
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral-at-metal catalysts: history, terminology, design, synthesis, and applications
Lilu
Zhang
and
Eric
Meggers
 *
*
Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse 4, 35043 Marburg, Germany. E-mail: meggers@chemie.uni-marburg.de
First published on 21st January 2025
Abstract
For decades, advances in chiral transition metal catalysis have been closely tied to the development of customized chiral ligands. Recently, however, an alternative approach to this traditional metal-plus-chiral-ligand method has emerged. In this new strategy, chiral transition metal catalysts are composed entirely of achiral ligands, with the overall chirality originating exclusively from a stereogenic metal center. This “chiral-at-metal” approach offers the benefit of structural simplicity. More importantly, by removing the need for chiral elements within the ligand framework, it opens up new possibilities for designing innovative catalyst architectures with unique properties. As a result, chiral-at-metal catalysis is becoming an increasingly important area of research. This review offers a comprehensive overview and detailed insights into asymmetric chiral-at-metal catalysis, encouraging scientists to explore new avenues in asymmetric transition metal catalysis and driving innovation in both fundamental and applied research.
 Lilu Zhang | Lilu Zhang received a Bachelor degree in Polymer Science from Nanjing University of Technology, P. R. China and a PhD degree in Inorganic Chemistry from Hong Kong Baptist University, Hong Kong. After postdoctoral research at the University of Texas at Austin and the University of Pennsylvania, USA, she started as an Assistant Professor in the Department of Chemistry and Biological Chemistry at Nanyang Technological University, Singapore. Since 2007, Lilu Zhang has been in the Department of Chemistry of the University of Marburg, Germany and currently holds the position of a staff scientist (Akademische Rätin). |
 Eric Meggers | Eric Meggers received a Diploma in Chemistry from the University of Bonn, Germany and a PhD degree in Organic Chemistry from the University of Basel, Switzerland. After postdoctoral research at the Scripps Research Institute, CA, USA, he started his independent career as Assistant Professor at the University of Pennsylvania, PA, USA. Since 2007, Eric Meggers has been Full Professor in the Department of Chemistry at the University of Marburg, Germany. He held a secondary appointment as Professor at the College of Chemistry and Chemical Engineering of Xiamen University, P. R. China from 2012 to 2016. His research program focuses on exploiting metal-centered stereochemistry for applications in medicine, chemical biology, and asymmetric catalysis. |
1. Introduction
1.1. Asymmetric catalysis
Asymmetric catalysis holds great promise as a highly cost-effective approach for producing enantiomerically pure chiral compounds, as a single chiral catalyst can amplify chiral information to produce many optically active chiral molecules.1,2 Chiral catalysts generally fall into three categories: biocatalysts (enzymes), chiral organic compounds (organocatalysts), and chiral metal complexes. It is unsurprising that chemists initially focused on developing synthetic chiral catalysts using transition metal complexes. The wide range of metals with varying oxidation states, diverse coordination geometries, and numerous ligand types allow transition metal catalysis to offer an extensive array of modes of activation and catalytic mechanisms.1.2. Chiral ligands for chiral transition metal catalysts
Asymmetric induction can be achieved when the metal complex itself is chiral. Traditionally, this chirality has been generated by combining metal salts or metal precursor complexes with one or more chiral ligands. This area of research, pioneered by Knowles,3 Noyori,4 Sharpless,5 Kagan,6 and others,2 has led to the development of numerous carefully tailored chiral ligands, including DuPhos, BINAP, PHOX, TADDOL, Salen, BOX, PyBOX, and Josiphos, among many others. These ligands, often referred to as “privileged chiral ligands,” have demonstrated exceptional versatility.7 As a result, chiral ligand design has been highly successful and central to asymmetric transition metal catalysis for over half a century (Fig. 1, left).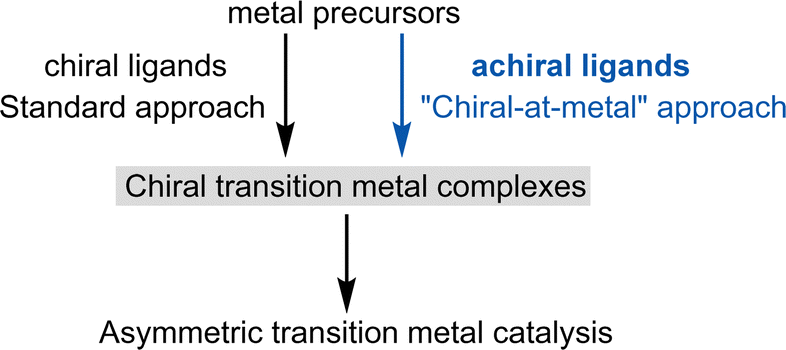 | ||
| Fig. 1 Chiral transition metal catalysis: chiral ligands versus achiral ligands for the design of chiral transition metal catalysts. | ||
1.3. Achiral ligands for chiral transition metal catalysts
However, since Alfred Werner's seminal work over a century ago,8 which first characterized chiral metal complexes and introduced the concept of metal-centered chirality, it has been well-established that chiral metal complexes can be formed entirely from achiral ligands. Despite this knowledge, chiral-at-metal complexes were largely overlooked in asymmetric transition metal catalysis until our group revealed their broad utility (Fig. 1, right).1.4. Terminology: “chirality-at-metal”
It is important to recognize that chiral metal complexes with chiral ligands often also feature a stereogenic metal center.9 To differentiate between chiral metal complexes that contain only achiral ligands and those that involve chiral ligands, we prefer the term “chiral-at-metal” (or more specifically, “chiral-at-iron”, “chiral-at-rhodium”, etc.). This terminology emphasizes that the overall chirality of the metal complex formally arises solely from the stereogenic metal center. Although the term “stereogenic-at-metal” has been used, we find it less precise, as a stereogenic metal center alone does not fully define the overall chirality.2. Early work
2.1. Seminal work of Alfred Werner (1911)
More than 100 years ago, Werner demonstrated that the octahedral coordination complex cis-[Co(en)2(NH3)Cl]Cl2 (en = 1,2-ethylenediamine) (M1) is chiral despite containing solely achiral ligands (Fig. 2a).10 Various terms have been used to describe this source of chirality, including “metal-centered chirality,” “metal centrochirality,” “chiral metal,” “chiral-at-metal,” and “stereogenic-at-metal”. However, it is important to note that chirality formally refers to the geometrical properties of an entire compound rather than a specific structural element. In Werner's complexes, chirality is a result of the helical arrangement of the two 1,2-ethylenediamine ligands, which form either a left-handed (Λ-configuration at the metal) or right-handed (Δ-configuration at the metal) helix.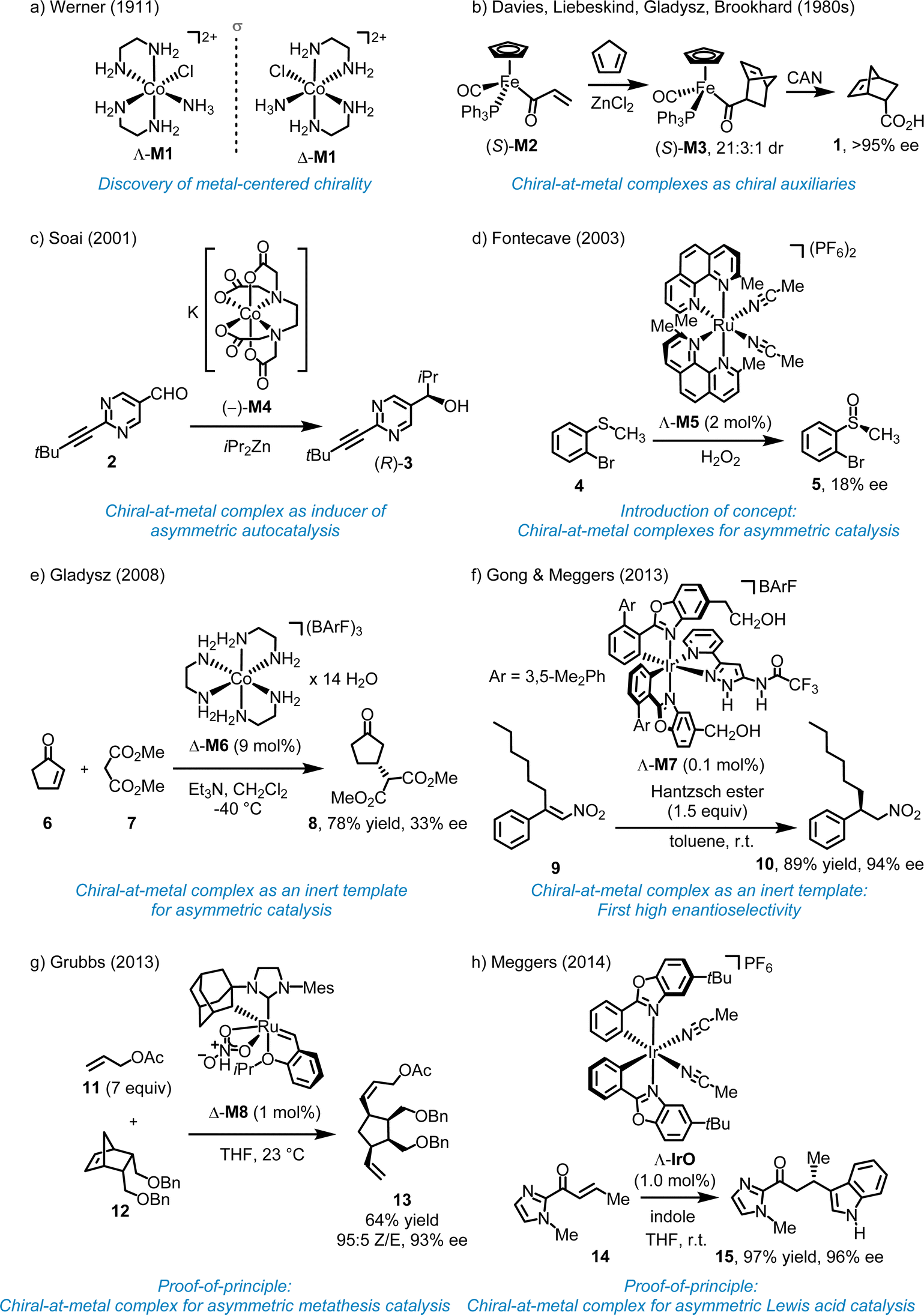 | ||
| Fig. 2 Early work on chiral-at-metal complexes and their applications. | ||
2.2. Chiral-at-metal complexes as chiral auxiliaries (1980s)
In 1969, Brunner reported the first optically active half-sandwich complex, featuring a central Mn ion asymmetrically coordinated to four distinct substituents in a pseudotetrahedral arrangement with sufficient configurational stability.11 In the 1980s, Davies, Gladysz, Brookhart, and Liebeskind initiated the first studies on related chiral-at-metal piano-stool half-sandwich complexes as tools for asymmetric organic synthesis, specifically using such chiral-at-metal complexes as chiral auxiliaries.12–17 For example, Davies demonstrated that the chiral-at-metal α,β-unsaturated acyl iron complex (S)-M2 can serve as a dienophile in the diastereoselective Diels–Alder reaction with cyclopentadiene, yielding the cycloaddition product (S)-M3.12 Following oxidative cleavage of the Fe–C bond using (NH4)2Ce(NO3)6 (CAN), endo-5-norbornene-2-carboxylic acid (1) was obtained with >95% enantiomeric excess (ee) (Fig. 2b). In this case, the enantiopure chiral-at-iron half-sandwich unit functioned as a chiral auxiliary for a diastereoselective cycloaddition.2.3. Chiral-at-metal complexes as inducer for asymmetric autocatalysis (2001)
Soai and coworkers reported that chiral-at-cobalt complexes, such as the EDTA complex (−)-M4, in which the chirality arises solely from the octahedral metal stereocenter, can act as chiral inducer in the enantioselective addition of diisopropylzinc to pyrimidine-5-carbaldehyde 2, producing the chiral alcohol (R)-3 with up to 94% ee (Fig. 2c).18 This reaction follows an autocatalytic mechanism, with the chiral cobalt complex functioning as an inducer rather than as a catalyst.2.4. First example of chiral-at-metal complex for asymmetric transition metal catalysis (2003)
In 2003, Fontecave proposed utilizing a reactive chiral-at-metal complex for asymmetric catalysis, with the metal acting simultaneously as the sole stereogenic center and the reactive site. Fontecave reported that cis-[Ru(2,9-Me2phen)2(MeCN)2] (PF6)2 (M5) catalyzes the oxidation of organic sulfides to sulfoxides (Fig. 2d).19 Although the ligands themselves are achiral, the cis-coordinated phenanthroline ligands induce helical chirality at the ruthenium center, resulting in either a Λ- or Δ-configured metal (left- or right-handed helix). However, the degree of asymmetric induction was very low, with the best result, the conversion 4 → 5, yielding only 18% ee, possibly due to a degradation of the ruthenium complex under the reaction conditions. As a result, while Fontecave introduced the concept of chiral-at-metal catalysis, the experiments did not serve as definitive proof-of-concept.2.5. Chiral-at-metal complexes as inert templates for asymmetric catalysis (since 2008)
In 2008, Gladysz reported that the Δ-enantiomer of the simple cobalt(III) complex [Co(en)3](BArF)3 (M6) with the large counterion tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF) catalyzes the Michael reaction of 2-cyclopenten-1-one (6) with dimethyl malonate (7) to provide the Michael adduct 8, although the enantioselectivity was low (33% ee) (Fig. 2e).20 Because of the inertness of the cobalt complex, catalysis must occur through interactions with the ligand sphere. In 2013, Gong and Meggers demonstrated the merit of this approach by introducing the chiral-at-metal complex Λ-M7 as a highly efficient catalyst for the enantioselective hydrogenation of nitro alkenes using Hantzsch ester (Fig. 2f).21 For instance, the β,β-disubstituted nitroalkene 9 was reduced to the corresponding nitroalkane 10 with 94% ee and 89% yield, using just 0.1 mol% of the chiral-at-iridium catalyst Λ-M7. Mechanistically, since the iridium complex is completely substitutionally inert, the metal center functions solely as a structural center, with catalysis occurring through the ligand sphere via hydrogen bonds and van der Waals interactions with the nitroalkene and Hantzsch ester. Subsequent publications further showcased the potential of octahedral chiral-at-metal complexes as inert templates for asymmetric catalysis.22 However, this approach does not represent asymmetric transition metal catalysis in the real sense and can be rather classified as asymmetric organocatalysis with an inert metal template. In this context, it is also worth mentioning the pioneering work by Fu and coworkers who used planar-chiral sandwich complexes for asymmetric nuclephilic catalysis.23 Although the metal does formally not serve as a stereogenic center, the metal is instrumental for generating planar chirality.2.6. First examples of efficient asymmetric catalysis with chiral-at-metal complexes (2013–2014)
The development of chiral-at-metal catalysts capable of achieving high enantioselectivities represents a relatively recent advancement. In 2013, Hartung and Grubbs unveiled the chiral-at-ruthenium catalyst M8, designed for diastereo- and enantioselective ring-opening/cross-metathesis reactions (Fig. 2g).24 For instance, 1 mol% of Δ-M8 catalyzed the reaction of a norbornene derivative (12) with an excess of allyl acetate (11), providing diene 13 in 64% yield, with 95% Z-selectivity and 93% ee. It is worth noting that, in addition to the ruthenium center of complex M8, the ruthenium-bound carbon atom of the adamantyl group also acts as a stereogenic center. However, this stereogenicity is intrinsically tied to the metal–carbon bond, and the ligand becomes achiral when this bond is broken. In 2014, Meggers and coworkers introduced the first chiral-at-metal complex for enantioselective Lewis acid catalysis (Fig. 2h).25 For instance, the conjugate addition of indole to α,β-unsaturated acyl imidazole 14 afforded the addition product 15 with 97% yield and 96% ee using 1.0 mol% of Λ-IrO. This work introduced a general design strategy which ultimately highlighted the broad applicability and effectiveness of chiral-at-metal catalysts within asymmetric catalysis.3. General design strategy for chiral-at-metal catalysts
3.1. Challenge in designing chiral-at-metal catalysts
The primary challenge in developing chiral-at-metal catalysts lies in ensuring that the metal center functions both as a configurationally stable stereocenter and as a reactive metal site. Unlike chiral metal complexes where chiral ligands often thermodynamically favor a single metal-centered configuration, chiral-at-metal complexes are prone to losing stereochemical information at the metal center due to ligand dissociation or isomerization.93.2. General design strategy
We developed a widely applicable structural framework for chiral-at-metal catalysts, centered around octahedral complexes with two cis-coordinated bidentate ligands and two monodentate ligands (Fig. 3).26–28 All ligands are achiral, and the overall chirality arises from the helical arrangement of the bidentate ligands, which formally creates a stereogenic metal center with either a Λ-configuration (left-handed helix) or Δ-configuration (right-handed helix). While these complexes are typically C2-symmetric, lower symmetry is also possible. | ||
| Fig. 3 General design strategy for chiral-at-metal catalysts with an octahedral coordination geometry. | ||
The foundation of the chiral-at-metal catalyst design involves incorporating inert bidentate ligands to establish and preserve the absolute stereochemistry at the metal center, alongside labile monodentate ligands that facilitate access to the metal center for substrates or reagents. This is achieved by leveraging three key effects: (1) the chelate effect, (2) high ligand field stabilization energy (LFSE),29 and (3) the trans-effect.30 While the chelate effect provides some extra stabilization for the bidentate ligands over the monodentate ones, this alone does not ensure inertness. In our experience, it is essential to combine the chelate effect with a strong ligand field, as the kinetic and thermodynamic properties of transition metal complexes are closely tied to the LFSE. This effect is particularly pronounced in octahedral low-spin 18-electron complexes. Additionally, tailored chelate ligands with strong σ-donating (raising the eg* orbital) and π-accepting (lowering the t2g orbitals) properties are employed to maximize ligand field splitting. The strong σ-donating coordination sites also play a critical role by being positioned trans to the monodentate ligands, thereby destabilizing them via the trans-effect. Thus, the σ-donating sites serve a dual function: enhancing the inertness of the bidentate ligand framework while simultaneously labilizing the monodentate ligands. This design offers a blueprint for combining configurational stability of the metal stereocenter with a high lability of the monodentate ligands.
The most popular implementation of this design strategy is illustrated in Fig. 3a. In this C2-symmetric standard design, the two bidentate ligands are identical and their strongly σ-donating groups are positioned trans to the monodentate ligands (typically MeCN), which results in the strong labilization of these monodentate ligands. A modified version of this design (variation #1) involves a non-C2-symmetric diastereomer in which only one monodentate ligand is labile (Fig. 3b). Another variation (variation #2) introduces different bidentate ligands. Lastly, in variation #3, only one of the bidentate ligands includes a strongly σ-donating coordination site. As shown in Fig. 3, these different ligand arrangements directly influence the lability of the monodentate ligands, with only those positioned trans to a strongly σ-donating group being labile.
4. Asymmetric catalysis with chiral-at-metal complexes
4.1. Chiral-at-iridium catalysts
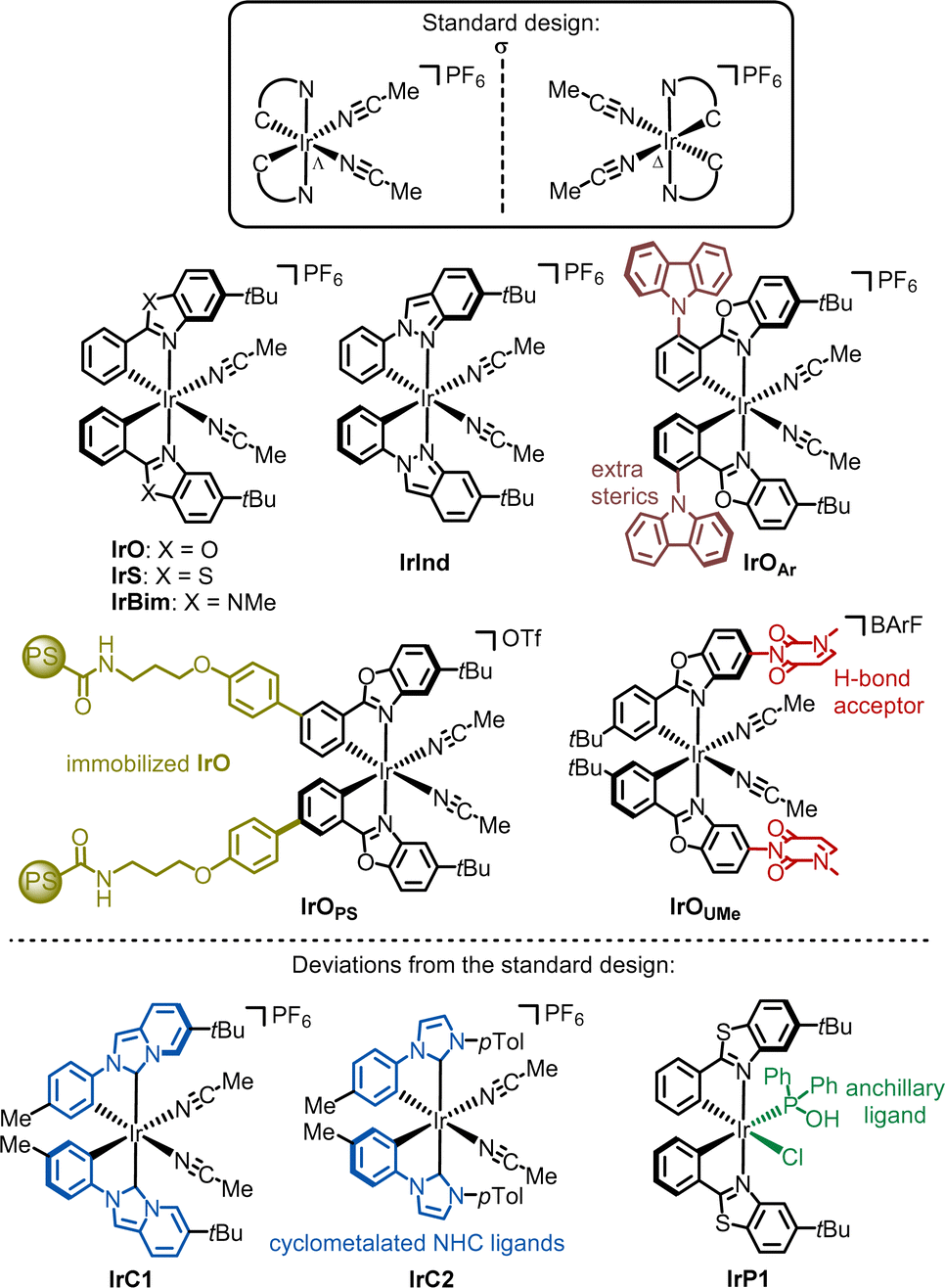 | ||
| Fig. 4 Overview of developed chiral-at-iridium catalysts. | ||
Deviations from the standard design have also been reported. For instance, IrC138 and IrC239 differ from the other chiral-at-iridium catalysts as they contain cyclometalated N-heterocyclic carbene (NHC) ligands. More recently, a bis-cyclometalated iridium catalyst featuring an anchillary phosphine oxide ligand (IrP1) was introduced.40
The synthesis of Λ- and Δ-IrS is shown as an example in Fig. 5.44 Accordingly, starting from IrCl3, the reaction with 2-phenylbenzothiazole (16) followed by the reaction with AgPF6 in MeCN provides rac-IrS (78%) in a diastereoselective manner as a racemate. The subsequent reaction of rac-IrS with the chiral thiazoline (S)-17 then produces the two diastereomeric complexes Λ-(S)-IrAux (45%) and Λ-(S)-IrAux (46%), which are separated using standard silica gel chromatography. These are then converted to virtually enantiopure Λ-IrS and Δ-IrS (92%, each >99% ee) by replacing the chiral auxiliary thiozoline ligand with two acetonitrile ligands through treatment with TFA in MeCN followed by NH4PF6 to obtain the hexafluorophosphate salts.
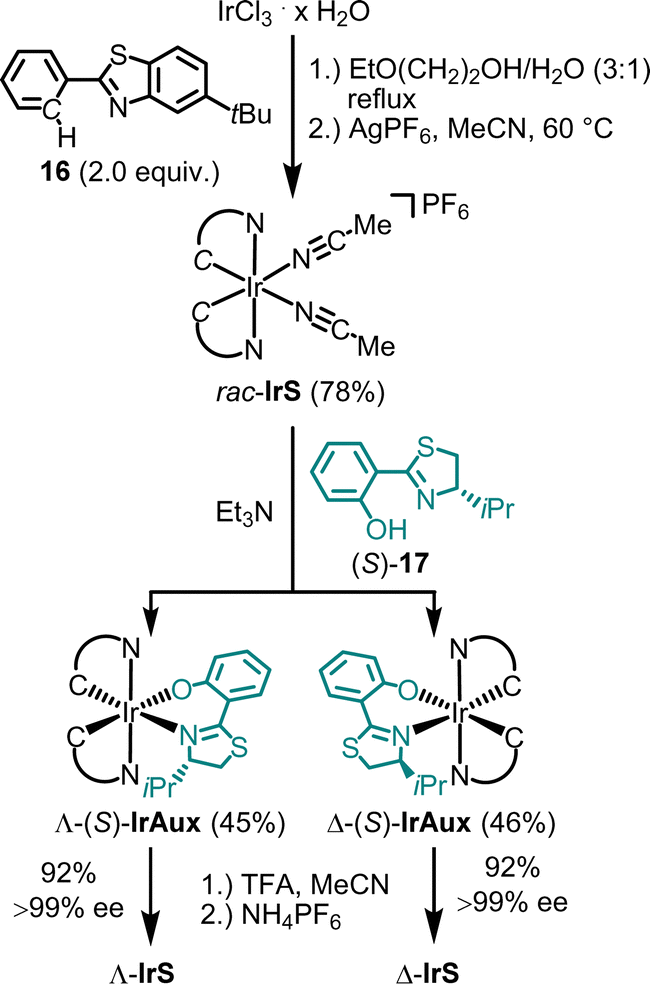 | ||
| Fig. 5 Chiral auxiliary-mediated synthesis of enantiomerically pure catalysts Λ-IrS and Δ-IrS. | ||
4.1.3.1. Lewis acid catalysis. In many of the reported cases, the chiral-at-iridium complexes function as chiral Lewis acids.26,31,32 What sets them apart from the wide variety of available chiral Lewis acid catalysts is their exceptional rigidity, which typically results in very high enantiomeric excess values. For instance, IrO, IrS, IrBim, and IrInd have been shown to act as chiral Lewis acid catalysts for the enantioselective conjugate addition of indoles to α,β-unsaturated 2-acyl imidazoles.25,34,45 In a specific example, the reaction of unsaturated 2-acyl imidazole 14 with indole, catalyzed by 1 or 2 mol% of chiral-at-iridium catalysts, yielded the Michael adduct 15 in 81–97% yield with 96–99% ee (Fig. 6a). Mechanistically, the bidentate N,O-coordination of the 2-acyl imidazole to the iridium center activates it toward attack by the nucleophilic indole (I). The chiral bis-cyclometalated iridium fragment effectively blocks the Re-face of the prochiral alkene (in the case of metal-centered Λ-configuration), directing the indole to the Si-face and providing strong asymmetric induction. In this and many other reported Michael reactions, using a variety of different Michael acceptors and nucleophiles, the benzothiazole-based catalysts Λ- and Δ-IrS consistently offer the best enantioselectivities. This can be attributed to the long C–S bond lengths in the benzothiazole, which position the tert-butyl groups somewhat closer to the catalytic site, enhancing steric hindrance. This often results in near-perfect asymmetric induction, with many examples achieving enantioselectivities of 99% ee.
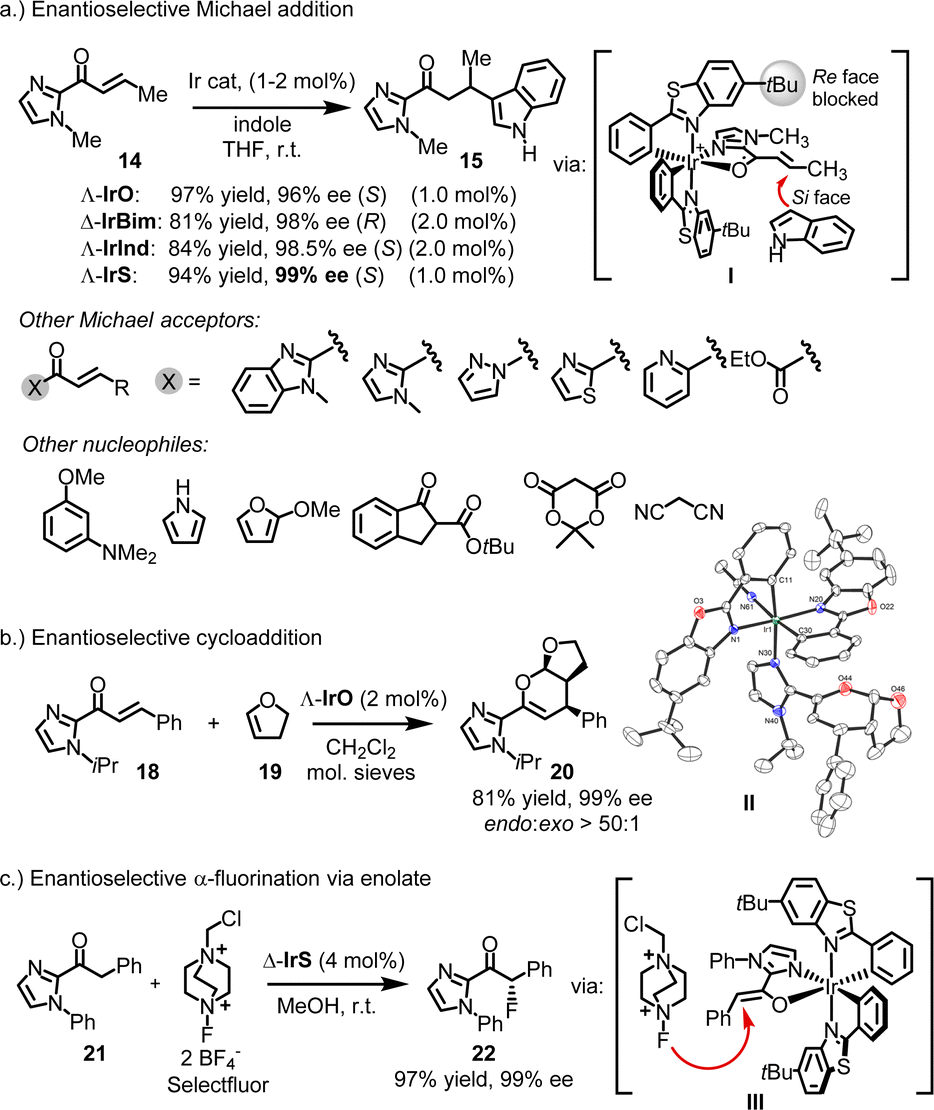 | ||
| Fig. 6 Chiral Lewis acid catalysis with chiral-at-iridium catalysts. This typically involves substrate activation through two-point binding by the iridium catalyst. | ||
There are instances where IrO outperforms IrS, such as in the hetero-Diels–Alder reaction between enone 18 and 1,2-dihydrofuran (19), producing the bicyclic product 20 with an 81% yield, excellent diastereoselectivity (endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :
:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exo > 50:1), and 99% ee (Fig. 6b).45 A crystal structure of the product coordinated to the catalyst could represent an intermediate in the catalytic cycle (II).
exo > 50:1), and 99% ee (Fig. 6b).45 A crystal structure of the product coordinated to the catalyst could represent an intermediate in the catalytic cycle (II).
Chiral-at-iridium complexes have also been employed in enolate chemistry. Xu reported remarkably high enantioselectivities in an enantioselective α-fluorination of 2-acyl imidazole 21 using Selectfluor, yielding the fluorinated product 22 with 97% yield and 99% ee.46 This reaction likely proceeds via an iridium enolate intermediate (III) (Fig. 6c).
Other reactions reported to be catalyzed by chiral-at-iridium complexes include asymmetric Nazarov cyclizations47 and the kinetic resolution of epoxides35 using CO2 to produce cyclic carbonates.
4.1.3.2. Asymmetric transfer hydrogenation. In addition to conventional chiral Lewis acid catalysis, bis-cyclometalated chiral-at-iridium complexes have been demonstrated to catalyze asymmetric hydrogenations48 and transfer hydrogenations.38,40,49,50 Mechanistically, these examples share the feature of an ancillary monodentate ligand coordinating to the iridium catalyst, facilitating hydrogen bonding with the substrate (see intermediates IV–VI in Fig. 7). For example, 2-acetyl benzothiophene (23) was reduced to its alcohol 24 with 93% yield and 99% ee, using ammonium formate as the reducing agent and only 0.2 mol% of the catalyst Λ-IrS (Fig. 7a).49 Remarkably, quantitative conversion with 98% ee was achieved even at a catalyst loading as low as 0.005 mol%. It is important to note that the presence of a pyrazole co-ligand (e.g.25) is essential for this catalysis, as it binds to the iridium catalyst, while reaction with ammonium formate generates an iridium hydride intermediate. This allows a Noyori–Ikariya-type transition state51 (IV), resulting in the concerted transfer of a hydride to the carbonyl carbon and a proton to the carbonyl oxygen.
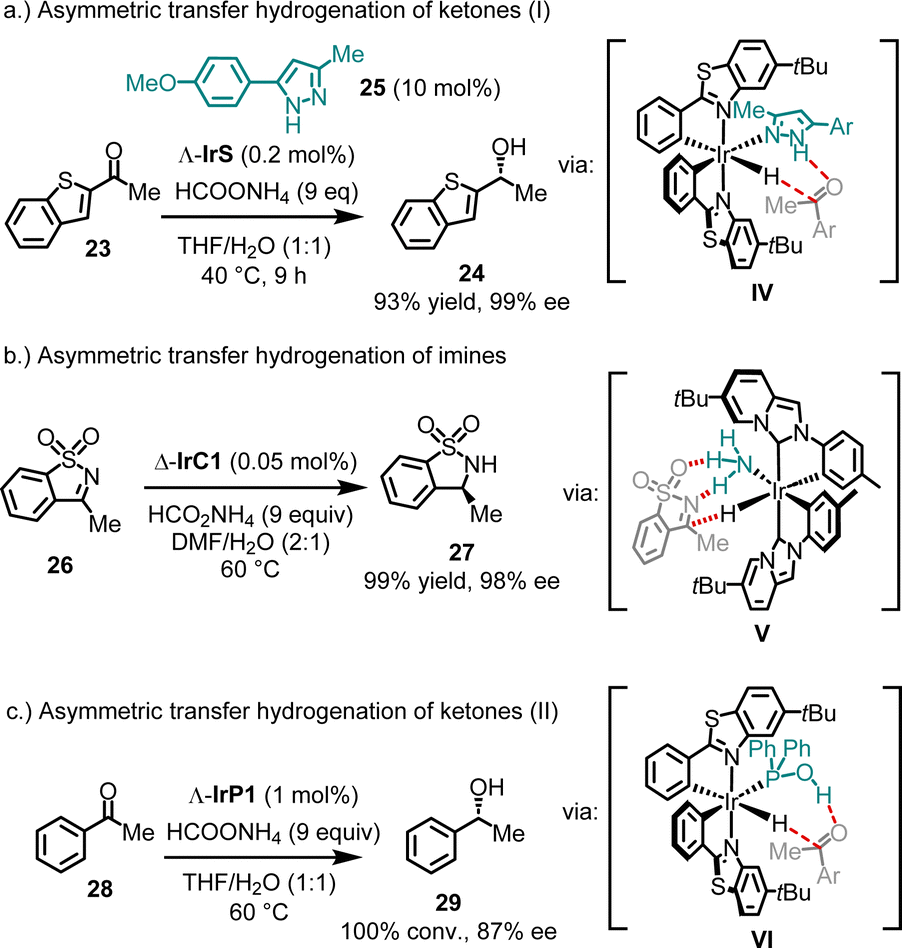 | ||
| Fig. 7 Asymmetric transfer hydrogenation with chiral-at-iridium complexes. | ||
In a related example, IrC1, bearing cyclometalated NHC ligands, was shown to catalyze the enantioselective reduction of sulfonylimines (Fig. 7b).38 For instance, using just 0.05 mol% of Δ-IrC1 with ammonium formate as the reducing agent, sulfonylimine 26 was converted to sultam 27 with 99% yield and 98% ee. Mechanistic studies indicated that in this system, NH3 acts as the ancillary ligand (V).
In contrast to these cases, where the ancillary ligand coordinates in situ, Pazos and Freixa reported that the complex IrP1, with a secondary phosphine oxide coordinated in its tautomeric hydroxy form, catalyzes the asymmetric transfer hydrogenation of acetophenone (28) to produce alcohol 29 with complete conversion and 87% ee (Fig. 7c).40 Here, the hydroxy group of the coordinated phosphine is proposed to form a hydrogen bond with the ketone in the transition state (VI).
4.1.3.3. Asymmetric photoredox catalysis. What truly distinguishes this class of chiral-at-iridium catalysts, however, is their capability to catalyze asymmetric photoreactions.26 Bis-cyclometalated iridium complexes are widely employed as photoredox catalysts or photosensitizers in contemporary organic photochemistry.52 In the realm of asymmetric photochemistry, these complexes are often paired with a chiral catalyst to achieve dual catalysis.53 Notably, we have documented several instances where IrO or IrS act as the sole catalyst for asymmetric photoredox catalysis.33,54–57 For example, visible light-induced radical α-alkylation of 2-acyl imidazole 30 with the electron-deficient benzyl bromide 31 afforded 32 quantitatively with 99% ee (via reaction of enolate intermediate VII with an electrophilic radical) (Fig. 8a),33 while the aminoalkylation of 2-acyl imidazole 33 with α-silylamine 34 under air afforded 35 with 92% yield and 97% ee (via the reaction of enolate intermediated VIII with an iminium ion) (Fig. 8b).54 In another case, IrS facilitated the reaction of trifluoromethylketones (e.g.36) with tertiary amines (e.g.37) to produce chiral 1,2-aminoalcohols (e.g.38) with 82% yield and 99% ee, through a photoredox-catalyzed radical–radical cross-coupling (intermediate IX) (Fig. 8c).57 In these asymmetric photoredox reactions, the chiral iridium catalyst functions both as a chiral Lewis acid and as a photocatalyst, following substrate binding.
 | ||
| Fig. 8 Asymmetric photoredox catalysis with chiral-at-iridium complexes. | ||
In all reported asymmetric photoreactions catalyzed by chiral-at-iridium complexes, it is noteworthy that the photoactive iridium species are generated in situ through substrate coordination, occasionally followed by deprotonation. The photochemical properties of these intermediates then govern the reaction pathways. The neutral iridium enolate intermediate (X) (Fig. 8a) functions as a strong photoactivated reductant. In contrast, the cationic iridium complex (XI), formed upon binding to a 2-acyl imidazole, acts as a weak photoactive oxidant (Fig. 8b). Meanwhile, coordination with more electron-deficient 2-trifluoroacetyl imidazoles (complex XII) generates a significantly stronger light-activated oxidant (Fig. 8c).
In conclusion, it is striking that the structurally simple chiral-at-metal iridium complexes, IrO and IrS, are capable of catalyzing such intricate visible-light-driven transformations, where asymmetric catalysis closely cooperates with photoredox chemistry. Furthermore, it is important to highlight that, in these reactions, the metal center assumes multiple roles: it serves as the sole source of chirality, acts as the Lewis acid center, and is a crucial component of the photoactive species generated in situ.
4.2. Chiral-at-rhodium catalysts
 | ||
| Fig. 9 Overview of developed chiral-at-rhodium catalysts. | ||
Since the cyclometalating ligands and their substitution patterns significantly influence catalytic performance and enantioselectivity, several other rhodium catalysts have been developed. Kang reported RhO derivatives with 3,5-Me2Ph or 3,5-(CF3)2Ph moieties on the phenyl groups of the phenylbenzoxazole ligands (RhOAr).99,100 In another modification, Kang and Du replaced the cyclometalated phenyl moiety with a naphthyl (RhONaphth).110
We further developed the chiral-at-Rh catalyst RhS, the lighter analogue of IrS, which features cyclometalated phenylbenzothiazole ligands instead of phenylbenzoxazoles.61
Importantly, the benzothiazole catalyst RhS often offers better enantioselectivities than its benzoxazole counterpart RhO. This can be attributed to the longer C–S bonds in the benzothiazole of RhS compared to the C–O bonds in the benzoxazole of RhO, bringing the tert-butyl groups of RhS closer to the active site (near the two labile acetonitrile ligands) and enhancing asymmetric induction (see Fig. 10 for superimposed RhS and RhO). The steric crowding at the active site can be further enhanced by substituting the tert-butyl groups with more bulky adamantyl (RhSAd)77 or trimethylsilyl (RhSTMS)85 groups. Another variant we developed is RhInd,87,93 the lighter congener of IrInd, where rhodium is cyclometalated by two phenylindazole ligands. We also introduced chiral-at-rhodium catalysts with two different cyclometalating ligands, such as RhMix, which incorporates both phenylbenzimidazole and phenylpyrazole ligands.86 Finally, the pre-catalyst RhO-βPhe disintegrates in situ to form a chiral-at-rhodium Lewis acid and β-phenylalanine to perform chiral Lewis acid/enamine co-catalysis.65
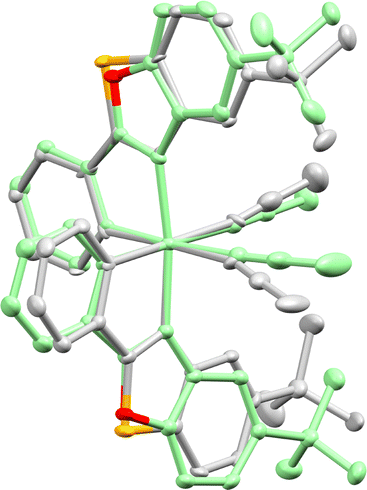 | ||
| Fig. 10 Superimposed crystal structure of RhS (grey, CCDC 1455731) and RhO (green, CCDC 1027145). Reproduced from ref. 61 with permission from The Royal Society of Chemistry, copyright 2016. | ||
All these chiral-at-rhodium complexes are air- and moisture-stable, tolerate silica gel chromatography, and remain fully configurationally stable in solution at room temperature.
:1 mixture of 2-ethoxyethanol and water under reflux afforded the rhodium dimer complex rac-RhOdim (62%). A subsequent reaction of rac-RhOdim with D-proline provided a mixture of diastereomers, Λ-(R)-RhProl and Δ-(R)-RhProl, which could not be separated by chromatography due to the limited stability of the complexes. However, we discovered that Δ-(R)-RhProl could be easily isolated in high purity with a 40% yield by washing the diastereomeric mixture with a CH2Cl2/diethyl ether solution, effectively exploiting the solubility difference between the two diastereomers. In an analogous fashion, the mirror-imaged complex Λ-(S)-RhProl can be synthesized using L-proline as the chiral auxiliary (36% yield). Exposure of the individual diastereomers to the weak acid NH4PF6 in MeCN at 50 °C, to labilize the prolinato ligand by protonation, resulted in a replacement of proline with two acetonitriles, thus affording Λ-RhO and Δ-RhO as single enantiomers (>99% ee). Thus, the abundant amino acid proline served as a convenient chiral auxiliary to generate enantiomerically pure chiral-at-rhodium catalysts Λ- and Δ-RhO.
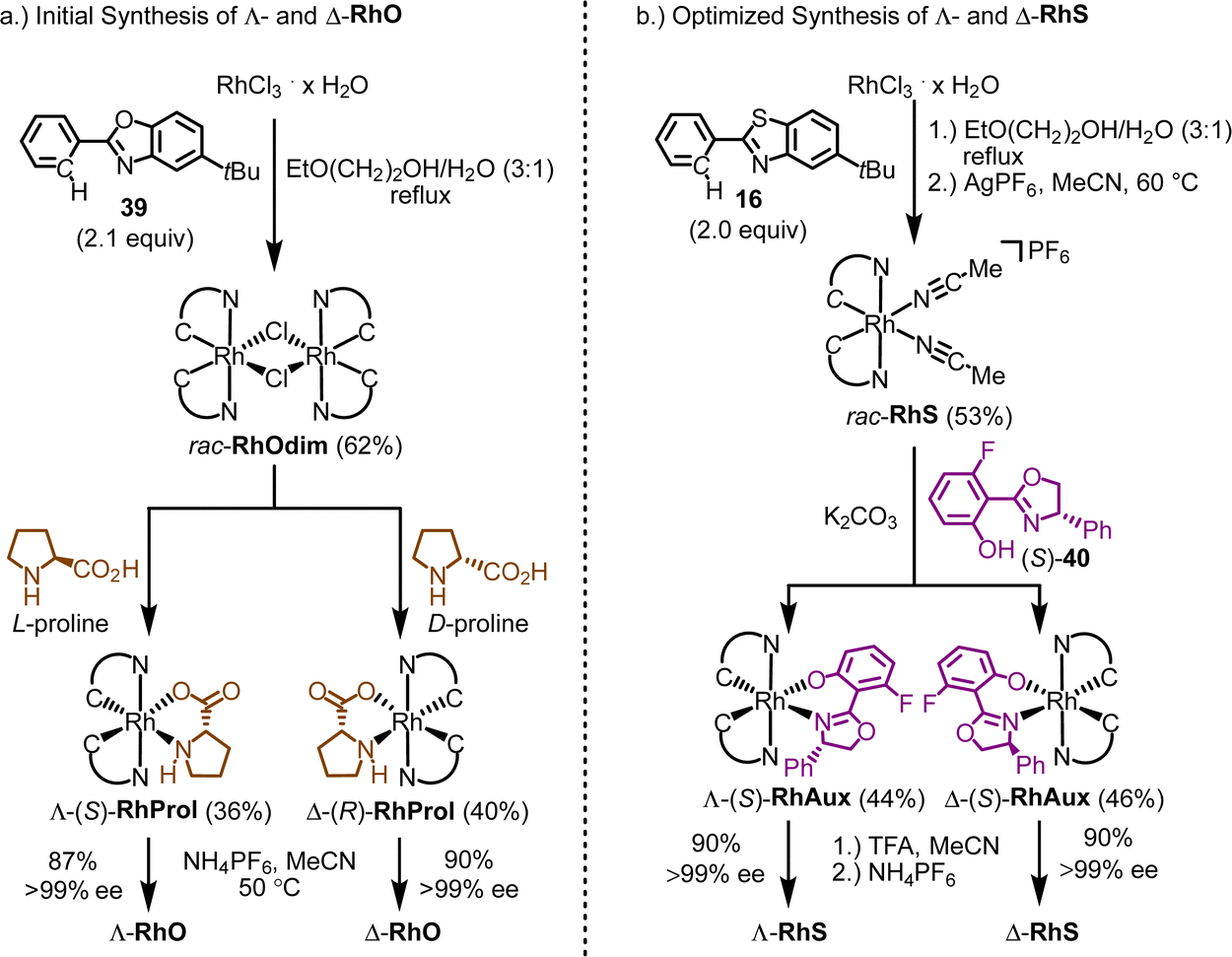 | ||
| Fig. 11 Chiral auxiliary-mediated synthesis of enantiomerically pure chiral-at-rhodium catalysts. (a) Λ- and Δ-RhO using L- and D-proline as chiral auxiliary ligands from the chiral pool. (b) Λ- and Δ-RhS using a fluorinated salicyloxazoline ligand. | ||
Finding a chiral-auxiliary-mediated method to generate enantiomerically pure RhS proved more challenging, as all initially tested chiral auxiliaries failed to afford intermediate rhodium auxiliary complexes with distinct solubility differences and lacked the stability required for separation by silica gel chromatography. Finally, a fluorinated salicyloxazoline turned out to be suitable (Fig. 11b).61 The auxiliary-mediated synthesis starts with rhodium trichloride hydrate which is first reacted with 5-tert-butyl-2-phenylbenzothiazole (16) followed by treatment with AgPF6 in MeCN to provide racemic RhS (73%). The complex rac-RhS is then reacted with the monofluorinated salicyloxazoline (S)-40 to provide a diastereomeric mixture of Λ-(S)-RhAux and Δ-(S)-RhAux. In contrast to RhProl, both diastereomers are sufficiently stable to be resolved into the single diastereomers (46% each) by silica gel chromatography. Alternatively, they can also be resolved based on their different solubilities in EtOH, or a combination of chromatography and exploiting solubility differences. Finally, starting with Λ-(S)-RhAux or Δ-(S)-RhAux, a TFA induced replacement of the coordinated auxiliary ligand with two acetonitriles under retention of configuration followed by treatment with NH4PF6 affords the individual enantiomers Λ-RhS (90%) and Δ-RhS (90%). A very detailed synthetic protocol is available.44
A crucial element of this auxiliary-mediated synthesis is the fluorinated salicyloxazoline ligand (S)-40 which was originally introduced by Ceroni and co-workers.130 The fluorine renders the coordinated phenolate less basic and thus stabilizes towards Lewis acid activation (such as silica gel) while the phenyl substituent of the oxazoline moiety undergoes π-stacking with the adjacent benzothiazole ligand (Fig. 12). These two aspects lead to an important improvement of the stability of the auxiliary complex so it can be handled and chromatographed without decomposition.
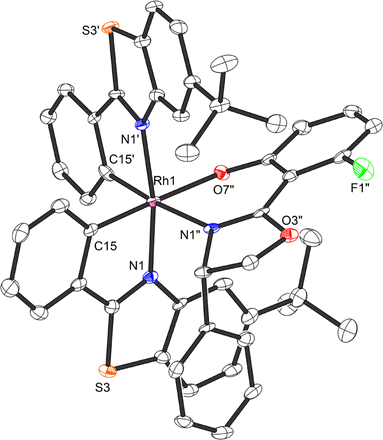 | ||
| Fig. 12 Crystal structure of the chiral auxiliary chiral-at-rhodium complex Λ-(S)-RhAux (CCDC 1455732). Reproduced from ref. 61 with permission from The Royal Society of Chemistry, copyright 2016. | ||
The first examples of chiral-at-rhodium catalysis were reported by our group in 2015 (Fig. 13a).58 For instance, Λ-RhO, at a catalyst loading of just 0.2 mol%, was shown to catalyze the enantioselective α-amination of 2-acyl imidazole 30 with dibenzyl azodicarboxylate (41), affording product 42 in 88% yield and 96% ee. In comparison, the heavier congener Λ-IrO required ten times the catalyst loading to achieve 86% yield and 92% ee. Mechanistically, the reaction is believed to proceed through a rhodium enolate intermediate (XIII), where the Si-face of the enolate's α-carbon is shielded by one of the tert-butyl groups, leading to an efficient asymmetric induction during the reaction of the rhodium enolate with the azodicarboxylate electrophile. Building on this initial work, chiral-at-rhodium complexes such as RhO, RhS, and their derivatives have been established by our group,58–95 as well as by Kang, Du, Su, Gong, Li and Xu,96–116,118–129 and others89,92,117 as highly versatile chiral Lewis acid catalysts for a broad range of reactions. In most catalytic processes, the substrate is activated through bidentate binding to the electrophilic rhodium center (e.g. see proposed catalytic intermediates XIII–XVII). Common substrates include 2-acyl imidazoles, N-acyl pyrazoles, 2-acyl pyridines, 1,3-dicarbonyl compounds, among others.
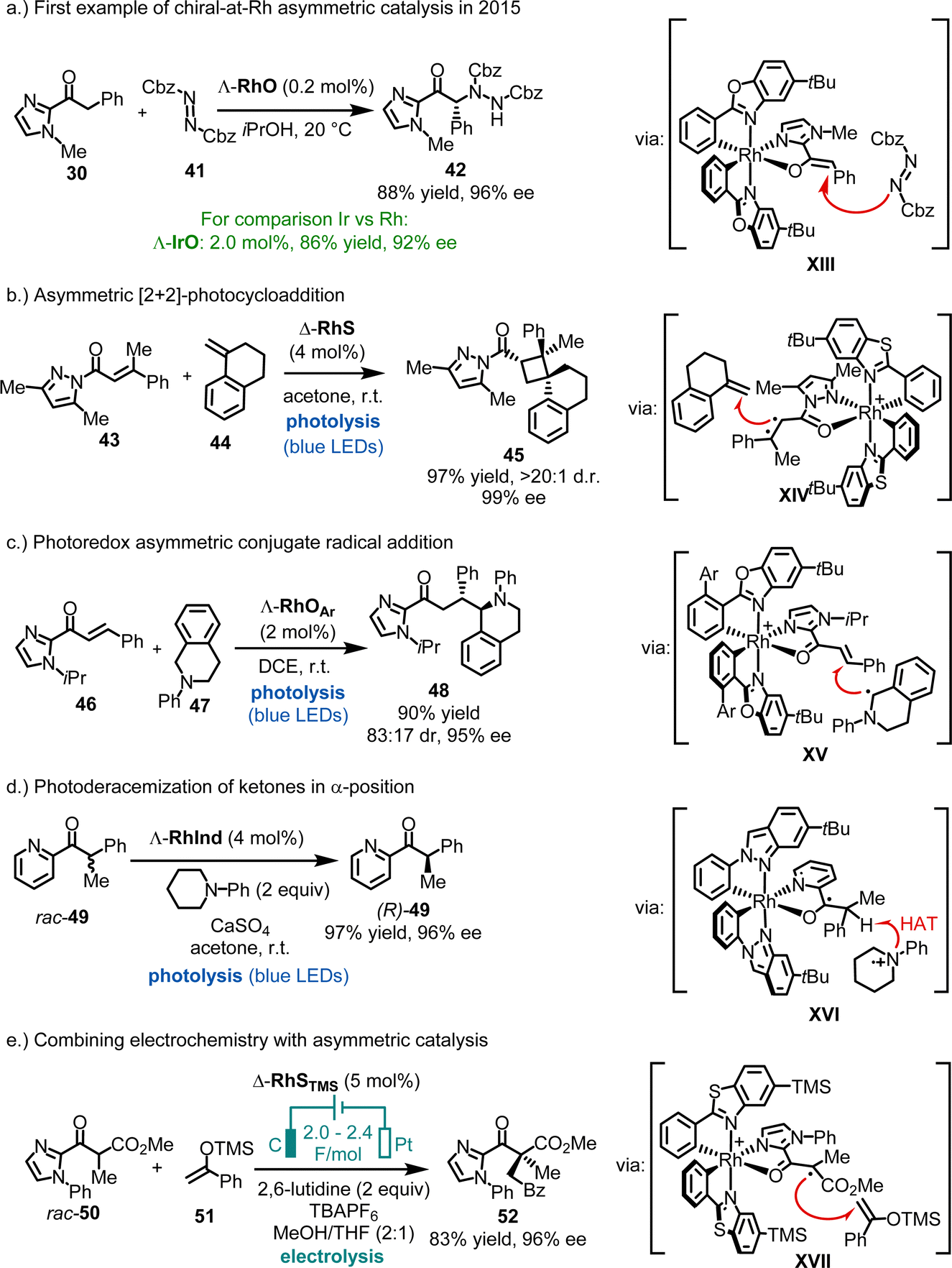 | ||
| Fig. 13 Catalytic applications of chiral-at-rhodium catalysts. | ||
Additionally, bis-cyclometalated rhodium complexes have proven to be excellent catalysts for asymmetric photochemistry by Meggers,27 Kang,102 and Alemán,117 whether employed in dual catalysis or as single catalysts. For example, we demonstrated highly enantioselective RhS-catalyzed [2+2] photocycloadditions under direct visible-light excitation (43 + 44 → 45) (Fig. 13b),73 while Kang reported a visible-light-induced asymmetric conjugate radical addition using RhOAr (Ar = 3,5-(CF3)2Ph) (46 + 47 → 48) (Fig. 13c).102 Recently, a bis-cyclometalated rhodium indazole complex (RhInd) was shown to catalyze the efficient α-photoderacemization of pyridylketone 49 (Fig. 13d).93 The manifold applications of bis-cyclometalated chiral-at-rhodium complexes for asymmetric photocatalysis was reviewed recently.27
Additionally, bis-cyclometalated rhodium complexes have proven to be effective catalysts for integrating electrochemistry with asymmetric catalysis.85,94 For instance, Meggers recently reported that RhSTMS catalyzes the oxidative cross-coupling of racemic 2-acyl imidazole rac-50 with silyl enol ether 51, providing a route to non-racemic 1,4-dicarbonyl compound 52 in 83% yield and 96% ee (Fig. 13e).85
In summary, since the first report in 2015,58 numerous studies have demonstrated the versatility of bis-cyclometalated chiral-at-rhodium complexes in asymmetric catalysis, particularly in reactions where the substrate binds via a two-point interaction. This versatility stems from their strong Lewis acid activity, in conjunction with photoactivity (triggered in situ upon substrate binding) and high chemical stability, making them suitable for a wide range of reaction types, including photoreactions, radical reactions, and redox processes.
4.3. Chiral-at-ruthenium and chiral-at-osmium catalysts
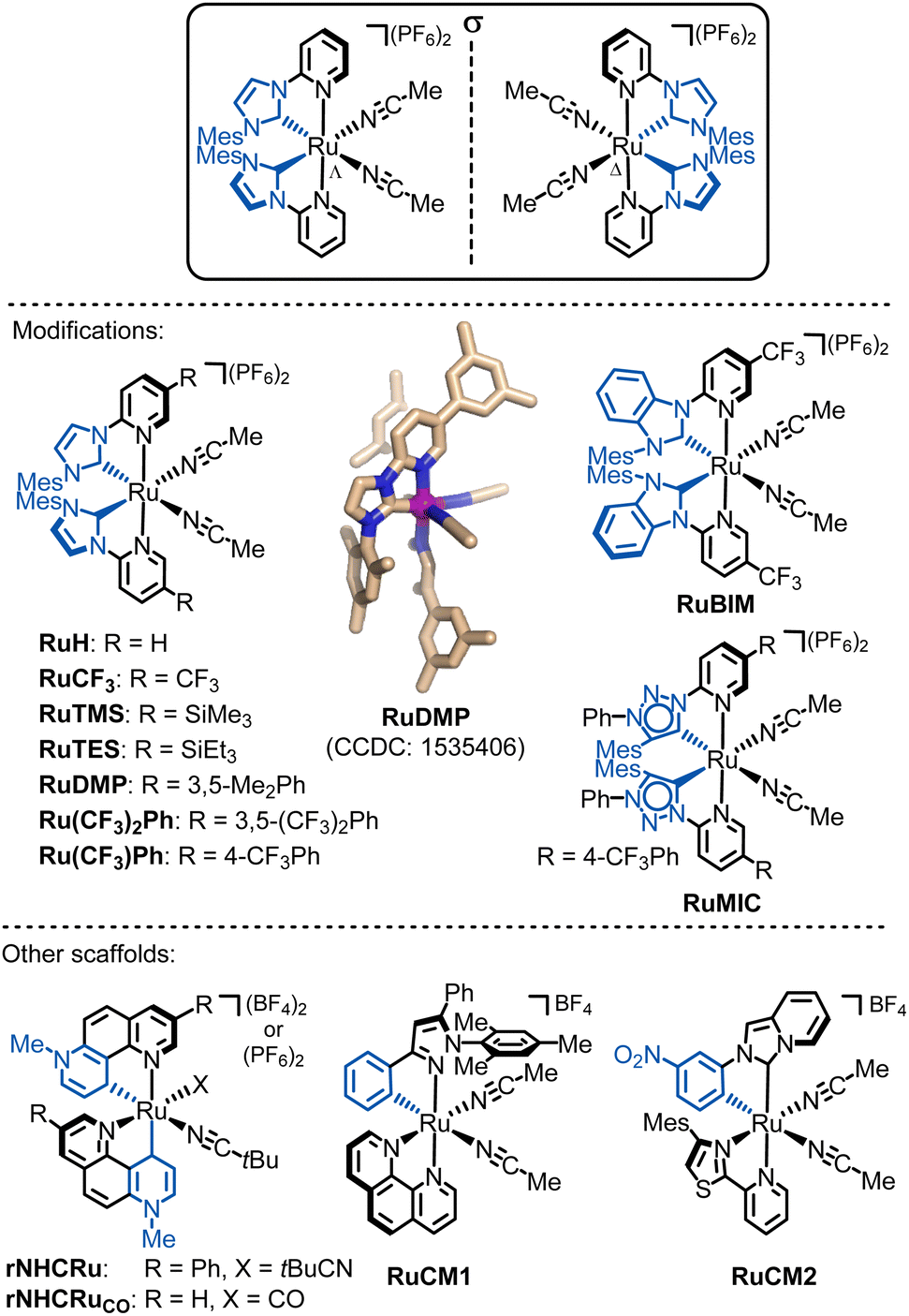 | ||
| Fig. 14 Overview of developed chiral-at-ruthenium catalyts. The shown crystal structure of RuDMP is reproduced from ref. 131 with permission from the American Chemical Society, copyright 2017. | ||
In a different design, our group in collaboration with Houk developed a chiral-at-ruthenium catalyst where ruthenium is cyclometalated by two 7-methyl-1,7-phenanthrolinium heterocycles in a non-C2-symmetric manner, forming chelating pyridylidene remote N-heterocyclic carbene ligands (rNHCs) (rNHCRu).146 Due to the lack of C2-symmetry, the two coordinated nitrile ligands are not equivalent. The strong σ-donating pyridylidene ligand renders the trans-positioned acetonitrile more labile, as evidenced by an elongated coordination bond. This catalyst demonstrated remarkable activity in the intramolecular C(sp3)–H amidation of 1,4,2-dioxazol-5-ones, leading to the formation of chiral γ-lactams. However, the C2-symmetric diastereomer promotes the undesired Curtius rearrangement. This example highlights the significance of both the relative stereochemistry (C2-symmetric vs. non-C2 symmetric diastereomer) and the absolute metal-centered stereochemistry (Λ versus Δ) in determining catalytic activity and enantioselectivity.
In addition to NHC, MIC, or rNHC groups as strongly σ-donating chelate ligands, incorporating even stronger σ-donating ligands, such as cyclometalating ligands that form metal-carbon σ-bonds, can increase the electron density at the ruthenium center, potentially yielding chiral-at-ruthenium catalysts with unique catalytic properties. For instance, the catalyst RuCM1,147 which features a cyclometalating phenylpyrazole ligand and coordinated 1,10-phenanthroline, is highly effective in converting diazoketones to chiral flavanones. Meanwhile, RuCM2,148 which features a cyclometalated N-(3-nitrophenyl)-imidazo[1,5-a]pyridinylidene ligand along with a bidentate 4-mesityl-2-(pyridin-2-yl)thiazole, catalyzes the enantioselective intramolecular cyclopropanation of trans-cinnamyl diazoacetate and an alkenyl diazoketone, leading to the formation of bicyclic cyclopropanes.
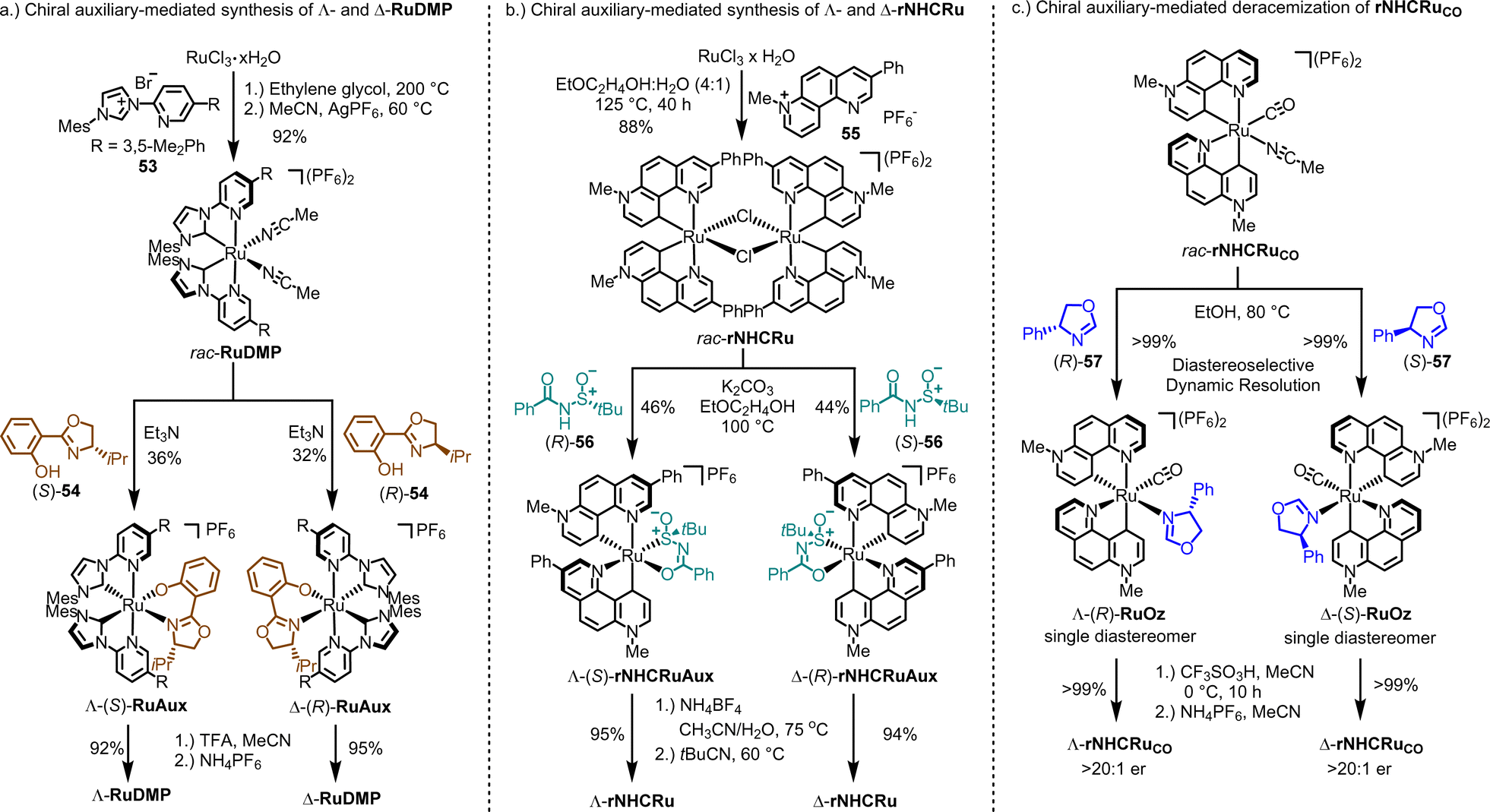 | ||
| Fig. 15 Chiral auxiliary-mediated synthesis of chiral-at-ruthenium catalysts. (a) Chiral auxiliary-mediated synthesis of Λ- and Δ-RuDMP. (b) Chiral auxiliary-mediated synthesis of Λ- and Δ-rNHCRu. (c) Synthesis of a non-racemic chiral-at-ruthenium catalyst Λ- and Δ-rNHCRuCO by chiral-auxiliary-mediated deracemization. | ||
The synthesis of enantiomerically pure Λ- and Δ-rNHCRu follows a similar overall strategy, but involves a different type of chiral auxiliaries and includes an unexpected isomerization.146 As depicted in Fig. 15b, reacting RuCl3 hydrate with 1,7-phenanthrolinium salt 55 in a 4:1 mixture of 2-ethoxyethanol and water at 125 °C results in the racemic chloro-bridged dimer complex rac-rNHCRu. Both ruthenium atoms are cyclometalated in a C2-symmetric manner by two 1,7-phenanthroline ligands, which can be described as chelating pyridyl pyridylidene ligands. In the subsequent step, the racemic mixture is reacted with either (R)- or (S)-N-benzoyl-tert-butanesulfinamide (56) in the presence of K2CO3, resulting in the formation of the single diastereomers, Λ-(S)-rNHCRuAux or Δ-(R)-rNHCRuAux, respectively. Remarkably, during the formation of these N-sulfinylcarboximidate complexes, an unexpected yet critical isomerization of the chelating pyridylidene ligands takes place. Finally, treatment with the weak acid NH4BF4, followed by reaction with pivalonitrile, yields the Λ-rNHCRu and Δ-rNHCRu catalysts.
All the methods discussed so far for generating chiral-at-metal catalysts have relied on chiral-auxiliary-ligand-mediated resolutions of racemic mixtures of the catalysts or its precursors. However, when the metal center exhibits lower configurational stability, such as at elevated temperature, alternative strategies become viable, allowing for the conversion of a racemic mixture into a single enantiomer of the catalyst. In line with this, we recently developed an auxiliary-mediated deracemization protocol for the synthesis of a non-C2-symmetric chiral-at-ruthenium catalyst (Fig. 15c).149 This catalyst features two cyclometalated 7-methyl-1,7-phenanthrolinium heterocycles, a CO ligand, and a labile MeCN ligand (rNHCRuCO, Fig. 14). When the monodentate chiral oxazoline ligand (R)-57 is coordinated in ethanol at 80 °C, the racemic mixture of the complex is converted into a single diastereomer, Λ-(R)-RuOz, with a quantitative yield through selective precipitation from the solution. At this elevated temperature, the two enantiomers of rNHCRuCO are in equilibrium, and coordination with (R)-57 induces selective precipitation of one diastereomer. After the oxazoline ligand is removed under acidic conditions, enantiomerically pure Λ-rNHCRuCO (>20:1 er) is obtained. The opposite enantiomer, Δ-rNHCRuCO, can be isolated using the chiral auxiliary ligand (S)-57 in an analogous fashion. While rNHCRuCO is not an especially versatile catalyst, it was shown to catalyze the enantioselective conversion of 1,2,4-dioxazol-5-ones to their chiral γ-lactams.149
4.3.3.1. Initial catalysis reports. The propeller-shaped Ru(PyNHC)2 catalysts were first employed in enantioselective alkynylation reactions involving trifluoromethyl ketones. For instance, at a catalyst loading of just 0.2 mol%, Λ-RuDMP efficiently catalyzes the alkynylation of 2,2,2-trifluoroacetophenone (58) with phenylacetylene (59), using catalytic amounts of the base Et3N, providing propargylic alcohol 60 with 98% yield and 99% ee (Fig. 16a).131 This approach was later utilized in the synthesis of key chiral intermediates for the AIDS drug Efavirenz.132 Mechanistically, experiments and DFT calculations by Chen and Houk are consistent with a mechanism in which the catalytic cycle proceeds via a ruthenium acetylide intermediate (XVIII), where the acetylide adds through an inner-sphere transfer to the ruthenium-bound trifluoromethyl ketone.133 The high rigidity of the propeller-shaped geometry contributes to the high enantioselectivity.
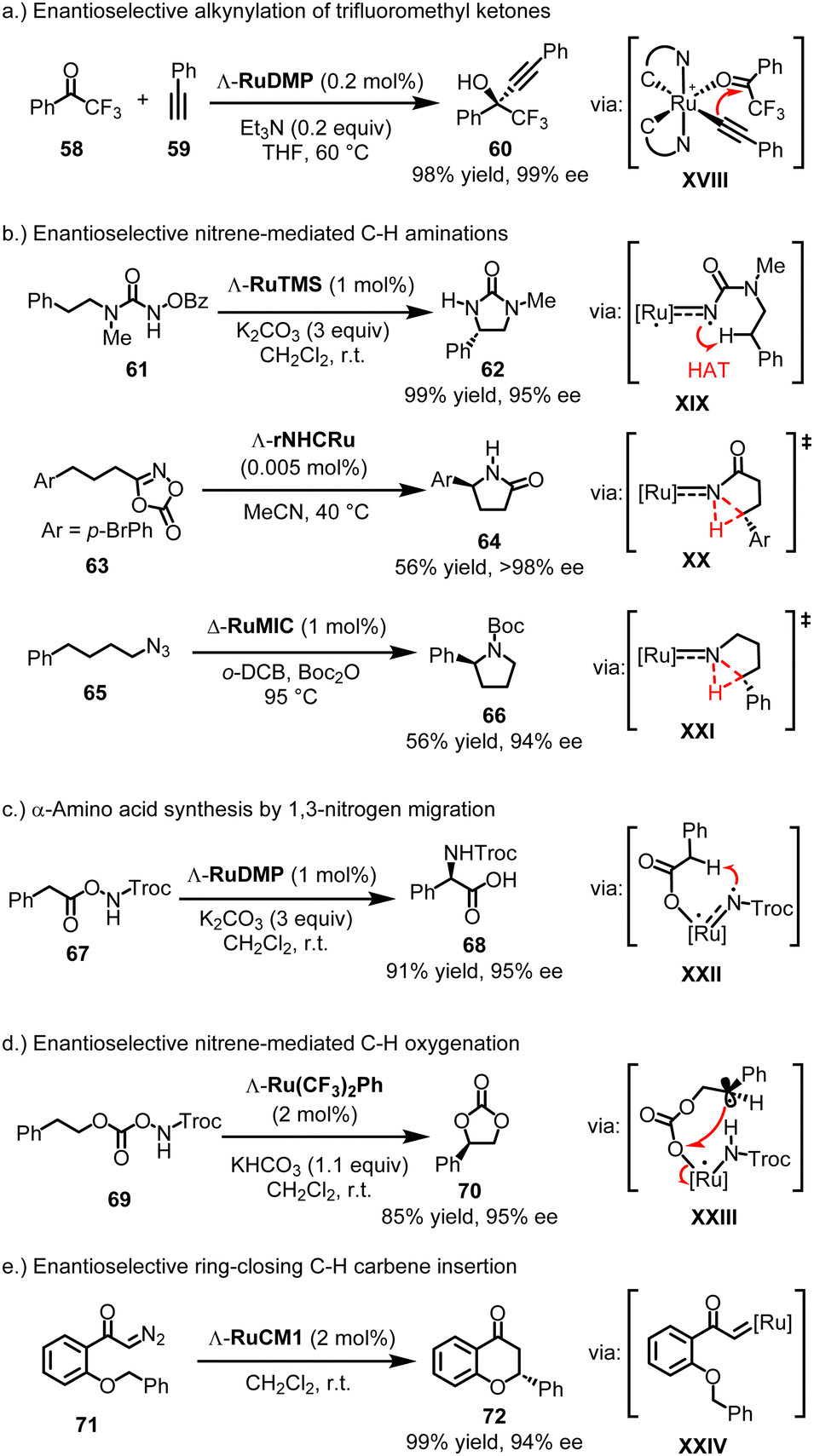 | ||
| Fig. 16 Catalytic applications of chiral-at-ruthenium catalysts. | ||
4.3.3.2. Nitrene-mediated C−H aminations. Subsequently, it was discovered that the Ru(PyNHC)2 complexes exhibit remarkable catalytic efficiency and stereocontrol in a large variety of nitrene-mediated asymmetric C(sp3)–H amination reactions.28 For example, Λ-RuTMS (1 mol%) catalyzes the ring-closing C–H amination of N-benzoyloxyurea 61, affording 2-imidazolidinone 62 in 99% yield and 95% ee, likely through a triplet ruthenium nitrene intermediate (XIX), followed by a 1,5-hydrogen atom transfer (HAT) and subsequent C–N bond formation (Fig. 16b).138 In contrast, the rNHC complex rNHCRu displays less versatility compared to the Ru(PyNHC)2 complexes. However, it shows exceptional catalytic performance in the ring-closing C–H amidation of dioxazolones.146 For instance, dioxazolone 63 was converted to chiral γ-lactam 64 in 56% yield and >98% ee using merely 0.005 mol% of Λ-rNHCRu. DFT calculations suggest a mechanism involving intramolecular C–H nitrene insertion via a ruthenium N-acylnitrene intermediate (XX). Regarding mesoionic chiral-at-ruthenium catalysts, the complex Δ-RuMIC (1 mol%) has been shown to catalyze the challenging ring-closing C–H amination of aliphatic azide 65, forming pyrrolidine 66 in 56% yield and 94% ee, likely through a ruthenium nitrene C–H insertion (XXI).145 Maybe most interesting is the discovery that the chiral-at-ruthenium Ru(PyNHC)2 complexes catalyze a novel 1,3-migratory nitrene C(sp3)–H insertion, which constitutes a highly straightforward and economic α-amino acid synthesis.143 For example, Λ-RuDMP (1 mol%) catalyzes the conversion of azanyl ester 67 to amino acid 68 in 91% yield and 95% ee (likely viaXXII) (Fig. 16c).
4.3.3.3. Nitrene-mediated C−H oxygenations. In addition to C–H aminations and amidations, chiral-at-ruthenium complexes have been reported to mediate uncommon nitrene-driven intramolecular C(sp3)–H oxygenations.137,142 For instance, Λ-Ru(CF3)2Ph (2 mol%) catalyzes the conversion of azanyl carbonate 69 to cyclic carbonate 70 with an 85% yield and 95% ee (likely viaXXIII) (Fig. 16d).142
4.3.3.4. Carbene C–H insertions. Finally, cyclometalated chiral-at-ruthenium catalysts have been reported to catalyze carbene chemistry such as enantioselective ring-closing C(sp3)–H carbene insertions.147,148 For example, Λ-RuCM1 (2 mol%) catalyzes the conversion of diazoketone 71 to flavanone 72 with a 99% yield and 94% ee (likely viaXXIV) (Fig. 16e).147
To summarize this section on chiral-at-ruthenium complexes, the cis-coordination of bidentate ligands to ruthenium results in a helical arrangement of the chelate ligands, creating a stereogenic metal center. Given the extensive development of ruthenium coordination chemistry and the numerous methods available for the stepwise or simultaneous introduction of ligands, a wide range of chiral-at-ruthenium catalysts have been created. Crucially, at least one of the bidentate ligands contains a strongly σ-donating group, such as a conventional NHC, a remote NHC, a mesoionic carbene, or a cyclometalating ligand. Among the various chiral-at-ruthenium catalysts, the C2-symmetric complexes [Ru(PyNHC)2(MeCN)2]2 (PF6)2 stand out due to their chemical robustness, high asymmetric induction, and strong catalytic performance, particularly in nitrene-mediated asymmetric reactions. This catalyst framework has been recently reviewed.28
 | ||
| Fig. 17 First example of a chiral-at-osmium catalysis. | ||
4.4. Chiral-at-iron catalysts
The development of reactive chiral-at-metal catalysts using earth-abundant metals is highly challenging due to the high lability of coordination bonds with 3d metals and the crucial need for a metal stereocenter that is configurationally stable. In 2019, our group reported the first chiral-at-iron catalyst, showcasing how ligand design can significantly influence both configurational stability and ligand lability.151 The catalyst employs the same ligand framework as related Ru(PyNHC)2 complexes.28 Specifically, iron is coordinated by two chelating N-(2-pyridyl)-substituted N-heterocyclic carbenes (PyNHC) in a C2-symmetrical arrangement, forming a helical structure with the iron center adopting either the Λ (left-handed) or Δ (right-handed) stereogenic configuration (Fig. 18). The coordination is completed by two acetonitrile molecules, resulting in an overall octahedral geometry. The dicationic iron complexes are typically used as hexafluorophosphate salts. | ||
| Fig. 18 Chiral-at-iron catalysis. | ||
Similar to the related chiral-at-ruthenium catalysts,28 the strongly σ-donating NHC moieties, combined with the σ-donating and π-accepting pyridyl ligands, provide a strong ligand field. This increases the stability of the helical arrangement of the two bidentate ligands, while the trans-effect of the σ-donating NHC ligands ensures high lability of the two acetonitrile ligands. Interestingly, it was shown that the CF3 groups have a positive effect on the stability of the iron complexes. Furthermore, the configurational stability depends on the nature of the σ-donating NHC ligands. For instance, replacing the imidazol-2-ylidene carbene groups of FeNHCMes with slightly more π-accepting benzimidazol-2-ylidenes (FeBIM)152 increases configurational robustness. Conversely, replacing them with more strongly σ-donating 1,2,3-triazol-5-ylidene mesoionic carbenes (FeMIC)153 completely eliminates configurational stability. Therefore, precise ligand design is essential for creating configurationally robust chiral-at-iron catalysts.
These chiral-at-iron complexes have proven to be suitable chiral Lewis acid catalysts for asymmetric hetero-Diels–Alder reactions. Notably, asymmetric induction in these reactions can be enhanced by replacing the 2,4,6-trimethylphenyl (Mes) groups at the NHC ligands (FeNHCMes) with bulkier 2,6-diisopropylphenyl (Dipp) groups (FeNHCDipp).154 These larger moieties interact directly with the catalytic site, influencing the asymmetric induction in inverse electron demand hetero-Diels–Alder reactions (see Fig. 18). For example, Λ-FeNHCDipp (3 mol%) catalyzes the reaction between the α,β-unsaturated α-ketoester 75 and 2,3-dihydrofuran (19) to afford 3,4-dihydro-2H-pyran 76 in 98% yield with 99:1 dr and 97% ee.
4.5. Other chiral-at-metal catalyst designs
A few chiral-at-metal catalysts have been reported that differ from the general design discussed in the previous section. Notably, Rodríguez, Passarelli and Carmona introduced an intriguing chiral-at-metal catalyst design through the use of a tripodal, tetradentate ligand featuring three distinct arms branching from a central coordinating amine moiety (Fig. 19a).155–162 Enantiomerically pure chiral-at-rhodium complexes (C)-RhTrip1 (clockwise absolute metal-centered configuration) and (A)-RhTrip1 (anticlockwise absolute metal-centered configuration) were synthesized using the amino acids (S)- or (R)-phenylglycine, respectively.157 The authors demonstrated that (A)-RhTrip1 functions as a chiral Lewis acid, efficiently catalyzing the Diels–Alder reaction between methacrolein (77) and cyclopentadiene (78) to provide the norbornene derivative 79 with high enantioselectivity (>99:1 er). DFT calculations support a mechanism in which the chiral Lewis acid (A)-RhTrip1 forms a well-defined chiral pocket. Methacrolein is then activated by coordinating to the aldehyde oxygen, while the diphenylphosphino arm of the tripodal ligand shields the Si-face, leaving the Re-face open for interaction with cyclopentadiene. Such tripodal chiral-at-rhodium complexes can also catalyze the enantioselective alkylation of indoles with nitrostyrenes,159 and 1,3-dipolar cycloadditions.160
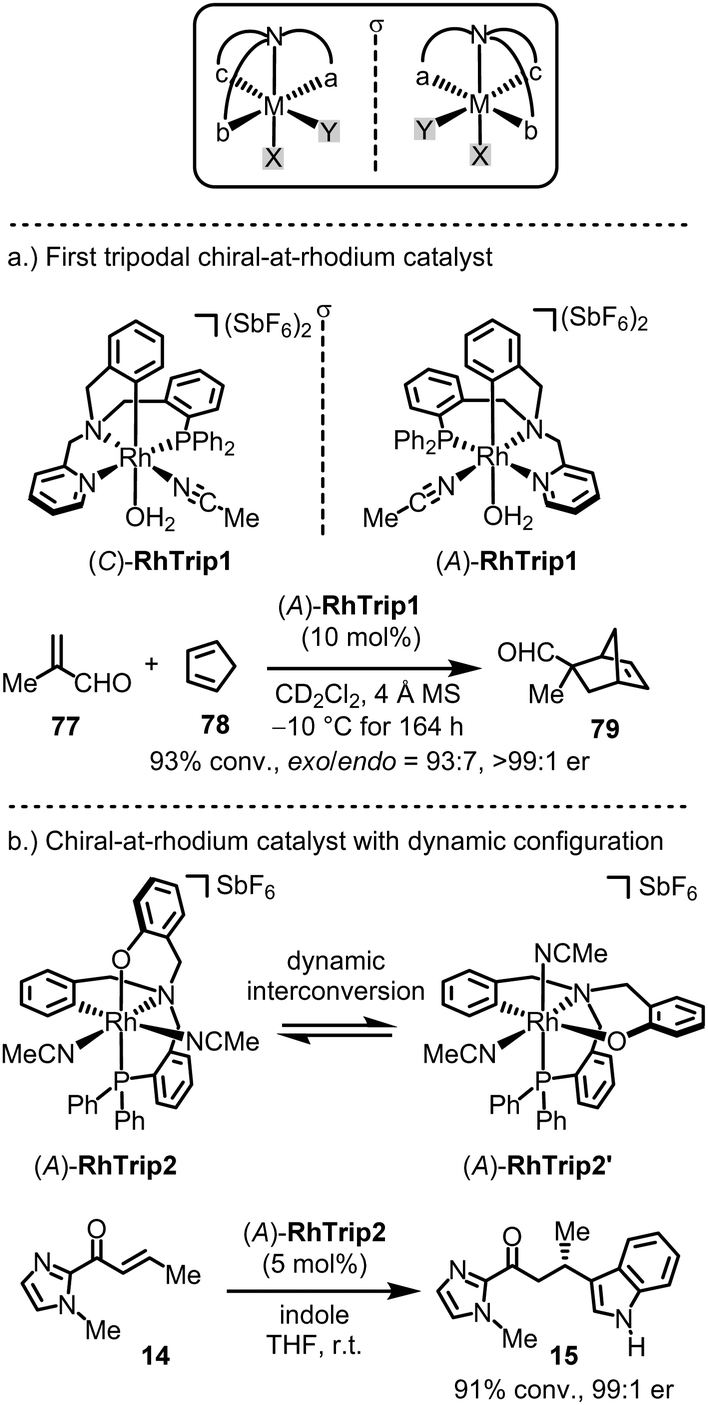 | ||
| Fig. 19 Chiral-at-rhodium catalyts based on a tripodal ligand design. | ||
Recently, the same group introduced a modified tripodal chiral-at-rhodium catalyst, RhTrip2, where the pyridyl moiety is substituted by a phenoxy group (Fig. 19b).162 Notably, RhTrip2 undergoes dynamic interconversion to its diastereomer, RhTrip2′, without racemization. Additionally, RhTrip2 was shown to catalyze the conjugate addition of indole (14 → 15) with high enantiomeric excess.
Shionoya's group reported the only known example of a tetrahedral, chiral-at-metal, asymmetric catalyst to date.163 They successfully synthesized a remarkably stable tetrahedral chiral-at-zinc complex, which was shown to catalyze an asymmetric oxa-Diels–Alder reaction by serving as a chiral Lewis acid. This was achieved through the use of the tailored tridentate ligand 80 (Fig. 20a). Upon reaction of ligand 80 with ZnEt2, the dimeric zinc complex rac-ZnInt1 was formed as a racemic mixture. The racemic complex was then treated with the chiral pyrrolidine (S)-dpp (81), serving as a monodentate ligand and leading to the formation of the complex (SZn,S)-ZnInt2 with a high diastereomeric ratio of 51:1 dr. NMR analysis revealed that, following coordination of (S)-dpp, the racemic zinc complex initially generated a nearly equal mixture of diastereomers, (SZn,S)- and (RZn,S)-ZnInt2. Over time, the mixture converted to the more thermodynamically stable (SZn,S)-diastereomer, suggesting a stereoinversion at the zinc center. The chiral auxiliary (S)-dpp was removed by treatment with tBuCN at room temperature for 7 days, resulting in the crystallization of (SZn)-ZnTet as a single enantiomer (>99% ee), with a 72% yield starting from ligand 80. The final complex, (SZn)-ZnTet, contains a stereogenic zinc center with four distinct coordinating groups. The configurational stability of (SZn)-ZnTet is likely influenced by the axial chirality of the biphenyl group in the backbone of the tridentate ligand, which becomes locked into a single conformation upon coordination with zinc. Interestingly, Shionoya recently also demonstrated that a related non-racemic nickel(II) complex could be obtained by spontaneous resolution to form conglomerate crystals, without the need for any chiral sources.164
 | ||
| Fig. 20 Chiral-at-zinc catalyt with tetrahedral coordination sphere. | ||
(SZn)-ZnTet (2 mol%) was shown to catalyze the oxa-Diels–Alder reaction between 1-naphthaldehyde (82) and the Danishefsky diene (83), affording dihydropyranone 84 in 98% yield with 87% ee (Fig. 20b). The authors obtained a crystal structure in which 1-naphthaldehyde is coordinated to the (racemic) zinc catalyst by replacing the labile tBuCN ligand. This structure suggests the mechanism of asymmetric induction, where the bulky mesityl group stacks with 1-naphthaldehyde, blocking the Si-face and forcing the diene to approach the carbonyl group from the Re-face in the transition state (XXV).
5. Summary and outlook
Alfred Werner first demonstrated metal-centered chirality over a century ago. However, until recently, asymmetric catalysis using chiral transition metal complexes has primarily depended on chiral ligands. Our group introduced a simple and general strategy that combines inert and labile ligands, enabling the metal center to function both as a stable metal stereocenter and as a reactive site for catalysis. Strong σ-donating ligands are central to this approach, which has been successfully applied to developing octahedral chiral-at-metal catalysts from metals like iridium, rhodium, ruthenium, osmium, and even iron. Recently, Shionoya extended this concept by designing a tetrahedral chiral-at-zinc catalyst.By eliminating the need for chiral motifs in the ligand sphere, untapped possibilities arise for designing chiral metal catalysts with unconventional ligand environments, opening the door to catalysts with novel properties. This has been demonstrated by us and others through a multitude of applications. In our opinion this is particularly well illustrated by bis-cyclometalated iridium(III) and rhodium(III) catalysts, which uniquely combine photochemical activation with asymmetric Lewis acid catalysis. The helical chirality of these catalysts, along with the configurational inertness of the iridium and rhodium centers, makes the use of chiral ligands unnecessary and even counterproductive.
A critical factor in designing such chiral-at-metal catalysts is the configurational stability of the metal stereocenter. This explains why most up to date reported chiral-at-metal catalysts are octahedral 18-electron complexes from noble metals with a low-spin d6 electron configuration, offering optimal ligand field stabilization and high configurational stability. The main challenge for these d6-metal complexes is balancing inert ligands, which stabilize the metal configuration, with labile ligands, which provide reactivity for substrate or reagent coordination. In many systems, this balance can be achieved by including very strong σ-donor ligands. These σ-donor ligands are typically part of a multidentate structure and labilize monodentate ligands in the trans-position via the trans-effect, while simultaneously increasing the ligand field, thereby enhancing the inertness of the multidentate ligands. This straightforward and general approach can even be extended to more labile 3d-metal complexes, such as chiral-at-iron catalysts.
Future research on chiral-at-metal catalysis will likely see more economic methods for synthesizing chiral-at-metal complexes,165 an extension to other metals including earth abundant and non-precious metals,166 the introdution of new ligand frameworks, the discovery of new catalytic transformations, and the application to the synthesis of pharmaceuticals.
We hope this review inspires new approaches for designing chiral-at-metal catalysts by exploring various other metals, ligand classes, and alternative coordination topologies.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to both current and former laboratory members, as well as collaborators, for their invaluable contributions to this project. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 883212).Notes and references
- B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5348–5355 CrossRef CAS.
- P. J. Walsh and M. C. Kozlowski, Fundamentals of asymmetric catalysis, University Science Books, Sausalito, California, 2009 Search PubMed.
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2024–2032 CrossRef CAS.
- D. Chusov, G. A. Molander, V. Ratovelomanana-Vidal, O. Riant and E. Schulz, ACS Catal., 2023, 13, 11494–11508 CrossRef CAS.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS.
- For a review on the scientific legacy of Alfred Werner, see: E. C. Constable and C. E. Housecroft, Chem. Soc. Rev., 2013, 42, 1429–1439 RSC.
- For reviews on different aspects of metal-centered chirality, see: (a) H. Brunner, Angew. Chem., Int. Ed., 1999, 38, 1194–1208 CrossRef CAS; (b) U. Knof and A. von Zelewsky, Angew. Chem., Int. Ed., 1999, 38, 302–322 CrossRef; (c) P. D. Knight and P. Scott, Coord. Chem. Rev., 2003, 242, 125–143 CrossRef CAS; (d) E. Meggers, Eur. J. Inorg. Chem., 2011, 2911–2926 CrossRef CAS; (e) E. B. Bauer, Chem. Soc. Rev., 2012, 41, 3153–3167 RSC; (f) E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637–1651 RSC; (g) A. Ehnbom, S. K. Ghosh, K. G. Lewis and J. A. Gladysz, Chem. Soc. Rev., 2016, 45, 6799–6811 RSC; (h) L. Zhang and E. Meggers, Chem. – Asian J., 2017, 12, 2335–2342 CrossRef CAS PubMed; (i) T. Cruchter and V. A. Larionov, Coord. Chem. Rev., 2018, 376, 95–113 CrossRef CAS; (j) P. S. Steinlandt, L. Zhang and E. Meggers, Chem. Rev., 2023, 123, 4764–4794 CrossRef CAS PubMed.
- A. Werner, Ber. Dtsch. Chem. Ges., 1911, 44, 1887–1898 CrossRef CAS.
- H. Brunner, Angew. Chem., Int. Ed. Engl., 1969, 8, 382–383 CrossRef CAS.
- S. G. Davies and J. C. Walker, J. Chem. Soc., Chem. Commun., 1986, 609–610 RSC.
- L. S. Liebeskind, M. E. Welker and R. W. Fengl, J. Am. Chem. Soc., 1986, 108, 6328–6343 CrossRef CAS.
- E. J. O'Connor, M. Kobayashi, H. G. Floss and J. A. Gladysz, J. Am. Chem. Soc., 1987, 109, 4837–4844 CrossRef.
- M. Brookhart, Y. Liu and R. C. Buck, J. Am. Chem. Soc., 1988, 110, 2337–2339 CrossRef CAS.
- M. Brookhart and Y. Liu, Organometallics, 1989, 8, 1572–1573 CrossRef CAS.
- S. G. Davies, Aldrichimica Acta, 1990, 23, 31–41 CAS.
- I. Sato, K. Kadowaki, Y. Ohgo, K. Soai and H. Ogino, Chem. Commun., 2001, 1022–1023 RSC.
- M. Chavarot, S. Ménage, O. Hamelin, F. Charnay, J. Pécaut and M. Fontecave, Inorg. Chem., 2003, 42, 4810–4816 CrossRef CAS.
- C. Ganzmann and J. A. Gladysz, Chem. – Eur. J., 2008, 14, 5397–5400 CrossRef CAS PubMed.
- L.-A. Chen, W. Xu, B. Huang, J. Ma, L. Wang, J. Xi, K. Harms, L. Gong and E. Meggers, J. Am. Chem. Soc., 2013, 135, 10598–10601 CrossRef CAS PubMed.
- For the merit of this chiral-at-metal catalysis with inert metal complexes, see also: (a) L.-A. Chen, X. Tang, J. Xi, W. Xu, L. Gong and E. Meggers, Angew. Chem., Int. Ed., 2013, 52, 14021–14025 CrossRef CAS; (b) J. Ma, X. Ding, Y. Hu, Y. Huang, L. Gong and E. Meggers, Nat. Commun., 2014, 5, 4531 CrossRef CAS PubMed; (c) L. Gong, L.-A. Chen and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10868–10874 CrossRef CAS; (d) W. Xu, M. Arieno, H. Löw, K. Huang, X. Xie, T. Cruchter, Q. Ma, J. Xi, B. Huang, O. Wiest, L. Gong and E. Meggers, J. Am. Chem. Soc., 2016, 138, 8774–8780 CrossRef CAS PubMed; (e) V. A. Larionov, B. L. Feringa and Y. N. Belokon, Chem. Soc. Rev., 2021, 50, 9715–9740 RSC.
- G. C. Fu, Acc. Chem. Res., 2000, 33, 412–420 CrossRef CAS.
- (a) J. Hartung and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10183–10185 CrossRef CAS PubMed; (b) J. Hartung, P. K. Dornan and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 13029–13037 CrossRef CAS PubMed; (c) J. Hartung and R. H. Grubbs, Angew. Chem., Int. Ed., 2014, 53, 3885–3888 CrossRef CAS PubMed.
- H. Huo, C. Fu, K. Harms and E. Meggers, J. Am. Chem. Soc., 2014, 136, 2990–2993 CrossRef CAS.
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS.
- X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed.
- C.-X. Ye and E. Meggers, Acc. Chem. Res., 2023, 56, 1128–1141 CrossRef CAS PubMed.
- R. H. Crabtree, The Organometallic Chemistry of Transition Metals, John Wiley & Sons Inc., Hoboken, New Jersey, 2014 Search PubMed.
- B. J. Coe and S. J. Glenwright, Coord. Chem. Rev., 2000, 203, 5–80 CrossRef CAS.
- Z.-Y. Cao, W. D. G. Brittain, J. S. Fossey and F. Zhou, Catal. Sci. Technol., 2015, 5, 3441–3451 RSC.
- P. Dey, P. Rai and B. Maji, ACS Org. Inorg. Au, 2022, 2, 99–125 CrossRef CAS.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- S. Brunen, Y. Grell, P. S. Steinlandt, K. Harms and E. Meggers, Molecules, 2021, 26, 1822 CrossRef CAS PubMed.
- J. Qin, V. A. Larionov, K. Harms and E. Meggers, ChemSusChem, 2019, 12, 320–325 CrossRef CAS.
- Z. Zhou, Y. Li, L. Gong and E. Meggers, Org. Lett., 2017, 19, 222–225 CrossRef CAS.
- V. A. Larionov, T. Cruchter, T. Mietke and E. Meggers, Organometallics, 2017, 36, 1457–1460 CrossRef CAS.
- Y. Li, M. Lei, W. Yuan, E. Meggers and L. Gong, Chem. Commun., 2017, 53, 8089–8092 RSC.
- Y. Tan, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2018, 2500–2504 CrossRef CAS.
- A. Pazos and Z. Freixa, ChemCatChem, 2024, 16, e202400585 CrossRef CAS.
- E. Meggers, Chem. Eur. J., 2010, 16, 752–758 CrossRef CAS.
- L. Gong, S. P. Mulcahy, K. Harms and E. Meggers, J. Am. Chem. Soc., 2009, 131, 9602–9603 CrossRef CAS PubMed.
- L. Gong, M. Wenzel and E. Meggers, Acc. Chem. Res., 2013, 46, 2635–2644 CrossRef CAS.
- J. Ma, X. Zhang, X. Huang, S. Luo and E. Meggers, Nat. Protoc., 2018, 13, 605–632 CrossRef CAS.
- X. Shen, H. Huo, C. Wang, B. Zhang, K. Harms and E. Meggers, Chem. – Eur. J., 2015, 21, 9720–9726 CrossRef CAS.
- G.-Q. Xu, H. Liang, J. Fang, Z.-L. Jia, J.-Q. Chen and P.-F. Xu, Chem. – Asian J., 2016, 11, 3355–3358 CrossRef CAS.
- T. Mietke, T. Cruchter, V. A. Larionov, T. Faber, K. Harms and E. Meggers, Adv. Synth. Catal., 2018, 360, 2093–2100 CrossRef CAS.
- X. Zhang, J. Qin, X. Huang and E. Meggers, Eur. J. Org. Chem., 2018, 571–577 CrossRef.
- C. Tian, L. Gong and E. Meggers, Chem. Commun., 2016, 52, 4207–4210 RSC.
- X. Zhang, J. Qin, X. Huang and E. Meggers, Org. Chem. Front., 2018, 5, 166–170 RSC.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- C. Wang, Y. Zheng, H. Huo, P. Röse, L. Zhang, K. Harms, G. Hilt and E. Meggers, Chem. – Eur. J., 2015, 21, 7355–7359 CrossRef CAS PubMed.
- H. Huo, C. Wang, K. Harms and E. Meggers, J. Am. Chem. Soc., 2015, 137, 9551–9554 CrossRef CAS PubMed.
- H. Huo, X. Huang, X. Shen, K. Harms and E. Meggers, Synlett, 2016, 749–753 CAS.
- C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685–688 CrossRef CAS PubMed.
- C. Wang, L.-A. Chen, H. Huo, X. Shen, K. Harms, L. Gong and E. Meggers, Chem. Sci., 2015, 6, 1094–1100 RSC.
- Y. Huang, L. Song, L. Gong and E. Meggers, Chem. – Asian J., 2015, 10, 2737–2742 Search PubMed.
- Y. Tan, W. Yuan, L. Gong and E. Meggers, Angew. Chem., Int. Ed., 2015, 54, 13045–13048 CrossRef CAS PubMed.
- J. Ma, X. Shen, K. Harms and E. Meggers, Dalton Trans., 2016, 45, 8320–8323 RSC.
- H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936–6939 CrossRef CAS PubMed.
- X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed.
- X. Shen, K. Harms, M. Marsch and E. Meggers, Chem. – Eur. J., 2016, 22, 9102–9105 CrossRef CAS.
- L. Song, L. Gong and E. Meggers, Chem. Commun., 2016, 52, 7699–7702 RSC.
- J. Ma, K. Harms and E. Meggers, Chem. Commun., 2016, 52, 10183–10186 RSC.
- Y. Zheng, K. Harms, L. Zhang and E. Meggers, Chem. – Eur. J., 2016, 22, 11977–11981 CrossRef CAS PubMed.
- C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed.
- L. Feng, X. Dai, E. Meggers and L. Gong, Chem. – Asian J., 2017, 12, 963–967 CrossRef CAS PubMed.
- X. Huang, X. Li, X. Xie, K. Harms, R. Riedel and E. Meggers, Nat. Commun., 2017, 8, 2245 CrossRef.
- X. Huang, S. Luo, O. Burghaus, R. D. Webster, K. Harms and E. Meggers, Chem. Sci., 2017, 8, 7126–7131 RSC.
- B. Tutkowski, E. Meggers and O. Wiest, J. Am. Chem. Soc., 2017, 139, 8062–8065 CrossRef CAS.
- X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS.
- J. Ma, A. R. Rosales, X. Huang, K. Harms, R. Riedel, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 17245–17248 CrossRef CAS PubMed.
- S. Chen, X. Huang, E. Meggers and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 17902–17907 CrossRef CAS PubMed.
- H. Lin, Z. Zhou, J. Cai, B. Han, L. Gong and E. Meggers, J. Org. Chem., 2017, 82, 6457–6467 CrossRef CAS PubMed.
- S. Luo, X. Zhang, Y. Zheng, K. Harms, L. Zhang and E. Meggers, J. Org. Chem., 2017, 82, 8995–9005 CrossRef CAS PubMed.
- W. Yuan, Z. Zhou, L. Gong and E. Meggers, Chem. Commun., 2017, 53, 8964–8967 RSC.
- Z. Zhou, Y. Li, B.-W. Han, L. Gong and E. Meggers, Chem. Sci., 2017, 8, 5757–5763 RSC.
- J. Ma, X. Xie and E. Meggers, Chem. – Eur. J., 2018, 24, 259–265 CrossRef CAS PubMed.
- X. Huang, J. Lin, T. Shen, K. Harms, M. Marchini, P. Ceroni and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 5454–5458 CrossRef CAS PubMed.
- N. Hu, H. Jung, Y. Zheng, J. Lee, L. Zhang, Z. Ullah, X. Xie, K. Harms, M.-H. Baik and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 6242–6246 CrossRef CAS.
- J. Ma, J. Lin, L. Zhao, K. Harms, M. Marsch, X. Xie and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 11193–11197 CrossRef CAS PubMed.
- F. F. de Assis, X. Huang, M. Akiyama, R. A. Pilli and E. Meggers, J. Org. Chem., 2018, 83, 10922–10932 CrossRef CAS PubMed.
- X. Huang, Q. Zhang, J. Lin, K. Harms and E. Meggers, Nat. Catal., 2019, 2, 34–40 CrossRef CAS.
- Y. Grell, Y. Hong, X. Huang, T. Mochizuki, X. Xie, K. Harms and E. Meggers, Organometallics, 2019, 38, 3948–3954 CrossRef CAS.
- P. S. Steinlandt, W. Zuo, K. Harms and E. Meggers, Chem. – Eur. J., 2019, 25, 15333–15340 CrossRef CAS.
- C. Zhang, S. Chen, C.-X. Ye, K. Harms, L. Zhang, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2019, 58, 14462–14466 CrossRef CAS PubMed.
- Y. Kuang, K. Wang, X. Shi, X. Huang, E. Meggers and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 16859–16863 CrossRef CAS PubMed.
- C.-X. Ye, S. Chen, F. Han, X. Xie, S. Ivlev, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2020, 59, 13552–13556 CrossRef CAS PubMed.
- Y. Grell, X. Xie, S. I. Ivlev and E. Meggers, ACS Catal., 2021, 11, 11396–11406 CrossRef CAS.
- H. Jung, M. Hong, M. Marchini, M. Villa, P. S. Steinlandt, X. Huang, M. Hemming, E. Meggers, P. Ceroni, J. Park and M.-H. Baik, Chem. Sci., 2021, 12, 9673–9681 RSC.
- C. Zhang, A. Z. Gao, X. Nie, C.-X. Ye, S. I. Ivlev, S. Chen and E. Meggers, J. Am. Chem. Soc., 2021, 143, 13393–13400 CrossRef CAS.
- P. Xiong, M. Hemming, S. I. Ivlev and E. Meggers, J. Am. Chem. Soc., 2022, 144, 6964–6971 CrossRef CAS PubMed.
- P. Xiong, S. I. Ivlev and E. Meggers, Nat. Catal., 2023, 6, 1186–1193 CrossRef CAS.
- J. Gong, K. Li, S. Qurban and Q. Kang, Chin. J. Chem., 2016, 34, 1225–1235 CrossRef CAS.
- G.-J. Sun, J. Gong and Q. Kang, J. Org. Chem., 2017, 82, 796–803 CrossRef CAS PubMed.
- T. Deng, G. K. Thota, Y. Li and Q. Kang, Org. Chem. Front., 2017, 4, 573–577 RSC.
- S.-W. Li, J. Gong and Q. Kang, Org. Lett., 2017, 19, 1350–1353 CrossRef PubMed.
- K. Li, Q. Wan and Q. Kang, Org. Lett., 2017, 19, 3299–3302 CrossRef CAS.
- J. Gong, S.-W. Li, S. Qurban and Q. Kang, Eur. J. Org. Chem., 2017, 3584–3593 CrossRef CAS.
- S.-X. Lin, G.-J. Sun and Q. Kang, Chem. Commun., 2017, 53, 7665–7668 RSC.
- P. P. Bora, G.-J. Sun, W.-F. Zheng and Q. Kang, Chin. J. Chem., 2018, 36, 20–24 CrossRef CAS.
- G. K. Thota, G. J. Sun, T. Deng, Y. Li and Q. Kang, Adv. Synth. Catal., 2018, 360, 1094–1098 CrossRef CAS.
- S.-W. Li, Q. Wan and Q. Kang, Org. Lett., 2018, 20, 1312–1315 CrossRef CAS PubMed.
- J. Gong, Q. Wan and Q. Kang, Org. Lett., 2018, 20, 3354–3357 CrossRef CAS.
- J. Gong, Q. Wan and Q. Kang, Adv. Synth. Catal., 2018, 360, 4031–4036 CrossRef CAS.
- S.-W. Li and Q. Kang, Chem. Commun., 2018, 54, 10479–10482 RSC.
- S. Qurban, J. Gong, Y. Du and Q. Kang, Org. Chem. Front., 2018, 5, 2870–2874 RSC.
- L. Hu, S. Lin, S. Li, Q. Kang and Y. Du, ChemCatChem, 2019, 12, 118–121 CrossRef.
- H. Liang, G.-Q. Xu, Z.-T. Feng, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2019, 84, 60–72 CrossRef CAS PubMed.
- S. Qurban, Y. Du, J. Gong, S.-X. Lin and Q. Kang, Chem. Commun., 2019, 55, 249–252 RSC.
- S. W. Li, Y. Du and Q. Kang, Org. Chem. Front., 2019, 6, 2775–2779 RSC.
- H. Zhang, S. Li, Q. Kang and Y. Du, Org. Chem. Front., 2019, 6, 3683–3687 RSC.
- Q. Wan, S. Li, Q. Kang, Y. Yuan and Y. Du, J. Org. Chem., 2019, 84, 15201–15211 CrossRef CAS PubMed.
- Q. Wan, L. Chen, S. Li, Q. Kang, Y. Yuan and Y. Du, Org. Lett., 2020, 22, 9539–9544 CrossRef CAS PubMed.
- R. I. Rodríguez, L. Mollari and J. Alemán, Angew. Chem., Int. Ed., 2021, 60, 4555–4560 CrossRef PubMed.
- S. Ming, S. Qurban, Y. Du and W. Su, Chem. – Eur. J., 2021, 27, 12742–12746 CrossRef CAS PubMed.
- Y. Ren, S. Lu, L. He, Z. Zhao and S.-W. Li, Org. Lett., 2022, 24, 2585–2589 CrossRef CAS PubMed.
- J. Pian, Q. Chen, Y. Luo, Z. Zhao, J. Liu, L. He and S.-W. Li, Org. Lett., 2022, 24, 5641–5645 CrossRef CAS PubMed.
- J. Yang, S. Ming, G. Yao, H. Yu, Y. Du and J. Gong, Org. Chem. Front., 2022, 9, 2759–2765 RSC.
- S. Lu, Z. Zhao, Y. Ren, G. Du, J. Zhao and S.-W. Li, Org. Chem. Front., 2022, 9, 3446–3451 RSC.
- S. Ming, J. Yang, S. Wu, G. Yao, H. Xiong, Y. Du and J. Gong, Org. Chem. Front., 2022, 9, 5147–5153 RSC.
- X. Chen, Y. An, G. Du, Y. Zhao, L. He, J. Zhao and S.-W. Li, J. Org. Chem., 2022, 87, 5497–5509 CrossRef CAS PubMed.
- X. Chen, Y. Zhao, C. Huang, Z. Zhao, W. Zhao and S.-W. Li, Chem. Commun., 2024, 60, 236–239 RSC.
- J. Pian, Z. Zhao, Y. Zhao, L. He, X.-L. Liu, Y. Li and S.-W. Li, J. Org. Chem., 2024, 89, 918–927 CrossRef CAS PubMed.
- Y. Zhao, X. Ji, X. Tian, Z. Zhao, J. Pian and S.-W. Li, Adv. Synth. Catal., 2024, 366, 1590–1594 CrossRef CAS.
- X. Ji, S. Zhu, Y. Li, Z. Zhao and S.-W. Li, Org. Chem. Front., 2024, 11, 5385–5389 RSC.
- X. Xie, Z. Zhao, J. Wang and S.-W. Li, Org. Lett., 2024, 26, 8144–8148 CrossRef CAS PubMed.
- E. Marchi, R. Sinisi, G. Bergamini, M. Tragni, M. Monari, M. Bandini and P. Ceroni, Chem. – Eur. J., 2012, 18, 8765–8773 CrossRef CAS.
- Y. Zheng, Y. Q. Tan, K. Harms, M. Marsch, R. Riedel, L. Zhang and E. Meggers, J. Am. Chem. Soc., 2017, 139, 4322–4325 CrossRef CAS.
- Y. Zheng, L. Zhang and E. Meggers, Org. Process Res. Dev., 2018, 22, 103–107 CrossRef CAS.
- S. Chen, Y. Zheng, T. Cui, E. Meggers and K. N. Houk, J. Am. Chem. Soc., 2018, 140, 5146–5152 CrossRef CAS PubMed.
- T. Cui, J. Qin, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2019, 195–198 CrossRef CAS.
- J. Qin, Z. Zhou, T. Cui, M. Hemming and E. Meggers, Chem. Sci., 2019, 10, 3202–3207 RSC.
- Z. Zhou, S. Chen, J. Qin, X. Nie, X. Zheng, K. Harms, R. Riedel, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2019, 58, 1088–1093 CrossRef CAS PubMed.
- Y. Tan, S. Chen, Z. Zhou, Y. Hong, S. Ivlev, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2020, 59, 21706–21710 CrossRef CAS PubMed.
- Z. Zhou, Y. Tan, T. Yamahira, S. Ivlev, X. Xie, R. Riedel, M. Hemming, M. Kimura and E. Meggers, Chem, 2020, 6, 2024–2034 CAS.
- Y. Tan, F. Han, M. Hemming, J. Wang, K. Harms, X. Xie and E. Meggers, Org. Lett., 2020, 22, 6653–6656 CrossRef CAS.
- Z. Zhou, Y. Tan, X. Shen, S. Ivlev and E. Meggers, Sci. China Chem., 2021, 64, 452–458 CrossRef CAS.
- E. Winterling, S. Ivlev and E. Meggers, Organometallics, 2021, 40, 1148–1155 CrossRef CAS.
- X. Nie, C.-X. Ye, S. I. Ivlev and E. Meggers, Angew. Chem., Int. Ed., 2022, 61, e202211971 CrossRef CAS.
- C.-X. Ye, X. Shen, S. Chen and E. Meggers, Nat. Chem., 2022, 14, 566–573 CrossRef CAS.
- X. Nie, C. W. Ritter, M. Hemming, S. I. Ivlev, X. Xie, S. Chen and E. Meggers, Angew. Chem., Int. Ed., 2023, 62, e202314398 CrossRef CAS PubMed.
- N. Demirel, P. Moths, S. I. Ivlev and E. Meggers, under revision.
- Z. Zhou, S. Chen, Y. Hong, E. Winterling, Y. Tan, M. Hemming, K. Harms, K. N. Houk and E. Meggers, J. Am. Chem. Soc., 2019, 141, 19048–19057 CrossRef CAS PubMed.
- F. Han, P. H. Choi, C.-X. Ye, Y. Grell, X. Xie, S. I. Ivlev, S. Chen and E. Meggers, ACS Catal., 2022, 12, 10304–10312 CrossRef CAS.
- E. Meggers, F. Han, Y. Xie, X. Xie and S. I. Ivlev, Synlett, 2022, 1403–1408 Search PubMed.
- D. Baran, S. I. Ivlev and E. Meggers, Organometallics, 2022, 41, 52–59 CrossRef CAS.
- G. Wang, Z. Zhou, X. Shen, S. Ivlev and E. Meggers, Chem. Commun., 2020, 56, 7714–7717 RSC.
- Y. Hong, L. Jarrige, K. Harms and E. Meggers, J. Am. Chem. Soc., 2019, 141, 4569–4572 CrossRef CAS PubMed.
- N. Demirel, J. Haber, S. I. Ivlev and E. Meggers, Organometallics, 2022, 41, 3852–3860 CrossRef CAS PubMed.
- N. Demirel, M. Dawor, G. Nadler, S. I. Ivlev and E. Meggers, Chem. Sci., 2024, 15, 15625–15631 RSC.
- Y. Hong, T. Cui, S. Ivlev, X. Xie and E. Meggers, Chem. – Eur. J., 2021, 27, 8557–8563 CrossRef CAS PubMed.
- M. Carmona, R. Rodríguez, I. Méndez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, Dalton Trans., 2017, 46, 7332–7350 RSC.
- M. Carmona, L. Tejedor, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, Chem. – Eur. J., 2017, 23, 14532–14546 CrossRef CAS.
- M. Carmona, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, J. Am. Chem. Soc., 2018, 140, 912–915 CrossRef CAS.
- J. Tellez, I. Mendez, F. Viguri, R. Rodriguez, F. J. Lahoz, P. Garcia-Orduna and D. Carmona, Organometallics, 2018, 37, 3450–3464 CrossRef CAS.
- M. Carmona, R. Rodríguez, V. Passarelli and D. Carmona, Organometallics, 2019, 38, 988–995 CrossRef CAS.
- A. G. Tejero, M. Carmona, R. Rodríguez, F. Viguri, F. J. Lahoz, P. García-Orduña and D. Carmona, RSC Adv., 2022, 12, 34704–34714 RSC.
- I. Barriendos, Í. Almárcegui, M. Carmona, A. G. Tejero, A. Soriano-Jarabo, C. Blas, Z. Aguado, D. Carmona, F. J. Lahoz, P. García-Orduña, F. Viguri and R. Rodríguez, ChemPlusChem, 2024, 89, e202400410 CrossRef CAS.
- A. G. Tejero, J. Castillo, F. Viguri, D. Carmona, V. Passarelli, F. J. Lahoz, P. García-Orduña and R. Rodríguez, Chem. – Eur. J., 2024, 30, e202303935 CrossRef CAS PubMed.
- K. Endo, Y. Liu, H. Ube, K. Nagata and M. Shionoya, Nat. Commun., 2020, 11, 6263 CrossRef CAS.
- Y. Liu, H. Ube, K. Endo and M. Shionoya, ACS Org. Inorg. Au, 2023, 3, 371–376 CrossRef CAS.
- Y.-P. Chu, X.-L. Yue, D.-H. Liu and J. Ma, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-krgpl-v2.
- K. Nagata, A. Hino, H. Ube, H. Sato and M. Shionoya, Dalton Trans., 2023, 52, 3295–3299 RSC.
| This journal is © The Royal Society of Chemistry 2025 |