
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural codes of organic electrode materials for rechargeable multivalent metal batteries
Quanquan
Guo†
ab,
Hao
Xu†
a,
Xingyuan
Chu†
a,
Xing
Huang
a,
Minghao
Yu
 *ab and
Xinliang
Feng
*ab
*ab and
Xinliang
Feng
*ab
aCenter for Advancing Electronics Dresden (cfaed) & Faculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany. E-mail: quanquan.guo@mpi-halle.mpg.de; hao.xu3@tu-dresden.de; xingyuan.chu@mailbox.tu-dresden.de; xing.huang@tu-dresden.de; minghao.yu@tu-dresden.de; xinliang.feng@tu-dresden.de
bMax Planck Institute of Microstructure Physics, Halle (Saale), 06120, Germany
First published on 18th March 2025
Abstract
Rechargeable multivalent metal batteries (MMBs) are considered as promising alternatives to Li-ion and Pb-acid batteries for grid-scale energy storage applications due to the multi-electron redox capability of metal anodes. However, the conventional inorganic cathodes used in MMBs face challenges with the sluggish diffusivity and poor storage of charge-dense multivalent cations in their crystal lattice. Organic electrode materials (OEMs), on the other hand, offer several advantages as MMB cathodes, including flexible structural designability, high resource availability, sustainability, and a unique ion-coordination storage mechanism. This review explores the intrinsic connection between the structural features of OEMs and their charge storage performance, aiming to unveil key design principles for organic molecules used in various MMB applications. We begin with an overview of the fundamental aspects of different MMBs (i.e., Zn/Mg/Ca/Al batteries), covering electrolyte selection, metal stripping/plating electrochemistry, and the fundamentals of cathode operation. From a theoretical understanding of redox activities, we summarize the properties of different redox sites and correlate the electrochemical properties of OEMs with various structural factors. This analysis further leads to the introduction of critical design considerations for different types of OEMs. We then critically review a wide range of organic compounds for MMBs, from small organic molecules to redox-active polymers and covalent-organic frameworks, focusing on their structure–property relationships, key electrochemical parameters, and strengths and shortcomings for multivalent ion storage. Finally, we discuss the existing challenges and propose potential solutions for further advancing OEMs in MMBs.
 Quanquan Guo | Dr Quanquan Guo received his PhD in December 2022 from the Sichuan University (China). He joined Prof. Feng's group as a Postdoc in January 2023. His current research focuses on the development of novel organic redox-active materials and functional artificial solid electrolyte interphases for sustainable energy storage devices. |
 Hao Xu | Dr Hao Xu received his bachelor's degree (June 2018) from Central South University (China) and PhD degree (March 2024) from Shanghai Jiao Tong University (China). He joined Prof. Feng's and Dr Yu's groups as a Postdoc in April 2024. His current research focuses on the electrode materials and electrolytes for magnesium batteries. |
 Xingyuan Chu | Xingyuan Chu is currently a PhD student at Technische Universität Dresden (TU Dresden). He received his bachelor's degree (2019) from Zhejiang University, China and MS degree from Imperial College London, UK. His research interests focus on multivalent metal batteries. |
 Xing Huang | Dr Huang Xing received his PhD in 2018 from the Institute of Chemistry at the Chinese Academy of Sciences under the guidance of Prof. Zhu Daoben. He pursued postdoctoral research at Rutgers University in collaboration with Professor Jing Li. He further advanced his research at the Max Planck Institute for Microstructure Physics and Dresden University of Technology, working alongside Professor Feng Xinliang. His research focuses on the design and development of novel conjugated coordination polymers (c-CPs) as high-mobility semiconductors and unconventional superconductors. His work explores the unique condensed matter physics of c-CPs and their potential applications in energy-related fields. |
 Minghao Yu | Dr Minghao Yu, PI, holds a research group (Materials & Electrochemistry for Sustainable Energy Storage) at Technische Universität Dresden. His research interests include (1) the development of novel 2D layered materials, (2) the investigation of advanced artificial interphases and electrolytes for next-generation batteries, (3) fundamental charge and ion dynamics during electrochemical energy storage processes, and (4) fabrication of sustainable energy storage devices, including supercapacitors, hybrid-ion capacitors, aqueous batteries, dual-ion batteries, and multivalent metal (Zn, Mg, and Al) batteries. He has published more than 130 scientific articles, which have attracted 22 |
 Xinliang Feng | Xinliang Feng is a full professor and has been the head of the Chair for Molecular Functional Materials at Technische Universität Dresden since 2014. His current scientific interests include organic synthetic methodology, organic synthesis, and supramolecular chemistry of π-conjugated systems, bottom-up synthesis of graphene and graphene nanoribbons, electrochemical exfoliation of 2D crystals, 2D polymers, and supramolecular polymers as well as 2D carbon-rich conjugated polymers for optoelectronic applications, energy storage and conversion, and new energy devices and technologies. He has published more than 778 research articles, which have attracted more than 95 |
1. Introduction
Rechargeable batteries, as essential electrochemical storage devices, play indispensable roles in storing intermittent electricity generated from renewable energy sources, such as water, wind, solar, tide, and geothermal power.1–4 Ever since the Sony company commercialized the first lithium-ion batteries (LIBs) in 1991, the passing three decades have witnessed this flagship technology becoming ubiquitous across various application domains, such as portable electronics and electric vehicles.5,6 However, several critical challenges have decelerated their booming development, including scarce Li and transition-metal (e.g., Co and Ni) resources, dendrite growth and flammable electrolyte-induced safety concerns.7 Given the importance of safety and sustainability, it is highly desirable to explore novel, reliable, safe, and green rechargeable battery technologies based on abundant raw materials. Rechargeable multivalent metal batteries (MMBs), which adopt multivalent metals as the anodes (i.e., Zn, Mg, Ca, and Al) and the corresponding multivalent ion-based species as the main charge carriers, have been identified as the promising next-generation technology candidates for the ‘post-Li’ age.8–11 In particular, the metal anodes involve multi-electron redox processes and offer large specific capacities, both gravimetrically and volumetrically, conferring MMBs with attractive theoretical energy densities. Additionally, these metal anodes are abundant and possess high autoignition temperatures, ensuring exceptional stability and inertness in air or conventional electrolytes. These characteristics mitigate safety concerns and facilitate practical handling for applications. In this regard, MMBs are particularly intriguing for the future large-scale or stationary energy storage, where cost, safety, and sustainability are crucial criteria.To fabricate full cells, reliable cathodes with high voltage and large specific capacity are other crucial components of MMBs.12–14 Following the historical development of LIB cathodes, a range of inorganic materials have been explored as the multivalent ion species-hosting cathodes, including intercalation-type (such as δ-MnO2, α-MoO3, MoS2, V2O5, and Prussian blue) and conversion-type cathodes (such as sulfur, selenium, bromine, iodine, and some transition metal chalcogenides).10,15–19 However, when transitioning from monovalent ion-based LIBs to multivalent ion-based MMBs, these inorganic materials frequently fall short of realizing their theoretical charge storage capacities, often exhibiting performance considerably below practical expectations. The primary explanation lies in the large and high charge densities of multivalent metal ions serving as charge carriers, which lead to the strong electrostatic interactions with the host inorganic materials and huge volume change of the inorganic cathode. These interactions significantly reduce the diffusivity of the ions within the electrode during electrochemical processes. Theoretical studies indicate that the diffusion barriers of multivalent ions are much higher than that of Li+ in the same host matrix. For instance, Li+ shows low migration energy barriers ranging from 400 to 600 meV in the spinel Mn2O4 electrodes, while the migration energy barriers of Zn2+, Mg2+, and Al3+ ions lie within the range of 850–1000, 600–800, and 900–1400 meV, respectively.20 Consequently, the performance of inorganic electrode materials for MMBs is substantially hindered by sluggish reaction kinetics, low power density, poor rate performance, limited reversibility, and inferior cycling life.
To circumvent the challenges faced by MMB cathodes, soft organic electrode materials (OEMs) with a unique ion-coordination mechanism have emerged with prospective opportunities for storing multivalent metal ions (Fig. 1a).21,22 The moderate coordination interactions between the organic host and multivalent ions, along with the ample intermolecular space and flexible molecular skeleton of organic compounds, enable the rapid diffusion of charge carriers with high charge densities and/or large sizes through OEMs. These characteristics also ensure that OEMs provide sufficient space and have capabilities to accommodate ions efficiently. Besides, OEMs offer additional benefits for MMBs such as abundant resources, flexible structural designs, excellent environmental tolerance, and sustainability. Comprising abundant and lightweight elements like carbon, hydrogen, oxygen, nitrogen, and sulfur, OEMs, some of which can be derived from biomass, are inherently eco-friendly. Furthermore, advancements in green organic chemistry and molecular engineering allow for precise control over the physicochemical and electrochemical properties of OEMs, enhancing their suitability as electroactive materials for cost-effective and sustainable energy storage applications (Fig. 1b).
 | ||
| Fig. 1 (a) Schematic illustration of a typical MMB device fabricated using an OEM cathode and a multivalent metal anode. (b) Potential applications of MMBs with OEM cathodes. (c) Comparison of different monovalent and multivalent metal anodes in terms of gravimetric capacity, volumetric capacity, cost, and abundance. (d) Comparison of various monovalent and multivalent cations with regard to atomic weight, ionic radius, and charge density. | ||
To date, numerous redox-active OEMs, ranging from small molecules to amorphous polymers and covalent organic frameworks (COFs), have been delicately designed for rechargeable MMBs.23 OEMs based on small molecules typically offer high specific capacities and enable efficient utilization of redox sites for accommodating multivalent ions. However, their dissolution in the electrolyte often leads to rapid capacity decay, while their inherently poor electronic conductivity results in suboptimal rate performance.24 Massive conductive agents are often required to increase the conductivity of OEM-based electrodes. However, the large ratio of conductive additives would compromise the overall energy density of the battery devices. To address this dilemma, macromolecules that polymerize redox-active building blocks into large chains or networks have been developed. After polymerization, the large molecular weight and strong intermolecular interaction of polymer chains or networks could inhibit their dissolution in the electrolyte. Meanwhile, the electronic conductivity of these polymer electrodes can be improved by constructing expanded conjugation systems. Starting from designing polymerizable redox-active monomers, a variety of polymer-based OEMs have been demonstrated for rechargeable MMBs, such as polyimides, polyimines, polytriphenylamine (PTPAn), nitroxide radical polymers, polyaniline (PANI), polypyrrole (PPY), polythiophene (PTh), poly(3,4-ethylenedioxythiophene) (PEDOT) and polypyrene.25,26 As a unique group of crystalline polymer-based OEMs, redox-active COFs have emerged as attractive cathode materials for MMBs, benefiting from their unique structural features like stable crystalline scaffolds, periodically organized redox-active sites, and ordered nanochannels.27,28 Specifically, the abundant functional groups distributed on the pore surface of COFs provide easily accessible redox-active sites for multivalent ion storage. Additionally, these atomically precise and flexible designs offer an ideal platform to explore redox mechanisms, aiding in the development of general design principles for OEM structures tailored to high-energy and high-power MMBs.
Given the extensive academic efforts dedicated to developing novel OEMs, only a few review articles have established explicit structure–property relationships for OEMs within specific types of MMBs.14,21,27 A comprehensive review that critically examines the structural design principles of high-performance OEMs across various MMBs is still lacking. In this review, we present the latest advancements in the utilization of OEMs for MMBs and elucidate the intrinsic link between their structural properties and charge storage performances, offering essential guidance for future OEM design in diverse energy storage applications. Beginning with an overview of the fundamental electrochemistry of different MMBs, including aqueous Zn/Al batteries and non-aqueous Mg/Ca/Al batteries, we briefly outline their key factors with regard to electrolyte selection, metal stripping/plating electrochemistry, and cathode fundamentals. Subsequently, we delve into the theoretical description of OEM redox properties, correlating key electrochemical attributes with various structural factors. We further review recent developments in OEM-based cathodes for MMBs, spanning from small organic molecules to redox-active polymers and COFs. Emphasis is placed on their respective structure–property relationships, key electrochemical parameters, and their advantages and shortcomings in MMBs. Finally, we conclude this review by highlighting the existing challenges that impede the practical applications of OEMs in MMBs and propose potential directions for future research efforts.
2. Promising MMBs and their basics
Multivalent metals like Zn, Mg, Ca, and Al have been identified as attractive anodes for constructing promising rechargeable batteries due to several key metrics, including abundance, cost, gravimetric capacity, and volumetric capacity (Fig. 1c). In the Earth's crust, these multivalent metal elements are relatively abundant (0.007% for Zn, 2.33% for Mg, 4.15% for Ca, and 8.23 for Al) in comparison with Li (0.002%). Consequently, their costs (2.2 USD kg−1 for Zn, 2.2 USD kg−1 for Mg, 2.28 USD kg−1 for Ca, and 1.9 USD kg−1 for Al) are considerably lower than that of Li (19.2 USD kg−1).13 Typically, metal anodes undergo a half-cell reaction shown in eqn (1). There are also special cases where complex ions serve as active species for metal stripping/plating, like AlCl4− and Al2Cl7− for non-aqueous Al batteries. Due to the multi-electron transfer characteristics of each multivalent metal atom, larger capacity can be realized by multivalent metal anodes (820 mA h g−1 for Zn, 2206 mA h g−1 for Mg, 1337 mA h g−1 for Ca, and 2980 mA h g−1 for Al) in comparison with the commercial graphite anode (372 mA h g−1) for LIBs. Additionally, benefiting from their high mass density, multivalent metal anodes can provide large volumetric capacities (5855 mA h cm−3 for Zn, 3834 mA h cm−3 for Mg, 2072 mA h cm−3 for Ca, and 8046 mA h cm−3 for Al), significantly outperforming graphite (840 mA h cm−3) and even Li metal (2061 mA h cm−3) anodes for LIBs.13 Another key advantage of multivalent metal anodes is their relatively low reactivity compared with Li, which enhances the safety of MMBs. Unlike the prevalent dendrite growth of Li metal, which can lead to short circuit and fire, Mg and Al exhibit dendrite-free deposition in some electrolytes.29 Despite the significant dendrite issue, Zn anodes are compatible with aqueous electrolytes, making Zn batteries even safer.30 Besides, recent efforts have also extended the use of aqueous electrolytes to Al batteries.31–33| M − ne− ⇌ Mn+ | (1) |
Following the success of LIBs, most reported MMBs adopt a similar ‘rocking chair’ configuration, in which cationic species typically act as charge carriers for both anodes and cathodes. This configuration introduces kinetics challenges associated with multivalent metal ions due to their higher charge density (112 C mm−3 for Zn2+, 120 C mm−3 for Mg2+, 52 C mm−3 for Ca2+ and 364 C mm−3 for Al3+) than Li+ (52 C mm−3). This high charge density originates from their higher charge and smaller size (0.75 Å for Zn2+, 0.72 Å for Mg2+, 1 Å for Ca2+ and 0.53 Å for Al3+, compared with 0.76 Å for Li+) (Fig. 1d).13 Because of their high charge density, multivalent cations have high de-solvation/dissociation energy with both solvent molecules and anions in electrolytes, resulting in considerable polarization on both the cathode and anode sides.34 Moreover, these cations often exhibit strong interactions with cathode materials, rendering insertion reactions kinetically unfavorable. Consequently, complex ions (e.g., MgCl+, AlCl2+, and AlCl4−) are frequently reported to be stored within electrodes, instead of neat multivalent cations.35,36 In MMBs employing aqueous electrolytes, H+ and its hydrated configuration (i.e., H3O+) have been extensively demonstrated to be partial charge carriers for cathodes. This is because H+ and H3O+ offer more favorable kinetics compared to multivalent cations owing to their smaller size (1.2 Å for H+ and 2.8 Å for H3O+), lighter mass (1.0 g mol−1 for H+ and 19.0 g mol−1 for H3O+), and rapid conduction in aqueous electrolytes governed by the Grotthuss mechanism.10 Besides, dual-ion configurations, where anions act as charge carriers for cathodes, have gained attention, as they bypass the large energy barrier associated with the solid diffusion of multivalent cations.3,37–40
2.1. Aqueous MMBs
Reversible Zn stripping/plating can be achieved in mild acidic aqueous electrolytes with various cheap and eco-friendly Zn salts as solutes, such as ZnCl2, Zn(SO4)2, Zn(NO3)2, Zn(CH3COO)2, Zn(ClO4)2, and Zn(CF3SO3)2.46 Recently, the concept of highly concentrated electrolytes for Zn batteries has emerged, such as 31 m ZnCl2 with 64 mol% H2O. All water molecules are located at the first solvation sheath, and the hydrogen bond network is significantly restricted in this electrolyte, leading to a largely widened electrochemical stability window. Nonetheless, Zn anodes face a number of challenges like dendrite growth, hydrogen evolution reaction (HER), and corrosion issues. Addressing Zn anode challenges represents an academic hotspot, and numerous effective strategies have been demonstrated with regard to electrolyte modification, interphase construction, separator functionalization, and anode structure engineering.30,47,48
At the cathode side, the anodic stability of water-based electrolytes limits the selection of cathode materials.49,50 Typically, Zn2+ acts as the main charge carrier for the Zn battery cathodes. In comparison with other multivalent cations, Zn2+ has been considered relatively ‘soft’ according to the hard and soft (Lewis) acids and bases theory.51 Nevertheless, compared with monovalent Li+ and Na+, Zn2+ still encounters kinetics penalties associated with desolvation and solid-state diffusion within cathodes, leading to low mass loading, poor rate capability, and large charge/discharge polarization issues.52 Moreover, conventional cathode materials with Mn-/V-based oxides as representatives suffer from obvious structural degradation due to the strong coulombic interaction between Zn2+ and lattice oxygen, as well as dissolution of transition cations (like Mn2+) in aqueous electrolytes during repeated charge and discharge. In mild-acid Zn-salt electrolytes, H+/H3O+ as kinetically favorable charge carriers have been frequently shown to be involved in cathode reactions, even with capacity contribution over 50%.53 Recently, we have shown that building hydrogen bond networking within cathode materials or the proton-filtering interphase on the cathode surface could promote the involvement of H+ charge carriers.10,54 However, the involvement of H+/H3O+ is a double-edged sword for AZBs. On one hand, the cathode kinetics are significantly improved, as well as rapid conduction in aqueous electrolytes governed by the Grotthuss mechanism. On the other hand, the acidic environment would degrade the electrochemical and chemical stability of cathode materials, accelerating the dissolution of active materials into the electrolyte.49
The majority of aqueous electrolytes employed for AABs are based on aqueous Al(OTF)3 solutions. Trivalent Al3+ in these aqueous electrolytes has the capability to generate a robust electric field in its vicinity, strongly bonding with water and effectively suppressing the water activity. However, the strong Al–O interaction would weaken the O–H bond, and thus H from water tends to become a free proton, rendering these electrolytes strongly acidic (pH = −0.5 for 5 m Al(OTF)3).59 As a result, inhibiting the HER still presents as a critical challenge, highlighting the need for the development of electrolytes with enhanced cathodic stability. Introducing organic additives represents a feasible strategy to expand the cathodic stability of AAB electrolytes. For example, Zhao et al. introduced polar pyridine-3-carboxylic acid into Al(OTF)3 electrolyte, optimizing the Al3+ solvation structure. The water activity was obviously suppressed, leading to better cathodic stability of the electrolyte. Regarding the cathodes used for AABs, only a few of inorganic materials have been employed, such as AlxMnO2.32,59 In most cases, the mass loading of these electrodes is quite low, typically less than 1 mg cm−2. The absence of high-loading cathodes could be primarily associated with the kinetics issues associated with Al3+. Besides, it remains an open question regarding the contribution of H+/H3O+ charge carriers to the charge storage of the demonstrated cathodes, given the acidic nature of the Al salt solutions.
2.2. Non-aqueous MMBs
Although the presence of Cl in these electrolytes could promote Mg stripping/plating by forming active species like (Mg2(μ-Cl)3·6THF)+, Cl-containing electrolytes were proved corrosive to current collectors and battery casings. Besides, their poor anodic stability (<3.2 V) limits the utilization of high-voltage cathode materials like Mn-based oxide (>3.3 V).66,67 Consequently, there is a pressing demand for Cl-free electrolytes that enable both reversible Mg stripping/plating and good compatibility with high-voltage cathodes. One approach to addressing this issue is the development of Mg salts with bulky and weakly coordinating anions, such as Mg(CB11H12)2 and Mg(B(hfip)4)2.68,69 However, these Mg salts are not commercially available, and their synthesis requires complex steps. Most recently, Lu et al. proposed a cation replacement route for the synthesis of Mg organoborate salts, which enables large-scale production of the targeted Mg salts (75 g) with high purity (99.76%) and yield (92%).70 By adjusting the chain length and F-substitution degree of the alcohols, different organoborates were obtained. The as-prepared Mg organoborate electrolytes enable high current endurance and enhanced electrochemical kinetics. Moreover, based on Mg(TFSI)2, Wang et al. revealed that fast and efficient Mg stripping/plating could be initiated by reorganizing the solvation structure of Mg2+ with methoxyethyl amine chelants.71 Compared with ether solvents, these chelants demonstrated a higher affinity for Mg2+. Moreover, the chelant-rich solvation sheaths bypass the energetically unfavorable desolvation process through reorganization, thus reducing the overpotential for both the anode and the cathode.
Owing to the kinetics issues associated with Mg2+, only a small range of inorganic materials exhibit the practical potential for Mg batteries. Chevrel-phase Mo6S8 as the first demonstrated cathode material was shown to enable facile Mg2+ intercalation/deintercalation in Cl-containing electrolytes, which benefits from its highly delocalized electron orbitals that mitigate the interactions with Mg2+ and facilitate Mg2+ diffusion. Moreover, surface Mo atoms were revealed to catalyze the dissociation of Mg2+ with surrounding Cl anions and solvent molecules,72 which also mitigate the energy barrier of charge transfer. However, the specific capacity of Mo6S8 is deemed unsatisfactory, achieving only 120 mA h g−1.61 In Cl-containing electrolytes, other metal chalcogenides, with either intercalation or conversion mechanism, were explored as cathode materials.36,73 In most intercalation cases, Mg–Cl species (such as MgCl+ and Mg2Cl3+) were identified as charge carrier species, bypassing the energetically unfavorable Mg–Cl dissociation step. These cathode materials were also explored with Cl-free electrolytes, where Mg2+ serves as a charge carrier.70,74 A special case is Mg0.15MnO2 operated in the methoxyethyl amine chelant-containing Mg(TFSI)2 electrolyte. The methoxyethyl amine chelants were found to co-intercalate into Mg0.15MnO2.71
Leveraging insights from experiences with Mg batteries, Ca salts with bulky and weakly coordinating anions have been explored to enable highly reversible Ca stripping/plating. For instance, calcium tetrakis(hexafluoroisopropyloxy)borate (Ca[B(hfip)4]2) in DME was shown to be compatible with the Ca metal anode, while a solid electrolyte interphase (SEI) capable of Ca2+ transport was detected on the Ca surface.70 In 2021, Wang et al. demonstrated that the strategies of reorganizing the solvation structure with methoxyethyl amine chelants for fast Mg stripping/plating were also applicable for Ca stripping/plating.71 Of note, all these demonstrated electrolytes employ ether solvents with good cathodic stability. Like Mg battery electrolytes, these electrolytes are limited in use for high-voltage cathodes due to the unsatisfactory anodic stability (≈3 V vs. Ca2+/Ca). Compared to Mg2+, Ca2+ exhibits a lower charge density, resulting in weaker interactions with solvent molecules and a few studies focusing on the desolvation of Ca2+. Despite this fact, the development of cathodes for Ca batteries has been hindered, with only a few relevant cases demonstrated (such as Mg0.15MnO2).71 A significant research space for the exploration of high-performance cathodes persists for Ca batteries.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) al. proposed an AlCl3-urea DES with an optimal molar ratio of 1.3. Raman spectral and Al nuclear magnetic resonance (NMR) spectral results revealed that the ionic species existing in the electrolyte are AlCl4−, Al2Cl7−, and [AlCl2·(urea)n]+, where [AlCl2·(urea)n]+ and Al2Cl7− are active for Al stripping/plating.81 Inorganic molten salts are another option for NABs, but high temperature is required in most cases (>200 °C). In 2022, Pang et al. proposed an alkali-chloroaluminate melt (NaCl–KCl–AlCl3) for NABs, which contains catenated AlnCl3n+1− species, such as Al2Cl7−, Al3Cl10− and Al4Cl13−.82 The electrolyte can operate at moderately elevated temperature (110 °C). Benefiting from the facile Al3+ desolvation kinetics of Al–Cl–Al linkages, this electrolyte enables fast-charging ability and resistance to dendritic shorting.
al. proposed an AlCl3-urea DES with an optimal molar ratio of 1.3. Raman spectral and Al nuclear magnetic resonance (NMR) spectral results revealed that the ionic species existing in the electrolyte are AlCl4−, Al2Cl7−, and [AlCl2·(urea)n]+, where [AlCl2·(urea)n]+ and Al2Cl7− are active for Al stripping/plating.81 Inorganic molten salts are another option for NABs, but high temperature is required in most cases (>200 °C). In 2022, Pang et al. proposed an alkali-chloroaluminate melt (NaCl–KCl–AlCl3) for NABs, which contains catenated AlnCl3n+1− species, such as Al2Cl7−, Al3Cl10− and Al4Cl13−.82 The electrolyte can operate at moderately elevated temperature (110 °C). Benefiting from the facile Al3+ desolvation kinetics of Al–Cl–Al linkages, this electrolyte enables fast-charging ability and resistance to dendritic shorting.| 4Al2Cl7− + 3e− ⇌ Al + 7 AlCl4− | (2) |
The mostly studied cathodes for NABs are based on carbonaceous materials, with graphene as the representative example. These carbonaceous cathodes make use of the AlCl4−-intercalation chemistry for charge storage as exemplified in eqn (3). Such an anion-intercalation chemistry allows for fast anion diffusion and intercalation with excellent cycling stability (7500 cycles). However, the limitation of carbonaceous cathodes lies in their limited specific capacity, resulting in a low energy density of <40 W h kg−1 for the whole cell.83 Inorganic metal compounds are another category of cathode materials for NABs. For example, Jayaprakash et al. found that V2O5 nanowires exhibited two plateaus in ionic-liquid electrolytes with a specific capacity of 305 mA h g−1.84 Al3+ served as a charge carrier as shown in eqn (4). However, due to the strong coulombic interaction between Al3+ and the cathode, only a few inorganic materials are suitable for the storage of Al3+. Meanwhile, the demonstrated ones typically suffer from poor cycling stability (<100 cycles). Conversion-type cathodes are also investigated for NABs. Among them, S is the most attracting one due to its low-cost and high capacity. However, S cathodes suffer from poor cycling stability and rate capability, due to ohmic polarization caused by the electron-insulating sulfur, electrochemical polarization on the cathode/electrolyte interface caused by sluggish charge transfer between Al and S, and slow diffusion in the solid-state reaction.85
| Cn + AlCl4− ⇌ Cn[AlCl4] + e− | (3) |
| Al3+ + V2O5 + 3e− ⇌ AlV2O5 | (4) |
3. Redox organic moieties for OEMs
The first study of OEMs for energy storage applications dates back to 1969, when Williams and colleagues pioneered the work that adopted dichloroisocyanuric acid as the cathode material in primary LIBs.86 Since then, rechargeable LIBs utilizing redox-active small molecule compounds and conductive polymers have gradually gained popularity in the history of energy storage.87 However, the successful implementation of inorganic electrode materials in the commercial LIB market diminished the competitiveness of OEMs and reduced the motivation to pursue their development, especially in the early 2000s. With the growing pursuit of sustainable and green energy storage devices, MMBs have gained worldwide attention as important alternatives to LIBs. Unlike monovalent ion batteries, rare inorganic materials show desirable practical application prospects in the MMB field, as discussed in Section 2. OEMs have been extensively studied as ideal redox-active materials for storing multivalent ions due to their unique coordination mechanisms and ample ion storage spaces. Moreover, OEMs add irreplaceable sustainability value to MMBs, as they are comprised of light elements and can be produced from biomass using green processes, which have minimal environmental footprints and enable sustainable production. In this section, we will analyze the intrinsic origins and fundamentals of redox moieties in OEMs, aiming to provide a molecular-level understanding of their charge storage mechanisms.3.1. Theoretical description of redox activity
The redox activity of organic compounds originates from the ability of certain functional groups to reversibly donate or accept electrons, enabling coordination with the corresponding anions or cations during electrochemical processes. Based on different ion storage mechanisms, these redox-active organic groups can be categorized into n-type (Fig. 2a), p-type (Fig. 2b), and bipolar-type moieties (Fig. 2c). From a thermodynamic description, all chemical or electrochemical reactions involve changes in Gibbs free energy. For instance, in an electrochemical reduction reaction, the change in Gibbs free energy for the reduction of an organic compound in the solution phase (ΔGred(S, sol)) can be calculated using eqn (5),88,89 where ΔGred(R, gas) is the Gibbs free energy variation in the gas phase (vacuum) at 298 K. ΔGsolv(R) and ΔGsolv(R−) represent the solvation free energy of the organic compound in neutral and anionic states, respectively. Accordingly, electron affinity (EA), defined as the Gibbs free energy required to add an electron to a neutral molecule in a vacuum, is a crucial parameter for determining the inherent redox activity of specific organic molecules. An organic material with a more negative EA is more reductive, leading to a higher redox potential.| ΔGred(S, sol) = ΔGred(R, gas) + ΔGsolv(R−) − ΔGsolv(R) | (5) |
 | ||
| Fig. 2 Electrochemical charge storage mechanism of different redox organic centers, including (a) n-type, (b) p-type, and (c) bipolar moieties. (d) The electron-donating groups with stronger electron-donating ability show a higher HOMO energy level and lower oxidation potential. (e) The electron-withdrawing groups with stronger electron-withdrawing ability show a higher LUMO energy level and lower reduction potential. | ||
From the perspective of molecular orbital theory, electronic properties such as the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels are closely related to EA. Generally, a lower HOMO energy corresponds to a higher oxidation potential for an organic molecule (Fig. 2d), while a lower LUMO energy indicates better electron affinity and a higher reduction potential (Fig. 2e). Structural factors like electron-withdrawing/-donating groups and the conjugation degree effectively tune the working potentials of organic materials by adjusting their HOMO and LUMO energy levels. Meanwhile, the HOMO/LUMO energy band gap (Eg) mainly influences electron transport behavior. A narrower Eg typically signifies higher electron conductivity, resulting in better rate performance. Since HOMO/LUMO energy levels are more convenient to be calculated than EA, they normally serve as an appropriate descriptor for predicting the redox activity of various organic molecule candidates. Using first-principles density functional theory (DFT), the redox potential (Ered) of a target organic compound can be estimated using eqn (6), where EH represents the redox potential of the absolute hydrogen electrode (i.e., 4.44 V), n represents the number of electrons involved, and F is the Faraday constant (9.65 × 104 C mol−1).
| Ered = −ΔGred(S, sol)/nF − EH | (6) |
It is important to note that the redox activity of OEMs is influenced not only by their chemical structures but also by the nature of the charge carriers and the electrolyte environment. When the organic material is reduced, it often coordinates with counter cations, which can alter the reaction energy during the ion insertion process. Therefore, the interaction between redox-active moieties and charge carriers plays a critical role in determining the final redox potential. For example, Lee and coworkers investigated the redox potential changes in disodium terephthalate (Na2TP) derivatives with electron-donating methyl (–CH3) and methoxy (–OCH3) groups and discussed how their interactions with Li+ and Na+ ions affect the redox behavior.90 The methyl substituent lowered the average discharge voltage of Na2TP by 60 mV in Li-ion cells, which was attributed to the higher LUMO energy level introduced by the electron-donating groups. Surprisingly, the Na2TP derivative with –OCH3 groups, despite being a stronger electron donor than –CH3, exhibited a positive shift in redox potential for Li+ storage. This finding contrasts with typical predictions for the redox potentials of organic materials. The unexpected increase in discharge voltage for the methoxy-modified Na2TP was due to the strong interaction between the charge-carrying Li+ cations and reduced host molecules. Specifically, Li+ ions form stable bonds with the electronegative oxygen atoms on the methoxy substituents, leading to the formation of stable six-membered ring configurations. This stable intermediate configuration significantly raises the discharge voltage. This report discloses that, in certain cases, strong interactions between charge carriers and redox-active moieties can become the dominant factor determining the redox potential.
3.2. n-Type organic moieties
n-Type redox organic moieties tend to undergo reduction from their neutral state by accepting electrons and forming negatively charged species (Fig. 2a). Accompanied by this reduction process, these moieties coordinate with cationic charge carriers from the electrolyte. During the reverse oxidation process, they are able to return their original neutral state via releasing the injected electrons and coordinated cations. One of the classic n-type examples is the carbonyl-based (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) moiety, which tends to accept one electron during the charge process and generate a radical monoanion (C–O˙−) through the enolization reaction. To date, a wide range of carbonyl derivatives have been demonstrated as the redox-active sites, such as quinones, carboxylates, imides, and anhydrides. Most of OEMs with carbonyl moieties show an average discharge potential of approximately −1.8 V vs. SHE. The carboxylate, imide, and anhydride compounds typically display a slightly lower potential of −2.0 V vs. SHE, which is relatively low as battery cathodes. Like carbonyl groups, thiocarbonyl (CS) moieties show comparable n-type charge-storage behaviors and redox activity, which could be explained by the fact that sulfur and oxygen are located in the same group in the periodic table of elements. Since sulfur is less electronegative than oxygen, thiocarbonyl moieties normally exhibit a higher redox potential (−1.0 V vs. SHE) and enhanced electronic conductivity.
O) moiety, which tends to accept one electron during the charge process and generate a radical monoanion (C–O˙−) through the enolization reaction. To date, a wide range of carbonyl derivatives have been demonstrated as the redox-active sites, such as quinones, carboxylates, imides, and anhydrides. Most of OEMs with carbonyl moieties show an average discharge potential of approximately −1.8 V vs. SHE. The carboxylate, imide, and anhydride compounds typically display a slightly lower potential of −2.0 V vs. SHE, which is relatively low as battery cathodes. Like carbonyl groups, thiocarbonyl (CS) moieties show comparable n-type charge-storage behaviors and redox activity, which could be explained by the fact that sulfur and oxygen are located in the same group in the periodic table of elements. Since sulfur is less electronegative than oxygen, thiocarbonyl moieties normally exhibit a higher redox potential (−1.0 V vs. SHE) and enhanced electronic conductivity.
Additionally, several electron-withdrawing groups, such as imine (CN), nitrile (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), azo (NN), and disulfide (C–S–S–C), have been proved as n-type redox moieties for charge storage. In detail, redox-active imine compounds generally consist of a heteroaromatic ring with nitrogen atoms in the conjugated structure. Representative examples of imine compounds are phenazine and triazine. With one imine moiety to accept one electron during electrochemical reduction, phenazine and triazine groups are able to allow multiple electron transfer owing to the high density of CN in their conjugated structures. Regarding redox nitrile moieties, 7,7,8,8-tetracyanoquinodimethane and its derivatives are mostly studied, probably because of the highest redox potential among all n-type OEMs (−2.0 to 0.2 V vs. SHE).91 The high redox potential of 7,7,8,8-tetracyanoquinodimethane is attributed to its strong electron-accepting characteristics originating from the four electron-withdrawing nitrile groups.
N), azo (NN), and disulfide (C–S–S–C), have been proved as n-type redox moieties for charge storage. In detail, redox-active imine compounds generally consist of a heteroaromatic ring with nitrogen atoms in the conjugated structure. Representative examples of imine compounds are phenazine and triazine. With one imine moiety to accept one electron during electrochemical reduction, phenazine and triazine groups are able to allow multiple electron transfer owing to the high density of CN in their conjugated structures. Regarding redox nitrile moieties, 7,7,8,8-tetracyanoquinodimethane and its derivatives are mostly studied, probably because of the highest redox potential among all n-type OEMs (−2.0 to 0.2 V vs. SHE).91 The high redox potential of 7,7,8,8-tetracyanoquinodimethane is attributed to its strong electron-accepting characteristics originating from the four electron-withdrawing nitrile groups.
Different from other n-type moieties, disulfide moieties (C–S–S–C), which contain single-bond-linked sulfur atoms, show a distinctive n-type redox mechanism. Two electrons can be accepted by the disulfide moieties with S–S bond cleavage to form R–S− anions at a redox potential of approximately −0.8 V vs. SHE, accompanied by the coordination with cationic charge carriers. Because of the two-electron redox per disulfide moiety, the theoretical specific capacity calculated based on one single disulfide moiety can reach a maximum value of 582.4 mA h g−1. However, the intrinsically sluggish kinetics originating from S–S bond breakage and reformation leads to a high overpotential. Moreover, the high solubility of the intermediates generated by the bond-breakage reaction leads to premature capacity decay associated with the loss of active materials. In addition, the family of azo compounds are also able to reversibly accept two electrons by coordinating with two cations, maintaining the high-capacity feature. Different from disulfide moieties, the azo compounds comprising double-bond-connected nitrogen atoms can form single N–N linkage after receiving two electrons, keeping the structure stable during electrochemical redox reactions. Therefore, the azo compounds normally show high specific capacity, excellent rate performances, and good capacity retention.
3.3. p-Type organic moieties
p-Type organic moieties prefer to donate electrons and participate in the redox process through a facile transformation between their neutral states and oxidized states (Fig. 2b). During this process, p-type moieties are reversibly coordinated/de-coordinated with anionic species (e.g., AlCl4−, CF3SO3−, PF6−, Cl−, ClO4−, and SO42−) from the electrolyte. Moieties like ether (C–O–C), thioether (C–S–C), tertiary amine, viologen, and thianthrene are typical examples that store charges via such a p-type mechanism. The anion coordination electrochemistry generally empowers p-type moieties with high redox potentials, which qualify them for use as battery cathodes.Ether moieties are the most representative examples with p-type anion storage behaviors. The redox-active ether moiety tends to lose one electron and generate an O+ cation during the electrochemical oxidation process, subsequently absorbing an anion from the electrolyte. This process delivers a high redox potential of −1 V vs. SHE. The representative thioether motif (C–S–C), which consists of two single bonds between sulfur and carbon atoms, also triggers a p-type reaction with a redox mechanism similar to that of the ether motif. The thioether moieties generally exhibit a higher redox potential than the ether-based ones due to the stronger electronegativity of sulfur atoms. For example, Zhan and coworkers reported that poly(ethene-1,1,2,2-tetrathiol) electrodes with plentiful thioether bonds showed a stable discharge specific capacity of 300 mA h g−1 and a high discharge voltage above −0.8 V vs. SHE for rechargeable LIBs.92 Moreover, the tertiary amine motif, such as triphenylamine, also permits p-type reactions to store anionic charge carriers, accompanied by the formation of N-cation intermediates.
Based on p-type elements (O, N, and S), heterocyclic compounds deliver high redox potentials over 0 V vs. SHE, such as dihydrophenazine, phenothiazine, and phenoxazine derivatives. The exploitation of these electrode materials for rechargeable batteries is often restricted by the electrochemical potential window of the electrolyte. Besides, these heterocyclic compound-based electrodes are capable of undergoing a two-electron redox reaction per molecule, offering high specific capacity and energy density. For instance, Li and coworkers designed a series of phenothiazine derivatives linked by alkyl chains of different lengths for rechargeable LIBs, all of which deliver a high redox potential over 0.5 V vs. SHE.93
3.4. Bipolar-redox moieties
There are a few bipolar-type redox organic moieties capable of exhibiting both n-type and p-type ion-storage behaviors, successfully combining the features and advantages in a single material system (Fig. 2c). Both cations and anions could be reversibly coordinated with bipolar-type moieties to achieve sequential dual-ion storage. Representative examples of this category are 2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)-based compounds containing nitroxide radical moieties (N–O˙).94 They are bipolar in nature and can be transformed to either a reduced state with a negative charge (N–O−) or an oxidized state (+NO). TEMPO-based moieties have been widely incorporated into polymers in order to suppress their dissolution in the electrolyte.95,96 Some conductive polymers, including polyacetylene, PANI, PPY, and PTh, are also classic electrode materials undergoing both n-type cation storage and p-type anion storage reactions.97 Moreover, the combination of separate n-type and p-type moieties in a polymerized structure through molecule engineering is also an effective strategy to build up new bipolar-type OEMs. For example, our group recently synthesized two types of two-dimensional (2D) polyimide-based COF electrodes, both of which present the redox-bipolar features by integrating n-type imide and p-type triazine moieties into one framework.35
Bipolar compounds are frequently employed as cathode materials by solely using the p-type reaction at a high redox potential (e.g., −2.4 V vs. SHE for TEMPO). In some cases, they are also suitable for serving as both cathodes and anodes for the construction of an all-organic full battery cell, by means of activating different redox-active sites over different voltage ranges. For example, Zhou and coworkers demonstrated the first dual-ion organic symmetric cell based on bipolar 4,4′-(phenazine-5,10-diyl)dibenzoate electrodes.98 The presence of the n-type benzoate moiety and the p-type phenazine moiety allowed this compound to store cations and anions at reduced and oxidized states, respectively. The large potential gap between two different redox moieties provided a high cell voltage of 2.5 V for the final symmetric cells.
4. Electrochemical properties of OEMs and affecting structural factors
In comparison to inorganic electrode materials, the unique advantage of flexible molecular design allows for the customization of OEMs’ charge-storage properties to suit different battery systems. Therefore, a comprehensive understanding of the structural factors that influence the electrochemical properties of OEMs is crucial. In this section, we analyze existing experience in correlating various structural factors of OEMs with their electrochemical properties, focusing on key parameters such as specific capacity, rate performance, redox potential, reversibility, and durability (Fig. 3). | ||
| Fig. 3 Schematic illustration summarizing ((a), Section 4) key electrochemical properties and ((b), Section 5) considerations of OEMs for high-performance MMB cathodes. | ||
4.1. Specific capacity
The type and density of redox moieties directly influence the theoretical quantity of charge transfer per OEM molecule, thereby co-determining the theoretical specific capacity of OEMs along with the molecular mass. Once the molecule structure of OEMs is resolved, the theoretical capacity (C, mA h g−1) can be estimated based on eqn (7),99 where n is the number of electrons participating in the redox reaction, M denotes the molecular weight in g mol−1, and F stands for Faraday's constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1). | (7) |
Most redox moieties are only capable of undergoing single-electron transfer reactions. Examples include n-type groups like carbonyl, thiocarbonyl, imine, and nitrile, as well as p-type groups such as ether, thioether, and tertiary amine. A few redox moieties can achieve two-electron reactions (e.g., azo, disulfide, dihydrophenazine, phenothiazine, phenoxazine, and TEMPO) or multi-electron reactions (e.g., 4,4′-(phenazine-5,10-diyl)dibenzoate). Incorporating dense multi-electron transfer redox moieties in OEMs represents an ideal direction for achieving high specific capacity in OEMs. For example, Li and coworkers studied three triimide compounds bearing three imide groups on benzene, triphenylene, and hexaazatrinaphthylene cores, respectively.100 They found that the nitrogen-rich heteroaromatic hexaazatrinaphthylene triimide with multiple active sites (imide and pyrazine) exhibited the highest specific capacity of 317 mA h g−1, compared with the other two materials. In addition to the intrinsic structures of OEMs, estimating the theoretical specific capacity should also consider the employed electrolyte. This consideration is important as the electrolyte determines whether the expected redox reaction can be fully achieved within its electrochemically stable potential window. For example, Zhou and coworkers reported a novel conjugated redox-active poly(diethyldihydrophenazine vinylene) polymer with a two-electron transfer process,101 whose electrochemical oxidation and reduction reactions take place in a high potential region ranging from 0.3 to 1.2 V vs. SHE. This means that the chosen electrolyte should have a high electrochemical stable potential window that can cover this high-potential range. Otherwise, the two-electron-transferring redox process would fail to proceed.
4.2. Rate performance
In reality, the rate performance of OEMs in a specific electrolyte is affected by a variety of kinetics issues. Since the electrochemical charge storage process involves the simultaneous release/storage of charges and ions, these kinetics issues can be analyzed from two aspects, namely electron transport and ion transport. There are two different mechanisms to describe electron transport in solid OEMs, including redox hopping and band-like charge transport.102,103 The redox hopping theory suggests that charge propagation occurs through electron hopping between adjacent redox sites. According to this theory, the density of redox sites and the conduction pathways are the primary factors that determine the overall electrical conductivity. On the other hand, the band-like charge transport mechanism involves electrons moving through a delocalized conjugated system. This mechanism is typical of most conductive polymer materials, which feature extended π-conjugated aromatic structures.104 The electrical conductivity of materials is determined by the Eg between the HOMO and LUMO energy levels. Materials with a band gap above 3.6 eV are considered insulators, while those with a band gap below 3.6 eV are classified as semiconductors. In metallic conductors, the conduction and valence bands overlap, with the Fermi level intersecting this band. Most organic materials fall into the categories of insulators or semiconductors. Therefore, molecular and material engineering approaches that narrow the Eg are highly effective in enhancing the electrical conductivity of organic materials. These approaches include incorporating more redox-active sites, designing large conjugated organic molecules, combining with dopants, and hybridizing with conductive fillers.The kinetics issues related to ion transport in OEMs, particularly in MMBs, remain insufficiently understood. Generally, crucial ion transport processes in battery electrodes include the charge transfer step at the electrolyte/electrode interface and solid-state ion diffusion within the electrode. During the interfacial charge transfer step, ion desolvation and/or anion–cation dissociation are always involved. Currently, there is a lack of experimental techniques or precise theoretical approaches to accurately discern the solvation configuration of charge carriers for OEMs. Often, neat ions or commonly considered ionic species in the selected electrolyte are assumed to be the charge carriers. However, such an assumption may not always hold true, particularly due to the ‘soft’ nature of OEM structures, which could accommodate the insertion of certain solvent or counter ions. Recently, Lukenhaus and workers studied the charge storage mechanism of non-conjugated radical polymers (poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl acrylamide)) (PTAm) in nine different aqueous electrolytes using advanced electrochemical quartz crystal microbalance (EQCM) characterization.105 They discovered that the chaotropic/kosmotropic properties and solvation states of both cations and anions are closely linked to the kinetics, swelling, and capacity of PTAm electrodes. Specifically, the kinetics display a clear relationship with the ionic size and chaotropic/kosmotropic characteristics. Electrolytes containing the smaller, more kosmotropic Cl− anion and Li+ cation exhibit the fastest kinetics. However, the Cl− anion is generally associated with a higher number of water molecules (30–38) compared to BF4− and OTf− anions (2–5) during insertion into the PTAm electrode, leading to reduced capacity and significant decay. Consequently, fully understanding the energy storage mechanism of organic electrodes is still challenging due to the complicated transfer process of combined electrons, ions, and solvent molecules.
Regarding ion diffusion within solid-state electrodes, the morphology of organic materials and their interactions with electrolytes are two crucial influencing factors. The ion diffusion time (τ) within the electrode depends on both the ion diffusion length (L) and the ion diffusion coefficient (D), according to eqn (8).106 Smaller particle sizes of organic materials shorten the diffusion length, and their favorable interactions with charge carriers improve the ion diffusion coefficient. Both factors contribute to shorter ion diffusion times and better rate performance.
 | (8) |
Sufficient intermolecular space within OEMs is essential for efficient ion transport and storage, making molecular curvature a valuable characteristic for promoting rapid ion movement in smaller molecules. For example, perylene diimide-hexaazatrinaphthylene (PHATN) designed using a molecular contortion strategy was shown to achieve a super-high pseudocapacitive performance.107 The PHATN molecule presents a tortile aromatic system structure with a contortion degree of 23°. This intriguing contorted feature enhances ion diffusivity and accessibility of redox-active moieties, resulting in rapid charging, high capacitance, and long-term stability. The resultant pseudocapacitor delivered a specific capacitance of 689 F g−1 at 0.5 A g−1 and maintained stability over 50000 cycles. Such designs are also effective in some OEMs, such as curved nanographene, concave dodecahedral nanocarbons, and larger polycyclic aromatic hydrocarbon fragments, improving their ion mobility and overall electrochemical performances.108,109
Additionally, polarized substituent groups around the redox moieties can enhance their rate properties by enabling co-coordination with charge carrier ions. For instance, non-conjugated carbonyl groups adjacent to the redox-active carbonyl groups in poly(perylenediimides) were revealed to facilitate adsorption and diffusion of Na+ and improve rate performance by co-coordinating with charge carrier cations.110 The poly(perylenediimides) with diketone groups show a high specific capacity of 130.8 mA h g−1 at 1 A g−1 with a capacity retention of 97% compared to the capacity at 0.1 A g−1 (134.9 mA h g−1), while the ones with single carbonyl groups only show a moderate capacity retention of 85.3% when the current density increased to 1 A g−1. Similarly, cyano groups with high dipole moments were found to promote the charge transfer and rate performance of n-type OEMs.111 As a representative example, cyano-substituted hexaazatrinaphthalene (HATN-3CN) exhibited a state-of-the-art rate capacity, with the capacity retention of 60.7% at a super-high current density of 20 A g−1. This enhancement was ascribed to the strong electron-withdrawing nature and extended π-conjugation provided by the cyano groups, which synergistically promote the dispersion of negative charges on the N atoms to stabilize the reactive intermediate (HATN-3CN6−) during the electrochemical process, thus accelerating reaction kinetics. Besides, the narrowed Eg between the HOMO and LUMO of HATN-3CN also contributes to a higher electrical conductivity, facilitating faster electron transfer in OEMs.
4.3. Redox potential
As discussed in Section 3.1, the intrinsic redox potential of OEMs is determined by their HOMO and LUMO levels. Generally, the organic molecule with a lower LUMO energy presents a good electron affinity and a higher reduction potential, while the one with a lower HOMO energy presents a higher oxidation potential.27 From early research experiences, the type of redox moiety sets the baseline for the redox potential of OEMs, while the chemical environment of redox moieties causes fluctuation in redox potential. The fluctuation factor for the electronic energy level is mainly the electron density in the redox center and around the redox center. Consequently, the redox potential can be tailored to a certain degree by substituent groups. Electron-withdrawing substituent groups (e.g., –CHO, –COOH, –CONH–, –SO3H, –F, –Cl, –Br, –NO2, –CN, and –NR3) could reduce the electron density of redox moieties, which are effective in raising the redox potential of OEMs by either lowering the LUMO level of n-type moieties or the HOMO level of p-type moieties. Conversely, electron-donating groups (e.g., –CH3, –CR3, phenyl, –OH, –NH2, –SH, and –OCH3) would lift the LUMO and HOMO levels of OEMs, thereby reducing the redox potential of OEMs. For example, a study reported by Chen et al. verified this principle by assessing the redox potential of n-type benzoquinone (BQ), tetramethylquinone (TMBQ) and tetrachlorobenzoquinone (TCBQ).112 The electron-withdrawing effect of methyl groups and the electron-donating effect of chlorine groups were validated by simulated LUMO levels using DFT calculations. The LUMO energy follows the order of TMBQ > BQ > TCBQ, while the redox potential follows the order of TCBQ > BQ > TMBQ. Due to the electron-donating effect of methyl groups, TMBQ exhibited the lowest reduction potential of 0.2 V vs. Ag/AgCl and TCBQ had the highest redox potential of 0.5 V vs. Ag/AgCl. Of course, one should notice that the introduction of redox-inactive substituent groups would sacrifice the specific capacity of OEMs, which could be a side-effect of potential regulation.OEMs with adjacent redox moieties could be particularly suitable for MMB applications, because these adjacent moieties can co-coordinate with a single multivalent metal ion. The number and relative position of substituents influence the redox potential of OEMs. A study on quinone compounds containing ortho-, para-, and meta-carbonyl groups supports this statement.113 Compounds with ortho-carbonyl groups exhibited higher voltages, higher capacities, and smaller overpotentials than para-quinones in AABs. The ortho-quinones showed higher output voltages due to their low LUMO energy levels. The smaller overpotential was attributed to the chelation of adjacent carbonyls with multivalent cations, enhancing the stability of discharged products and facilitating the reaction of ortho-quinone molecules. In contrast, meta-carbonyl compounds often produce unstable radial anions after accepting electrons, leading to low capacity and poor cycle stability. This is likely why anhydride and imide derivatives exhibit lower capacity compared to quinone compounds, which typically only accept two electrons for their four carbonyl groups. Conversely, para-quinone and cyclohexanehexone can reversibly accept two and six electrons, respectively, thereby demonstrating higher capacities. Moreover, HATN-3CN exhibited a higher discharge voltage of 0.72 V vs. Zn/Zn2+ compared to HATN without cyano groups (approximately 0.53 V) in AZBs.111 By increasing the number of substituent groups, the discharge voltage can further improve and approach around 1.0 V for organic tetracyanoquinodimethane materials.91
4.4. Reversibility and durability
Reversibility and durability are essential electrochemical properties for the practical applications of OEMs. The instability of redox intermediates is a key factor that negatively affects the cycling stability of OEMs. During the redox process, OEMs often generate radicals that must be stabilized to prevent the formation or breaking of electron-pair single bonds. Reactive radicals pose a significant challenge for OEMs, as they can lead to irreversible radical coupling among small molecules, resulting in the formation of dimers or oligomers. This issue is often evidenced by the low coulombic efficiencies of OEMs. To ensure the generation of stable radicals during redox reactions, aromatic π-extension is desired for OEMs to delocalize spin and charge density. For instance, the electrochemical performance comparison of 1,4-naphthoquinone (1,4-NQ), 9,10-anthraquinone (9,10-AQ), and 5,7,12,14-pentacenetetrone (PT) in aqueous zinc batteries demonstrates the effectiveness of this π-extension strategy.114,115 Thanks to the highly π-conjugated system, the PT electrodes showed superior cycling stability compared to the smaller quinones, achieving a high capacity retention of over 95% after 5000 cycles at a current density of 2.5 A g−1. Substituent groups can also contribute to stabilizing radical structures through steric protection. For example, quaternized amines are able to store anions via the formation of nitrogen radical cations (R3N˙+ or R4N+) during electrochemical oxidation. These radical intermediates are highly reactive and can easily couple with each other, losing redox activities. In our recent study, we found that aromatic phenyl substituents could efficiently stabilize the formed N radical cations by electron delocalization, maintaining the redox activity of amine molecules.116 This stabilization suppressed the oligomerization and dissolution of reactive amine molecules into electrolytes, resulting in highly reversible and durable amine electrodes for anion storage.In addition to redox reversibility, the solubility and structural degradation of OEMs are critical concerns that impact charge storage reversibility and durability. Small molecule-based OEMs, particularly in polar solvent electrolytes, often show substantial capacity loss within the first few cycles due to pronounced material dissolution. Structural factors, such as extensive aromatization and specific substituent groups, can effectively reduce the dissolution of small molecules. For example, calix[4]quinone (C4Q) featuring a macrocyclic structure exhibited superior capability to coordinate with multivalent metal ions into its large cavity.113 The resultant cathodes showed a high specific capacity of 400 mA h g−1 for Al(OTF)2+ cation storage, corresponding to 89.7% utilization of redox centers based on their theoretical capacity of 446 mA h g−1. Additionally, these electrodes showed improved stability, with a capacity retention of 81% and a CE of above 98% per cycle, compared to other quinone-based cathodes. This enhanced performance benefitted from the macrocyclic structure's extensive aromatization and abundant redox-active groups, which facilitated coordination with multivalent metal ions and improved electrochemical properties. Hybridization with insoluble substrates such as carbon nanotubes (CNTs), carbon black, graphene, and MXenes can also reduce the dissolution of OEMs in electrolytes.15 The strong interactions between the substrate and organic molecules play a decisive role in improving cycling performance. Covalent grafting of organic molecules onto insoluble matrices is more effective than physisorption. For instance, Park and coworkers grafted 2,3-diamino-1,4-naphthoquinone onto carboxyl group-modified porous current collectors via a simple amidation reaction.117 The obtained composite cathode achieved the capacity retention of 99% after 500 cycles at 0.2C, proving the pronounced effect of the covalent grafting strategy. Polymerization of soluble redox-active molecules can also inhibit dissolution and loss of active materials owing to the large molecular weight. The polymerization degree, molecular weight, and interactions with the electrolyte of redox-active polymers are critical factors influencing the stability of polymer electrodes. However, synthesized polymer electrodes may still face swelling issues, causing a certain degree of performance decay. 2D COFs with robust linkages offer a promising solution to this issue due to their high surface area, porous structure, rigid and robust crystalline frameworks.
5. Design considerations for different types of OEMs
Ideal OEM cathodes for MMBs should exhibit large specific capacity, excellent rate capability, high redox potential, long-term reversibility and durability. Various types of OEMs, including small molecules, amorphous polymers, and crystalline COFs, have been investigated to meet these criteria. Beyond the general design principles outlined earlier, specific considerations are necessary when designing different types of OEMs based on their distinct structural characteristics (Fig. 3).5.1. Small organic molecules
Benefiting from rich design versatility, dense redox moieties, and facial synthesis, small organic molecules have been widely studied in electrochemical energy storage applications. Compared with high-molecular-weight polymer materials with repeated structural units, the small organic molecules normally show a high content of redox-active moieties and efficient utilization of these sites due to the absence of redox-inactive units. Moreover, it is easy to synthesize small organic molecules with specific functional groups and accurate molecular structure, or modify them with electron-withdrawing or electron-donating groups. Despite their numerous advantages, redox-active small organic molecules often face significant challenges, particularly severe dissolution during repeated electrochemical cycles, which undermines their long-term cycling stability. As mentioned earlier, molecule engineering strategies, such as introducing functional substituent groups and extending conjugated structures, have proven effective in suppressing this problem. Moreover, the poor electrical conductivity of small organic molecules is another intrinsic drawback. To mitigate this issue, a large number of conductive additives are required, leading to unsatisfactory volumetric and gravimetric energy densities of MMBs at the device level. Hybridization with conductive fillers has proved to be an efficient approach to improve their conductivities and stabilities. For example, Park and coworkers reported a self-standing and flexible nanohybrid organic electrode based on flavin molecules and conductive single-walled carbon nanotubes (SWCNTs).118 This hybrid material exhibited good capacity retention (no decay after 100 cycles) and excellent power capability for lithium storage. The aromatic structure of flavin molecules allows them to strongly anchor to the hydrophobic surfaces of conductive SWCNT scaffolds via π–π interactions, which contribute to both improved conductivity and anti-dissolution properties without compromising their redox-active properties. Additionally, enhancing yield rates and purity while reducing production costs are also crucial issues for the practical applications of these redox-active small organic molecules.5.2. Amorphous polymers
Redox-active polymers have attracted increasing attention for MMBs due to their flexibility, tunability, processability, and thermal and electrochemical stability. Generally, depending on the polymerizable groups of the monomers, there are two main approaches to synthesize redox-active amorphous polymers, including condensation polymerization and free radical polymerization. Designing polymerizable monomers with suitable redox-active sites is crucial for achieving polymer materials with desirable electrochemical performance. However, the redox-inactive linkages or substituents often reduce the specific capacity of redox-active polymers. To improve the specific capacity and energy density of polymer-based batteries, introducing multiple redox centers and increasing the redox-site density per repeat unit are always pursued. Moreover, according to the location of functional sites, the redox-active polymers could be classified into backbone-type and side-chain type polymers. Conductive polymers are typical backbone-type redox-active polymers. Ever since the first PPY-based organic Al batteries were reported by Osteryoung et al. in 1984, various conductive polymers including PANI, PPY, PEDOT, and polypyrene have been widely studied for MMBs.119 However, for achieving ideal performance, they require a high doping level (above 20%) to ensure desirable conductivities, which significantly reduces the mass of active materials in the electrodes, resulting in practical capacities typically below 150 mA h g−1. This issue has motivated the community to explore alternative conductive polymers with high intrinsic conductivity. Additionally, if polymer materials could incorporate redox-active sites on both the backbone and side chains, the density of redox-active sites and energy storage performance could be theoretically enhanced. For example, Strietzel and coworkers reported naphthoquinone- and hydroquinone-modified PEDOT as electrodes.120 The conductive PEDOT main chain not only facilitates electron transport and reduces material solubility, but also undergoes electrochemical reduction and oxidation processes. Simultaneously, the quinone groups located at the side chains of PEDOT also provide additional redox-active sites. Such a design enables the efficient use of redox sites, offering a high specific capacity, good cycling stability, and rapid charging capacities.Furthermore, the slow diffusion of multivalent ions along polymer chains poses a significant challenge for most redox-active polymer electrodes. Due to the large steric hindrance and strong interactions between multivalent ions and polymer chains, these electrodes typically suffer from poor ionic diffusivity and rate performance. However, it is worth noting that an extended π-conjugated structure can create an expansive molecular plane and provide abundant open 2D ionic channels. At the same time, the presence of abundant delocalized π-electrons within the structure further facilitates ion diffusion during electrochemical redox reactions. Inspired by this concept, a linear π-conjugated quinone-based polymer (poly-(phenazine-alt-pyromellitic anhydride) (PPPA)) was synthesized via condensation polymerization. This polymer features an ultrahigh Zn2+ diffusion coefficient of 1.2 × 10−7 cm s−1.121 The extended π-conjugated structure and dense π-conjugated interlayer stacking within the PPPA chains provide abundant delocalized π-electrons and open 2D channels. These structural features enable fast electron and ion transport, leading to superior ion diffusivity compared to other organic cathode materials.
5.3. Redox-active COFs
COFs are crystalline porous polymers created by linking organic units into ordered 2D or 3D networks through covalent bonds. Structurally, COF materials are composed of at least two types of building blocks connected by different covalent linkages. Dynamic covalent chemistry is a crucial tool in the synthesis of COF materials by controlling their crystal nucleation and growth. To date, substantial efforts have been devoted to developing novel COF materials for energy storage. Currently reported linkages for redox-active COFs are dominated by imine bonds formed via the Schiff base reaction,122,123 imide linkages by amidation reaction,124 pyrazine linkages by nucleophilic aromatic substitution or condensation reaction,125 and sp2-carbon (CC) by Knoevenagel condensation or aldol condensation.126,127 The reversibility of dynamic covalent bonds under thermodynamic control allows COFs to correct synthesis errors, transforming initially formed amorphous intermediates into thermodynamically favored crystalline networks. COFs feature large specific surface areas, customizable pore chemistry, well-defined redox-active sites, excellent electrochemical stability, and good ion accessibility. However, precisely synthesizing COFs with designed structures and functions remains challenging due to the influence of both thermodynamic and kinetic factors on the formation of highly crystalline COFs. This complexity is one reason why research on their application in MMBs is still in its early stage.
The development of COFs for ion storage applications is also constrained by their low specific capacity, which limits the energy density of the resultant batteries. This limitation stems from the fact that many components in COFs are redox-inactive. Consequently, increasing the density of redox-active sites per structural unit is crucial when designing COF structures for battery applications. For example, Alshareef and coworkers designed a quinone-incorporated 1,4,5,8,9,12-hexaazatriphenylene-based COF (HAQ-COF) with dense redox-active centers.128 During discharge in an aqueous Mg salt solution, Mg2+ coordinated to both CN and CO, with a maximum of 12 Mg2+ ions coordinated within one inner hexagonal COF structural unit. Moreover, due to their well-defined and regular ion diffusion channels, redox-active COFs are expected to exhibit a better rate performance compared to amorphous polymer materials. However, their rate performance remains unsatisfactory, primarily due to their inherently poor electronic conductivity. Developing COFs with fully in-plane conjugation could be a promising approach to enhance their rate performance. For example, Shi and coworkers reported a nitrogen-rich 2D conjugated COF with multiple carbonyl groups (TB-COF) for Ca2+ ion storage.129 Thanks to the multiple redox-active sites in its skeleton, the TB-COF electrode can store a maximum of nine Ca2+ ions per TB-COF repetitive unit, enabling a high reversible capacity of 253 mA h g−1 at 1.0 A g−1. The high density of nitrogen atoms and the π-conjugated structure endowed the TB-COF with high electrical conductivity (5.9 × 10−9 S cm−1) and high-rate capacity. Additionally, in order to improve their electrochemical stability, it is advisable to design COFs with robust linkages, such as imide and sp2-carbon linkages. These linkages can remain stable under repeated charging and discharging processes.
Apart from the above-mentioned issues, defects and grain boundaries in bulk COFs often impede ion transport, increasing mass transport resistance and hindering charge carriers from accessing deeply buried active sites. In contrast, 2D COF nanosheets, consisting of single or a few atomic layers, can reduce the ionic transport path and expose more ion-accessible active sites. Therefore, exfoliating layered COFs into nanosheets can significantly enhance their maximum charge storage performance.130 For example, Wang and coworkers delaminated bulk anthraquinone-based COFs into few-layer redox-active nanosheets through a facile and scalable mechanical milling method.131 Compared with pristine COFs featuring multiple stacking structures, the exfoliated COFs showed a shorter ion/electron migration length and faster ionic/electronic diffusion, as well as a large portion of ion storage sites. As a result, the exfoliated COF-based LIBs showed significantly improved capacity, electrochemical stabilities, and rate performances, compared to their monomer and pristine COF-based counterparts.
6. Progress in OEM electrodes for MMBs
With the growing demand for abundant and environmentally friendly alternatives to traditional inorganic electrodes, a significant amount of interest in OEMs for MMBs has been sparked in the last decade (Fig. 4a). This section provides a comprehensive overview of recent developments in this field. The scope includes various organic materials, including small molecules, polymers and 2D COFs, and investigation of their potential to show high capacity, flexibility, and reduced environmental impact. Their respective electrochemical properties, such as voltage, specific capacity and energy density, are summarized in Fig. 4b and c. | ||
| Fig. 4 (a) Key progress and timeline for the development of OEMs for MMBs. Performance comparison regarding specific capacity, working voltage, and energy density among different (b) organic small molecules, (c) amorphous polymers, and crystalline COF electrodes. | ||
Theoretical simulations provide a powerful framework for understanding and predicting the electrochemical performance of OEMs, offering significant insights into their electronic structure and electrochemical properties. Among different simulation methods, DFT is the most widely used due to its ability to accurately describe the electronic structure of molecules and materials with relatively high computational efficiency. The foundation of DFT lies in solving the Kohn–Sham equations, which approximate the many-electron Schrödinger equation. In DFT, the ground state energy of a system is calculated by minimizing the total energy functional with respect to the electron density. This electron density is derived self-consistently and provides a direct pathway to determine key electronic properties, such as HOMO/LUMO energies, electrostatic potential (ESP), and so on. HOMO and LUMO energies are determined by solving the eigenvalues of the Kohn–Sham Hamiltonian. These eigenvalues correspond to the molecular orbitals’ energy levels, with the HOMO being the highest occupied orbital and the LUMO being the lowest unoccupied orbital. Eg, a key indicator of electronic properties, indicates the difference between the HOMO and LUMO energies. Additionally, ESP is a derived property calculated from the electron density and nuclear charge distribution. It represents the Coulomb potential experienced by a unit positive charge at any point in space and is often visualized using contour maps or 3D surface projections to indicate electron-rich and electron-deficient regions.
In addition, the choice of software and computational method depends on the nature of the calculated structures (finite vs. periodic). For small molecules (finite system), where localized orbitals and discrete electronic states are prevalent, software programs such as Gaussian or ORCA are often used. These programs implement localized atomic orbital basis sets (e.g., 6-31G and cc-pVDZ) to represent wavefunctions. However, these software programs are less suited for systems requiring periodicity or delocalization. For polymers and COFs (infinite systems with repeating units), modeling using plane-wave-based DFT programs, such as Vienna Ab initio Simulation Package (VASP), Quantum ESPRESSO, or CP2K, is required. These programs support periodic boundary conditions and can simulate the delocalized electronic states and interlayer interactions that are characteristic of infinite systems.
This review is logically divided into subsections, with an introduction to the basic characteristics of organic electrode materials and a thorough analysis of their stability and electrochemical performance coming first. The parts that follow concentrate on the creative design techniques meant to improve conductivity, capacity, and cycle life. Moreover, this review gives a clear account of the progress by highlighting the movement from fundamental material properties to sophisticated performance characteristics. The need for ongoing research and innovation is highlighted in this section by addressing the issues that still need to be addressed, despite the substantial progress that has been accomplished. These challenges include decreasing electrode dissolution and enhancing material stability for commercial applications.
6.1. Small organic molecules
Among all reported OEMs for MMBs, the predominant focus remains on small molecules due to the availability of numerous commercially accessible redox compounds, including both naturally derived and synthetic options.51,132–135 Naturally derived small molecules with low cost and good sustainability hold the potential to overcome the drawbacks of conventional electrode materials for batteries. Additionally, recent synthetic efforts in the lab have further enriched the diversity of redox-active small molecules for MMBs. Here, as displayed in Fig. 5, we summarize these advancements by categorizing them into four categories: carbonyl-, amine-, imine- and nitrile-based small molecules. Their unique characteristics, charge storage methods, and electrochemical performances will be deeply analyzed in this section.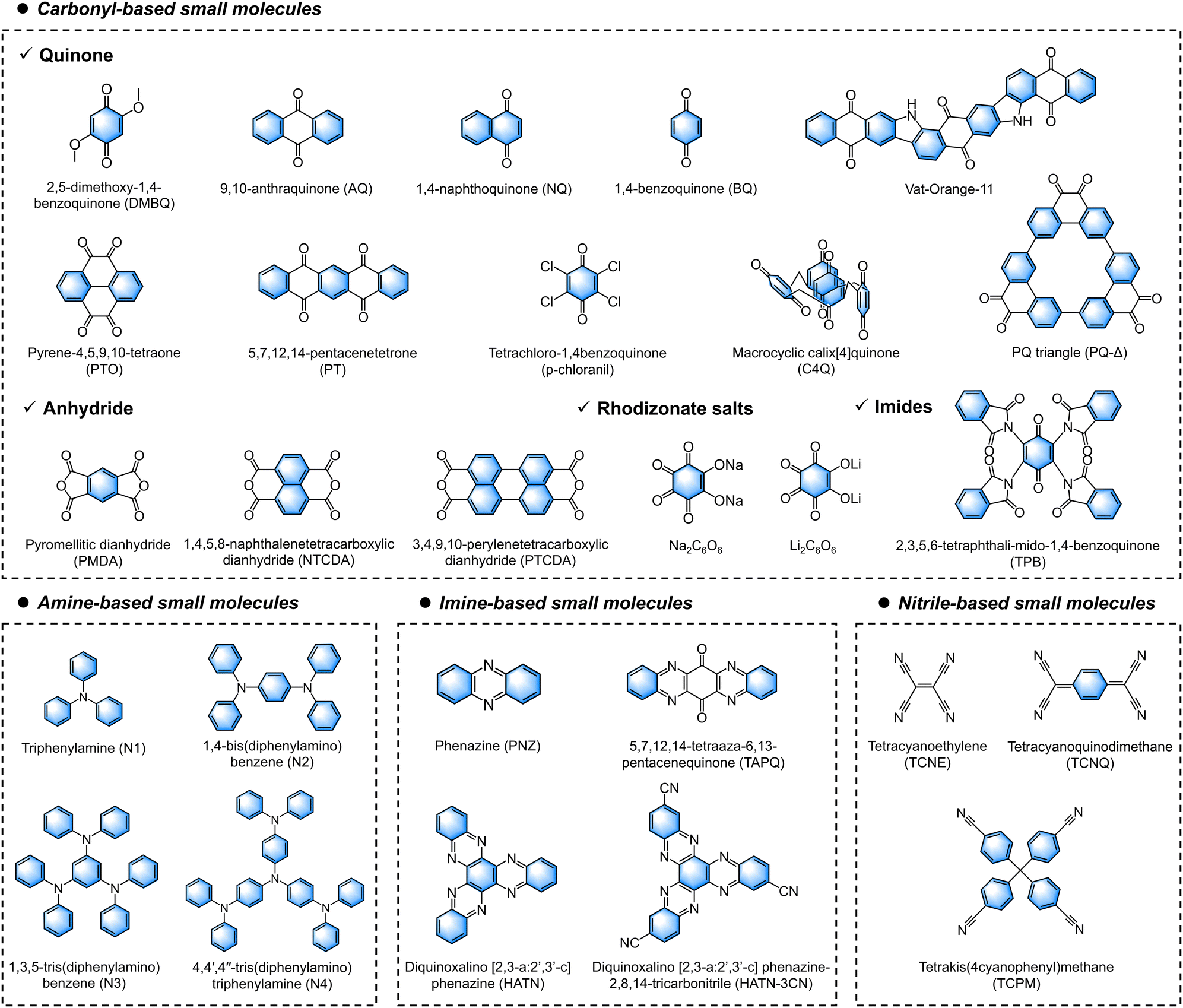 | ||
| Fig. 5 Molecular structures of carbonyl-, amine-, imine-, and nitrile-based small molecules. | ||
O) could take electrons to form enolate anions (C–O−),136–141 which coordinate with cationic species from the electrolyte to form the corresponding enolate salts.142,143 During discharging, CO transforms to C–O−. During charging, connected cations can be released, allowing for the recovery of the CO structure.144 Carbonyl-containing small molecules are the most extensively studied redox molecules for batteries given their efficient charge-storage capability. According to different chemical structures, carbonyl-based molecules briefly include quinones, anhydrides, and rhodizonate salts.
6.1.1.1. Quinone-based small molecules. Quinones are derivatives of aromatic compounds formed by converting an even number of –CH
groups into –C(O)– groups. This transformation introduces two ketone functionalities into the aromatic ring, which can be positioned in either the para-(1,4-) or ortho-(1,2-) configurations. The quinone family contains several types, with variations in the positioning of the carbonyl groups and the structure of the aromatic ring. As the simplest quinone, 1,4-benzoquinone (p-quinone) features a benzene ring where two hydrogen atoms are replaced by two oxygen atoms at the 1 and 4 positions, directly opposite to each other. In contrast, 1,2-benzoquinone (o-quinone) has its carbonyl groups positioned at the 1 and 2 locations on the benzene ring, adjacent to each other, resulting in a compound that is often less stable than its 1,4-isomer. Additionally, naphthoquinone is derived from naphthalene (two fused benzene rings), with carbonyl groups substituting C–H on the rings. In these structures, the carbonyl groups enhance the aromaticity of the benzene ring, creating a planar and conjugated system.
The early application of quinone-based small molecules was demonstrated in NMBs using 2,5-dimethoxy-1,4-benzoquinone (DMBQ) cathodes.145 DMBQ is a commercially available quinone derivative where the basic p-quinone structure is modified by substituting methoxy groups (–OCH3) at the 2 and 5 positions of the benzene ring. Besides, DMBQ has a high theoretical capacity (319 mA h g−1) based on a two-electron redox reaction. Sano et al. examined the charge storage characteristics of the DMBQ cathode in the electrolyte of 0.5 M Mg(ClO4)2)/γ-butyrolactone.146 The DMBQ cathode showed a discharge capacity of 260 mA h g−1, exceeding 50% of the theoretical value. It was demonstrated that the crystal structure was reversibly restored when Mg2+ was extracted from the DMBQ electrode, although Mg2+ insertion could transform the DMBQ molecular crystal into an amorphous-like structure. Additionally, the discharge and charge processes were only reversible for five cycles, with the discharge capacities gradually decreasing during cycling. Another work revealed that the electrolyte composition could significantly affect the charge storage capability of the DMBQ electrode. The impact of various sulfone electrolyte additives was explored on the electrochemical performance of DMBQ cathodes for Mg batteries.147 Specifically, the DMBQ cathodes displayed initial discharge capacities of 78, 162, and 126 mA h g−1 in the magnesium bis(trifluoromethanesulfonyl)amide (Mg(TFSA)2)/sulfolane (SL), Mg(TFSA)2/ethyl-i-propyl sulfone (EiPS), and Mg(TFSA)2/di-n-propyl sulfone (DnPS) electrolytes, respectively. The DMBQ cathodes using these three types of electrolytes showed dramatically decreased discharge capacities, which could be explained by the loss of active materials during cycling. After ten cycles, the capacities of DMBQ cathodes in Mg(TFSA)2/SL and Mg(TFSA)2/DnPS electrolytes were still higher than 60 mA h g−1, while the capacity of the DMBQ cathode in the Mg(TFSA)2/EiPS electrolyte dropped to only 10% of the theoretical value. Along cycling, the apparent color change was noticed in the Mg(TFSA)2/EiPS electrolyte, indicating that the DMBQ could be mostly likely to leach from the electrode into the Mg(TFSA)2/EiPS electrolyte.
The electrochemical performance of the DMBQ cathode in the full cell is also tightly associated with Mg stripping and plating. Specifically, the increased Mg stripping and plating over-potentials reduce the discharge voltage and increase the charge voltage of the full cells, thereby exacerbating battery polarization. Pan et al. showed that the DMBQ cathode-based full cells fabricated using the Mg(TFSI)2–MgCl2/DME electrolyte displayed higher discharge plateaus than those fabricated using the Mg(TFSI)2/diglyme electrolyte, as the MgCl2-containing electrolytes facilitated better Mg stripping and plating.148,149 Upon discharge, a new signal at 63.1° was detected in the X-ray diffraction (XRD) pattern of the DMBQ electrode. This signal disappeared in the subsequent charging process, and the XRD pattern recovered to that of the pristine electrode, indicating the good reversibility of the DMBQ electrode. Moreover, the discharge profiles of the DMBQ cathode using the 0.5 M Mg(TFSI)2–2MgCl2/DME electrolyte at different rates all showed discharge plateaus at around 2 V vs. Mg. However, the DMBQ cathode maintained a low discharge capacity of 74 mA h g−1 after 30 cycles (32.7% of the first discharge capacity), indicating the significant capacity loss during cycling as a result of the electrode dissolution in the electrolyte.
The structural tunability of organic electrodes holds immense potential for future functionalization and optimization. Beyond DMBQ cathodes, three other quinone-based organic cathodes,150 including anthraquinone (AQ), 1,4-naphthoquinone (NQ), and 1,4-benzoquinone (BQ), have also been investigated. In light of their lower molecular weights, NQ and BQ exhibit theoretical specific capacities of 339 mA h g−1 and 496 mA h g−1, respectively, which are dramatically higher than that of AQ (257 mA h g−1). On the other hand, the organics with lower molecular weight frequently show higher solubility in the electrolyte, which could cause faster capacity decay during battery cycling. Here, the AQ cathodes maintained a discharge capacity of 80 mA h g−1 after 10 cycles, which were higher than those of NQ (30 mA h g−1) and BQ (55 mA h g−1) cathodes. Moreover, the NQ and BQ cathodes showed 0.2 V and 0.5 V higher discharge plateaus than the AQ cathode in the Mg(TFSI)2–2MgCl2/DME electrolyte, respectively. Of note, the dissolution issue of these quinone-based organic cathodes cannot be completely inhibited. To mitigate electrode dissolution, efforts have also focused on enlarging the conjugated structure and increasing the molecular weight of quinone-based compounds. For instance, vat dyes (such as vat green 8, vat brown BR, and vat olive T) have been considered as potential cathode materials for NMBs.151 Vat Orange 11, a specific type of anthraquinone dye, features a structure based on the anthraquinone framework with benzene rings fused together and carbonyl groups on opposite sides of the central ring. Vat Orange 11, with its large molecular size, can be easily synthesized in the lab or extracted from plants. Debashis et al. reported the use of commercial Vat-Orange-11 as a cathode material for NMBs with APC and Mg(HMDS)2–4MgCl2/THF/PP13TFSI electrolytes.152 The electrode achieved a specific capacity of about 80 mA h g−1 at a current density of 100 mA g−1 with an average discharge voltage of 1.5 V vs. Mg. Moreover, the electrode can be charged/discharged for 1000 cycles with reversible transformation between the carbonyl and enolate groups, indicating that the extended conjugated structure significantly improved the electrochemical stabilities of quinone-based cathodes.
Another typical example of quinone-based molecules with an extended conjugated structure is pyrene-4,5,9,10-tetraone (PTO). PTO is a derivative of pyrene, featuring four carbonyl groups at the 4, 5, 9, and 10 positions, which facilitate rapid and reversible redox processes when used as cathode materials. Yao et al. evaluated PTO cathodes for NMBs fabricated using an Mg(CB11H12)2-based electrolyte.153 To understand the magnesium storage behavior, the PTO electrode was dismantled at various discharged states and immersed in the DME solvent. The electrode, after discharging to 2.0 V vs. Mg (with Mg-PTO as the discharge intermediate), was highly soluble in the DME solvent, resulting in a purple solution. In contrast, the fully discharged electrode at 0.9 V vs. Mg (with 2Mg-PTO as the discharge product) was insoluble in the DME solvent. These findings indicate that PTO undergoes a sequential conversion to an electrolyte-soluble Mg-PTO and an electrolyte-insoluble 2Mg-PTO, suggesting a heterogeneous liquid–solid reaction mechanism. A functionalized separator with graphene oxide (GO) was employed to effectively mitigate the issue of electrode dissolution in the electrolyte. The assembled Mg//PTO cell exhibited good cyclability for over 500 cycles at 0.2C (1C = 408 mA g−1), retaining 84% of its initial capacity (315 mA h g−1). Even at high rates of 20C and 50C, the cell attained high specific capacities of 278 and 210 mA h g−1, respectively. The fast-charging capability enabled the Mg//PTO cell to achieve a large specific power of 30 kW kg−1 and a high material-level specific energy of 313 W h kg−1, representing the state of the art among reported magnesium batteries.
Quinone-based molecules have also gained considerable attention as cathode materials for NABs. Kim et al. evaluated three phenanthrenequinone (PQ)-based OEMs, namely a monomer, a linear trimer, and a molecular triangle, as cathodes for NABs.154 Among them, the rigid triangular macrocycle (PQ-Δ) significantly enhanced both specific capacity and cyclability. Previous studies have revealed that soluble halides (MX3) in solution undergo asymmetric cleavage, producing ions of [MX2]+ and [MX4]−.155,156 Each quinone oxygen of PQ-Δ can coordinate with one AlCl2+, resulting in tetracoordinated Al centers. Therefore, a single PQ-Δ molecule is able to accommodate three AlCl2+ cations.157–159 The PQ-Δ cathode exhibited exceptional cyclability, sustaining up to 5000 cycles with a reversible specific capacity of 94 mA h g−1. This superior cycling performance was attributed to the sterically hindered structure of PQ-Δ, which enabled high structure flexibility to withstand the molecular strain introduced by AlCl2+ insertion and extraction. Moreover, the creation of a PQ-Δ/graphite hybrid (denoted PQ-Δ-HY) greatly improved electrical conductivity,154 allowing for bipolar anion and cation storage, i.e., AlCl2+ storage through coordination with PQ-Δ and AlCl4− storage through intercalation into graphite. The PQ-Δ-HY electrode demonstrated outstanding cycling stability, retaining a high capacity of 114 mA h g−1 (94%) after 500 charge/discharge cycles at 0.2 A g−1. Furthermore, when the current density was increased to 2.5, 5, and 10 times the initial rate, the capacity retention of the PQ-Δ-HY electrode was 94%, 83%, and 81%, respectively, confirming its excellent rate capability.
As illustrated in Fig. 6a and b, Kao et al. investigated a series of quinone derivatives for NABs in the AlCl3/EMIMCl electrolyte, including BQ, AQ, PQ, and 2,6-dichloro-1,4-benzoquinone (DCBQ).160 During the discharge process, EMI+ cations acted as charge carriers, coordinating with carbonyl oxygen atoms of these quinone derivatives. The BQ cathode showed the best discharge capacity of 177 mA h g−1, while the AQ and PQ cathodes achieved specific capacities of 170 mA h g−1 and 160 mA h g−1, respectively. The DCBQ cathode had the lowest discharge capacity of 79 mA h g−1. However, due to their high solubility in ionic liquid electrolytes, these quinone derivatives were not stable during cycling. To address the dissolution problem, 2,3,5,6-tetraphthalimido-1,4-benzoquinone (TPB) was synthesized for NABs using the AlCl3/urea electrolyte. The assembled Al//TPB cell achieved a good specific capacity of 175 mA h g−1 over 250 cycles at a current density of 100 mA g−1 with a CE of over 90% (Fig. 6c). Moreover, the Al//TPB cell was used to power a red LED with high illumination, demonstrating its practical application. At a high current density of 1 A g−1, the Al//TPB cell exhibited a discharge capability of 50 mA h g−1, attributed to its structural flexibility and stability. Further improvement was achieved by Zhou et al., who applied a molybdenum foil current collector instead of carbon fiber paper, enhancing the electrochemical performance of AQ cathodes in NABs using the AlCl3/EMIMCl electrolyte.161 The assembled Al//TPB cell demonstrated a reversible specific capacity of 215 mA h g−1 with long cycling stability over 200 cycles.
 | ||
| Fig. 6 (a) The first discharge curves of various small molecule quinone derivatives. (b) Specific capacities and discharge voltages of different small molecule derivatives. (c) Long-term cycling performance of the Al//TPB cell cycled at 0.1 A g−1 over 250 cycles. The inset shows that a red LED with high illumination was powered by an Al//TPB cell under bending strain conditions. Reproduced with permission.160 Copyright 2020, American Chemical Society. Investigation of the redox-active center of the C4Q cathode. (d) GCD curve of the Al//C4Q battery at 0.2 A g−1. (e) In situ FTIR spectra of the C4Q electrode. (f) Liquid 13C NMR spectra of C4Q electrodes at selected states (left) and the corresponding structural variations at three selected states (right). Reproduced with permission.113 Copyright 2021, John Wiley & Sons, Inc. | ||
Additionally, quinone-based small molecules were studied as potential cathode materials for Ca2+ storage, including 2,5-dichloro-1,4-benzoquinone (2,5-dichloro BQ), 2,5-dimethoxybenzo-1,4-quinone (DMQ), 1,5-dichloro-anthraquinone (1,5-dichloro AQ), phenanthrenequinone (9,10-PQ), PTCDA, and 5,7,12,14-pentacenetetrone (PT).162 Among these, 2,5-dichloro BQ and DMQ cathodes exhibited relatively higher discharge voltages due to the presence of less electron-donating phenyl groups. However, the high charge density of Ca2+ limited their actual capacities because of strong electrostatic repulsion between Ca2+ and the organic molecules. In light of the coplanar stacked layered structure stabilized by van der Waals forces and the abundant one-dimensional channels oriented perpendicular to the molecular layers, the PT cathode exhibited distinctive Ca2+ storage properties. These structural characteristics facilitated the reversible uptake and removal of Ca2+ in PT cathodes resulting in a decent Ca2+ diffusion coefficient of 10−8–10−11 S cm−1. The PT electrode demonstrated attractive Ca2+-storage performance with a high discharge capacity of 150 mA h g−1 at 5 A g−1 and excellent rate capability, retaining a capacity of 86 mA h g−1 at 100 A g−1. Charge storage in the PT cathode relied on the reversible coordination with protons and Ca2+ from the electrolyte. Theoretical computations suggested that π–π stacking interactions lead to charge delocalization in the Ca2+-coordinated enolate intermediate across the stacks. One Ca2+ ion was stabilized by interactions with four carbonyl groups from neighboring PT molecules.
In addition to non-aqueous electrolytes, quinone-based electrodes have also been investigated in aqueous electrolytes for application in aqueous multivalent metal batteries. Increasing the number of carbonyl groups within individual molecules has proven to be an effective method for improving the electrochemical performance of these electrodes. Li and colleagues investigated six multi-carbonyl quinone compounds as cathode materials, including PTO, 1,4-NQ, 1,2-NQ, AQ, phenanthrenequinone (9,10-PQ), and calix[4]quinone (C4Q), with an aluminum anode and an aqueous Al(OTF)3 electrolyte.113 Specifically, C4Q is a macrocyclic compound belonging to the calixarene family, derived from calix[4]arene through oxidation.163 The diazo coupling reaction first occurred between calix[4]arene and p-aminobenzoic acid to afford 5,11,17,23-tetrakis[(p-carboxyphenyl)azo]-25,26,27,28-tetrahydroxy calix[4]arene (compound 1). After an azo reduction reaction, compound 1 was transformed into 5,11,17,23-tetraamino-25,26,27,28-tetrahydroxy calix[4]arene (compound 2). Finally, compound 2 was further oxidized to C4Q. The molecule consists of a cyclic arrangement of four phenolic units (aromatic rings) linked by methylene bridges, with each phenolic unit oxidized to a quinone form, featuring two ketone substitutions at the para positions. This macrocyclic structure facilitates electron acceptance due to the lower LUMO energy, resulting in a higher reduction potential.164 The average discharge voltages of the quinone compounds followed the sequence: AQ < 9,10-PQ < 1,4-NQ < 1,2-NQ < PTO < C4Q, aligning with the theoretically computed LUMO energy trend.
The macrocyclic C4Q, with its wide cavity, facilitated effective ion coordination and the high utilization of carbonyl centers. Among the studied quinone compounds, C4Q demonstrated the highest specific capacity of 400 mA h g−1 at 100 mA g−1, the lowest polarization of 42 mV, and good capacity retention of 81% after 50 cycles. Even at −20 °C, the C4Q cathode retained a competitive capacity of 224 mA h g−1. The typical Galvanostatic discharge and charge (GCD) profiles of the Al-C4Q battery at 200 mA g−1 are displayed in Fig. 6d. In situ FTIR was used to monitor the charging and discharging products in real time to look into the structural change in the quinone electrode (Fig. 6e). The reversible reaction of carbonyl groups is demonstrated by the –CO bond intensity (stretching vibration: 1655 cm−1), which steadily decreases during discharging and increases upon charging. Ex situ liquid 13C NMR spectroscopy of various states was also analyzed (Fig. 6f). In the pristine stage, four peaks at 187.7 (A), 185.8 (B), 146.4 (C), and 134.1 (D) ppm correspond to the four distinct carbon atoms in C4Q. During discharging, new peaks at 151.2 (E) and 141.7 (F) ppm emerge, while peaks (A) and (B) disappear, indicating the transformation from –CO to –C–O–. Concurrently, peaks (C) and (D) are gradually replaced by peaks (C′) and (D′) due to the injection of negative charges into the conjugated system. The charging process shows a reverse trend, with the NMR spectra nearly identical to the pristine state when charged to 1.4 V. These results indicate that carbonyl groups are involved in every step of the electrochemical reaction. AZBs fabricated using quinone-based cathodes offer numerous advantages, including enhanced safety, low cost, minimal environmental impact, sufficient energy density, extended cycle life, rapid charging capability, thermal stability, and versatility in applications.165–167 Despite these advantages, the electrochemical performance of quinone-based molecules in aqueous zinc batteries also faced some challenges. One major issue is the low voltage plateaus, which limit the energy output of the batteries. Another significant problem is the dissolution of active materials in the aqueous electrolyte, which leads to capacity fade and reduced battery lifespan. Kundu et al. investigated the use of tetrachloro-1,4-benzoquinone (TCBQ) as a cathode material for AZBs utilizing an aqueous Zn(OTf)2 electrolyte.168 CV curves of the TCBQ electrode revealed facile and reversible coordination with Zn2+, a process facilitated by its flexible lattice structure that permits molecular reorientation (Fig. 7a). The TCBQ cathode exhibited minimal voltage polarization (50 mV) and a flat plateau at around 1.1 V, achieving a high capacity of >200 mA h g−1 and an energy efficiency of ≈95% at 43.4 mA g−1 (Fig. 7b). Despite these promising metrics, the capacity of the TCBQ cathode declined sharply to 70 mA h g−1 after 50 cycles. This capacity fading was attributed to the formation of large microstructure particles from discharged and charged products, which led to a loss of electrolyte contact with the electrode over time. A distinct phase transition mechanism between TCBQ and Zn2+-inserted TCBQ was unveiled by multiple characterization methods. DFT calculations indicated that molecular column rotation in TCBQ facilitated Zn2+ insertion with a minimal volume change of −2.7% (Fig. 7c). To mitigate structural instability during cycling, embedding TCBQ molecules within the mesoporous nanochannels of CMK-3 carbon was further proposed. This approach enhanced electrochemical rechargeability and cyclability by improving the electronic conductivity of the cathodes and curtailing the growth of discharge and charge products. The CMK-3/TCBQ composite cathodes demonstrated an initial capacity of 170 mA h g−1 at 43.4 mA g−1, retaining a reversible capacity of 90 mA h g−1 after 100 cycles. The confinement effect provided by the mesoporous nanochannels of CMK-3 proved crucial in enhancing the electrochemical cyclability of quinone-based cathodes for AZBs.
 | ||
| Fig. 7 (a) CV curves of the Zn anode (black line) against the TCBQ (red line) in 1 M Zn(OTf)2 aqueous electrolyte at 5 and 0.1 mV s−1, respectively. (b) GCD profiles of the p-chloranil cathode at a 0.2C rate (1C = 217 mA g−1). (c) Structural models of p-chloranil and Zn-p-chloranil obtained after DFT structural optimization. Reproduced with permission.168 Copyright 2018, American Chemical Society. (d) Schematic illustration of the preparation procedure of C4Q. (e) Discharge/charge voltages and capacities of selected quinone compounds (1,2-NQ, 1,4-NQ, 9,10-PQ, 9,10-AQ, and C4Q) in aqueous RZBs. (f) GCD curves of Zn-C4Q batteries at 20 mA g−1. The upper x axis represents the uptake number of Zn2+ ions. One Zn2+ with two-electron transfers generates a specific capacity of 112 mA h g−1. (g) Cycling performance of Zn-C4Q batteries with the Nafion membrane or filter paper as a separator at the current density of 100 mA g−1. Reproduced with permission.169 Copyright 2018, Science. | ||
Quinone-based compounds with carbonyl groups in the para-position and in the ortho-position were further evaluated by Zhao et al., which were used as cathode materials for AZBs in an aqueous Zn(CF3SO3)2 electrolyte.169 The compounds C4Q, 9,10-AQ, and 1,4-NQ have carbonyls in the para-position, and the compounds 9,10-PQ, and 1,2-NQ have carbonyls in the ortho-position. Amongst them, C4Q was synthesized using raw materials of calix[4]arene and p-aminobenzoic acid (Fig. 7d).163,170 The study revealed that quinone compounds with para-position carbonyls exhibited higher capacities and reduced charge/discharge polarization compared to those with ortho-position carbonyls. The steric hindrance associated with ortho carbonyls adversely affects electrochemical performance. Notably, C4Q, with its eight carbonyl groups and an open bowl structure, showed the best performance among the tested materials (Fig. 7e). Specifically, C4Q delivered a flat operating voltage of 1.0 V, a large capacity of 335 mA h g−1, and a high energy efficiency of 93% at a low current density of 20 mA g−1. Additionally, the C4Q cathode demonstrated excellent cycling stability, retaining 87% of its capacity over 1000 cycles at 500 mA g−1, with a minimal voltage difference between discharge and charge (70 mV), which contributed to the high energy efficiency (Fig. 7f). To prevent electrode dissolution, a cation-exchange membrane (Nafion) was adopted as a separator, ensuring better cycling stability of the C4Q cathode at 20 mA g−1 (Fig. 7g). The Nafion separator also prevented the zinc anode from contacting with dissolved quinone, further enhancing the overall electrochemical performance.
Due to their high electron delocalization, inherent insolubility in aqueous electrolyte, and large theoretical energy density, PTO cathodes are also suitable for application in AZBs. Guo et al. investigated AZBs utilizing PTO cathodes with a ZnSO4 electrolyte.171 The PTO cathode demonstrated excellent electrochemical performance, with rapid kinetics and remarkable reversibility, achieving a high specific capacity of 336 mA hg−1 at a low current density of 40 mA g−1. As a result, the Zn//PTO full cell exhibited an impressive cycle life, extending beyond 1000 cycles. It also delivered a high energy density of 187 W h kg−1 and exhibited a power performance akin to a supercapacitor, with a power density of 22 W kg−1. Additionally, a flexible Zn//PTO battery in a belt form was fabricated, showcasing its potential applications in wearable electronics.
Additionally, an anti-freezing dual-cation electrolyte composed of 3.5 M Mg(ClO4)2 and 1 M Zn(ClO4)2 was employed in conjunction with PTO cathodes to develop low-temperature AZBs.172 The inclusion of hydrogen-ligand ClO4− and oxygen-ligand Mg2+ significantly reduces the hydrogen-bonding ratio in water molecules, resulting in an exceptionally low freezing point of −121 °C. This organic additive-free aqueous-salt hydrate exhibited favorable properties such as high ionic conductivity, low viscosity, and reduced activation energy at −70 °C. The binding energy of PTO with metal cations was compared, showing that PTO binds more strongly with two Zn2+ (−9.41 eV, Zn2PTO) than with two Mg2+ (−10.46 eV, Mg2PTO) or a combination of one Zn2+ and one Mg2+ (−9.47 eV, ZnMgPTO) as illustrated in Fig. 8a. However, considering the de-solvation energy required for metal cations to bind to PTO, Zn2+ has a greater hydration energy (−1.72 eV) than Mg2+ (−4.14 eV). After adjusting for de-solvation energy, the recalculated binding energy for Zn2PTO is −5.97 eV, which is lower than those of ZnMgPTO (−3.61 eV) and Mg2PTO (−2.18 eV). This finding reveals the preferential binding of PTO to Zn2+ ions, as shown in Fig. 8b. Consequently, the Zn//PTO battery with this dual-cation electrolyte achieved a high discharge capacity of 101.5 mA h g−1 at 200 mA g−1 and −70 °C (Fig. 8c). It also demonstrated stable cycling performance over 100 cycles with minimal capacity loss, underscoring its excellent performance at low temperatures.
 | ||
| Fig. 8 (a) Binding energies between PTO and Zn2+ or Mg2+. (b) The corrected binding energy levels of Zn2PTO, ZnMgPTO, and Mg2PTO. (c) GCD curves of the Zn-PTO battery at different temperatures. Reproduced with permission.172 Copyright 2021, Springer Nature Limited. (d) Schematic illustration of the aqueous rechargeable Zn-PQ-Δ cell. (e) Structural formula of PQ-Δ. (f) Electrochemical redox chemistry of PQ-Δ in an aqueous rechargeable zinc battery. GCD profiles and cycling performance of PQ-Δ cathodes at current densities of (g) 30 mA g−1 and (h) 150 mA g−1. Reproduced with permission.173 Copyright 2020, American Chemical Society. | ||
Compared to the PTO molecule, the PQ-Δ molecule features a more extended conjugation structure, which offers the potential for enhanced stability during battery cycling. Nam et al. drew inspiration from the successful application of the redox-active PQ-Δ in NABs.154 They applied PQ-Δ with a layered superstructure and rigid geometry to AZBs using an aqueous Zn(CF3SO3)2 electrolyte (Fig. 8d–f).173 The PQ-Δ cathodes demonstrated a voltage plateau around 0.84 V and a high specific capacity of 203 mA h g−1 at a current density of 30 mA g−1, with a CE of 99.6%. This capacity reflects the acceptance of six electrons per PQ-Δ molecule, translating to two electrons per phenanthrenequinone unit (Fig. 8g). The Zn//PQ-Δ cells showed remarkable stability, retaining a specific capacity of 210 mA h g−1 after 500 cycles at 150 mA g−1 (Fig. 8h). Co-insertion of H2O together with Zn2+ was indicated for the PQ-Δ cathode, which mitigated interfacial charge-transfer resistance primarily by reducing the desolvation energy penalty associated with hydrated Zn2+ ions.
As for AZBs, H+ inevitably present in the aqueous electrolyte (e.g., aqueous ZnSO4 electrolyte) can also react with the carbonyl groups of quinone-based cathodes concurrently with the storage of Zn2+, which might aggravate the poor cycle life arising from the inherent instability and solubility of electrode materials in the electrolyte. The sulfur heterocyclic quinone dibenzo[b,i]thianthrene-5,7,12,14-tetraone (DTT), known for its low solubility, was proposed by Wang et al. as a cathode material for AZBs.174 The DDT molecule was synthesized by reaction of 2,3-dichloro-1,4-naphthoquinone and sodium sulfide. The Zn//DTT cells demonstrated rapid charge storage kinetics and a high reversible capacity of 211 mA h g−1 at a low current density of 50 mA g−1, calculated based on the four carbonyl groups and the molecular weight of DTT. This result indicates that only about 75% of the carbonyl groups could be used for charge storage. Computational and experimental results suggested that DTT electrodes could simultaneously store Zn2+ and H+. The most favorable structure of the discharged product was DTT2(H+)4(Zn2+), where two neighboring DTT molecules were bound by one Zn2+ with the enhanced stability and reduced electrode dissolution. The Zn//DTT full cell using a thick separator was cycled for up to 23000 times at a high current density of 2 A g−1, indicating its potential for long-term grid-scale energy storage. In addition, the flexible Zn//DTT battery showed a stable cycle performance (120 mA h g−1) over 120 cycles at 0.5 A g−1 while bent from flat to 180°, indicating its potential applications in wearable and flexible electronic devices.
6.1.1.2. Anhydride-based small molecules. Anhydride-based molecules represent another prominent category of carbonyl-based compounds. These molecules feature two acyl groups bonded to the same oxygen atom, with the general formula (RCO)2O. Anhydrides can be either cyclic or acyclic, and they are highly reactive due to the electrophilic nature of the carbonyl carbons. Among various anhydride-based molecules, the well-known perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA), also recognized as the organic pigment “red 224”, features a central aromatic ring and two anhydride groups. It is widely utilized in molecular metal batteries. PTCDA crystals form by stacking layers through π–π interactions. Each PTCDA molecule can transfer two electrons during the charge and discharge processes.
In theory, Mg2+-storage organic structures should possess substantial conjugation to effectively accommodate bivalent Mg2+ and buffer charge density changes during repeated magnesiation and demagnesiation. Conjugated dianhydrides, known for their affordability and accessibility, are particularly appealing candidates for this purpose.142,143,175 As shown in Fig. 9a, three conjugated dianhydrides, including pyromellitic dianhydride (PMDA), 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA), and PTCDA, were assessed by Yang et al. to compare their Mg2+ storage capabilities.176 Despite having the same number of carbonyl groups, these dianhydrides exhibited different levels of conjugation and theoretical capacities (246, 200, and 137 mA h g−1 for PMDA, NTCDA, and PTCDA, respectively). The study revealed that the extent of conjugation decisively affected the reversible capacity, rate capability, and cycle stability. Specifically, the reversible Mg2+ storage capacity followed the order of PMDA (45 mA h g−1) < NTCDA (90 mA h g−1) < PTCDA (160 mA h g−1). Despite its lowest theoretical capacity, PTCDA offered the highest reversible capacity, verifying its superior ability to withstand electron density changes during Mg2+ storage and its low solubility in the electrolyte. The enolization of carbonyl groups served as the foundation for Mg2+ storage, as demonstrated by mechanistic investigations. FTIR and ultraviolet-visible (UV-vis) spectroscopy techniques were used to directly monitor and evaluate the behavior of dissolution inhibition, which demonstrated strong structural reversibility in the potential range of 0.6 to 2.5 V. Moreover, X-ray photoelectron spectroscopy (XPS) demonstrated that the electrochemical Mg2+ storage mechanism of PTCDA involved the conversion of carbonyl and enolate groups (Fig. 9b). The assembled Mg//PTCDA cell with the Mg(HMDS)2–MgCl2–AlCl3/tetraglyme electrolyte showed a reversible capacity of 160 mA h g−1 at 50 mA g−1 after 100 cycles, indicating that the large conjugated structure was of special importance for the high-performance anhydride-based cathodes for NMBs.
 | ||
| Fig. 9 (a) The relationship between the theoretical capacities and conjugated structures of PMDA, NTCDA, and PTCDA. (b) Ex situ Mg 2p, C 1s, and O 1s XPS spectra of PTCDA electrodes. Reproduced with permission.176 Copyright 2021, John Wiley & Sons, Inc. (c) GCD profiles of a two-electrode cell. The Roman numeral V in red represents a fully magnesiated electrode. (d) Ex situ FT-IR spectra and (e) ex situ XRD patterns of PTCDA corresponding to the selected points in (c). P stands for the pristine electrode; PW denotes a pristine electrode soaked in the saturated (4.8 M) Mg(NO3)2 aqueous electrolyte, and GF stands for a glass-fiber separator. (f) Schematic images of the PTCDA crystal lattice showing the coordination of three PTCDA molecules per Mg2+ ion. (g) Schematic images showing the packing of the PTCDA molecules, and how the Mg2+ ions locate within the crystal stacks. Vector fields showing the mean displacement of atoms upon magnesium insertion, depicting a clear rotation of the individual PTCDA molecules in opposite directions. Reproduced with permission.177 Copyright 2017, American Chemical Society. | ||
When different electrolytes are applied for NMBs, the PTCDA cathode exhibits varying electrochemical performance. In the work of Cui et al.,178 the PTCDA cathode coupled with the APC electrolyte achieved an initial capacity of 130 mA h g−1 at 100 mA g−1. However, after 50 cycles, the reversible capacity decreased to 75 mA h g−1, due to PTCDA disintegration in the APC electrolyte. To address this issue, a salt dissolution inhibition approach with LiCl (APC-LiCl electrolyte) was employed. It resulted in an initial capacity of 126 mA h g−1 at 200 mA g−1 for the PTCDA electrode, and the reversible capacity remained 100 mA h g−1 at 200 mA g−1 even after 150 cycles, with a CE of 98%. FTIR and UV-vis spectra confirmed robust chemical structure stability, while XPS spectra revealed the transformation between carbonyl and enolate groups during electrochemical Mg2+ storage. The idea of inhibiting cathode dissolution was extended to other methods by adding soluble salts like KCl and NaCl to the APC electrolyte. These methods also significantly enhance the reversibility of the PTCDA electrode. For example, the PTCDA cathodes coupled with the APC-NaCl and APC-KCl electrolytes could show reversible capacities of 104 and 90 mA h g−1 at 200 mA g−1 after 65 cycles, respectively.
Furthermore, it was found by Rodríguez-Pérez et al. that crystalline PTCDA with a van der Waals structure was particularly suitable for hosting Mg2+ in an aqueous Mg(NO3)2 electrolyte.177 Upon insertion into the PTCDA structure, Mg2+ ions could induce a distinctive squeezing deformation, leading to both compression and expansion of the electrode structure across various crystallographic orientations. The PTCDA electrode exhibited an initial discharge capacity of 136 mA h g−1 at 20 mA g−1 and demonstrated excellent rate capability, achieving 75 mA h g−1 at 500 mA g−1. Using PTCDA as the negative electrode and excess activated carbon as the positive electrode, the first-cycle charge/discharge potential profiles for the assembled NMBs are depicted in Fig. 9c. Ex situ FTIR spectra revealed that the insertion of Mg2+ ions into PTCDA involved an enolization process of at least two carbonyl groups to accommodate one Mg2+ ion (Fig. 9d). Due to steric constraints, the coordination of each Mg2+ ion should be with at least two neighboring PTCDA molecules. Ex situ XRD patterns (Fig. 9e) proved that pure PTCDA had a β-form crystal structure belonging to the monoclinic P21/c space group.138 Notably, the shift of the (011) peak to greater 2θ values, indicating significant constriction along one dimension of the PTCDA structure, suggested structural alteration upon Mg2+ insertion (Fig. 9f). The negative charge of the enolates is delocalized via the stacks due to the π–π stacking architecture of the crystal, allowing a single Mg2+ ion to coordinate with three PTCDA molecules. The rotation of PTCDA molecules in different directions away from the herringbone arrangement toward a more linear configuration is facilitated by reduced carbonyl repulsion and coordination with a Mg2+ ion (Fig. 9g). Vector fields illustrating the mean displacement resulting from Mg2+ insertion make the rotation of the molecules easy to visualize, with individual PTCDA molecules rotating in opposing directions. This discovery of ion storage in aqueous systems may provide critical insights into anhydride-based MMBs using the non-aqueous electrolytes.
Compared with Mg, Ca has a relatively lower stripping/plating potential.179 However, the Ca anode was rarely reported to couple with small molecule cathodes, which could be ascribed to the irreversible Ca stripping and plating. The developed small molecule materials for aqueous/nonaqueous Ca ion storage could be suitable for fabricating promising reversible Ca metal batteries. Benefitting from the weak van der Waals forces, quinone-based molecules, when used in Ca batteries, provide more flexible solid structures with low repulsion for Ca2+ diffusion. As an example, the PTCDA electrode exhibited quite promising calcium storage performances with a highly reversible capacity around ∼158 mA h g−1 at 10 mA g−1 in the saturated Ca(ClO4)2/PC electrolyte. Saturated nonaqueous electrolytes were found to suppress PTCDA dissolution due to the limited availability of free solvent molecules, while also promoting a highly active interfacial reaction with PTCDA.
6.1.1.3. Rhodizonate salts. Sustainable rhodizonate salts, such as lithium rhodizonate (Li2C6O6) and sodium rhodizonate (Na2C6O6), are promising candidates for battery applications due to their four-electron-transfer mechanism. Rhodizonate salts contain the rhodizonate anion, C6O62−, featuring a highly conjugated, aromatic six-membered ring with alternating double bonds and oxygen atoms, contributing to its stability and distinctive coloration. With theoretical capacities of 580 mA h g−1 for Li2C6O6 and 501 mA h g−1 for Na2C6O6, these salts offer attractive potential.180–185 Tian et al. investigated three rhodizonate salts as cathode materials for NMBs in the APC-LiCl electrolyte, including bulk phase Na2C6O6, as-precipitated Na2C6O6, and nanocrystalline Li2C6O6.186 Nanocrystalline Na2C6O6 exhibited an initial discharge capacity of approximately 450 mA h g−1 at 50 mA g−1, retaining a reversible capacity of up to 200 mA h g−1 after 50 cycles. Despite the fact that Li+ was the primary charge carrier for the Na2C6O6 cathode reaction, the Mg–O–C residue resulting from Mg co-storage contributed to mitigating the exfoliation of C6O6 layers, enhancing cycling performance. The Mg//Na2C6O6 cell retained a capacity of 200 mA h g−1 even at a high current density of 1 A g−1, retaining a reversible capacity of 125 mA h g−1 after 600 cycles. The output energy and power densities of 525 W h kg−1 and 4490 W kg−1 surpassed those of conventional high-voltage inorganic cathodes.
Our group pioneered the exploration of amine molecules for NABs in the AlCl3/EMIMCl electrolyte. We investigated triphenylamine (N1), 1,4-bis(diphenylamino)benzene (N2), 1,3,5-tris(diphenylamino)benzene (N3), and 4,4′,4′′-tris(diphenylamino)triphenylamine (N4), which have different numbers of nitrogen centers and theoretical capacities of 109, 130, 139, and 144 mA h g−1, respectively (Fig. 10a).116 The N1, N2, and N3 cathodes demonstrated inferior electrochemical performance compared with the N4 cathode, primarily due to the severe dissolution of the active material into the electrolyte. In contrast, an activation process was recognized for N4, resulting in significant capacity enhancement during the initial 25 cycles (Fig. 10b). The capacities stabilized at 135 mA h g−1 at 0.1 A g−1 with a CE of 99.9%. High-molecular-weight N4 oligomers (up to 5220 with 7 repeating units) were detected for the cycled electrode (Fig. 10c), indicating that the N4 monomer underwent in situ oligomerization during cycling to form (N4)n. This oligomerization could enhance the conductivity of the cathode and mitigate the electrode dissolution. Since the oligomerization of N4 was irreversible, the subsequent electrochemical reaction of the (N4)n electrode was proposed to occur between the neutral (N4)n and fully charged (N44+)n states, involving quinone diiminium and the benzidine dication during the multi-electron transfer process (Fig. 10d). The constructed Al-(N4)n cell after conditioning cycles presented a reversible capacity of 116 mA h g−1 at 1 A g−1 after 4000 cycles.
 | ||
| Fig. 10 (a) Working principle of the Al–amine battery and the proposed aromatic amine molecules. Ph represents phenyl groups. (b) GCD curves of the Al–N4 cell at different cycles. (c) Matrix-assisted laser desorption/ionization time of flight mass spectrum of a cycled N4 electrode. (d) The proposed energy storage process of (N4)n. Anion is omitted in the reaction.116 Copyright 2022, John Wiley & Sons, Inc. | ||
N). The nitrogen atom in these compounds is typically bonded to either a hydrogen atom or an organic substituent. Imine-based small molecules can exhibit both aromatic and non-aromatic characteristics, and their high electronegativity imparts desirable redox activity. π-Conjugated aromatic molecules containing imine groups can function as neutral organic bases, capable of chelating Lewis acids through coordination reactions.187
Tie et al. demonstrated the use of HATN as a cathode material in AZBs with ZnSO4 electrolyte.188 HATN was obtained by reacting hexaketocyclohexane octahydrate with o-phenylenediamine in glacial acetic acid. As illustrated in Fig. 11a, the positive molecular ESP values, which indicate nucleophilic centers, are highlighted in red on the van der Waals surface of the HATN molecule, while the negative MESP values, representing the electrophilic centers, are shown in blue.189 The LUMO and HOMO energies of HATN were calculated to be −2.78 and −6.46 eV, respectively, resulting in an Eg of 3.68 eV (Fig. 11b). This Eg value is lower than those of most organic molecules, suggesting that HATN has superior inherent electronic conductivity.117 Furthermore, the pyrazine nitrogen atoms in HATN act as active sites for H+ ion uptake, as determined from the minimum energy principle (Fig. 11c). The CV curves of the Zn//HATN cell displayed three pairs of redox peaks, corresponding to the reversible structural transitions from HATN to HATN-2H, HATN-4H, and HATN-6H, respectively (Fig. 11d). The Zn//HATN cell demonstrated exceptional rate performance and high capacity, attributed to its π-conjugated aromatic structure and rapid H+ coordination/incoordination reaction kinetics (Fig. 11e). At a high current density of 20 A g−1, the Zn//HATN cell maintained a capacity of 123 mA h g−1, retaining 33.2% of the capacity observed at 0.1 A g−1. After 150 cycles, the specific capacity could be restored to 320 mA h g−1 when the current density was reduced back to 0.1 A g−1. Additionally, the high-performance Zn//HATN cell exhibited reversible capacities of 140 mA h g−1 at 5 A g−1 after 5000 cycles. These findings suggest that proton insertion chemistry holds significant promise for improving the electrochemical performance of aqueous Zn-organic batteries.
 | ||
| Fig. 11 (a) Calculated ESP distribution of the HATN molecule. (b) Optimized structure and calculated HOMO/LUMO energy of the HATN molecule. (c) Calculated structural evolution and the protonation pathway during the discharging process. (d) The CV curves of aqueous Zn//HATN batteries (0.01 mV s−1). (e) Rate capabilities of Zn//HATN batteries at various current densities. Reproduced with permission.188 Copyright 2020, John Wiley & Sons, Inc. (f) Calculated HOMO/LUMO energy and the energy gap of HATN and HATN-3CN molecules. (g) Cycle performance of HATN-3CN cathodes at 0.3 A g−1. (h) Ex situ1H solid-state NMR spectra of the HATN-3CN electrode at marked points. Reproduced with permission.111 Copyright 2021, Elsevier B.V. (i) LUMO/HOMO energy levels of HATN and HMHATN, as well as optimized structures obtained using the DFT method. (j) Cycle stability of the HMHATN electrode at 2 A g−1. (k) GCD curves at the bending degree of 180° and the inset is the corresponding optical image of the flexible aqueous battery. Reproduced with permission.190 Copyright 2022, Elsevier B.V. | ||
To further increase the operation voltage of HATN cathodes for MMBs, Ye et al. successfully incorporated strong electron-withdrawing and conjugating –CN groups into HATN, creating a HATN-3CN cathode material for AZBs in ZnSO4 electrolyte through a simple condensation reaction.97,111,191 As shown in Fig. 11f, the theoretical operating voltage of HATN-3CN increased as the LUMO energy decreased. Additionally, by reducing the Eg between the HOMO and LUMO, electronic conductivity was enhanced, facilitating faster electron transport and reaction rates for improved rate performance. The HATN-3CN cathode, featuring the rich pyrazine moiety, exhibited a notable capacity of 320 mA h g−1 at 0.05 A g−1. Furthermore, HATN-3CN displayed remarkable cycling stability with 99.2% capacity sustained after 150 cycles at 0.3 A g−1 (Fig. 11g). Moreover, the Zn//HATN-3CN cell demonstrated impressive cycling stability, retaining a reversible capacity of around 280 mA h g−1 at 5 A g−1 after 5800 cycles. Mechanistic studies revealed the fluctuation of the –CN redox center in conjunction with H+/Zn2+ co-participation during charging/discharging, which was verified by the ex situ1H solid-state NMR spectral results (Fig. 11h), shedding light on the relationship between the electrochemical process and molecular orbitals. Thus, the introduction of the –CN substituent to modify the molecular orbitals resulted in improved electrochemical performance of resultant batteries.
In addition to the –CN group, hexafluorohexaazatrinaphthylene (HFHATN) was synthesized by introducing strong electron-withdrawing F atoms into HATN. It was investigated as the cathode material for AZBs in the aqueous ZnSO4 electrolyte.192 The LUMO energy level of HFHATN was −3.03 eV, lower than that of HATN (−2.13 eV), indicating the higher electron affinity of HFHATN. Consequently, the Zn//HFHATN cell displayed a capacity of 461 mA h g−1 at 0.04 A g−1 after 10 cycles. The HFHATN cathodes also showed a capacity of 191 mA h g−1 at 5 A g−1 after 1000 cycles. This excellent electrochemical performance was attributed to its multiple redox active sites and insolubility in electrolytes and the F atom introduction. The F atom decreases the Eg between the LUMO and HOMO, improving electrical conductivity and enhancing electron transfer speed on the cathode.
Apart from electron-withdrawing groups like –CN and –F, Sun et al. designed and synthesized hexamethoxy hexaazatrinaphthylene (HMHATN) by introducing electron-donating –OCH3 groups as cathode materials for AZBs.190 Theoretical calculations presented in Fig. 11i indicated that HMHATN showed a higher LUMO level energy than HATN, revealing that HATN had a higher reduction potential and greater electron affinity than HMHATN. Besides, the introduction of –OCH3 decreased the Eg for HMHATN, improving its electrochemical performance. The HMHATN and HATN electrodes delivered discharge capacities of 196 and 233 mA h g−1 at 2 A g−1 and retained capacities of 191 and 229 mA h g−1 after 1000 cycles, respectively (Fig. 11j). The capacity retention rates for HMHATN and HATN electrodes were 97.4% and 98.3%, respectively, with coulombic efficiencies close to 100%. Thus, while the introduction of electron-donating groups like –OCH3 did not significantly alter cycling stability, it reduced actual capacities due to increased molecular weight. In addition, the HMHATN and HATN electrodes could also be used to assemble flexible Zn batteries to lighten the LED light (Fig. 11k), showing good foldability and flexibility.
The electrochemical performances of phenazine (PNZ) and 2,3-diamine-phenazine (DAP) cathodes were also investigated for AZBs.193 In the aqueous ZnSO4 electrolyte, the Zn//PNZ cell demonstrated an initial capacity of 204 mA h g−1 at 0.1 A g−1, retaining 89% of the initial capacity after 100 cycles. In contrast, the Zn//DAP cell demonstrated a much lower reversible capacity of 28 mA h g−1 at 0.1 A g−1 after 100 cycles. This difference was attributed to the hydrophilic NH2 groups in DAP, which increase its solubility in the aqueous electrolyte via hydrogen bonding, leading to poor cycling stability. The hydrophilic nature of DAP facilitated its dissolution in the electrolyte, while PNZ, due to steric hindrances from nearby hydrogen atoms, was less prone to hydration and dissolution. Gao et al. introduced a novel organic cathode material, 5,7,12,14-tetraaza-6,13-pentacenequinone (TAPQ), for AZBs in the ZnSO4 electrolyte, integrating quinone and pyrazine structures.194 Synthesized in two steps using 2,5-dihydroxy-1,4-benzoquinone and o-phenylenediamine, TAPQ cathodes exhibited a reversible capacity of 443 mA h g−1 at 50 mA g−1, representing 86% of its theoretical capacity (515 mA h g−1) based on a six-electron reaction. Operating within a balanced voltage range of 0.5–1.6 V vs. Zn/Zn2+, the Zn//TAPQ cell showed a reversible capacity of 270 mA h g−1 at 50 mA g−1 after 250 cycles, with an average CE of 99.96%, showing great potential in practical applications.
Nevertheless, imine-based organic electrodes face challenges due to their low intrinsic conductivity and solubility in aqueous Zn electrolytes. To address these issues, creating high-symmetry molecules can lower solubility and reduce the dipole moment. Additionally, extending π conjugation by constructing stiff aromatic rings can enhance ordered organization and π–π interactions among organic molecules. Niu et al. synthesized a high-symmetry, Y-shaped molecule with a large π-conjugated plane, triphenazino[2,3-b] (1,4,5,8,9,12-hexaazotriphenylene) (TPHATP), through a facile and low-cost route for use in AZBs.195 The Zn//TPHATP cell showed an initial capacity of 318 mA h g−1 at 0.1 A g−1, retaining a reversible capacity of 254 mA h g−1 after 170 cycles. Furthermore, the TPHATP cathode delivered a capacity of 109 mA h g−1 at 10 A g−1 after 5000 cycles, with 97.44% capacity retention and a decay rate of 0.000512% per cycle. The study confirmed that the energy storage mechanism in TPHATP cathodes was primarily H+-dominant, excluding the involvement of Zn2+. This exclusion was attributed to the rapid hydrolysis of Zn-nitride-type discharge products.
| Molecule | Electrolyte | Battery type | Average discharge potential | Specific capacity | Cycle life | Rate performance | Ref. |
|---|---|---|---|---|---|---|---|
| 2,5-Dimethoxy-1,4-benzoquinone (DMBQ) | 0.5 M Mg(ClO4)2/γ-butyrolactone | Nonaqueous Mg battery | 1.1 and 0.8 V vs. Mg/Mg2+ | 250 mA h g−1 at 20 mA g−1 | 210 mA h g−1 at 20 mA g−1 (the 5th cycle) | 146 | |
| 0.5 M Mg(TFSA)2/SL | 0.8 and 0.4 V vs. Mg/Mg2+ | 78 mA h g−1 at 10 mA g−1 | 60 mA h g−1 at 10 mA g−1 (the 10th cycle) | 147 | |||
| 0.5 M Mg(TFSA)2/EiPS | 0.8 and 0.4 V vs. Mg/Mg2+ | 162 mA h g−1 at 10 mA g−1 | 60 mA h g−1 at 10 mA g−1 (the 10th cycle) | ||||
| 0.5 M Mg(TFSA)2/DnPS | 0.8 and 0.4 V vs. Mg/Mg2+ | 126 mA h g−1 at 10 mA g−1 | 30 mA h g−1 at 20 mA g−1 (the 10th cycle) | ||||
| 0.5 M Mg(TFSI)2–MgCl2/DME | 2 V vs. Mg/Mg2+ | 226 mA h g−1 at 64 mA g−1 | 74 mA h g−1 at 64 mA g−1 (the 30th cycle) | 150 mA h g−1 at 320 mA g−1 | 145 | ||
| Anthraquinone (AQ) | 0.6 M Mg(TFSI)2–2MgCl2/DME | 0.65 and 0.25 V vs. Mg/Mg2+ | 152 mA h g−1 at 100 mA g−1 | 10 mA h g−1 at 100 mA g−1 (the 30th cycle) | 200 | ||
| 1,4-Naphthoquinone (NQ) | 0.6 M Mg(TFSI)2–2MgCl2/DME | 0.65 and 0.25 V vs. Mg/Mg2+ | 138 mA h g−1 at 100 mA g−1 | 12 mA h g−1 at 100 mA g−1 (the 40th cycle) | |||
| 1,4-Benzoquinone (BQ) | 0.6 M Mg(TFSI)2–2MgCl2/DME | 0.65 and 0.25 V vs. Mg/Mg2+ | 130 mA h g−1 at 100 mA g−1 | 50 mA h g−1 at 100 mA g−1 (the 40th cycle) | |||
| Vat-Orange-11 | APC | 1.5 V vs. Mg/Mg2+ | 260 mA h g−1 at 100 mA g−1 | 80 mA h g−1 at 100 mA g−1 (the 100th cycle) | 26 and 40 mA h g−1 at 2 and 0.5 A g−1, respectively | 152 | |
| Mg(HMDS)2–MgCl2/THF and PP13TFSI | 1.6 V vs. Mg/Mg2+ | 81 mA h g−1 at 100 mA g−1 | 80 mA h g−1 at 100 mA g−1 (the 100th cycle), 25 mA h g−1 at 500 mA g−1 (the 500th cycle) | 15 mA h g−1 at 1 A g−1 | |||
| Pyrene-4,5,9,10-tetraone (PTO) | 0.5 mol kg−1 MMC | Nonaqueous Mg battery | 2.1 V vs. Mg/Mg2+ | 307 mA h g−1 at 408 mA g−1 | 230 mA h g−1 at 2040 mA g−1 (the 500th cycle) | 291, 278, and 210 mA h g−1 at 2.04, 8.16, and 20.4 A g−1, respectively | 201 |
| 2 M ZnSO4/H2O | Aqueous Zn battery | 0.8 V vs. Zn/Zn2+ | 336 mA h g−1 at 40 mA g−1 | 145 mA h g−1 at 3000 mA g−1 (1000th cycle) | 300, 213, and 162 mA h g−1 at 0.1, 1, and 5 A g−1, respectively | 171 | |
| 3.5 M Mg(ClO4)2 + 1 M Zn(ClO4)2 | 0.8 V vs. Zn/Zn2+ | 101 mA h g−1 at 200 mA g−1 | 100 mA h g−1 at 0.2 A g−1 (the 100th cycle) | 71 mA h g−1 at 1.2 mA g−1 | |||
| PQ triangle (PQ-Δ) | AlCl3/EMIMCl | Nonaqueous Al battery | 1.5 V vs. Al/Al3+ | 81 mA h g−1 at 200 mA g−1 | 82 mA h g−1 at 200 mA g−1 (the 200th cycle), 53 mA h g−1 at 2000 mA g−1 (the 5000th cycle) | 110 and 70 mA h g−1 at 0.1 and 10 A g−1, respectively | 154 |
| 3 M Zn(CFSO3)2/H2O | Aqueous Zn battery | 0.78 V vs. Zn/Zn2+ | 225 mA h g−1 at 30 mA g−1 | 210 mA h g−1 at 150 mA g−1 (500th cycle) | 173 | ||
| 2,3,5,6-Tetraphthali-mido-1,4-benzoquinone (TPB) | AlCl3/urea | Nonaqueous Al battery | 2, 1.5, 1 V vs. Al/Al3+ | 178 mA h g−1 at 100 mA g−1 | 175 mA h g−1 at 100 mA g−1 (the 250th cycle) | 50 mA h g−1 at 1 A g−1 | 160 |
| 9,10-Anthraquinone | 1 M Al2(SO4)3/H2O | - | 0.4 V vs. SCE | 87 mA h g−1 at 1 A g−1 | 168 mA h g−1 at 1 A g−1 (the 500th cycle) | 202 | |
| AlCl3/EMIMCl | Nonaqueous Al battery | 1.1 V vs. SCE | 215 mA h g−1 at 100 mA g−1 | 192 mA h g−1 at 100 mA g−1 (the 200th cycle) | 229, 219, and 166 mA h g−1 at 20, 50, and 500 mA g−1, respectively | 161 | |
| Macrocyclic calix[4]quinone (C4Q) | 1 M Al(OTF)3/H2O | Aqueous Al battery | 1.1 V vs. Al/Al3+ | 400 mA h g−1 at 100 mA g−1 | 269 mA h g−1 at 200 mA g−1 (the 50th cycle) | 371, 334, and 300 mA h g−1 at 0.2, 0.4, 0.8 A g−1, respectively | 203 |
| 3 M Zn(CFSO3)2/H2O | Aqueous Zn battery | 1.0 V vs. Zn/Zn2+ | 335 mA h g−1 at 20 mA g−1 | 215 mA h g−1 at 0.5 A g−1 (the 100th cycle), | 333, 220, and 172 mA h g−1 at 0.05, 0.25, 1 A g−1, respectively | 169 | |
| 5,7,12,14-Pentacenetetrone (PT) | CaCl2/H2O | — | −0.5 V vs. Ag/AgCl | 150 mA h g−1 at 5 A g−1 | 85 mA h g−1 at 30000 mA g−1 (the 3000th cycle) |
142, 115, and 96 mA h g−1 at 10, 50, and 90 A g−1, respectively | 162 |
| Tetrachloro-1,4-benzoquinone (p-chloranil) | 1 M Zn(OTf)2/H2O | Aqueous Zn battery | 1.1 V vs. Zn/Zn2+ | 170 mA h g−1 at 43.4 mA g−1 | 90 mA h g−1 at 43.4 mA g−1 (the 100th cycle), 83 mA h g−1 at 217 mA g−1 (the 200th cycle) | 118 mA h g−1 at 217 mA g−1 | 168 |
| Dibenzo[b,i]thianthrene-5,7,12,14-tetraone (DTT) | 2 M Zn(SO4)2/H2O | 0.8 V vs. Zn/Zn2+ | 211 mA h g−1 at 50 mA g−1 | 188 mA h g−1 at 0.1 A g−1 (150th cycle), 80 mA h g−1 at 2A g−1 (23000th cycle) |
97 mA h g−1 at 2 A g−1 | 174 | |
| Pyromellitic dianhydride (PMDA) | 0.2 M Mg(HMDS)2–MgCl2–2AlCl3/tetraglyme | Nonaqueous Mg battery | 0.8 V vs. Mg/Mg2+ | 110 mA h g−1 at 50 mA g−1 | 45 mA h g−1 at 50 mA g−1 (the 100th cycle) | 176 | |
| 1,4,5,8-Naphthalenetetracarboxylic dianhydride (NTCDA) | 0.2 M Mg(HMDS)2–MgCl2–2AlCl3/tetraglyme | 0.8 V vs. Mg/Mg2+ | 130 mA h g−1 at 50 mA g−1 | 90 mA h g−1 at 50 mA g−1 (the 100th cycle) | |||
| 3,4,9,10-Perylenetetracarboxylic dianhydride (PTCDA) | 0.2 M Mg(HMDS)2–MgCl2–2AlCl3/tetraglyme | Nonaqueous Mg battery | 1.6 V vs. Mg/Mg2+ | 200 mA h g−1 at 50 mA g−1 | 160 mA h g−1 at 50 mA g−1 (the 100th cycle) | 152, 124, and 73 mA h g−1 at 0.1, 0.2 and 0.5 mA g−1, respectively | |
| 4.8 M Mg(NO3)2 | — | −0.2 V vs. Zn/Zn2+ | 136 mA h g−1 at 20 mA g−1 | 75 mA h g−1 at 0.5 A g−1 | 177 | ||
| Na2C6O6 | 0.25 M APC + 1 M LiCl | Nonaqueous Mg battery | 1.0 V vs. Mg/Mg2+ | 250 mA h g−1 at 50 mA g−1 | 200 mA h g−1 at 50 mA g−1 (the 50th cycle) | 200, 140, and 100 mA h g−1 at 1, 2.5, and 5 A g−1, respectively | 186 |
| Li2C6O6 | 0.25 M APC + 1 M LiCl | 1.0 V vs. Mg/Mg2+ | 240 mA h g−1 at 50 mA g−1 | 150 mA h g−1 at 50 mA g−1 (the 50th cycle) | 25 mA h g−1 at 5 A g−1 | ||
| Diquinoxalino [2,3-a:2′,3′-c] phenazine (HATN) | 2 M ZnSO4 | Nonaqueous Zn battery | 0.75 V vs. Zn/Zn2+ | 405 mA h g−1 at 100 mA g−1 | 140 mA h g−1 at 5 A g−1 (the 5000th cycle) | 123 mA h g−1 at 20 A g−1 | 188 |
| Diquinoxalino [2,3-a:2′,3′-c]phenazine-2,8,14-tricarbonitrile (HATN-3CN) | 2 M ZnSO4 | 0.87 and 0.58 V vs. Zn/Zn2+ | 320 mA h g−1 at 50 mA g−1 | 318 mA h g−1 at 0.3 A g−1 (the 150th cycle), 270 mA h g−1 at 5 A g−1 (the 5800th cycle) | 190 mA h g−1 at 20 A g−1 | 111 | |
| 5,7,12,14-Tetraaza-6,13-pentacenequinone (TAPQ) | 1 M ZnSO4 | 0.9 V vs. Zn/Zn2+ | 443 mA h g−1 at 50 mA g−1 | 282 mA h g−1 at 50 mA g−1 (the 100th cycle) | 260, 246, 222, and 182 mA h g−1 at 0.2, 0.5, 1, and 2 A g−1, respectively | 194 | |
| Phenazine (PNZ) | 2 M ZnSO4 | 0.85 V vs. Zn/Zn2+ | 265 mA h g−1 at 20 mA g−1 | 204 mA h g−1 at 100 mA g−1 (the 100th cycle) | 204 | ||
| Tetracyanoethylene (TCNE) | AlCl3/EMIMCl | Nonaqueous Al battery | 58 mA h g−1 at 500 mA g−1 | 35 mA h g−1 at 500 mA g−1 (the 100th cycle) | 199 | ||
| Tetracyanoquinodimethane (TCNQ) | AlCl3/EMIMCl | 1.75 and 1 V vs. Zn/Zn2+ | 180 mA h g−1 at 500 mA g−1 | 115 mA h g−1 at 500 mA g−1 (the 2000th cycle) | |||
| Tetrakis(4-cyanophenyl)methane (TCPM) | AlCl3/EMIMCl | 82 mA h g−1 at 500 mA g−1 | 42 mA h g−1 at 500 mA g−1 (the 100th cycle) | ||||
6.2. Amorphous polymers
While small-molecule OEMs offer significant advantages over their inorganic counterparts, they often suffer from rapid capacity loss in MMBs due to their high solubility or the unstable redox intermediates in MMB electrolytes. Polymerization of redox-active monomers into macromolecular chains or networks has emerged as an effective strategy to stabilize solid-state OEMs. The strong intermolecular interactions and high molecular weight of redox-active polymers contribute to their improved stability, mitigating the dissolution issue of OEMs during repeated electrochemical processes. Additionally, certain redox-active polymers with extended π-conjugated chains and abundant mobile electrons exhibit enhanced electrical conductivity, specific capacity, superior rate performance, and energy density compared to small-molecule OEMs. According to the incorporated redox moieties, these electroactive polymers can be classified into six categories: carbonyl-, imine-, amine-, sulfur-, nitroxide radical-containing polymers and conductive polymers (Fig. 12). In this section, we will delve into their distinctive features, charge storage mechanisms, and electrochemical performances in detail.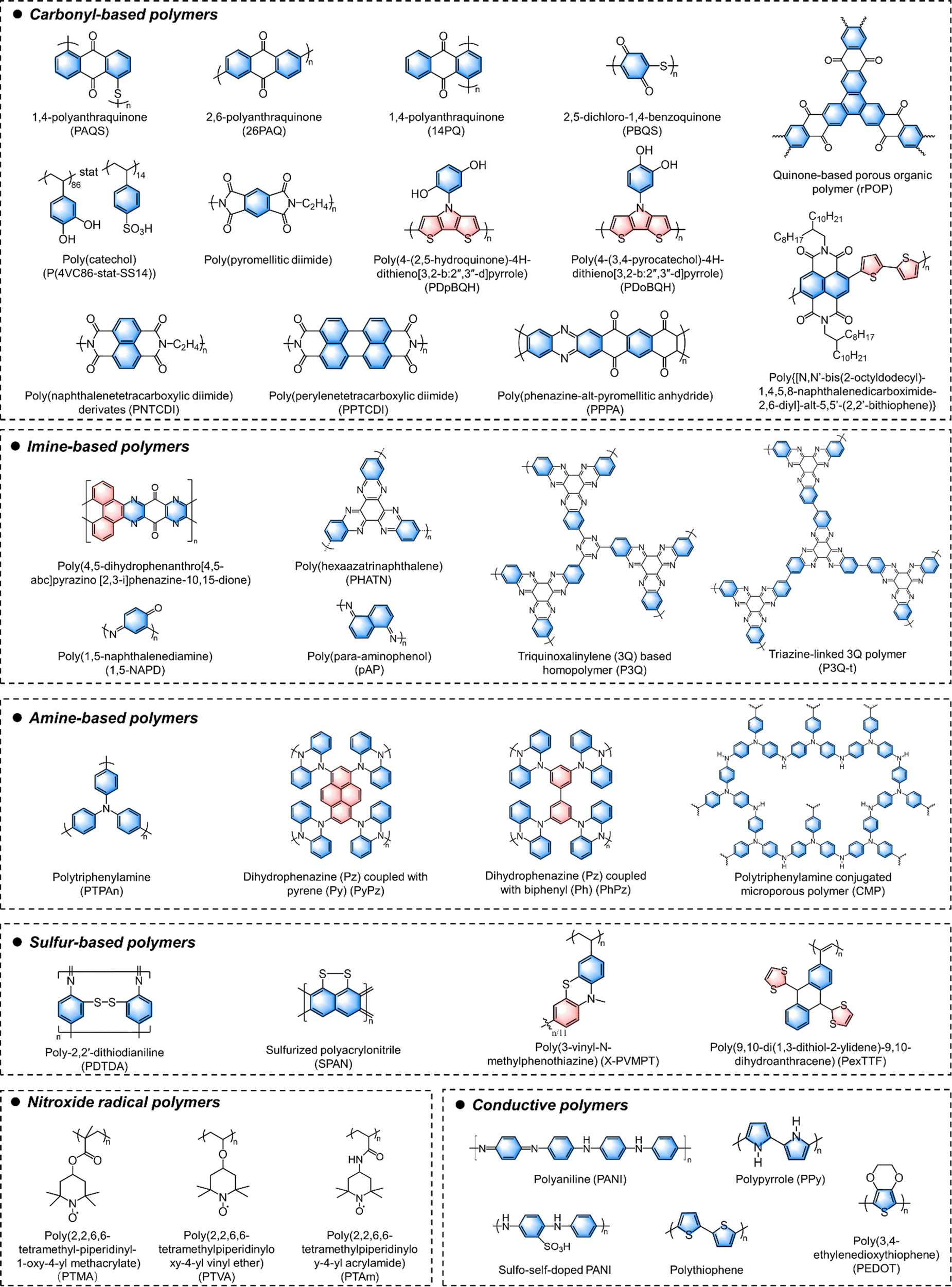 | ||
| Fig. 12 Molecular structures of redox-active amorphous polymers reported for MMBs. | ||
While electrochemical polymer electrodes can effectively address the dissolution issue faced by their monomer counterparts, their actual electrochemical performance still falls short in terms of capacity and rate capability. This challenge arises from the intrinsic structural feature of polymers with twisted molecular chains, which exhibit poor molecular planarity, leading to sluggish ion diffusion. Enhancing the diffusion kinetics of multivalent ions is crucial to overcome the performance bottleneck of quinone-based polymers but remains a significant challenge. In this regard, Hu and colleagues designed PPPA with an extended planar π-conjugated framework for AZBs via a one-step polycondensation reaction with electroactive phenazine and pyromellitic anhydride (PMDA) monomers.121 The enlarged π-conjugated structure of PPPA not only provided abundant open 2D channels for fast Zn2+ transport but also supplied plentiful delocalized π-electrons, synergistically facilitating ion diffusion during electrochemical redox reactions. Based on a reversible Zn2+-coordination mechanism, the prepared PPPA electrode delivered a discharge capacity of 210.2 mA h g−1 at 50 mA g−1 and lifespan stability over 20000 cycles with 70.6% capacity retention at 5 A g−1 in a 2.0 M Zn(OTf)2 aqueous electrolyte (Fig. 13a and b). Impressively, an ultrahigh Zn2+ diffusion coefficient of 1.2 × 10−7 cm2 s−1 was achieved, surpassing conventional cathode materials and organic compounds for AZBs. As revealed by DFT calculations (Fig. 13c), the Eg between the HOMO and LUMO gradually reduced with an increasing π-conjugated plane in the PPPA samples, effectively accelerating intramolecular electron transfer during the electrochemical process and thereby improving overall ion diffusion kinetics and rate performance. This work provides an effective and rational design of a planar π-conjugated structure to realize ultrahigh ion transport kinetics for polymer electrodes.
 | ||
| Fig. 13 (a) GCD curves at 50 mA g−1 and (b) rate-performance of the PPPA cathode. (c) HOMO and LUMO energy levels of different PPPA unit configurations by DFT simulation. Reproduced with permission.121 Copyright 2022 John Wiley & Sons Inc. (d) Illustration of PI@CNT synthesis. (e) Comparison of rate performance of the PI@CNT composite as Li, Mg, and Al ion cathodes. (f) Reduction potentials and the corresponding complexes obtained from quantum chemistry calculations for Li-PI and Mg-PI systems. Copyright 2022 John Wiley & Sons Inc.213 Copyright 2022 John Wiley & Sons Inc. | ||
Compared with para-quinone polymers, ortho-carbonyl polymers exhibit several advantageous properties. These include higher redox potential, improved reversibility, and reduced catecholates (1,2-positions), which create a favorable environment for the reversible coordination complex formation with multivalent cations. This phenomenon is reminiscent of catechol-cation interactions observed in various biological processes, such as biological energy transduction and Fe3+ uptake in microbial siderophores.214 Recently, Tang and colleagues grafted hydroquinone or pyrocatechol redox pendants onto a conductive conjugated ladder-type polymer backbone.215 The synthesized poly(4-(2,5-hydroquinone)-4H-dithieno[3,2-b:2′′,3′′-d]pyrrole) (PDpBQH) and poly(4-(3,4-pyrocatechol)-4H-dithieno[3,2-b:2′′,3′′-d]pyrrole) (PDoBQH) were applied as cathodes for AZBs in 2 M ZnSO4 aqueous electrolytes. The PDpBQH electrode achieved a specific capacity of 120 mA h g−1 at 0.1 A g−1 and a capacity retention of 52.5% at 5 A g−1. Besides, a high capacity retention over 79% was observed for the PDpBQH electrode after 500 electrochemical cycles. In contrast, the pyrocatechol-derived PDoBQ electrode showed much worse redox stability with a capacity retention of only 53% after 500 cycles. This cyclability difference was attributed to the good molecular planarity and uniform ESP distribution of the hydroquinone-derived PDpBQ cathode, which allowed for fast charge transfer and outstanding redox reaction reversibility for Zn2+ hosting.
In addition, Marcilla and coworkers discovered that the redox-active polymers bearing catechol pendant groups could serve as universal hosts for reversibly storing numerous cations, including H+, Li+, Na+, K+, Ca2+, Mg2+, Zn2+, Ce3+, and Al3+.216 The overall electrochemical performance of the electrode was determined by the strength of the catechol–cation interactions. Stronger complexes (monovalent ions < divalent ions < trivalent ions) typically exhibited higher redox potentials and slower diffusion kinetics. These results indicated that the electrochemical properties of catechol polymers could be tailored through rational macromolecular structure engineering. Furthermore, the same group demonstrated a poly(catechol) redox copolymer (P(4VC86-stat-SS14)) as the cathode for AZBs.217 In a 4 M Zn(TFSI)2 aqueous solution, the assembled Zn//P(4VC86-stat-SS14) cell simultaneously delivered remarkable capacities of 324 and 98 mA h g−1 at 1 and 450C, respectively, along with extremely high cyclability, exhibiting only a 0.00035% capacity fading rate per cycle over 48000 cycles at 30C. Interestingly, this battery was able to operate at an extremely low temperature (−35 °C) while still maintaining good capacity values (178 mA h g−1) owing to the low glass-transition temperature of the polymer chains.
Polyimides (PIs), synthesized via the polycondensation of aromatic dianhydrides and diamines, represent another group of carbonyl-containing polymers for MMBs that are promising for large-scale production and synthesis. The repeating imide units, consisting of two carbonyl groups connected by a nitrogen atom, enable polyimides to undergo at least 2-electron-transfer redox reactions and exhibit n-type ion storage behaviors. Typical examples of polyimides for MMBs include pyromellitic diimide (PMDI)-based, naphthalenetetracarboxylic diimide (NTCDI)-based, and perylenetetracarboxylic diimide (PTCDI)-based polymer electrodes.26,218–220 In 2018, Wang and colleagues reported a poly(pyromellitic diimide)@CNT (PPMDI@CNT) cathode, which demonstrated good versatility for storage of different ions, including Li+, Mg2+, and Al3+ (Fig. 13d).213 Benefiting from the ion-coordination charge storage mechanism, the cathode could reversibly store these charge carriers at a fast rate. Specifically, the PPMDI@CNT cathode demonstrated highly reversible capacities of 180, 130, and 110 mA h g−1 at 0.15 A g−1 for Li+, Mg2+, and Al3+, respectively. Moreover, full cells assembled by coupling PPMDI@CNT cathodes and metal anodes delivered fast charging capability (>3 A g−1) and long cycle life (>10000 cycles) (Fig. 13e and f). These results suggest that this low-cost polyimide electrode is a promising universal cathode for different types of batteries. Moreover, Lu and coworkers reported a hybrid poly(perylenetetracarboxylic diimide)/ZIF-8 (PPTCDI/MOF) material as a cathode material in NABs.221 This hybrid material delivered a capacity of 83 mA h g−1 at 1 A g−1 with nearly 100% CE and remained stable after 1800 cycles. The study revealed that cationic chloroaluminates (specifically AlCl2+) interacted with reduced carbonyl bonds in PPTCDI during discharge. This insight helps to better understand the working mechanism of PPTCDI in complex NABs with multiple possible charge carriers (i.e., [AlClx]3−x).
Polyimide electrodes have also been explored for Ca2+ and Zn2+ storage. Yao and colleagues reported a poly(naphthalenetetracarboxiimide) (PNTCDI) electrode for Ca2+ storage.222 The PNTCDI electrode displayed a high capacity of 160 mA h g−1 through a two-step and two-electron enolization reaction for Ca2+ storage, exhibiting excellent capacity retention (88%) and CE (99%) after 1000 cycles at 0.4 A g−1. Recently, Wang and colleagues studied the electrochemical performances of three types of polyimides for Zn2+ storage, including PPMDI, PNTCDI, and PTCDI derivatives.223 Among the three polyimides, the PPTCDI-based electrode demonstrated remarkable performance in a 1.5 M Zn(OTf)2 electrolyte, delivering ultrahigh loading (≈50 mg cm−2), theoretical-close capacity (128 mA h g−1 at 50 mA g−1), high CE (≈100%), and exceptional stability for up to one million cycles. All these representative examples imply that redox-active polyimide-derived electrodes with appropriate structural design show decent potentials for multivalent ion accommodation, especially considering their good electronic conductivity, large π-conjugated structure, and superior electrochemical stability.
N–) in redox-active polymer chains exhibit limited redox activity and poor redox reversibility due to the instability of radical intermediates generated during electrochemical reduction. To address this issue, functional pyridine, pyrazine, and triazine groups, which possess stable aromatic structures, are used as redox-active moieties for ion storage. These aromatic rings containing imine groups can delocalize electrons and stabilize the formed radical intermediates, ensuring their stability during repeated reduction and oxidation cycles. HATN is such a representative aromatic center, which is characterized by an electron-deficient, rigid, and planar aromatic discotic system with six imine functional groups. Wang and colleagues discovered that poly(hexaazatrinaphthalene) (PHATN) with abundant reversible multi-electron-deficient pyrazine sites in the main chains can serve as a universal high-rate and stable cathode material for storing monovalent and multivalent metal ions.224 The PHATN was synthesized by a condensation reaction between cyclohexanehexone and amines. In Na-ion batteries, PHATN delivered a reversible capacity of 220 mA h g−1 at 50 mA g−1, and maintained 100 mA h g−1 after 50000 cycles at 10 A g−1. Taking advantages of superfast de-/insertion kinetics through large conjugated polymer chains, this PHATN electrode is also qualified for storing divalent Mg2+ and trivalent Al3+ ions. In NMBs, the PHATN electrodes can show a reversible capacity of 110 mA h g−1 after 200 cycles and a good rate performance of 60 mA h g−1 even at a high current density of 10C. In rechargeable Al batteries, the PHATN electrodes showed a capacity of 92 mA h g−1 after 100 cycles. Moreover, the configuration of redox-active polymers also influences the ion storage capacities and ion diffusion kinetics. This statement can be well supported by a recent study conducted by Tang and colleagues, who designed two types of polymers based on triquinoxalinylene (3Q): a homopolymer (P3Q) and a triazine-linked 3Q polymer (P3Q-t).225 They observed that the P3Q polymer with poor planarity could accommodate both Zn2+ and H+, whereas P3Q-t selectively coordinated with Zn2+ only. Furthermore, the P3Q-t electrode exhibited faster reaction dynamics and lower Zn2+ transfer resistance, attributed to its highly conjugated planarity and electronegative fused-ring pathways resulting from intermolecular and intramolecular effects. As a result, P3Q-t demonstrated a high capacity of 237 mA h g−1 at 0.3 A g−1 and retained 45% of its capacity at 15 A g−1, with an average capacity loss of only 19% over 1500 cycles. These findings underscore the potential of pyrazine-based polymer electrodes for developing environmentally friendly, high-energy-density, fast, and ultra-stable rechargeable batteries.
In addition to the aromatic pyrazine-based polymers mentioned above, the fused imine motifs within polymer chains are also favorable for enhancing the overall electrochemical performance. Luo and colleagues explored a heteroatom π-conjugated polymer featuring synergistic CO and CN active centers, poly(4,5-dihydrophenanthro[4,5-abc]pyrazino [2,3-i]phenazine-10,15-dione) (PYTQ) (Fig. 14a and b).226 The fused CN sites in the polymer promoted good π-conjugation and concurrently enhanced the redox reactivity of CO sites, leading to a significant improvement in the utilization efficiency of redox-active sites. In a low-cost AlCl3/urea electrolyte for NABs, the resulting cathode, capable of storing four AlCl2(urea)2+ per repeating unit, delivered a high discharge capacity of 295 mA h g−1 at 0.1 A g−1 and retained a capacity of 85 mA h g−1 after 4000 cycles. The resultant NABs exhibited an excellent specific energy density of 413 W h kg−1 (Fig. 14c and d). Moreover, Li and colleagues synthesized a multilayer polymer composite cathode by in situ electrodepositing stable poly(1,5-naphthalenediamine) poly(1,5-NAPD) as an interlayer and conductive poly(para-aminophenol (pAP) as a coating onto nanoporous carbon.227 In this hybrid material design, the poly(1,5-NAPD) interlayer not only facilitated poly(pAP) deposition to enhance the reaction kinetics, but also interconnected with poly(pAP) to improve the structural integrity of the polymer cathode. Thanks to the synergistic effect of the dual organic layers, the synthesized polymer electrode exhibited a high capacity (348 mA h g−1), excellent rate capability (132 mA h g−1 at 40 A g−1), and long lifespan over 5000 cycles even at a practical mass loading (10.2 mg cm−2).
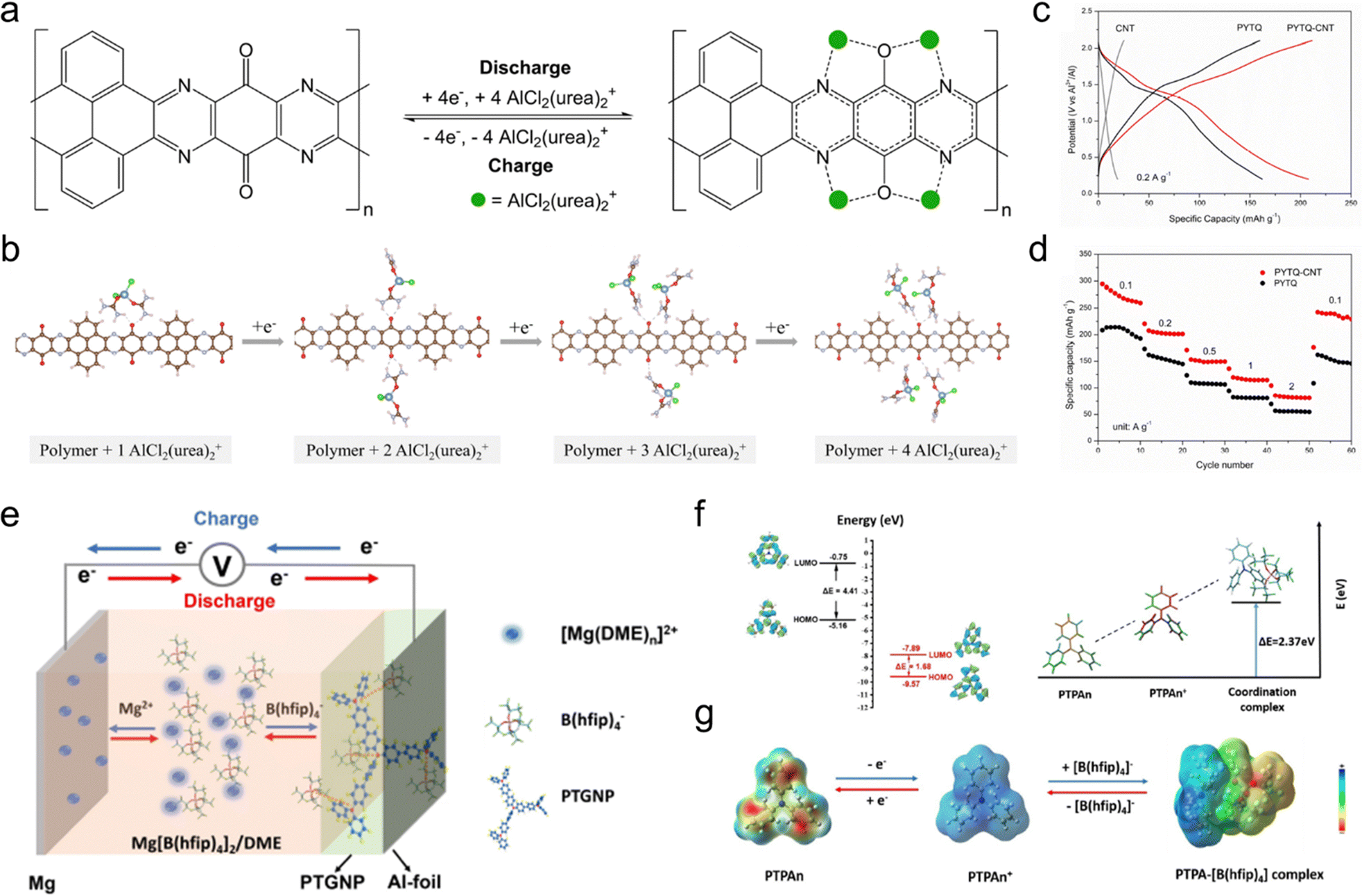 | ||
| Fig. 14 (a) The proposed redox mechanism of the PYTQ polymer as a cathode material for the Al battery with AlCl3/urea electrolyte. (b) Configuration revolution of the PYTQ polymer upon multi-step AlCl2(urea)2+ accommodation. (c) GCD profiles of PYTQ-CNT, PYTQ, and CNT electrodes at 0.2 A g−1. (d) Rate capability at different current densities. Reproduced with permission. Copyright 2022 John Wiley & Sons Inc.226 (e) Schematic illustration of the Mg//PTGNP dual-ion cell configuration and working principle. (f) Energy level diagram of pristine PTPAn (left) and PTPAn+ (right) by DFT calculations and the corresponding formation energy changes, ΔE. (g) ESP maps of a PTPAn molecule and the formed coordination complex during the electrochemical process. Reproduced with permission. Copyright 2023 John Wiley & Sons Inc.228 | ||
Redox-active polymer materials with ion-accessible microstructure design would benefit the maximum realization of their theoretical compromise. For instance, Liu and colleagues reported a poriferous PTPAn-based conjugated microporous polymer (PTPAn-CMP) cathode for AZBs.230 The synthesized PTPAn-CMP combined the advantages of extended π-conjugation along the long-range ordered skeleton and permanent 3D microporosity, which enhanced the exposure of amine functional groups and improved anion-diffusion kinetics. The electrode exhibited a pseudocapacitive-dominated behavior with Cl− anions acting as dominant charge carriers. Transitioning from nonporous conjugated PTPAn to porous PTPAn-CMP significantly increased the utilization efficiency of active N sites from 39.4% to 83.2%, resulting in a dramatically enhanced energy density of 236 W h kg−1 and maximum power density of 6.8 kW kg−1. The resultant electrode can also maintain a superior capacity retention of 87.6% after 1000 charge–discharge cycles. Moreover, Jiang and co-workers developed two conjugated microporous polymers, PyPz and PhPz, as AlCl4−-hosting cathodes for NABs, where redox-active dihydrophenazine (Pz) was coupled with pyrene (Py) or biphenyl (Ph), respectively.231 The planar Py junction endowed PyPz with an extended conjugated skeleton and a higher surface area than PhPz with a twisted linkage, resulting in higher redox activity for anion storage. Consequently, the PyPz cathode exhibited a high capacity of 231 mA h g−1 and exceptional cyclability over 100000 cycles. Moreover, the fast kinetics for AlCl4− storage enabled PyPz to deliver a high areal capacity of 2.53 mA h cm−2 and operate at low temperatures (−30 °C). These results demonstrate that integrating the dual-ion cell configuration with anion-storage electrochemistry provides a promising approach toward advanced multivalent metal batteries.
The other p-type organosulfur polymers are those based on thioether. Schubert et al. demonstrated a novel poly(acetylene)-based polymer, poly(9,10-di(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene) (PexTTF), as the cathode material in AZBs,233 which was synthesized via rhodium (Rh)-catalyzed polymerization of 2-ethynyl(exTTF). The sulfur groups on the thioether side chain tend to lose two electrons and form radial cation, resulting in the absorption of anions from the electrolyte. It exhibited a high capacity of 128 mA h g−1 at 20C with an average discharge voltage of 1.1 V, excellent cycling stability (14% loss) over 1000 cycles at 120C, and superior rate capability up to 120C (47 mA h g−1). Despite their excellent electrochemical performance, more efforts are still needed to enhance the specific capacity of p-type polymers by reducing the molar mass of repeating units or incorporating more redox-active centers. In addition, Esser and colleagues reported a cross-linked poly(3-vinyl-N-methylphenothiazine)-based cathode material (X-PVMPT) for NABs with a AlCl3/EMIMCl ionic-liquid electrolyte (Fig. 15a).234 With the two-electron redox process of phenothiazine, X-PVMPT can reversibly store AlCl4− or Al2Cl7− ions at potentials of 0.81 and 1.65 V vs. Al, respectively (Fig. 15b and c). The resultant electrode showed a specific capacity of 167 mA h g−1 in Al batteries and maintained a desirable capacity of 64 mA h g−1 at 100C. After repeated charging and discharging for 5000 cycles at 10C, it can show a good capacity retention of 88%. This study reported the first example of a reversible two-electron redox process for a phenothiazine-based electrode material, paving the way for utilizing multi-electron-transfer OEMs for NABs.
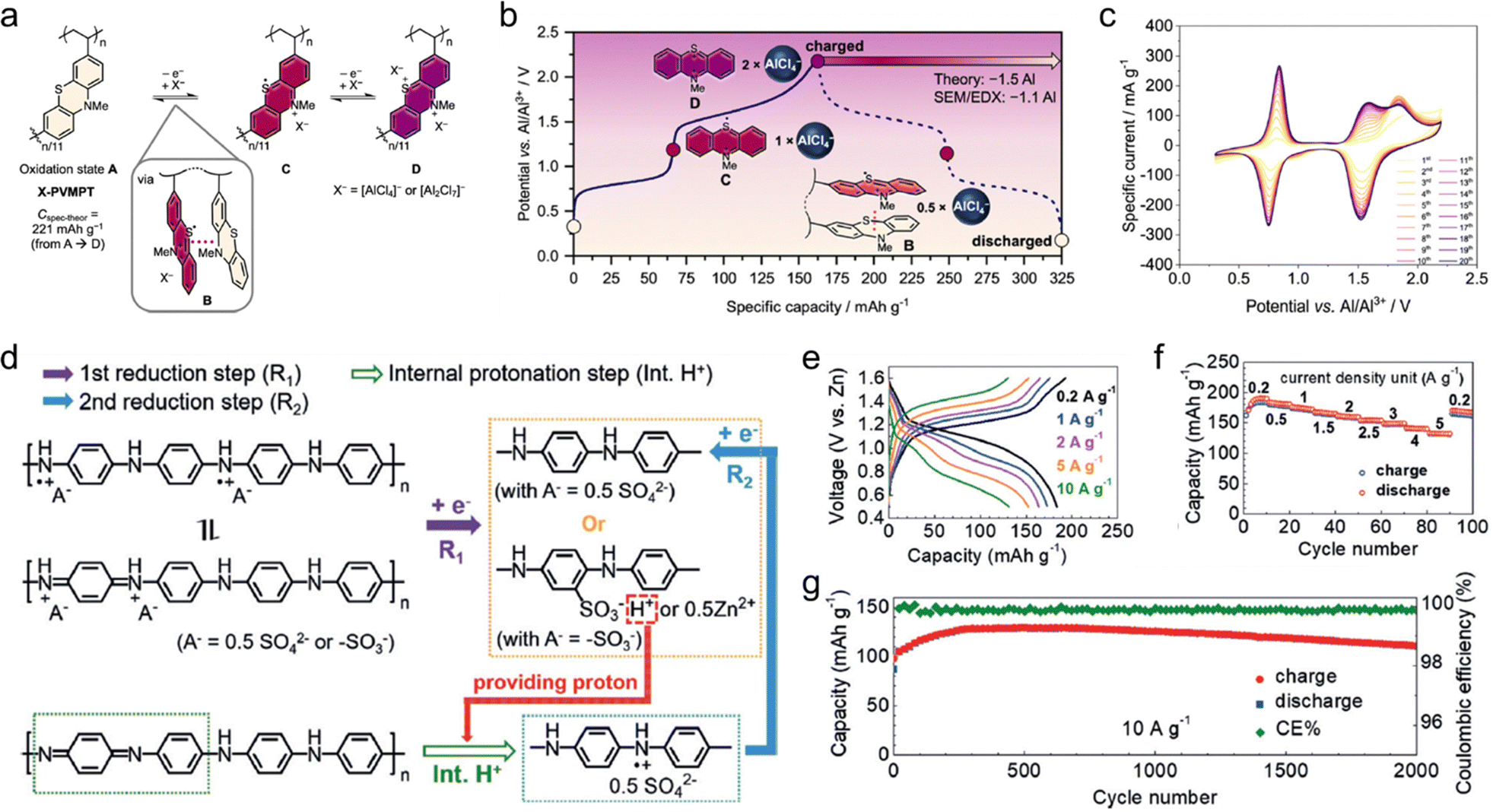 | ||
| Fig. 15 (a) Redox processes in the phenothiazine-based polymer X-PVMPT with different oxidation states. (b) GCD profiles (at a 0.5C rate, after 50 cycles of preconditioning) with correlated oxidation states of the PT units and required (AlCl4)− ions for counterbalancing of charges. (c) CV evolution of X-PVMPT-based electrodes during the first 20 cycles (0.2 mV s−1). Reproduced with permission. Copyright 2023 Royal Society of Chemistry.234 (d) Proposed reduction processes, (e) GCD curves, (f) rate performances, and (g) cycling stability of the resultant sulfo-self-doped PANI cathode. Reproduced with permission. Copyright 2018 John Wiley & Sons Inc.235 | ||
To enhance electrochemical stability, grafting functional nitroxide radicals onto insoluble substrates is a viable approach. For instance, polyacrylamide, a well-known and widely used hydrophilic material, can be functionalized with TEMPO groups for AZBs. Nishide et al. studied a hydrophilic radical polymer, poly(2,2,6,6-tetramethylpiperidinyloxy-4-yl vinyl ether) (PTVE), as a cathode material in a 0.1 M ZnCl2/NH4Cl aqueous electrolyte.94 The PTVE-based cathode demonstrated a discharge capacity of 131 mA h g−1. After cycling for 500 cycles, it could retain 65% of its capacity. To further improve the hydrophilicity and electrochemical performance in aqueous electrolytes, the same research group designed a water-swellable polyacrylamide-based radical polymer, PTAm, for AZBs.95 The PTAm-based electrode delivered an output voltage of 1.2 V, excellent cycle life with 14% capacity loss after 2000 cycles, and fast charge capability (60C). Future work should focus on reducing the solubility of nitroxide free radical polymers and enhancing their electrochemical stability over long-term cycling.
groups to –NH+– groups.243 Protonation endowed PANI with more active sites, enhanced conductivity, and reversible AlCl2+ storage. As a result, the Al//PANI(H+) battery exhibited a high discharge capacity of 200 mA h g−1 at 1 A g−1 and a stable cycling life with only 0.003% capacity decay per cycle over 8000 cycles.
When PANI is utilized as the battery electrode material, doping PANI with counter-ions is required to improve electrical conductivity and promote redox processes. For AZBs, PANI offers better electrochemical redox activity at high proton concentrations. However, such a highly acidic environment is not compatible with the Zn metal anode, causing the corrosion problem and the generation of hydrogen gas. As a result, the Zn//PANI cell demonstrated in early studies often encounter fast capacity fade due to the lack of a continuous H+ supply, even with the addition of pH buffer additives. To address this issue, Sun and coworkers developed a sulfo-self-doped PANI cathode via a facile electrochemical copolymerization process using aniline and metanilic acid monomers.235 Interestingly, the introduced SO3− group not only acted as a self-dopant but also served as an internal proton reservoir, retaining a high local H+ concentration on the polymer backbone. Since the pKa value of metanilic acid (3.7) is lower than the pH value of the ZnSO4 electrolyte (4.2), the SO3− group in the PANI chains was fully ionized in the ZnSO4 electrolyte. This effect contributed to maintaining high redox activity even in a mildly acidic environment (Fig. 15d). In a full Zn battery, the self-doped PANI cathode provided a high capacity of 184 mA h g−1 at 0.2 A g−1, excellent rate performance (70% capacity retention at 10 A g−1), and a long cycle life of over 2000 cycles with nearly 100% CE (Fig. 15e–g). This self-doped PANI leveraged the smart pH adjustment ability of self-dopants to overcome the discrepancies in active conditions between the cathode and anode, paving the way for the use of conducting polymers as cathode materials for stable AZBs.
Similarly, conductive polymers based on PPy, PTh, polyacetylene, and polyphenylene have been studied as cathode materials for MMBs.244–247 Doping these polymer electrodes with counter-ions enables redox reactions, theoretically leading to high capacities. However, achieving the predicted capacities requires high doping levels, which limit cycling stability due to side reactions associated with the dopants. Additionally, conductive polymers face challenges in fully realizing the utilization of active sites. Combining these polymers with carbon materials via in situ growth or mixing is an effective strategy to enhance active site utilization and conductivity. Commonly used carbon materials include ordered mesoporous carbon (OMC), CNTs, and GO.
Another representative electroactive conductive polymer is the inexpensive conjugated polypyrene. Its building block, pyrene (C16H10), consists of four condensed aromatic rings and undergoes a p-type redox mechanism at relatively high redox potentials. Specifically, pyrene can gain up to one positive charge per four aromatic rings to generate a radical cation during electrooxidation, with the positive charge counterbalanced by an anion from the electrolyte. Kovalenko et al. demonstrated a compelling polypyrene-based cathode for reversibly storing AlCl4− anions in NABs, which delivered a capacity of 70 mA h g−1 with an average voltage of 1.7 V.248 Interestingly, the electrochemical properties of polypyrenes could be fine-tuned through appropriate chemical derivatization. This statement was exemplified by poly(nitropyrene-co-pyrene), which increased capacity to 100 mA h g−1 and showed excellent cyclic stability over 1000 cycles in AlCl3/EMIMCl electrolyte. Given the vast possibilities of polymeric aromatic hydrocarbon compounds in terms of compositional and structural tunability, low cost, high abundance, and production scalability, this work provides further inspiration for the development of rechargeable Al batteries. Moreover, the utilization of the 3D conductive polymer PEDOT as a cathode material for rechargeable AABs was also demonstrated by Schoetz and coworkers. The PEDOT electrode achieved a specific energy of 50–64 W h kg−1, specific power of 32–40 W kg−1, and stable operation over 100 cycles.249
| Compound | Electrolyte | Battery type | Average discharge potential | Specific capacity | Cycle life | Rate performance | Ref. |
|---|---|---|---|---|---|---|---|
| 1,4-Polyanthraquinone (PAQS) | 0.37 M MgCl2–0.15 M Mg(TFSI)2 in a mixture of THF and glyme | Nonaqueous Mg battery | 1.2–1.6 V vs. Mg/Mg2+ | 180 mA h g−1 at 0.05 A g−1 | 20% capacity retention after 100 cycles at 0.05 A g−1 | 152, 141, and 100 mA h g−1 at 0.1, 0.2, and 0.5 A g−1, respectively | 208 |
| Ca[B(hfip)4]2 salt dissolved in DME | Nonaqueous Ca battery | 2.1 V vs. Ca/Ca2+ | 169.3 mA h g−1 at 0.5C (122.5 mA g−1) | 114.2 mA h g−1 after 1000 cycles | — | 210 | |
| 0.3 M Mg(HMDS)2–4MgCl2 | Nonaqueous Mg battery | 1.5–2.0 V vs. Mg/Mg2+ | 100 mA h g−1 at 0.5C (122 mA g−1) | 30 mA h g−1 after 100 cycles at 0.5C | — | 211 | |
| 2,6-Polyanthraquinone (26PAQ) | 1.71 and 1.52 V vs. Mg/Mg2 | 122 mA h g−1 at 130 mA g−1 | 112 mA h g−1 after 100 cycles at 130 mA g−1 | 90, 75, and 45 mA h g−1 at 260, 560 and 1120 mA g−1, respectively | |||
| 1,4-Polyanthraquinone (14PQ) | 1.57 V and 1.48 V vs. Mg/Mg2 | 132.7 mA h g−1 at 130 mA g−1 | 104.9 mA h g−1 after 100 cycles at 130 mA g−1 | 86, 71, and 48 mA h−1 at 260, 560 and 1120 mA g−1, respectively | |||
| 2,5-Dichloro-1,4-benzoquinone (PBQS) | 3 M Aqueous Zn(CF3SO3)2 | Aqueous Zn battery | 0.95 V vs. Zn/Zn2+ | 203 mA h g−1 at 20 mA g−1 | 150 mA h g−1 after 50 cycles at 0.2C (86%) | 166, 144, 139, 135, and 126 mA h g−1 at 40, 100, 200, 400 and 1000 mA g−1, respectively | 207 |
| Quinone-based porous organic polymer (rPOP) | 1 M Aqueous ZnSO4 | Aqueous Zn battery | 0.6–1.0 V vs. Zn/Zn2+ | 120 mA h g−1 at 0.1 A g−1 | 95% capacity retention after 1000 cycles at 1.0 A g−1 | 89, 69, and 58 mA h g−1 at 0.2, 0.5, 1 A g−1, respectively | 212 |
| 66% capacity retention after 30000 cycles at 2.0 A g−1 |
|||||||
| Poly(phenazine-alt-pyromellitic anhydride) (PPPA) | 2.0 M Aqueous Zn(OTf)2 | Aqueous Zn battery | 0.75 V vs. Zn/Zn2+ | 210.2 mA h g−1 at 50 mA g−1 | 70.6% capacity retention at 5 A g−1 after 20000 cycles |
191, 182.1, 170.9, 162.3, 154.4 and 139.7 mA h g−1 at 0.1, 0.2, 0.5, 1, 2 and 5 A g−1, respectively | 121 |
| Poly(4-(2,5-hydroquinone)-4H-dithieno[3,2-b:2′′,3′′-d]pyrrole) (PDpBQH) | 1 M Aqueous ZnSO4 | Aqueous Zn battery | 1.16 V vs. Zn/Zn2+ | 120 mA h g−1 at 0.1 A g−1 | 79% capacity retention after 500 cycles | 110, 91, 86, and 63 mA h g−1 at 0.5, 1, 2, and 5 A g−1, respectively | 215 |
| Poly(4-(3,4-pyrocatechol)-4H-dithieno[3,2-b:2′′,3′′-d]pyrrole) (PDoBQH) | 1.3 V vs. Zn/Zn2+ | 90 mA h g−1 at 0.5 A g−1 | 53% capacity retention after 500 cycles | 88, 75, 56, 50 mA h g−1 at 0.5, 1, 2, and 5 A g−1, respectively | |||
| Poly(catechol) (P(4VC86-stat-SS14)) | 4 M Aqueous Zn(TFSI)2 | Aqueous Zn battery | 1.1 V vs. Zn/Zn2+ | 327 mA h g−1 at 1C (1C = 344 mA g−1) | 85% capacity retention after 12500 cycles at 10C |
318, 296, 270, 240, 195, 148, 65 mA h g−1 at 2, 5, 10, 30, 60, and 150 and 1350C, respectively | 217 |
| Poly{[N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} | 0.2 M Mg(TFSI)2 in diglyme | Nonaqueous Mg battery | 1.50 V vs. Mg/Mg2+ | 52.8 mA h g−1 at 0.011 A g−1 | 88% capacity retention after 2500 cycles at 0.3 A g−1 | 70% at 50C (1C = 54 mA g−1) | 219 |
| Poly(pyromellitic diimide) (PPMDI) | Phenyl magnesium chloride (PhMgCl) with AlCl3 in THF | Nonaqueous Al battery | 1.30 V vs. Mg/Mg2+ | 130 mA h g−1 at 0.15 A g−1 | 87% capacity retained after 200 cycles at 0.15 A g−1 | 55% at 3 A g−1 | 213 |
| AlCl3/EMIMCl | Nonaqueous Al battery | 0.7 V vs. Al/Al3+ | 110 mA h g−1 at 0.15 A g−1 | 85% capacity retained after 100 cycles at 0.15 A g−1 | 55% at 3 A g−1 | ||
| Poly(perylenetetracarboxylic diimide) (PPTCDI) | AlCl3/EMIMCl | Nonaqueous Al battery | 1.2 V vs. Al/Al3+ | 83 mA h g−1 at 1 A g−1 | A capacity decay of 0.0067% per cycle after 1800 cycles at 1 A g−1 | 65, 55, 48, and 40 mA h g−1 at 2, 5, 8, and 10 A g−1, respectively | 221 |
| Poly(naphthalenetetracarboxylic diimide) derivatives (PNTCDI) | Aqueous 1.5 M Zn(OTf)2 | Aqueous Zn battery | 0.2–0.4 V vs. Zn/Zn2+ | 128 mA h g−1 at 50 mA g−1 | 88% capacity retention after 50000 cycles |
101 mA h g−1 at 5 mA g−1 | 223 |
| Poly(4,5-dihydrophenanthro[4,5-abc]pyrazino [2,3-i]phenazine-10,15-dione) | AlCl3/urea (1.3:1) |
Nonaqueous Al battery | 1.3 V vs. Al/Al3+ | 295 mA h g−1 at 1 mA g−1 | 85 mA h g−1 at 1 A g−1 after 4000 cycles | 208, 153, 120, and 85 mA h g−1 at 0.2, 0.5, 1, and 2 A g−1, respectively | 226 |
| 146 mA h g−1 at 2 A g−1 after 3500 cycles | |||||||
| Poly(hexaazatrinaphthalene) (PHATN) | — | Nonaqueous Mg battery | 1.50 V vs. Mg/Mg2+ | 125 mA h g−1 at 20 mA g−1 | 110 mA h g−1 for 200 cycles at 20 mA g−1 | 104, 82, and 60 mA h g−1 at 40, 100, and 200 mA g−1, respectively | 224 |
| AlCl3/EMIMCl | Nonaqueous Al battery | 0.8 V vs. Al/Al3+ | 125 mA h g−1 at 50 mA g−1 | A capacity of 92 mA h g−1 after 100 cycles at 50 mA g−1 | — | ||
| Triquinoxalinylene (3Q) based homopolymer (P3Q) | 2 M ZnSO4 aqueous solution | Aqueous Zn battery | 0.6 V vs. Zn/Zn2+ | 115 mA h g−1 at 0.3 A g−1 | 10% capacity retention at 3 A g−1 | 80, 75, 45, 30, 10, and 0 at 0.5, 1, 2, 5, 10, and 20 A g−1, respectively | 225 |
| Triazine-linked 3Q polymer (P3Q-t) | 0.7 V vs. Zn/Zn2+ | 237 mA h g−1 at 0.3 A g−1 | 11% of capacity loss over 1500 cycles at 3 A g−1 | 220, 200, 170, 150, 120, and 60 at 0.5, 1, 2, 5, 10, and 20 A g−1, respectively | |||
| Poly(1,5-naphthalenediamine, 1,5-NAPD) and poly(para-aminophenol, pAP) hybrid materials | 2 M ZnSO4 aqueous solution | Aqueous Zn battery | 1.1 V vs. Zn/Zn2+ | 348 mA h g−1 at 0.1 A g−1 | 90.1% even after 5000 cycles at 5 A g−1 | 300, 270, 250, 230, 200, 180, 160, and 132 mA h g−1 at 0.2, 0.5, 1, 2, 5, 10, 20, and 40 A g−1, respectively | 227 |
| Polytriphenylamine (PTPAn) | 0.5 M Mg[B(hfip)4]2/DME electrolyte | Nonaqueous Mg battery | 3 V vs. Mg/Mg2+ | 105 mA h g−1 at 545 mA g−1 | 53 mA h g−1 after 500 cycles at 1090 mA g−1 (0.86% per cycle) | 105 mA h g−1 at 1090 mA g−1 | 228 |
| 0.25 M Ca[B(hfip)4]2/DME electrolyte | Nonaqueous Ca battery | 2.8 V vs. Ca/Ca2+ | 85 mA h g−1 at 545 mA g−1 | 50 mA h g−1 after 3000 cycles at 1090 mA g−1 | — | ||
| Polytriphenylamine conjugated microporous polymer (CMP) | 2 m ZnCl2 aqueous electrolyte | Aqueous Zn battery | 1.15 V vs. Zn/Zn2+ | 210.7 mA h g−1 at 0.5 A g−1 | 87.6% capacity retention after 1000 cycles at 6 A g−1 | 151, 141, 125, 122, and 107.5 mA h g−1 at 1, 2, 3, 4, and 6 A g−1, respectively | 230 |
| Dihydrophenazine (Pz) coupled with pyrene (Py) (PyPz) | AlCl3/1-ethyl-3-methylimidazolium chloride | Nonaqueous Al battery | 0.75 vs. Al/Al3+ | 231 mA h g−1 at 0.1 A g−1 | 93.9% capacity retention after 1000 cycles at 0.2 A g−1 | 212, 205, 201, 198, 195, 185, 175, 116 mA h g−1 at 0.2, 0.5, 1, 2, 5, 10, 20, and 30 A g−1, respectively | 231 |
| 90.3% capacity retention after 50000 cycles at 5 A g−1 |
|||||||
| 78.1% capacity retention after 100000 cycles at 10 A g−1 |
|||||||
| Dihydrophenazine (Pz) coupled with biphenyl (Ph) (PhPz) | 0.65 vs. Al/Al3+ | 137 mA h g−1 at 0.1 A g−1 | 74.7% capacity retention after 1000 cycles at 0.2 A g−1 | 128, 123, 110, 102, 86, 61 mA h g−1 at 0.2, 0.5, 1, 2, 5, and 10 A g−1, respectively | |||
| Poly-2,2′-dithiodianiline (PDTDA) | 0.25 M Mg(AlCl2BuEt)2/THF electrolyte | Nonaqueous Al battery | 1.4 V vs. Mg/Mg2+ | 78 mA h g−1 | Slow capacity degradation over 30 cycles | — | 232 |
| Sulfurized polyacrylonitrile (SPAN) | 1.2 V vs. Mg/Mg2+ | 117.3 mA h g−1 | 60% retained after 22 cycles | — | |||
| Poly(9,10-di(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene) (PexTTF) | 1 M aqueous solution of zinc tetrafluoroborate hexahydrate | Aqueous Zn battery | 1.1 V vs. Zn/Zn2+ | 128 mA h g−1 at 20C | 81 mA h g−1 after 10000 cycles at 10C |
47 mA h g−1 at 120C | 233 |
| Poly(3-vinyl-N-methylphenothiazine) (X-PVMPT) | AlCl3/EMIMCl ionic liquid electrolyte | Nonaqueous Al battery | 0.81 and 1.65 V vs. Al/Al3+ | 167 mA h g−1 at 0.11 A g−1 | 85% retention after 10000 cycles at 2.2 A g−1 |
105, 64, 47 mA h g−1 at 11, 22 and 26.4 A g−1, respectively | 234 |
| Poly(2,2,6,6-tetramethylpiperidinyloy-4-yl acrylamide) (PTAm) | — | Aqueous Zn battery | 1.2 V vs. Zn/Zn2+ | 114 mA h g−1 at 68 μA cm−2 | 14% capacity loss after 2000 cycles | — | 95 |
| Sulfo-self-doped PANI | 1 M ZnSO4 | Aqueous Zn battery | 1.1 V vs. Zn/Zn2+ | 184 mA h g−1 at 0.2 A g−1 | 110 mA hg−1 capacity at 10 A g−1 over 2000 cycles |
172, 163, 151, and 130 mA hg−1 at 1, 2, 5, and 10 Ag−1, respectively |
235 |
6.3. Crystalline polymers: covalent-organic frameworks
Although amorphous polymers with redox-active sites can overcome the shortcomings of the corresponding small precursors related to dissolution and conductivity issues, they still suffer from poor diffusivity of multivalent ions. This limitation originates from the large steric hindrance and strong interactions between host polymers and target ions. Charge carriers have difficulty freely transporting across polymer chains by the hopping mechanism, and a certain degree of redox moieties cannot be effectively activated. Moreover, the complicated polymerization process makes it challenging to synthesize polymers with precise structures, posing significant difficulties in accurately understanding the structure–performance relationship of redox-active polymers. In this regard, crystalline COFs are ideal electrode materials for accommodating multivalent metal ions. The high rigidity and large polymerization degree of COFs endow redox moieties with strong resistance to dissolution, promising long cycling durability. Additionally, the molecularly designable porosity and precisely defined topology of COFs enable efficient access of redox moieties by charge carrier ions, thus enhancing the efficient utilization of these redox moieties. Furthermore, the adjustable pore configuration, coupled with the functionalization of frameworks through pre-customization or post-synthesis strategies, allows for precise manipulation of the chemical structures. This flexible and precise designability provides a novel perspective for deepening the understanding of the fundamental problems of organic electrode materials. Based on these merits, COF-based OEMs have been primarily demonstrated in relatively mature battery devices, such as Li-ion and Na-ion batteries. Their development in rechargeable MMBs is still not fully explored, and the respective structure–property relationships need more in-depth investigation. This section summarizes recent research on COF-based OEMs for different MMBs. Based on different redox moieties, we divide them into three categories: COFs with carbonyl groups, COFs with N-heterocycles, and multi-redox-site COFs (Fig. 16).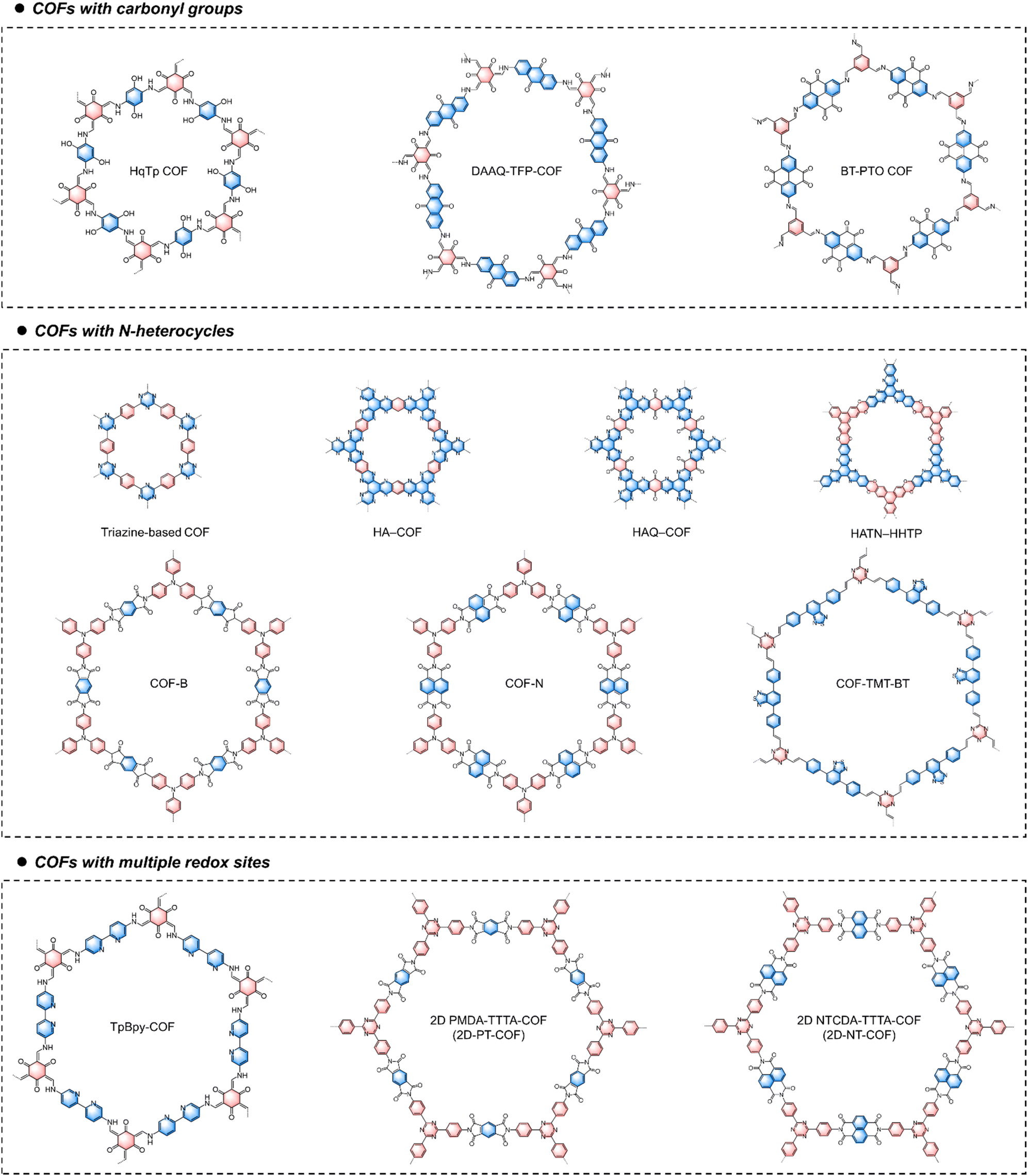 | ||
| Fig. 16 Molecular structures of reported redox-active crystalline COFs for MMBs. | ||
O⋯Zn and Zn⋯N–H between adjacent HqTp layers and charge carriers contributed to an excellent discharge capacity of 276 mA h g−1 at 125 mA g−1. Recently, Li and coworkers used the same COF material to store Ca2+ ions. The COF electrode exhibited a high specific capacity of 119.5 mA h g−1 at 1 A g−1 and maintained 78.7 mA h g−1 even at an extremely high current of 50 A g−1.252
Building blocks with a high density of carbonyl groups have been specially designed to prepare COF-based OEMs with improved charge storage capacity and excellent stability, such as PTO and anthraquinone.253 Tao and coworkers designed a novel orthoquinone COF (BT-PTO) with plentiful carbonyl groups for AZBs.122 The BT-PTO materials were prepared via the Schiff-based solvothermal condensation reaction between a benzenetricarboxaldehyde node and PTO active monomer. This COF showed a unique co-insertion mechanism, where Zn2+ inserted first, followed by H+ insertion. Thanks to the extended conjugated conductive plane, regular pore channels, and carbonyl-rich active sites, this COF delivered a high reversible capacity of 225 mA h g−1 at 0.1 A g−1 and exceptional long-term cyclability (with a retention rate of 98.0% at 5 A g−1 after 10000 cycles). Significantly, this co-insertion behavior evolves into enriched H+ insertion at high current densities, empowering the COF with ultra-fast kinetics to achieve an unprecedented specific power of 184 kW kg−1 and a high energy density of 92.4 W h kg−1.
Besides quinone-based COFs, imide-based COFs with abundant carbonyl distribution also show great potential for MMB applications. Our group reported a 2D polyarylimide COF (PI-COF) for Zn2+ storage (Fig. 17a).124 The PI-COF was synthesized via the condensation reaction between NTCDA and tris (4-aminophenyl) amine. The well-organized pore channels of the PI-COF allowed high accessibility to the built-in redox-active carbonyl groups and efficient ion diffusion with a low energy barrier. The constructed PI-COF electrode exhibited a specific capacity of 92 mA h g−1 at 0.7 A g−1, high rate capability of 79.8% at 7 A g−1, and long cycle life with 85% capacity retention over 4000 cycles (Fig. 17b). To study the chemical structure evolution during the electrochemical process, DFT simulations were performed to calculate the most stable configurations for the Zn2+-storage reaction. We found that the ion storage process occurred in two steps, as shown in Fig. 17c. In the first step, Zn2+ is able to coordinate with the carbonyl O of the pristine PI-COF, requiring an average energy of 1.36 eV per Zn. In the second step, the energy needed for the other Zn2+ binding slightly increases to 1.91 eV. For the fully charged Zn2+-storage product, each Zn2+ is bonded with two carbonyl O atoms and is located in the 1D nanochannel between two adjacent PI-COF layers. Additionally, in situ Raman spectro-electrochemistry was carried out to reveal the two Zn2+-storage mechanism of the PI-COF (Fig. 17d). At the pristine state, the PI-COF showed the characteristic peaks of C–N and CC from 1300 to 1650 cm−1. In State I, Zn2+ would interact with the carbonyl O of the PI-COF via forming O–Zn–O bridges, which is evidenced by the downshift of Raman bands from 1148 to 1132 cm−1 and from 1406 to 1383 cm−1. In State II, the appearance of the major band at 1439 cm−1 proved the formation of two C–O bonds in one naphthalene diimide unit. The PI-COF with dual Zn2+-bonded naphthalene diimide units would result in a new symmetric molecular system. This study demonstrates the feasibility of using the PI-COF as a Zn2+-storage electrode and shows promising prospects for constructing reliable aqueous energy storage devices. Moreover, Xu and coworkers reported two polyimide COF cathodes for Mg batteries using benzenetetracarboxylic anhydride (PMDA) and NTCDA as the main redox-active monomers.254 Benefitting from the large conjugated structure and smaller particle size, the NTCDA-based COF showed a better Mg2+ storage performance compared to the PMDA-based one. The resultant COF demonstrated a high Mg2+-storage capacity of 120 mA h g−1 and considerable rate performance with 50 mA h g−1 at 2 A g−1, as well as good cyclability with 99% capacity retention after 300 cycles.
 | ||
| Fig. 17 (a) Top and side views of the modeled atomic structure of a PI-COF. (b) GCD profiles at different current densities. (c) The Zn2+-storage mechanism of the PI-COF simulated by DFT calculations. (d) Raman spectra of the PI-COF electrode at potentials of 0 (initial state), −0.5 (State I), and −0.9 V (State II). Reproduced with permission. Copyright 2020 American Chemical Society.124 (e) Schematic illustration of the reaction mechanism of triazine-based COF cathodes in chloride-free electrolyte. (f) Rate performance and (g) possible electrochemical redox mechanism of a triazine-based COF. Reproduced with permission. Copyright 2020 American Chemical Society.255 | ||
000 cycles. This work demonstrates that molecular engineering of the COF structure is an effective approach to boost practical charge storage performance.
Triazine with high CN density is also a promising redox N-heterocycle. In 2020, Wang and coworkers demonstrated an environmentally benign, low-cost, and sustainable triazine-COF cathode for Mg2+ storage (Fig. 17e).255 The triazine-COF was synthesized by the polymerization of 1,4-dicyanobenzene through annealing with a ZnCl2 catalyst in quartz ampules at 400 °C. This resulted in a 2D layered structure comprising benzene and triazine rings. The triazine-COF electrode delivered a high power density of 2.8 kW kg−1, a high specific energy density of 146 W h kg−1, and an ultralong cycle life of 3000 cycles with a very slow capacity decay rate of 0.0196% per cycle (Fig. 17f). Comprehensive electrochemical analysis demonstrated that the triazine ring sites in the COF acted as redox centers for reversible reactions with Mg2+, and the ultrafast reaction kinetics was primarily attributed to pseudocapacitive behavior (Fig. 17g). Moreover, Ciesielski and coworkers reported a novel and structurally robust olefin-linked COF (COF-TMT-BT) for AZBs.126 COF-TMT-BT was synthesized via the aldol condensation between 2,4,6-trimethyl-1,3,5-triazine (TMT) and 4,4′-(benzothiadiazole-4,7-diyl)dibenzaldehyde (BT). Both benzothiadiazole and triazine are electrochemically active groups, capable of reversibly coordinating with Zn2+ through controlled ion complexation and release. This COF-TMT-BT electrode delivered a high capacity of 283.5 mA h g−1 at a current density of 0.1 A g−1, an outstanding maximum energy density of 219.6 W h kg−1, and a power density of 23.2 kW kg−1. Impressively, the irreversible covalent CC linkages endowed COF-TMT-BT with improved stability in aqueous electrolytes, retaining a specific capacity of 186.8 mA h g−1 after 800 cycles.
C, CO, and CN sites. According to DFT calculations, a maximum of nine Ca2+ ions can be stored per HAQ-COF repetitive unit. Besides, the HAQ-COF exhibited a high-rate capacity of 104 mA h g−1 at 70 A g−1 and a capacity decay of only 0.01% per cycle after 3000 cycles at 5 A g−1. These accumulated findings are anticipated to provide new insights into designing and manufacturing COF electrodes for high-performance MMBs. Overall, a thorough performance comparison of these reported COF materials for MMBs is provided in Table 3.
 | ||
| Fig. 18 (a) Design and synthetic routes of a redox-bipolar 2D-PT-COF and 2D-NT-COF with dual-ion storage capability. (b) Imide moieties after injecting one or two electrons and triazine moieties after extracting one electron. (c) Solid-state 27Al MAS NMR spectra of the electrolyte, electrolyte-soaked electrode (2D-NT-COF30-OCV), fully charged electrode (2D-NT-COF30-charge), and (d) fully discharged electrode (2D-NT-COF30-discharge). Inset shows the deconvolution of the 27Al spectrum. (e) GCD curves and (f) rate performance of the 2D-PT-COF30 and the 2D-NT-COF30 electrodes. (g) Cycling performance (capacity decay rate per cycle) comparison of our electrodes with the reported organic RAB cathodes. Reproduced with permission. Copyright 2023 John Wiley & Sons Inc.35 | ||
| Compound | Electrolyte | Battery type | Average discharge potential | Specific capacity | Cycle life | Rate performance | Ref. |
|---|---|---|---|---|---|---|---|
| HqTp COF | Aqueous 3 M ZnSO4 | Aqueous Zn battery | 0.8 V vs. Zn/Zn2+ | 276 mA h g−1 at 125 mA g−1 | 95% retention after 1000 cycles | 200, 154, 125, and 85 mA h g−1 at 312.5, 625, 1250, and 3750 mA g−1, respectively | 123 |
| DAAQ-TFP-COF | 0.6 M MgTFSI2 – 1.2 M MgCl2 DME | Nonaqueous Mg battery | 1.3 V vs. Mg/Mg2+ | 130 mA h g−1 at 0.02 A g−1 | 79% capacity retention after 2000 cycles at 500 mA h g−1 | 90, 75, 50, 40, 30, 25, and 18 mA h g−1 at 0.05, 0.1, 0.2, 0.5, 1, 2, and 3 A g−1, respectively | 253 |
| BT-PTO COF | 3 M Zn(CF3SO3)2 | Aqueous Zn battery | 0.8 V vs. Zn/Zn2+ | 220 mA h g−1 at 0.5 A g−1 | 98.0% retention at 5 A g−1 after 10000 cycles |
213, 206, 197, 190, 182, 179, 174, and 172 mA h g−1 at 1, 2, 5, 10, 20, 30, 50, and 85 A g−1, respectively | 122 |
| COF-B | Mg(TFSI)2–MgCl2 in DME | Nonaqueous Mg battery | 1.3 V vs. Mg2+/Mg | 92 mA h g−1 at 50 mA g−1 | 57 mA h g−1 after 100 cycles at 50 mA g−1 | — | 254 |
| COF-N | 1.5 V vs. Mg2+/Mg | 120 mA h g−1 at 50 mA g−1 | 101 mA h g−1 after 300 cycles at 200 mA g−1 | 118, 114, 97, 75 and 50 mA h g−1 at 0.1, 0.2, 0.5, 1 and 2 A g−1, respectively | |||
| HATN–HHTP | 0.5 M Mg(TFSI)2 in DME | Nonaqueous Mg battery | 1.24 V vs. Mg/Mg2+ | 124 mA h g−1 at 10 mA g−1 | 150 cycles with a capacity retention of 83.5% | 118, 108, and 100 mA h g−1 at 20, 40, and 100 mA g−1, respectively | 256 |
| AlCl3/EMIMCl | Nonaqueous Al battery | 0.9 V vs. Al/AlCl2+ | 128 mA h g−1 at 20 mA g−1 | Retains 80% capacity after 350 cycles at 100 mA g−1 | 116, 114, 112, and 110 mA h g−1 at 20, 40, 100, and 200 mA g−1, respectively | ||
| HAQ-COF | Aqueous 2.0 M ZnSO4 | Aqueous Zn battery | 0.84 V vs. Zn/Zn2+ | 339 mA h g−1 at 0.1 A g−1 | 187.5 mA h g−1 after 200 cycles at 1 A g−1 | 317, 279, 255, 228, 170, and 95.6 mA h g−1 at 0.2, 0.5, 1, 2, 5, and 10 A g−1, respectively | 257 |
| 128 mA h g−1 after 10000 cycles at 5 A g−1 |
|||||||
| HA-COF | 0.53 V vs. Zn/Zn2+ | 164 mA h g−1 at 0.1 A g−1 | 90 mA h g−1 after 200 cycles at 1 A g−1 | 136, 111, 90.2, 70.8, 50.4, and 35.4 mA h g−1 at 0.2, 0.5, 1, 2, 5 and 10 A g−1, respectively | |||
| 61 mA h g−1 after 10000 cycles at 5 A g−1 |
|||||||
| Triazine-based COF | 0.5 M Mg(TFSI)2/DME | Nonaqueous Mg battery | 1.6 V vs. Mg/Mg2+ | 107 mA h g−1 at 0.2C (1C = 114 mA h g−1) | 74 mA h g−1 can be retained after 100 cycles at 0.5C | 90, 80, 74.9, 60, 55.6, and 40 mA h g−1 at 0.5, 1, 2, 5, 10, and 20C, respectively | 255 |
| COF-TMT-BT | 2 M aqueous Zn(CF3SO3)2 | Aqueous Zn battery | 1.27 V vs. Zn/Zn2+ | 283.5 mA h g−1 at 0.1 A g−1 | 186.8 mA h g−1 after 2000 cycles at 0.1 A g−1 | 261.2, 232.6, 191.4, and 146.2 mA h g−1 at 0.2, 0.5, 1, and 2 A g−1, respectively | 126 |
| TpBpy-COF | AlCl3/EMIMCl | Nonaqueous Al battery | 1.2 V vs. Zn/Zn2+ | 307 mA h g−1 at 0.1 A g−1 | 150 mA h g−1 after 13000 cycles at 2 A g−1 |
245, 162, 125, and 113 mA h g−1 at 0.5, 1, 2, and 5 A g−1, respectively | 258 |
| 2D PMDA-TTTA-COF (2D-PT-COF) | AlCl3/EMIMCl | Nonaqueous Al battery | 1.2 V vs. Al/Al3+ | 90 mA h g−1 at 0.1 A g−1 | >97% capacity retention after 4000 cycles at 1 A g−1 | 81, 63, 54, and 48 mA h g−1 at 0.2, 0.4, 0.8, and 1.6 A g−1, respectively | 35 |
| 2D NTCDA-TTTA-COF (2D-NT-COF) | 1.3 V vs. Al/Al3+ | 132 mA h g−1 at 0.1 A g−1 | >97% capacity retention after 4000 cycles at 1 A g−1 (0.0007% capacity decay per cycle) | 123, 105, 88, and 79 mA h g−1 at 0.2, 0.4, 0.8, and 1.6 A g−1, respectively |
7. Summary and perspectives
Developing Li-free and Co-free battery technologies based on low-cost and abundant multivalent metal anodes and organic/polymer cathodes has promising potential to enhance the sustainability, affordability, and environmental friendliness of energy storage devices. In this regard, the search for high-potential, high-capacity, and high-stability OEMs is of significance for the development and application of MMBs. In this review, we highlighted the recent progress in electrochemically active OEMs for MMBs, focusing on their structure–property relationships and the origins of their redox activity. This review briefly analyzed the fundamental electrochemistry of different MMBs and outlined their respective key features. After that, a complete understanding of redox-active moieties at the molecular level was delicately discussed for building the accurate structure–performance relationships of OEMs. Furthermore, we analyzed the respective electrochemical properties of OEMs and the corresponding affecting structural factors in detail. Various redox-active organic compounds, spanning from small molecules to amorphous polymers and crystalline COFs, were next thoroughly summarized in terms of their advantages, design considerations, reaction mechanisms, electrochemical performance, and optimization strategies. This comprehensive overview aims to provide a detailed molecule-level understanding of OEMs for MMB cathodes.Despite substantial advancements in OEMs over the past few years, this field is still in its infancy, and several critical challenges remain. Future research should focus on exploring new materials and redox electrochemistry for OEMs, emphasizing the design and synthesis of high-redox-potential, large-capacity, long-cycling OEMs to achieve low-cost, high-energy, safe, stable, and sustainable MMBs. From the fundamental aspects of organic molecular design, there are still many challenges and questions that remain to be solved for high-performance OEMs.
(1) Given that a significant number of OEMs have been explored as promising cathodes for MMBs, their energy densities still fall short compared to commercially available Li-ion batteries. Voltage and capacity are the two key factors determining a battery's energy density. Therefore, material design strategies that widen the working potential and improve specific capacity are crucial for achieving higher energy density. For large-scale practical applications, an ideal OEM should have a large specific capacity (>200 mA h g−1), a high working potential (>2 V vs. metal anode), and long cyclability (>1000 cycles). Regarding reaction voltage, p-type organic cathodes based on anion insertion reactions exhibit a higher voltage compared to the n-type ones. Introducing p-type redox-active moieties is promising for achieving high-reaction potentials in OEMs. However, the reversible capacity of p-type organic cathodes is relatively low and is often accompanied by the consumption of the electrolyte. Incorporating electron-withdrawing functional groups into OEMs is another viable approach to enhance the electrochemical reaction potential. To improve capacity, one promising solution is to incorporate multiple electroactive centers into a single organic structure and reduce the weight of electrochemically inactive components. This approach increases the density of active redox centers per organic compound or repeating unit in the polymer. OEMs with adjacent redox-active centers and strong intermolecular interactions are particularly suitable for high-capacity MMBs due to their ability to co-coordinate with multivalent metal ions, potentially enabling the exclusive storage of these ions. Additionally, the well-defined porous structure and large surface area of polymeric materials are highly advantageous for MMBs, as they enhance the accessibility of fused electroactive sites and maximize the theoretical capacity. This design shortens the diffusion pathways for multivalent cations and facilitates reaction kinetics within the batteries.
(2) The intrinsic low electronic conductivity of OEMs often results in poor rate performance and limited charge storage capacity, significantly restricting their practical applications in energy storage. To enhance electronic conductivity, large amounts of conductive carbon (over 30 wt%) are typically added to organic electrodes. However, this common strategy sacrifices energy density due to the large ratio of inactive materials in the electrode. To address this issue, in situ growth of organic materials on the surfaces of graphene and CNTs through π–π stacking interactions is an effective method. The high conductivity and insolubility of carbon materials not only promote electron transfer but also mitigate the dissolution of OEMs. Additionally, improving electron transport within OEMs can be achieved by extending the π-conjugated structure of the materials. Another major drawback of OEMs is their high solubility in electrolytes. Many carbonyl-based compounds suffer from limited cycling performance due to their high electrolyte-philic nature. Polymerizing these organic moieties into high molecular-weight and insoluble polymers can greatly extend their cycle life. Furthermore, replacing liquid electrolytes with solid-state electrolytes that have limited solubility and polarizability can also address the dissolution issue. However, fabricating solid-state electrolytes with high transfer numbers and ionic conductivity for multivalent charge carriers remains a significant challenge for MMBs.
(3) An in-depth understanding of the electrochemical mechanisms and structural evolution of OEMs during the charge and discharge processes is essential for guiding the future design of high-performance MMBs. Unlike the relatively straightforward rocking-chair mechanism in Li-ion batteries, the working mechanisms of OEMs in MMBs are much more complex. To fully understand the electrochemical processes, it is necessary to identify the specific charge carriers stored (e.g., AlCl4−, AlCl2+, MgCl+, and H+/Zn2+ co-storage), co-insertion of solvent molecules, bond cleavage and formation, ion-storage dynamics, formation of the solid electrolyte interphase, and intermediate structures. Advanced electrochemical characterization techniques play a crucial role in elucidating these mechanism fundamentals. Many studies use ex situ techniques such as X-ray XPS, Raman spectroscopy, XRD, and energy-dispersive X-ray spectroscopy (EDX) to analyze electrochemical performance. However, these ex situ characterization techniques can’t avoid exposing samples to the ambient environment, leading to structural changes and ambiguous results. Moreover, theoretical calculations and simulations can provide valuable insights into charge transport (electronic or ionic conduction) and mass transport. Techniques such as DFT, molecular dynamics (MD), and multiphysics simulations like COMSOL are instrumental in gaining a deeper understanding of these processes. Some cutting-edge simulation approaches, such as artificial intelligence, high throughput calculations, and machine learning, can also be involved for the accelerated structure screening of OEMs. Integrating these advanced characterization methods with theoretical models will significantly enhance our ability to establish a feedback loop for designing advanced OEM structures for MMBs.
(4) The topological architecture and nanoengineering of active materials with appropriate chemical structures are also crucial for enhancing the electrochemical performance of OEMs. For instance, porous polymers, such as conjugated microporous polymers and COFs, can significantly improve electrochemical performance due to the good accessibility of active sites by the electrolyte. Exfoliating layered COFs into nanosheets has been found to expose more active centers, facilitate reaction kinetics, and maximize the utilization of active materials. Additionally, taking advantage of high-density redox sites of organic materials, the hybridization with other inorganic electrode materials (such as MXenes, MnO2, MoO3, V2O5, MoS, etc.) can be an effective strategy to combine the respective advantages. These components can form hybrid composites through approaches such as physical mixing, covalent grafting, and in situ growth. In this regard, the selection of hybrid components and the construction of interfacial interactions would be the key factor that can determine the final electrochemical performances.
(5) The composition of electrolytes is crucial for optimizing the electrochemical performance of OEMs in MMBs. The electrolyte not only affects the plating/stripping behavior of metal anodes but also influences the electrochemical window, reaction kinetics, cycling stabilities, and electrochemical reactions of OEMs. It is essential to search for safe, non-toxic, and stable electrolytes that exhibit high ionic conductivity and enable efficient, dendrite-free metal stripping/deposition and charge carrier intercalation. For OEMs, choosing an appropriate electrolyte also involves considering compatibility with OEMs, electrochemical stability windows, and the solubility of OEMs in the electrolytes. For example, in non-aqueous MMBs, commonly used electrolytes are air-sensitive and high-cost ionic liquids or chloride-containing electrolytes. Although these electrolytes can facilitate efficient, dendrite-free metal stripping/deposition and charge carrier intercalation, they are highly corrosive to OEMs and can decompose their molecular structure. Additionally, some OEMs, such as dihydrophenazine, exhibit high redox potential, which necessitates electrolytes with high-potential stability.
(6) Although significant progress has been made, there is still a long way to go before achieving large-scale applications of OEMs for various MMB applications. Beyond the aforementioned solubility and conductivity issues, several other challenges restrict their further development. First, organic materials, composed of light elements, tend to have relatively low volumetric energy density. Second, while p-type organic materials can store anions, their redox reactions consume the electrolyte salts, which can lead to changes in electrolyte concentration. Third, redox-active polymer electrodes can be swollen by the electrolyte, resulting in unstable electrochemical performance. Lastly, most high-performance organic materials are synthesized through complex processes and conditions, inevitably increasing their production costs and limiting their practical applications. If these above-mentioned issues and challenges would be properly addressed, this would open a bright window for the commercial applications of organic MMBs in portable electronics, electric vehicles, grid storage systems, smart robotics, aerospace technology and other cost-effective energy storage fields.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by European Union's Horizon Europe research and innovation programme (ERC Starting Grant, BattSkin, 101116722), European Union's Horizon 2020 research and innovation programme (LIGHT-CAP 101017821), the ERC Consolidator Grant (T2DCP, 819698), German Research Foundation (DFG) within the Cluster of Excellence, CRC 1415 (Grant No. 417590517), and Polymer-based Batteries (SPP 2248, RACOF-NMA).References
- Z. Zhu, T. Jiang, M. Ali, Y. Meng, Y. Jin, Y. Cui and W. Chen, Chem. Rev., 2022, 122, 16610–16751 Search PubMed.
- Y. Liang and Y. Yao, Nat. Rev. Mater., 2023, 8, 109–122 Search PubMed.
- G. Wang, M. Yu and X. Feng, Chem. Soc. Rev., 2021, 50, 2388–2443 Search PubMed.
- J. Ding, H. Bin Yang, X.-L. Ma, S. Liu, W. Liu, Q. Mao, Y. Huang, J. Li, T. Zhang and B. Liu, Nat. Energy, 2023, 8, 1386–1394 Search PubMed.
- H. Wan, J. Xu and C. Wang, Nat. Rev. Chem., 2024, 8, 30–44 Search PubMed.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 Search PubMed.
- W. Wu, W. Luo and Y. Huang, Chem. Soc. Rev., 2023, 52, 2553–2572 Search PubMed.
- G. Brunklaus, P. Lennartz and M. Winter, Nat. Rev. Electr. Eng., 2024, 1, 79–92 Search PubMed.
- K. Qin, J. Huang, K. Holguin and C. Luo, Energy Environ. Sci., 2020, 13, 3950–3992 Search PubMed.
- Q. Guo, W. Li, X. Li, J. Zhang, D. Sabaghi, J. Zhang, B. Zhang, D. Li, J. Du, X. Chu, S. Chung, K. Cho, N. N. Nguyen, Z. Liao, Z. Zhang, X. Zhang, G. F. Schneider, T. Heine, M. Yu and X. Feng, Nat. Commun., 2024, 15, 2139 Search PubMed.
- H. Xu, T. Xie, Y. Li, F. Sun, C. Zhang, Z. Li, Y. Yao, Y. Li, Y. Zhan, X. Zou, C. Shi, Z. Wu, R. Laine and J. Zou, Adv. Energy Mater., 2024, 14, 2401154 Search PubMed.
- J. Kim, M. Kim, J. Lee, J. An, S. Yang, H. C. Ahn, D.-J. Yoo and J. W. Choi, Chem. Soc. Rev., 2024, 53, 8878–8902 Search PubMed.
- Y. Liu, G. He, H. Jiang, I. P. Parkin, P. R. Shearing and D. J. L. Brett, Adv. Funct. Mater., 2021, 31, 2010445 Search PubMed.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 Search PubMed.
- D. Sabaghi, J. Polčák, H. Yang, X. Li, A. Morag, D. Li, A. S. Nia, S. Khosravi H, T. Šikola, X. Feng and M. Yu, Adv. Energy Mater., 2024, 14, 2302961 Search PubMed.
- M. Yu, R. Dong and X. Feng, J. Am. Chem. Soc., 2020, 142, 12903–12915 Search PubMed.
- M. Yu, H. Shao, G. Wang, F. Yang, C. Liang, P. Rozier, C.-Z. Wang, X. Lu, P. Simon and X. Feng, Nat. Commun., 2020, 11, 1348 Search PubMed.
- A. Morag, X. Chu, M. Marczewski, J. Kunigkeit, C. Neumann, D. Sabaghi, G. Z. Żukowska, J. Du, X. Li, A. Turchanin, E. Brunner, X. Feng and M. Yu, Angew. Chem., Int. Ed., 2024, 63, e202401818 Search PubMed.
- J. Du, Y. Zhao, X. Chu, G. Wang, C. Neumann, H. Xu, X. Li, M. Löffler, Q. Lu, J. Zhang, D. Li, J. Zou, D. Mikhailova, A. Turchanin, X. Feng and M. Yu, Adv. Mater., 2024, 36, 2313621 Search PubMed.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964–974 Search PubMed.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54–70 Search PubMed.
- Z. Ju, T. Zheng, B. Zhang and G. Yu, Chem. Soc. Rev., 2024, 53, 8980–9028 Search PubMed.
- Y. Chen, K. Fan, Y. Gao and C. Wang, Adv. Mater., 2022, 34, 2200662 Search PubMed.
- S. Lee, J. Hong and K. Kang, Adv. Energy Mater., 2020, 10, 2001445 Search PubMed.
- J. Xie and Q. Zhang, Small, 2019, 15, 1805061 Search PubMed.
- A. Nimkar, G. Bergman, E. Ballas, N. Tubul, N. Levi, F. Malchik, I. Kukurayeve, M. S. Chae, D. Sharon, M. Levi, N. Shpigel, G. Wang and D. Aurbach, Angew. Chem., Int. Ed., 2023, 62, e202306904 Search PubMed.
- S. Haldar, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2023, 145, 13494–13513 Search PubMed.
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 Search PubMed.
- M. Li, J. Lu, X. Ji, Y. Li, Y. Shao, Z. Chen, C. Zhong and K. Amine, Nat. Rev. Mater., 2020, 5, 276–294 Search PubMed.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 Search PubMed.
- C. Yan, C. Lv, B.-E. Jia, L. Zhong, X. Cao, X. Guo, H. Liu, W. Xu, D. Liu, L. Yang, J. Liu, H. H. Hng, W. Chen, L. Song, S. Li, Z. Liu, Q. Yan and G. Yu, J. Am. Chem. Soc., 2022, 144, 11444–11455 Search PubMed.
- Q. Ran, H. Shi, H. Meng, S.-P. Zeng, W.-B. Wan, W. Zhang, Z. Wen, X.-Y. Lang and Q. Jiang, Nat. Commun., 2022, 13, 576 Search PubMed.
- C. Yan, C. Lv, L. Wang, W. Cui, L. Zhang, K. N. Dinh, H. Tan, C. Wu, T. Wu, Y. Ren, J. Chen, Z. Liu, M. Srinivasan, X. Rui, Q. Yan and G. Yu, J. Am. Chem. Soc., 2020, 142, 15295–15304 Search PubMed.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 Search PubMed.
- Y. Liu, Y. Lu, A. Hossain Khan, G. Wang, Y. Wang, A. Morag, Z. Wang, G. Chen, S. Huang, N. Chandrasekhar, D. Sabaghi, D. Li, P. Zhang, D. Ma, E. Brunner, M. Yu and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202306091 Search PubMed.
- A. Morag, X. Chu, C. Neumann, D. Pohl, M. Borrelli, D. Sabaghi, M. Löffler, Z. Sofer, A. Turchanin, M. Yu and X. Feng, Energy Storage Mater., 2022, 53, 435–443 Search PubMed.
- D. Sabaghi, Z. Wang, P. Bhauriyal, Q. Lu, A. Morag, D. Mikhailovia, P. Hashemi, D. Li, C. Neumann, Z. Liao, A. M. Dominic, A. S. Nia, R. Dong, E. Zschech, A. Turchanin, T. Heine, M. Yu and X. Feng, Nat. Commun., 2023, 14, 760 Search PubMed.
- W. Li, H. Ma, W. Tang, K. Fan, S. Jia, J. Gao, M. Wang, Y. Wang, B. Cao and C. Fan, Nat. Commun., 2024, 15, 9533 Search PubMed.
- Z. Huang, X. Li, Z. Chen, P. Li, X. Ji and C. Zhi, Nat. Rev. Chem., 2023, 7, 616–631 Search PubMed.
- X. Wu, Y. Xu, C. Zhang, D. P. Leonard, A. Markir, J. Lu and X. Ji, J. Am. Chem. Soc., 2019, 141, 6338–6344 Search PubMed.
- H. C. Bolton, A select bibliography of chemistry, 1492–1892, Smithsonian institution, 1893 Search PubMed.
- Y. Shi, Y. Chen, L. Shi, K. Wang, B. Wang, L. Li, Y. Ma, Y. Li, Z. Sun, W. Ali and S. Ding, Small, 2020, 16, 2000730 Search PubMed.
- S. P. Perlepes, T. Kabanos, V. Lazaridou and J. M. Tsangaris, Inorg. Chim. Acta, 1986, 117, 27–37 Search PubMed.
- C. Xu, H. Du, B. Li, F. Kang and Y. Zeng, Electrochem. Solid-State Lett., 2009, 12, A61 Search PubMed.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 Search PubMed.
- T. Zhang, Y. Tang, S. Guo, X. Cao, A. Pan, G. Fang, J. Zhou and S. Liang, Energy Environ. Sci., 2020, 13, 4625–4665 Search PubMed.
- Z. Yi, G. Chen, F. Hou, L. Wang and J. Liang, Adv. Energy Mater., 2021, 11, 2003065 Search PubMed.
- X. Zhang, J.-P. Hu, N. Fu, W.-B. Zhou, B. Liu, Q. Deng and X.-W. Wu, InfoMat, 2022, 4, e12306 Search PubMed.
- G. Li, L. Sun, S. Zhang, C. Zhang, H. Jin, K. Davey, G. Liang, S. Liu, J. Mao and Z. Guo, Adv. Funct. Mater., 2024, 34, 2301291 Search PubMed.
- J. Ding, F. Li, X. Ren, Y. Liu, Y. Li, Z. Shen, T. Wang, W. Wang, Y.-G. Wang, Y. Cui, H. Yang, T. Zhang and B. Liu, Nat. Commun., 2024, 15, 3641 Search PubMed.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 Search PubMed.
- M. Zhang, R. Liang, T. Or, Y.-P. Deng, A. Yu and Z. Chen, Small Struct., 2021, 2, 2000064 Search PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 Search PubMed.
- H. Zhang, W. Wu, Q. Liu, F. Yang, X. Shi, X. Liu, M. Yu and X. Lu, Angew. Chem., Int. Ed., 2021, 60, 896–903 Search PubMed.
- P. Benjamin, The Voltaic Cell: Its Construction and Its Capacity, J. Wiley, 1893 Search PubMed.
- G. W. Heise, E. A. Schumacher and N. C. Cahoon, J. Electrochem. Soc., 1948, 94, 99 Search PubMed.
- P. R. Gifford and J. B. Palmisano, J. Electrochem. Soc., 1988, 135, 650 Search PubMed.
- G. R. Pastel, Y. Chen, T. P. Pollard, M. A. Schroeder, M. E. Bowden, A. Zheng, N. T. Hahn, L. Ma, V. Murugesan, J. Ho, M. Garaga, O. Borodin, K. Mueller, S. Greenbaum and K. Xu, Energy Environ. Sci., 2022, 15, 2460–2469 Search PubMed.
- C. Wu, S. Gu, Q. Zhang, Y. Bai, M. Li, Y. Yuan, H. Wang, X. Liu, Y. Yuan, N. Zhu, F. Wu, H. Li, L. Gu and J. Lu, Nat. Commun., 2019, 10, 73 Search PubMed.
- H. Xu, D. Zhu, W. Zhu, F. Sun, J. Zou, R. M. Laine and W. Ding, Chem. Eng. J., 2022, 428, 131031 Search PubMed.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265 Search PubMed.
- L. W. Gaddum and H. E. French, J. Am. Chem. Soc., 1927, 49, 1295–1299 Search PubMed.
- A. Brenner, J. Electrochem. Soc., 1971, 118, 99 Search PubMed.
- T. D. Gregory, R. J. Hoffman and R. C. Winterton, J. Electrochem. Soc., 1990, 137, 775 Search PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 Search PubMed.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 Search PubMed.
- H. Xu, Y. Li, D. Zhu, Z. Li, F. Sun, W. Zhu, Y. Chen, J. Zhang, L. Ren and S. Zhang, Adv. Energy Mater., 2022, 12, 2270159 Search PubMed.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 Search PubMed.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900–7904 Search PubMed.
- S. Li, J. Zhang, S. Zhang, Q. Liu, H. Cheng, L. Fan, W. Zhang, X. Wang, Q. Wu and Y. Lu, Nat. Energy, 2024, 9, 285–297 Search PubMed.
- S. Hou, X. Ji, K. Gaskell, P.-F. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 Search PubMed.
- L. F. Wan, B. R. Perdue, C. A. Apblett and D. Prendergast, Chem. Mater., 2015, 27, 5932–5940 Search PubMed.
- V. Duffort, X. Sun and L. F. Nazar, Chem. Commun., 2016, 52, 12458–12461 Search PubMed.
- D. Wang, X. Du, G. Chen, F. Song, J. Du, J. Zhao, Y. Ma, J. Wang, A. Du, Z. Cui, X. Zhou and G. Cui, Angew. Chem., Int. Ed., 2023, 62, e202217709 Search PubMed.
- R. J. Staniewicz, J. Electrochem. Soc., 1980, 127, 782 Search PubMed.
- D. Aurbach, R. Skaletsky and Y. Gofer, J. Electrochem. Soc., 1991, 138, 3536 Search PubMed.
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Nat. Mater., 2016, 15, 169–172 Search PubMed.
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2018, 17, 16–20 Search PubMed.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324–328 Search PubMed.
- S. Wang, K. V. Kravchyk, A. N. Filippin, U. Müller, A. N. Tiwari, S. Buecheler, M. I. Bodnarchuk and M. V. Kovalenko, Adv. Sci., 2018, 5, 1700712 Search PubMed.
- M. Angell, C.-J. Pan, Y. Rong, C. Yuan, M.-C. Lin, B.-J. Hwang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 834–839 Search PubMed.
- Q. Pang, J. Meng, S. Gupta, X. Hong, C. Y. Kwok, J. Zhao, Y. Jin, L. Xu, O. Karahan, Z. Wang, S. Toll, L. Mai, L. F. Nazar, M. Balasubramanian, B. Narayanan and D. R. Sadoway, Nature, 2022, 608, 704–711 Search PubMed.
- E. Faegh, B. Ng, D. Hayman and W. E. Mustain, Nat. Energy, 2021, 6, 21–29 Search PubMed.
- N. Jayaprakash, S. K. Das and L. A. Archer, Chem. Commun., 2011, 47, 12610–12612 Search PubMed.
- H. Yang, H. Li, J. Li, Z. Sun, K. He, H.-M. Cheng and F. Li, Angew. Chem., Int. Ed., 2019, 58, 11978–11996 Search PubMed.
- D. L. Williams, J. J. Byrne and J. S. Driscoll, J. Electrochem. Soc., 1969, 116, 2 Search PubMed.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 Search PubMed.
- D. G. Truhlar, C. J. Cramer, A. Lewis and J. A. Bumpus, J. Chem. Educ., 2004, 81, 596 Search PubMed.
- P. Winget, C. J. Cramer and D. G. Truhlar, Theor. Chem. Acc., 2004, 112, 217–227 Search PubMed.
- S. Lee, J. E. Kwon, J. Hong, S. Y. Park and K. Kang, J. Mater. Chem. A, 2019, 7, 11438–11443 Search PubMed.
- Y. Zhong, Y. Li, J. Meng, X. Lin, Z. Huang, Y. Shen and Y. Huang, Energy Storage Mater., 2021, 43, 492–498 Search PubMed.
- J. Zhang, Z. Song, L. Zhan, J. Tang, H. Zhan, Y. Zhou and C. Zhan, J. Power Sources, 2009, 186, 496–499 Search PubMed.
- Y. Gong, W. Zhang, Z. Liu, M. Fang, J. Yang, Y. Wang, M. Gao, J. Zhang, Q.-H. Yang and Z. Li, Adv. Mater., 2024, 36, 2312486 Search PubMed.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Macromol. Chem. Phys., 2009, 210, 1989–1995 Search PubMed.
- K. Koshika, N. Chikushi, N. Sano, K. Oyaizu and H. Nishide, Green Chem., 2010, 12, 1573–1575 Search PubMed.
- T. P. Nguyen, A. D. Easley, N. Kang, S. Khan, S.-M. Lim, Y. H. Rezenom, S. Wang, D. K. Tran, J. Fan, R. A. Letteri, X. He, L. Su, C.-H. Yu, J. L. Lutkenhaus and K. L. Wooley, Nature, 2021, 593, 61–66 Search PubMed.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 Search PubMed.
- G. Dai, Y. He, Z. Niu, P. He, C. Zhang, Y. Zhao, X. Zhang and H. Zhou, Angew. Chem., Int. Ed., 2019, 58, 9902–9906 Search PubMed.
- J. F. Parker, J. S. Ko, D. R. Rolison and J. W. Long, Joule, 2018, 2, 2519–2527 Search PubMed.
- Z. Wang, Q. Qi, W. Jin, X. Zhao, X. Huang and Y. Li, ChemSusChem, 2021, 14, 3858–3866 Search PubMed.
- X. Zhao, X. Qiu, H. Xue, S. Liu, D. Liang, C. Yan, W. Chen, Y. Wang and G. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202216713 Search PubMed.
- F. B. Kaufman, A. H. Schroeder, E. M. Engler, S. R. Kramer and J. Q. Chambers, J. Am. Chem. Soc., 1980, 102, 483–488 Search PubMed.
- C. N. Gannett, L. Melecio-Zambrano, M. J. Theibault, B. M. Peterson, B. P. Fors and H. D. Abruña, Mater. Rep.: Energy, 2021, 1, 100008 Search PubMed.
- R. Saha, K. Gupta and C. J. Gómez García, Cryst. Growth Des., 2024, 24, 2235–2265 Search PubMed.
- T. Ma, C.-H. Li, R. M. Thakur, D. P. Tabor and J. L. Lutkenhaus, Nat. Mater., 2023, 22, 495–502 Search PubMed.
- H. Xia, W. Zhang, S. Cao and X. Chen, ACS Nano, 2022, 16, 8525–8530 Search PubMed.
- J. C. Russell, V. A. Posey, J. Gray, R. May, D. A. Reed, H. Zhang, L. E. Marbella, M. L. Steigerwald, Y. Yang, X. Roy, C. Nuckolls and S. R. Peurifoy, Nat. Mater., 2021, 20, 1136–1141 Search PubMed.
- Y. Zhang, Y. Zhu, D. Lan, S. H. Pun, Z. Zhou, Z. Wei, Y. Wang, H. K. Lee, C. Lin, J. Wang, M. A. Petrukhina, Q. Li and Q. Miao, J. Am. Chem. Soc., 2021, 143, 5231–5238 Search PubMed.
- H. Liu, Y. Zhang, J. Dong, T. Ye, J. Hao, Y. Yang, X. Jiang, X. Kang, Y. Bando and X. Wang, J. Mater. Chem. A, 2018, 6, 14894–14902 Search PubMed.
- X. Gao, Y. Chen, C. Gu, J. Wen, X. Peng, J. Liu, Z. Zhang, Q. Zhang, Z. Liu and C. Wang, J. Mater. Chem. A, 2020, 8, 19283–19289 Search PubMed.
- Z. Ye, S. Xie, Z. Cao, L. Wang, D. Xu, H. Zhang, J. Matz, P. Dong, H. Fang, J. Shen and M. Ye, Energy Storage Mater., 2021, 37, 378–386 Search PubMed.
- X. Yang, Y. Ni, Y. Lu, Q. Zhang, J. Hou, G. Yang, X. Liu, W. Xie, Z. Yan, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202209642 Search PubMed.
- Y. Li, L. Liu, Y. Lu, R. Shi, Y. Ma, Z. Yan, K. Zhang and J. Chen, Adv. Funct. Mater., 2021, 31, 2102063 Search PubMed.
- C. Mirle, V. Medabalmi and K. Ramanujam, ACS Appl. Energy Mater., 2021, 4, 1218–1227 Search PubMed.
- C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, J. Mater. Chem. A, 2019, 7, 23378–23415 Search PubMed.
- G. Wang, E. Dmitrieva, B. Kohn, U. Scheler, Y. Liu, V. Tkachova, L. Yang, Y. Fu, J. Ma, P. Zhang, F. Wang, J. Ge and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202116194 Search PubMed.
- J. Lee, H. Kim and M. J. Park, Chem. Mater., 2016, 28, 2408–2416 Search PubMed.
- M. Lee, J. Hong, H. Kim, H.-D. Lim, S. B. Cho, K. Kang and C. B. Park, Adv. Mater., 2014, 26, 2558–2565 Search PubMed.
- P. G. Pickup and R. A. Osteryoung, J. Am. Chem. Soc., 1984, 106, 2294–2299 Search PubMed.
- C. Strietzel, M. Sterby, H. Huang, M. Strømme, R. Emanuelsson and M. Sjödin, Angew. Chem., Int. Ed., 2020, 59, 9631–9638 Search PubMed.
- F. Ye, Q. Liu, H. Dong, K. Guan, Z. Chen, N. Ju and L. Hu, Angew. Chem., Int. Ed., 2022, 61, e202214244 Search PubMed.
- S. Zheng, D. Shi, D. Yan, Q. Wang, T. Sun, T. Ma, L. Li, D. He, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117511 Search PubMed.
- A. Khayum M, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 Search PubMed.
- M. Yu, N. Chandrasekhar, R. K. M. Raghupathy, K. H. Ly, H. Zhang, E. Dmitrieva, C. Liang, X. Lu, T. D. Kühne, H. Mirhosseini, I. M. Weidinger and X. Feng, J. Am. Chem. Soc., 2020, 142, 19570–19578 Search PubMed.
- C. Wang, R. Li, Y. Zhu, Y. Wang, Y. Lin, L. Zhong, H. Chen, Z. Tang, H. Li, F. Liu, C. Zhi and H. Lv, Adv. Energy Mater., 2024, 14, 2302495 Search PubMed.
- H. Peng, S. Huang, V. Montes-García, D. Pakulski, H. Guo, F. Richard, X. Zhuang, P. Samorì and A. Ciesielski, Angew. Chem., Int. Ed., 2023, 62, e202216136 Search PubMed.
- H. Lyu, C. S. Diercks, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 6848–6852 Search PubMed.
- G. Zou, Z. Tian, V. S. Kale, W. Wang, S. Kandembeth, Z. Cao, J. Guo, J. Czaban-Jóźwiak, L. Cavallo, O. Shekhah, M. Eddaoudi and H. N. Alshareef, Adv. Energy Mater., 2023, 13, 2203193 Search PubMed.
- S. Zhang, Y.-L. Zhu, S. Ren, C. Li, X.-B. Chen, Z. Li, Y. Han, Z. Shi and S. Feng, J. Am. Chem. Soc., 2023, 145, 17309–17320 Search PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 Search PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 Search PubMed.
- A. Manthiram and J. B. Goodenough, Nat. Energy, 2021, 6, 844–845 Search PubMed.
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, 1065 Search PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 Search PubMed.
- X. Yang and A. L. Rogach, Adv. Energy Mater., 2020, 10, 2000288 Search PubMed.
- Y. Chen and C. Wang, Acc. Chem. Res., 2020, 53, 2636–2647 Search PubMed.
- X. Wang, W. Tang, Y. Hu, W. Liu, Y. Yan, L. Xu and C. Fan, Green Chem., 2021, 23, 6090–6100 Search PubMed.
- W. Luo, M. Allen, V. Raju and X. Ji, Adv. Energy Mater., 2014, 4, 1400554 Search PubMed.
- A. Abouimrane, W. Weng, H. Eltayeb, Y. Cui, J. Niklas, O. Poluektov and K. Amine, Energy Environ. Sci., 2012, 5, 9632–9638 Search PubMed.
- X. Han, C. Chang, L. Yuan, T. Sun and J. Sun, Adv. Mater., 2007, 19, 1616–1621 Search PubMed.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 Search PubMed.
- Y. Lu, Y. Cai, Q. Zhang and J. Chen, Adv. Mater., 2022, 34, 2104150 Search PubMed.
- L. Zhu, G. Ding, L. Xie, X. Cao, J. Liu, X. Lei and J. Ma, Chem. Mater., 2019, 31, 8582–8612 Search PubMed.
- X. Gao, X. Qi and X. Zhao, Energy Fuels, 2024, 38, 12221–12227 Search PubMed.
- B. Pan, D. Zhou, J. Huang, L. Zhang, A. K. Burrell, J. T. Vaughey, Z. Zhang and C. Liao, J. Electrochem. Soc., 2016, 163, A580 Search PubMed.
- H. Sano, H. Senoh, M. Yao, H. Sakaebe and T. Kiyobayashi, Chem. Lett., 2012, 41, 1594–1596 Search PubMed.
- H. Senoh, H. Sakaebe, H. Sano, M. Yao, K. Kuratani, N. Takeichi and T. Kiyobayashi, J. Electrochem. Soc., 2014, 161, A1315 Search PubMed.
- D. Z. Baofei Pan, J. Huang, L. Zhang, A. K. Burrell, J. T. Vaughey, Z. Zhang and C. Liao, J. Electrochem. Soc., 2016, 163, A580 Search PubMed.
- H. Xu, X. Zhang, T. Xie, Z. Li, F. Sun, N. Zhang, H. Chen, Y. Zhu, X. Zou and C. Lu, Energy Storage Mater., 2022, 46, 583–593 Search PubMed.
- J. Bitenc, T. Pavcnik, U. Kosir and K. Pirnat, Materials, 2020, 13, 506 Search PubMed.
- W. Ai, W. Zhou, Z. Du, C. Sun, J. Yang, Y. Chen, Z. Sun, S. Feng, J. Zhao, X. Dong, W. Huang and T. Yu, Adv. Funct. Mater., 2017, 27, 1603603 Search PubMed.
- T. Debashis, H. M. Viswanatha, M. N. K. Harish and S. Sampath, J. Electrochem. Soc., 2020, 167, 070561 Search PubMed.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 Search PubMed.
- D. J. Kim, D.-J. Yoo, M. T. Otley, A. Prokofjevs, C. Pezzato, M. Owczarek, S. J. Lee, J. W. Choi and J. F. Stoddart, Nat. Energy, 2019, 4, 51–59 Search PubMed.
- E. S. Klimov, A. V. Lobanov and G. A. Abakumov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1981, 30, 1664–1666 Search PubMed.
- W. Kaim, Acc. Chem. Res., 1985, 18, 160–166 Search PubMed.
- G. A. Razuvaev, G. A. Abakumov, E. S. Klimov, E. N. Gladyshev and P. Y. Bayushkin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1977, 26, 1034–1037 Search PubMed.
- G. Van Koten, J. T. B. H. Jastrzebski and K. Vrieze, J. Organomet. Chem., 1983, 250, 49–61 Search PubMed.
- P. E. Barker, A. Hudson and R. A. Jackson, J. Organomet. Chem., 1981, 208, C1–C2 Search PubMed.
- Y.-T. Kao, S. B. Patil, C.-Y. An, S.-K. Huang, J.-C. Lin, T.-S. Lee, Y.-C. Lee, H.-L. Chou, C.-W. Chen, Y. J. Chang, Y.-H. Lai and D.-Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 25853–25860 Search PubMed.
- L. Zhou, Z. Zhang, L. Cui, F. Xiong, Q. An, Z. Zhou, X.-F. Yu, P. K. Chu and K. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100354 Search PubMed.
- C. Han, H. Li, Y. Li, J. Zhu and C. Zhi, Nat. Commun., 2021, 12, 2400 Search PubMed.
- W. Huang, Z. Zhu, L. Wang, S. Wang, H. Li, Z. Tao, J. Shi, L. Guan and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 9162–9166 Search PubMed.
- S. Er, C. Suh, M. P. Marshak and A. Aspuru-Guzik, Chem. Sci., 2015, 6, 885–893 Search PubMed.
- Y. Li, H. Xu, H. Zhao, J. Li, T. Lu, M. Xu and L. Pan, Coord. Chem. Rev., 2024, 518, 216097 Search PubMed.
- H. Qiu, R. Hu, X. Du, Z. Chen, J. Zhao, G. Lu, M. Jiang, Q. Kong, Y. Yan, J. Du, X. Zhou and G. Cui, Angew. Chem., Int. Ed., 2022, 61, e202113086 Search PubMed.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 Search PubMed.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 Search PubMed.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 Search PubMed.
- B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893–962 Search PubMed.
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741 Search PubMed.
- T. Sun, S. Zheng, H. Du and Z. Tao, Nano-Micro Lett., 2021, 13, 204 Search PubMed.
- K. W. Nam, H. Kim, Y. Beldjoudi, T.-W. Kwon, D. J. Kim and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 2541–2548 Search PubMed.
- Y. Wang, C. Wang, Z. Ni, Y. Gu, B. Wang, Z. Guo, Z. Wang, D. Bin, J. Ma and Y. Wang, Adv. Mater., 2020, 32, 2000338 Search PubMed.
- H.-g Wang and X.-b Zhang, Chem. – Eur. J., 2018, 24, 18235–18245 Search PubMed.
- H. Yang and F. Xu, ChemPhysChem, 2021, 22, 1455–1460 Search PubMed.
- I. A. Rodríguez-Pérez, Y. Yuan, C. Bommier, X. Wang, L. Ma, D. P. Leonard, M. M. Lerner, R. G. Carter, T. Wu, P. A. Greaney, J. Lu and X. Ji, J. Am. Chem. Soc., 2017, 139, 13031–13037 Search PubMed.
- L. Cui, L. Zhou, K. Zhang, F. Xiong, S. Tan, M. Li, Q. An, Y.-M. Kang and L. Mai, Nano Energy, 2019, 65, 103902 Search PubMed.
- Y. Y. Yao, Y. Zhan, X. Y. Sun, Z. Li, H. Xu, R. M. Laine and J. X. Zou, Batteries, 2023, 9, 203 Search PubMed.
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J.-M. Tarascon, ChemSusChem, 2008, 1, 348–355 Search PubMed.
- H. Kim, D.-H. Seo, G. Yoon, W. A. Goddard, III, Y. S. Lee, W.-S. Yoon and K. Kang, J. Phys. Chem. Lett., 2014, 5, 3086–3092 Search PubMed.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861–868 Search PubMed.
- T. Yamashita, H. Momida and T. Oguchi, Electrochim. Acta, 2016, 195, 1–8 Search PubMed.
- Y. Wang, Y. Ding, L. Pan, Y. Shi, Z. Yue, Y. Shi and G. Yu, Nano Lett., 2016, 16, 3329–3334 Search PubMed.
- C. Wang, Y. Fang, Y. Xu, L. Liang, M. Zhou, H. Zhao and Y. Lei, Adv. Funct. Mater., 2016, 26, 1777–1786 Search PubMed.
- J. Tian, D. Cao, X. Zhou, J. Hu, M. Huang and C. Li, ACS Nano, 2018, 12, 3424–3435 Search PubMed.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 Search PubMed.
- Z. Tie, L. Liu, S. Deng, D. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 4920–4924 Search PubMed.
- R. Guo, Y. Wang, S. Heng, G. Zhu, V. S. Battaglia and H. Zheng, J. Power Sources, 2019, 436, 226848 Search PubMed.
- G. Sun, B. Yang, X. Chen, Y. Wei, G. Yin, H. Zhang and Q. Liu, Chem. Eng. J., 2022, 431, 134253 Search PubMed.
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 Search PubMed.
- X. Chen, H. Su, B. Yang, G. Yin and Q. Liu, Sustainable Energy Fuels, 2022, 6, 2523–2531 Search PubMed.
- Q. Wang, Y. Liu and P. Chen, J. Power Sources, 2020, 468, 228401 Search PubMed.
- Y. Gao, G. Li, F. Wang, J. Chu, P. Yu, B. Wang, H. Zhan and Z. Song, Energy Storage Mater., 2021, 40, 31–40 Search PubMed.
- S. Niu, Y. Wang, J. Zhang, Y. Wang, Y. Tian, N. Ju, H. Wang, S. Zhao, X. Zhang, W. Zhang, C. Li and H.-B. Sun, Small, 2024, 20, 2309022 Search PubMed.
- A. Casey, S. D. Dimitrov, P. Shakya-Tuladhar, Z. Fei, M. Nguyen, Y. Han, T. D. Anthopoulos, J. R. Durrant and M. Heeney, Chem. Mater., 2016, 28, 5110–5120 Search PubMed.
- H. Cha, H. N. Kim, T. K. An, M. S. Kang, S.-K. Kwon, Y.-H. Kim and C. E. Park, ACS Appl. Mater. Interfaces, 2014, 6, 15774–15782 Search PubMed.
- K. Moriyama, M. Kuramochi, K. Fujii, T. Morita and H. Togo, Angew. Chem., Int. Ed., 2016, 55, 14546–14551 Search PubMed.
- F. Guo, Z. Huang, M. Wang, W.-L. Song, A. Lv, X. Han, J. Tu and S. Jiao, Energy Storage Mater., 2020, 33, 250–257 Search PubMed.
- J. Bitenc, T. Pavčnik, U. Košir and K. Pirnat, Materials, 2020, 13, 506 Search PubMed.
- Z. Chen, Y. Zhao, F. Mo, Z. Huang, X. Li, D. Wang, G. Liang, Q. Yang, A. Chen, Q. Li, L. Ma, Y. Guo and C. Zhi, Small Struct., 2020, 1, 2000005 Search PubMed.
- L. Yan, X. Zeng, S. Zhao, W. Jiang, Z. Li, X. Gao, T. Liu, Z. Ji, T. Ma, M. Ling and C. Liang, ACS Appl. Mater. Interfaces, 2021, 13, 8353–8360 Search PubMed.
- Q. Zou, Y. Sun, Z. Liang, W. Wang and Y. C. Lu, Adv. Energy Mater., 2021, 11, 2101552 Search PubMed.
- J. Sun, C. Deng, Y. Bi, K.-H. Wu, S. Zhu, Z. Xie, C. Li, R. Amal, J. Luo, T. Liu and D.-W. Wang, ACS Appl. Energy Mater., 2020, 3, 2516–2525 Search PubMed.
- Y. Zhang, Y. Liang, H. Dong, X. Wang and Y. Yao, J. Electrochem. Soc., 2020, 167, 070558 Search PubMed.
- J. Bitenc, K. Pirnat, G. Mali, B. Novosel, A. Randon Vitanova and R. Dominko, Electrochem. Commun., 2016, 69, 1–5 Search PubMed.
- G. Dawut, Y. Lu, L. Miao and J. Chen, Inorg. Chem. Front., 2018, 5, 1391–1396 Search PubMed.
- J. Bitenc, K. Pirnat, T. Bančič, M. Gaberšček, B. Genorio, A. Randon-Vitanova and R. Dominko, ChemSusChem, 2015, 8, 4128–4132 Search PubMed.
- D. Bier, Z. Li, S. Klyatskaya, N. Sbei, A. Roy, S. Riedel, M. Fichtner, M. Ruben and Z. Zhao-Karger, ChemSusChem, 2023, 16, e202300932 Search PubMed.
- J. Bitenc, A. Scafuri, K. Pirnat, M. Lozinšek, I. Jerman, J. Grdadolnik, B. Fraisse, R. Berthelot, L. Stievano and R. Dominko, Batteries Supercaps, 2021, 4, 214–220 Search PubMed.
- B. Pan, J. Huang, Z. Feng, L. Zeng, M. He, L. Zhang, J. T. Vaughey, M. J. Bedzyk, P. Fenter, Z. Zhang, A. K. Burrell and C. Liao, Adv. Energy Mater., 2016, 6, 1600140 Search PubMed.
- O. Buyukcakir, R. Yuksel, F. Begar, M. Erdogmus, M. Arsakay, S. H. Lee, S. O. Kim and R. S. Ruoff, ACS Appl. Energy Mater., 2023, 6, 7672–7680 Search PubMed.
- X. Fan, F. Wang, X. Ji, R. Wang, T. Gao, S. Hou, J. Chen, T. Deng, X. Li, L. Chen, C. Luo, L. Wang and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 7146–7150 Search PubMed.
- Y. J. Kim, W. Wu, S.-E. Chun, J. F. Whitacre and C. J. Bettinger, Adv. Mater., 2014, 26, 6572–6579 Search PubMed.
- X. Wang, J. Xiao and W. Tang, Adv. Funct. Mater., 2022, 32, 2108225 Search PubMed.
- N. Patil, A. Mavrandonakis, C. Jérôme, C. Detrembleur, J. Palma and R. Marcilla, ACS Appl. Energy Mater., 2019, 2, 3035–3041 Search PubMed.
- N. Patil, C. de la Cruz, D. Ciurduc, A. Mavrandonakis, J. Palma and R. Marcilla, Adv. Energy Mater., 2021, 11, 2100939 Search PubMed.
- A. Nimkar, F. Malchick, B. Gavriel, M. Turgeman, G. Bergman, T. Fan, S. Bublil, R. Cohen, M. Weitman, N. Shpigel, M. D. Levi and D. Aurbach, ACS Energy Lett., 2021, 6, 2638–2644 Search PubMed.
- H. Dong, Y. Liang, O. Tutusaus, R. Mohtadi, Y. Zhang, F. Hao and Y. Yao, Joule, 2019, 3, 782–793 Search PubMed.
- T. Bančič, J. Bitenc, K. Pirnat, A. Kopač Lautar, J. Grdadolnik, A. Randon Vitanova and R. Dominko, J. Power Sources, 2018, 395, 25–30 Search PubMed.
- J. Zhou, X. Yu, J. Zhou and B. Lu, Energy Storage Mater., 2020, 31, 58–63 Search PubMed.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 Search PubMed.
- Z. Xu, M. Li, W. Sun, T. Tang, J. Lu and X. Wang, Adv. Mater., 2022, 34, 2200077 Search PubMed.
- M. Mao, C. Luo, T. P. Pollard, S. Hou, T. Gao, X. Fan, C. Cui, J. Yue, Y. Tong, G. Yang, T. Deng, M. Zhang, J. Ma, L. Suo, O. Borodin and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 17820–17826 Search PubMed.
- X. Wang, J. Tang and W. Tang, Adv. Funct. Mater., 2022, 32, 2200517 Search PubMed.
- X. Peng, Y. Xie, A. Baktash, J. Tang, T. Lin, X. Huang, Y. Hu, Z. Jia, D. J. Searles, Y. Yamauchi, L. Wang and B. Luo, Angew. Chem., Int. Ed., 2022, 61, e202203646 Search PubMed.
- Y. Zhao, Y. Huang, F. Wu, R. Chen and L. Li, Adv. Mater., 2021, 33, 2106469 Search PubMed.
- Y. Xiu, A. Mauri, S. Dinda, Y. Pramudya, Z. Ding, T. Diemant, A. Sarkar, L. Wang, Z. Li, W. Wenzel, M. Fichtner and Z. Zhao-Karger, Angew. Chem., Int. Ed., 2023, 62, e202212339 Search PubMed.
- R. Zhou, Z. Hou, K. Fan, C. K. Wun, Q. Liu, T. W. Benedict Lo, H. Huang and B. Zhang, J. Power Sources, 2023, 569, 232995 Search PubMed.
- H. Zhang, L. Zhong, J. Xie, F. Yang, X. Liu and X. Lu, Adv. Mater., 2021, 33, 2101857 Search PubMed.
- W. Ma, L.-W. Luo, X. Huang, P. Dong, Y. Chen, C. Zhang, F. Huang, J.-X. Jiang and Y. Cao, Adv. Energy Mater., 2023, 13, 2203253 Search PubMed.
- Y. NuLi, Z. Guo, H. Liu and J. Yang, Electrochem. Commun., 2007, 9, 1913–1917 Search PubMed.
- B. Häupler, C. Rössel, A. M. Schwenke, J. Winsberg, D. Schmidt, A. Wild and U. S. Schubert, NPG Asia Mater., 2016, 8, e283–e283 Search PubMed.
- G. Studer, A. Schmidt, J. Büttner, M. Schmidt, A. Fischer, I. Krossing and B. Esser, Energy Environ. Sci., 2023, 16, 3760–3769 Search PubMed.
- H.-Y. Shi, Y.-J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 Search PubMed.
- H. W. Kim, H.-J. Kim, H. Byeon, J. Kim, J. W. Yang, Y. Kim and J.-K. Kim, J. Mater. Chem. A, 2020, 8, 17980–17986 Search PubMed.
- Y. Xie, K. Zhang, Y. Yamauchi, K. Oyaizu and Z. Jia, Mater. Horiz., 2021, 8, 803–829 Search PubMed.
- S. Jiang, Y. Xie, Y. Xie, L.-J. Yu, X. Yan, F.-G. Zhao, C. J. Mudugamuwa, M. L. Coote, Z. Jia and K. Zhang, J. Am. Chem. Soc., 2023, 145, 14519–14528 Search PubMed.
- Q. Chen, Y. N. Nuli, J. Yang, J. L. Wang and Y. G. Guo, Acta Phys.-Chim. Sin., 2013, 29, 2295–2299 Search PubMed.
- K. Zhang, Y. Xie, B. B. Noble, M. J. Monteiro, J. L. Lutkenhaus, K. Oyaizu and Z. Jia, J. Mater. Chem. A, 2021, 9, 13071–13079 Search PubMed.
- S.-Y. Ha, Y.-W. Lee, S. W. Woo, B. Koo, J.-S. Kim, J. Cho, K. T. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 4063–4073 Search PubMed.
- J. Tang and F. Xu, Mater. Lett., 2022, 320, 132365 Search PubMed.
- S. Wang, S. Huang, M. Yao, Y. Zhang and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 11800–11807 Search PubMed.
- S. Li, K. Shu, C. Zhao, C. Wang, Z. Guo, G. Wallace and H. K. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 16679–16686 Search PubMed.
- V. Raju, J. V. Rani and P. Basak, Electrochim. Acta, 2020, 361, 137097 Search PubMed.
- A. Lahiri, L. Yang, G. Li and F. Endres, ACS Appl. Mater. Interfaces, 2019, 11, 45098–45107 Search PubMed.
- N. S. Hudak, J. Phys. Chem. C, 2014, 118, 5203–5215 Search PubMed.
- M. Walter, K. V. Kravchyk, C. Böfer, R. Widmer and M. V. Kovalenko, Adv. Mater., 2018, 30, 1705644 Search PubMed.
- T. Schoetz, B. Craig, C. Ponce de Leon, A. Bund, M. Ueda and C. T. J. Low, J. Energy Storage, 2020, 28, 101176 Search PubMed.
- Y. Wang, H. Cui, R. Li, C. Yue, H. Pan, Z. Tang, X. Wang, Y. Lin, H. Li, C. Han, D. Nan, C. Zhi and H. Lv, Energy Storage Mater., 2024, 65, 103102 Search PubMed.
- A. B. Ikhe, N. Naveen, K.-S. Sohn and M. Pyo, Electrochim. Acta, 2018, 283, 393–400 Search PubMed.
- L. Li, G. Zhang, X. Deng, J. Hao, X. Zhao, H. Li, C. Han and B. Li, J. Mater. Chem. A, 2022, 10, 20827–20836 Search PubMed.
- O. Lužanin, R. Dantas, R. Dominko, J. Bitenc and M. Souto, J. Mater. Chem. A, 2023, 11, 21553–21560 Search PubMed.
- H. Gui and F. Xu, Mater. Lett., 2023, 346, 134549 Search PubMed.
- R. Sun, S. Hou, C. Luo, X. Ji, L. Wang, L. Mai and C. Wang, Nano Lett., 2020, 20, 3880–3888 Search PubMed.
- S. Li, Y. Liu, L. Dai, S. Li, B. Wang, J. Xie and P. Li, Energy Storage Mater., 2022, 48, 439–446 Search PubMed.
- W. Wang, V. S. Kale, Z. Cao, Y. Lei, S. Kandambeth, G. Zou, Y. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 Search PubMed.
- H. Lu, F. Ning, R. Jin, C. Teng, Y. Wang, K. Xi, D. Zhou and G. Xue, ChemSusChem, 2020, 13, 3447–3454 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |