controlling the loading of alkali and its relative position with respect to the adjacent sites should also be considered, which can impose additional tuning effects to the bindings of reaction intermediates. Ideally, the alkali–support interface should be arranged in a way that maximizes the interplay among ionic, covalent and electrostatic tunings to optimize catalytic activity and selectivity.
(4) Evaluating stability. Due to the ionic tuning effect, the deposition of alkali metal tends to interact strongly with the O-anchored open-shell intermediates, such as *HCOO and *CO3 species in CO/CO2 hydrogenations, via significant surface-to-adsorbate charge transfer and thus strong O–K/Cs interactions. Experimentally, both *HCOO and *CO3 species are frequently observed on alkali-modified catalysts as abundant surface species.26,33,126–132 Such strong binding at the alkali sites can help to facilitate the adsorption of CO2 and thus overall CO2 conversion, while in some cases these species are over-stabilized and form a self-assembled K/Cs+–CO32− or K/Cs+–HCOO− island, which block or decompose the active sites at the interface.15 To avoid such potential problems, kinetic studies including the multiple pathways running in parallel are necessary, making sure that catalyst deactivation over time does not occur under the reaction conditions. The theoretical kinetic studies should also be supplemented by a long-term stability test to determine the stability of alkali promoters under reaction temperatures and pressures.133
Data availability
No primary research results, software or code were included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge the financial support provided by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Science of the US Department of Energy (DOE) under contract no. DE-SC0012704 through BNL-FWP CO-040.References
- M. Luo, R. J. O’Brien, S. Bao and B. H. Davis, Appl. Catal., A, 2003, 239, 111–120 Search PubMed.
- K. Liu, A. Wang and T. Zhang, ACS Catal., 2012, 2, 1165–1178 Search PubMed.
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef PubMed.
- G. Preda, G. Pacchioni, M. Chiesa and E. Giamello, Phys. Chem. Chem. Phys., 2009, 11, 8156–8164 RSC.
- D. O. Uner, Ind. Eng. Chem. Res., 1998, 37, 2239–2245 CrossRef.
- G. Ertl, M. Weiss and S. B. Lee, Chem. Phys. Lett., 1979, 60, 391–394 Search PubMed.
- Q. Wang, J. Guo and P. Chen, Chem, 2021, 7, 3203–3220 Search PubMed.
- A. Ozaki, Acc. Chem. Res., 1981, 14, 16–21 Search PubMed.
- J. H. Pazmiño, M. Shekhar, W. Damion Williams, M. Cem Akatay, J. T. Miller, W. Nicholas Delgass and F. H. Ribeiro, J. Catal., 2012, 286, 279–286 Search PubMed.
- P. Panagiotopoulou, Appl. Catal., B, 2018, 236, 162–170 CrossRef.
- T. Wu, J. Lin, Y. Cheng, J. Tian, S. Wang, S. Xie, Y. Pei, S. Yan, M. Qiao, H. Xu and B. Zong, ACS Appl. Mater. Interfaces, 2018, 10, 23439–23443 Search PubMed.
- B. Liang, H. Duan, T. Sun, J. Ma, X. Liu, J. Xu, X. Su, Y. Huang and T. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 925–932 CrossRef.
- J. A. Rodriguez, E. R. Remensal, P. J. Ramirez, I. Orozco, Z. Liu, J. Graciani, S. D. Senanayake and J. Fernández Sanz, ACS Catal., 2019, 9, 10 Search PubMed.
- N. Fischer, R. Henkel, B. Hettel, M. Iglesias, G. Schaub and M. Claeys, Catal. Lett., 2016, 146, 509–517 CrossRef.
- I. Waluyo, K. Mudiyanselage, F. Xu, W. An, P. Liu, J. A. Boscoboinik, J. A. Rodriguez and D. J. Stacchiola, J. Phys. Chem. C, 2019, 123, 8057–8066 CrossRef.
- J. Ren, Y. Wang, J. Zhao, S. Tan and H. Petek, J. Am. Chem. Soc., 2019, 141, 4438–4444 Search PubMed.
- B. A. Rohr, A. R. Singh and J. K. Nørskov, J. Catal., 2019, 372, 33–38 CrossRef.
- M. Zhou and B. Liu, ChemCatChem, 2015, 7, 3928–3935 Search PubMed.
- F. Zasada, P. Stelmachowski, G. Maniak, J.-F. Paul, A. Kotarba and Z. Sojka, Catal. Lett., 2009, 127, 126–131 Search PubMed.
- N. Yang, X. Liu, A. S. Asundi, J. K. Nørskov and S. F. Bent, Catal. Lett., 2018, 148, 289–297 CrossRef.
- Y.-X. Wang and G.-C. Wang, Phys. Chem. Chem. Phys., 2018, 20, 19850–19859 RSC.
- Y.-X. Wang and G.-C. Wang, ACS Catal., 2019, 9, 2261–2274 Search PubMed.
- C. Liu and P. Liu, ACS Catal., 2015, 5, 1004–1012 CrossRef.
- Y.-X. Wang and G.-C. Wang, J. Phys. Chem. C, 2018, 122, 15474–15484 CrossRef.
- B. Zhao and G.-C. Wang, J. Phys. Chem. C, 2019, 123, 17273–17282 CrossRef.
- G. Varvoutis, M. Lykaki, E. Papista, S. A. C. Carabineiro, A. C. Psarras, G. E. Marnellos and M. Konsolakis, J. CO2 Util., 2021, 44, 101408 CrossRef.
- Z. Shi, H. Yang, P. Gao, X. Chen, H. Liu, L. Zhong, H. Wang, W. Wei and Y. Sun, Chin. J. Catal., 2018, 39, 1294–1302 CrossRef.
- R. Shi, W. Liao, P. J. Ramírez, I. Orozco, M. Mahapatra, J. Kang, A. Hunt, I. Waluyo, S. D. Senanayake, P. Liu and J. A. Rodriguez, Angew. Chem., Int. Ed., 2022, 61, e202208666 CrossRef PubMed.
- Y.-Y. Song and G.-C. Wang, Catal. Sci. Technol., 2022, 12, 1487–1498 RSC.
- M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef PubMed.
- C. T. Campbell and B. E. Koel, Surf. Sci., 1987, 186, 393–411 CrossRef.
- J. A. Rodriguez, E. R. Remesal, P. J. Ramírez, I. Orozco, Z. Liu, J. Graciani, S. D. Senanayake and J. F. Sanz, ACS Catal., 2019, 9, 10751–10760 CrossRef.
- B. Liang, H. Duan, X. Su, X. Chen, Y. Huang, X. Chen, J. J. Delgado and T. Zhang, Catal. Today, 2017, 281, 319–326 CrossRef.
- R. Hamlyn, M. Mahapatra, I. Orozco, A. Hunt, I. Waluyo, M. G. White, S. D. Senanayake and J. Rodriguez, J. Chem. Phys., 2020, 152, 044701 CrossRef PubMed.
- R. M. Liu, C. M. Chen, W. Chu and W. J. Sun, Materials, 2022, 15, 13 Search PubMed.
- R. W. Gurney, Phys. Rev., 1935, 47, 479–482 CrossRef.
- J. P. Muscat and D. M. Newns, Surf. Sci., 1978, 74, 355–364 Search PubMed.
- N. D. Lang, Phys. Rev. B: Condens. Matter Mater. Phys., 1971, 4, 4234–4244 Search PubMed.
- F. M. Hoffmann and R. A. de Paola, Phys. Rev. Lett., 1984, 52, 1697–1700 CrossRef.
- H. P. Bonzel, Surf. Sci. Rep., 1988, 8, 43–125 Search PubMed.
- H. Li, T. Chen and G. Wang, Appl. Catal., A, 2022, 639, 118607 Search PubMed.
- L. Proaño, E. Tello, M. A. Arellano-Trevino, S. Wang, R. J. Farrauto and M. Cobo, Appl. Surf. Sci., 2019, 479, 25–30 CrossRef.
- L. Lopez, V. Montes, H. Kušar, S. Cabrera, M. Boutonnet and S. Järås, Appl. Catal., A, 2016, 526, 77–83 Search PubMed.
- A. M. Hilmen, M. T. Xu, M. J. L. Gines and E. Iglesia, Appl. Catal., A, 1998, 169, 355–372 Search PubMed.
- C. Zhang, S. Li, L. Zhong and Y. Sun, J. Phys. Chem. C, 2021, 125, 6061–6072 CrossRef.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 Search PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 5 CrossRef PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 Search PubMed.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 Search PubMed.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- Y. Yang, M. G. White and P. Liu, J. Phys. Chem. C, 2012, 116, 248–256 CrossRef.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546 CrossRef PubMed.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 Search PubMed.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CAS.
- J. Wang, G. Zhang, J. Zhu, X. Zhang, F. Ding, A. Zhang, X. Guo and C. Song, ACS Catal., 2021, 11, 1406–1423 CAS.
- W. Liao, C. Tang, H. Zheng, J. Ding, K. Zhang, H. Wang, J. Lu, W. Huang and Z. Zhang, J. Catal., 2022, 407, 126–140 CAS.
- J. Nunan, K. Klier, C.-W. Young, P. B. Himelfarb and R. G. Herman, J. Chem. Soc., Chem. Commun., 1986, 193–195, 10.1039/C39860000193.
- J. A. Rodriguez, W. D. Clendening and C. T. Campbell, J. Phys. Chem., 1989, 93, 5238–5248 CAS.
- J. A. Rodriguez, W. D. Clendening, J. M. Campbell, W. Min and C. T. Campbell, J. Vac. Sci. Technol., A, 1989, 7, 2118–2120 CAS.
- J. Nakamura, J. M. Campbell and C. T. Campbell, J. Chem. Soc., Faraday Trans., 1990, 86, 2725–2734 Search PubMed.
- K. Klier, R. G. Herman, J. G. Nunan, K. J. Smith, C. E. Bogdan, C. W. Young and J. G. Santiesteban, in Stud. Surf. Sci. Catal., ed. D. M. Bibby, C. D. Chang, R. F. Howe and S. Yurchak, Elsevier, 1988, vol. 36, pp. 109–125 Search PubMed.
- J. G. Nunan, C. E. Bogdan, K. Klier, K. J. Smith, C.-W. Young and R. G. Herman, J. Catal., 1988, 113, 410–433 CAS.
- J. Sun, S. Wan, F. Wang, J. Lin and Y. Wang, Ind. Eng. Chem. Res., 2015, 54, 7841–7851 CAS.
- J. Sun, Q. Cai, Y. Wan, S. Wan, L. Wang, J. Lin, D. Mei and Y. Wang, ACS Catal., 2016, 6, 5771–5785 CAS.
- D. Xu, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, ACS Catal., 2020, 10, 5250–5260 CAS.
- X. Wang, P. J. Ramírez, W. Liao, J. A. Rodriguez and P. Liu, J. Am. Chem. Soc., 2021, 143, 13103–13112 CrossRef CAS PubMed.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Appl. Catal., A, 2003, 238, 55–67 CrossRef.
- G. R. Sheffer and T. S. King, J. Catal., 1989, 115, 376–387 CrossRef.
- G. R. Sheffer and T. S. King, J. Catal., 1989, 116, 488–497 CrossRef.
- I. Chorkendorff and H. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed.
- W. D. Mross, Catal. Rev., 1983, 25, 591–637 CrossRef.
- M. P. Kiskinova, Stud. Surf. Sci. Catal., 1991, 70, 1–345 CrossRef.
- W. An, F. Xu, D. Stacchiola and P. Liu, ChemCatChem, 2015, 7, 3865–3872 CrossRef.
- W. Liao and P. Liu, ACS Catal., 2020, 10, 5723–5733 CrossRef.
- R. Shi, W. Liao, P. J. Ramírez, I. Orozco, M. Mahapatra, J. Kang, A. Hunt, I. Waluyo, S. D. Senanayake, P. Liu and J. A. Rodriguez, Angew. Chem., Int. Ed., 2022, 61, e202208666 CrossRef PubMed.
- V. Mehar, W. Liao, M. Mahapatra, R. Shi, H. Lim, I. Barba-Nieto, A. Hunt, I. Waluyo, P. Liu and J. A. Rodriguez, ACS Nano, 2023, 17, 22990–22998 CrossRef PubMed.
- J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2012, 14, 427–438 RSC.
- R. Shi, P. J. Ramírez, R. Rosales, M. Mahapatra, N. Rui and J. A. Rodriguez, J. Phys. Chem. C, 2024, 128, 3260–3268 CrossRef.
- P. Liu and J. A. Rodriguez, J. Chem. Phys., 2007, 126, 164705 Search PubMed.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757–1760 CAS.
- S. P. Mehandru and A. B. Anderson, Surf. Sci., 1989, 219, 68–76 CAS.
- J. R. B. Gomes and J. A. N. F. Gomes, Surf. Sci., 1999, 432, 279–290 CAS.
- R. R. Contreras and A. J. Aizman, Int. J. Quantum Chem., 1990, 38, 89–96 Search PubMed.
- A. Kakekhani, L. T. Roling, A. Kulkarni, A. A. Latimer, H. Abroshan, J. Schumann, H. AlJama, S. Siahrostami, S. Ismail-Beigi, F. Abild-Pedersen and J. K. Nørskov, Inorg. Chem., 2018, 57, 7222–7238 CAS.
- H. P. Bonzel, J. Vac. Sci. Technol., A, 1984, 2, 866–872 CAS.
- O. B. Christensen and J. K. Nørskov, Chem. Phys. Lett., 1993, 214, 443–446 CAS.
- E. Broclawik, J. Datka, B. Gil, W. Piskorz and P. Kozyra, Top. Catal., 2000, 11, 335–341 Search PubMed.
- J. Datka, P. Kozyra and E. Kukulska-Zając, Catal. Today, 2004, 90, 109–114 CAS.
- E. Kukulska-Zaj
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) c and J. Datka, J. Phys. Chem. C, 2007, 111, 3471–3475 CrossRef.
c and J. Datka, J. Phys. Chem. C, 2007, 111, 3471–3475 CrossRef. - E. Broclawik, J. Załucka, P. Kozyra, M. Mitoraj and J. Datka, Catal. Today, 2011, 169, 45–51 CrossRef CAS.
- H. Yang, Y. Chen, X. Cui, G. Wang, Y. Cen, T. Deng, W. Yan, J. Gao, S. Zhu, U. Olsbye, J. Wang and W. Fan, Angew. Chem., Int. Ed., 2018, 57, 1836–1840 CrossRef CAS PubMed.
- W. An, A. E. Baber, F. Xu, M. Soldemo, J. Weissenrieder, D. Stacchiola and P. Liu, ChemCatChem, 2014, 6, 2364–2372 CrossRef CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403 CrossRef CAS PubMed.
- Y.-P. Ma and G.-C. Wang, J. Mol. Model., 2023, 29, 375 CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- R. C. E. Hamlyn, M. Mahapatra, I. Orozco, I. Waluyo, A. Hunt, J. A. Rodriguez, M. G. White and S. D. Senanayake, J. Phys. Chem. C, 2020, 124, 3107–3121 CrossRef CAS.
- W. Li, G. Zhang, X. Jiang, Y. Liu, J. Zhu, F. Ding, Z. Liu, X. Guo and C. Song, ACS Catal., 2019, 9, 2739–2751 CAS.
- Y. Zang, Z. Zhang, J. Qu, F. Gao, J. Gu, T. Wei and X. Lin, J. Colloid Interface Sci., 2024, 658, 167–178 CAS.
- S. Li, Y. Xu, H. Wang, B. Teng, Q. Liu, Q. Li, L. Xu, X. Liu and J. Lu, Angew. Chem., Int. Ed., 2023, 62, e202218167 CAS.
- P. R. Khangale, Catal. Lett., 2022, 152, 2745–2755 CAS.
- Z. Zhang, X. Hu, Y. Wang, S. Hu, J. Xiang, C. Li, G. Chen, Q. Liu, T. Wei and D. Dong, Fuel, 2019, 237, 566–579 CAS.
- P. R. Khangale, R. Meijboom and K. Jalama, Catal. Lett., 2021, 151, 3396–3403 CAS.
- H. Jo, H.-J. Chun, J. R. Sugiarto, M. K. Khan, M. Irshad, W. Yoon, S. K. Kim and J. Kim, Appl. Catal., B, 2024, 359, 124457 CAS.
- X. Chen, R. Gao, Q. Wang, K. Hu, F. Wang, C. Deng, L. Xu, C. Zhang, K.-W. Jun, S. Ki Kim, T. Zhao, H. Wan and G. Guan, Fuel, 2024, 364, 131061 CAS.
- J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, T. P. Senftle, W. Liu, J. Wang, Y. Wang, A. Zhang, Q. Fu, C. Song and X. Guo, Sci. Adv., 2022, 8, eabm3629 CAS.
- Y.-X. Wang and G.-C. Wang, J. Phys. Chem. C, 2022, 126, 17579–17588 CAS.
- C. Ye, F. Dattila, X. Chen, N. López and M. T. M. Koper, J. Am. Chem. Soc., 2023, 145, 19601–19610 CAS.
- X. W. Nie, L. L. Meng, H. Z. Wang, Y. G. Chen, X. W. Guo and C. S. Song, Phys. Chem. Chem. Phys., 2018, 20, 14694–14707 CAS.
- M. S. Duyar, C. Tsai, J. L. Snider, J. A. Singh, A. Gallo, J. S. Yoo, A. J. Medford, F. Abild-Pedersen, F. Studt, J. Kibsgaard, S. F. Bent, J. K. Nørskov and T. F. Jaramillo, Angew. Chem., Int. Ed., 2018, 57, 15045–15050 CAS.
- A. Andersen, S. M. Kathmann, M. A. Lilga, K. O. Albrecht, R. T. Hallen and D. Mei, Catal. Commun., 2014, 52, 92–97 CAS.
- S. C. Ammal and A. Heyden, ACS Catal., 2019, 9, 7721–7740 CAS.
- H. Zhang and P. Liu, Chem. Catal., 2025, 5, 101156 CAS.
- B. Eren, D. Zherebetskyy, L. L. Patera, C. H. Wu, H. Bluhm, C. Africh, L.-W. Wang, G. A. Somorjai and M. Salmeron, Science, 2016, 351, 475–478 CAS.
- L. Xu, K. G. Papanikolaou, B. A. J. Lechner, L. Je, G. A. Somorjai, M. Salmeron and M. Mavrikakis, Science, 2023, 380, 70–76 CAS.
- D. Cheng, Z. Wei, Z. Zhang, P. Broekmann, A. N. Alexandrova and P. Sautet, Angew. Chem., Int. Ed., 2023, 62, e202218575 CrossRef CAS PubMed.
- S. Jensen, R. Cheula, M. Hedevang, M. Andersen and J. V. Lauritsen, Angew. Chem., Int. Ed., 2024, 63, e202405554 CAS.
- Z. Zhang, W. Gee, P. Sautet and A. N. Alexandrova, J. Am. Chem. Soc., 2024, 146, 16119–16127 CrossRef CAS PubMed.
- D. B. Clarke and A. T. Bell, J. Catal., 1995, 154, 314–328 CAS.
- G. J. Millar, C. H. Rochester and K. C. Waugh, J. Catal., 1995, 155, 52–58 CAS.
- L. Barberis, C. I. Versteeg, J. D. Meeldijk, J. A. Stewart, B. D. Vandegehuchte and P. E. de Jongh, ACS Catal., 2024, 14, 9188–9197 CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CAS.
- X. Rong, H.-J. Wang, X.-L. Lu, R. Si and T.-B. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CAS.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CAS.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CAS.
- H. Wang, X. Nie, Y. Chen, X. Guo and C. Song, J. CO2 Util., 2018, 26, 160–170 CAS.
- A. Bansode, B. Tidona, P. R. von Rohr and A. Urakawa, Catal. Sci. Technol., 2013, 3, 767–778 CAS.
- S. Das, M. Sengupta and A. Bordoloi, ChemCatChem, 2017, 9, 1845–1853 CAS.
- S. J. Ahlers, M. M. Pohl, M. Holena, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2016, 6, 2171–2180 CAS.
- J. Wang, T. Wang, Y. Xi, G. Gao, P. Sun and F. Li, Angew. Chem., Int. Ed., 2023, 62, e202311335 CAS.
- P. Tian, M. Gu, R. Qiu, Z. Yang, F. Xuan and M. Zhu, Ind. Eng. Chem. Res., 2021, 60, 8705–8713 CrossRef CAS.
- R. Liu, D. Leshchev, E. Stavitski, M. Juneau, J. N. Agwara and M. D. Porosoff, Appl. Catal., B, 2021, 284, 119787 CrossRef CAS.
- M. E. Gálvez, S. Ascaso, P. Stelmachowski, P. Legutko, A. Kotarba, R. Moliner and M. J. Lázaro, Appl. Catal., B, 2014, 152–153, 88–98 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |