
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignin-based porous carbon adsorbents for CO2 capture
Daniel
Barker-Rothschild
 a,
Jingqian
Chen
a,
Zhangmin
Wan
a,
Scott
Renneckar
b,
Ingo
Burgert
cd,
Yong
Ding
*cd,
Yi
Lu
*a and
Orlando J.
Rojas
*abe
a,
Jingqian
Chen
a,
Zhangmin
Wan
a,
Scott
Renneckar
b,
Ingo
Burgert
cd,
Yong
Ding
*cd,
Yi
Lu
*a and
Orlando J.
Rojas
*abe
aBioproducts Institute, Department of Chemical and Biological Engineering, The University of British Columbia, 2360 East Mall, Vancouver, BC V6T 1Z3, Canada. E-mail: yi.lu@ubc.ca; orlando.rojas@ubc.ca
bDepartment of Wood Science, University of British Columbia, 2424 Main Mall, Vancouver, BC V6T 1Z4, Canada
cWood Materials Science, Institute for Building Materials, ETH Zürich, 8093 Zürich, Switzerland. E-mail: yoding@ethz.ch
dWoodTec Group, Cellulose & Wood Materials, Empa, 8600 Dübendorf, Switzerland
eDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada
First published on 11th November 2024
Abstract
A major driver of global climate change is the rising concentration of atmospheric CO2, the mitigation of which requires the development of efficient and sustainable carbon capture technologies. Solid porous adsorbents have emerged as promising alternatives to liquid amine counterparts due to their potential to reduce regeneration costs. Among them, porous carbons stand out for their high surface area, tailorable pore structure, and exceptional thermal and mechanical properties, making them highly robust and efficient in cycling operations. Moreover, porous carbons can be synthesized from readily available organic (waste) streams, reducing costs and promoting circularity. Lignin, a renewable and abundant by-product of the forest products industry and emerging biorefineries, is a complex organic polymer with a high carbon content, making it a suitable precursor for carbon-based adsorbents. This review explores lignin's sources, structure, and thermal properties, as well as traditional and emerging methods for producing lignin-based porous adsorbents. We examine the physicochemical properties, CO2 adsorption mechanisms, and performance of lignin-derived materials. Additionally, the review highlights recent advances in lignin valorization and provides critical insights into optimizing the design of lignin-based adsorbents to enhance CO2 capture efficiency. Finally, it addresses the prospects and challenges in the field, emphasizing the significant role that lignin-derived materials could play in advancing sustainable carbon capture technologies and mitigating climate change.
 Daniel Barker-Rothschild | Daniel Barker-Rothschild is a PhD student in the Department of Chemical and Biological Engineering at The University of British Columbia (Vancouver, Canada) working under the supervision of Professor Orlando Rojas. He obtained his BSc (2020) and MSc (2022) in Chemical Engineering at the University of Alberta (Edmonton, Canada). His research focuses on using data-driven approaches to better understand complex and variable lignocellulosic feedstocks and to support their strategic upgrading via structure–property–performance relationships. Daniel has focused these efforts on lignin valorization via colloidal particle formation and the synthesis of lignin-derived carbons for CO2 capture and energy storage materials. |
 Jingqian Chen | Jingqian Chen received her PhD in Chemical and Biological Engineering at the University of British Columbia in 2020. She joined Prof. Orlando J. Rojas's group as a postdoctoral research fellow in 2021. Her research interests include top-down bio-polymer fractionation (hemicelluloses, lignin and cellulose), bottom-up bio-colloidal particle assembly and related material applications. The recent focus of her work is lignin nanoparticle size fractionation, photonic color assembly, and lignin-derived carbon for energy and environmental applications. |
 Zhangmin Wan | Zhangmin Wan is a PhD student at the University of British Columbia in Canada, under the supervision of Professor Orlando J. Rojas. His research primarily investigates the fundamental properties of nanopolysaccharide, such as cellulose and chitin nanocrystals (nanofibrils), and lignin molecules. By employing computational modeling, atomic force microscopy (AFM), and X-ray scattering/diffraction techniques, he explores their mechanical properties and the origin of bio-recalcitrance. |
 Scott Renneckar | Professor Scott Renneckar is a Canada Research Chair (Tier II) at The University of British Columbia and Program Director of the Bioeconomy Sciences and Technology Program in the Faculty of Forestry. His research group, Advanced Renewable Materials, contains a dynamic team of researchers working on projects of lignin and heteropolysaccharide valorization, biomass fractionation, nanocomposites and advanced carbon-based materials. Dr Renneckar is an elected Fellow of the International Academy of Wood Science and former Chair of the Cellulose and Renewable Materials Division of the American Chemical Society. |
 Ingo Burgert | Prof. Dr Ingo Burgert Ingo has been a Professor of Wood Materials Science at ETH Zurich and Empa, since 2011. He studied wood science and technology at the University of Hamburg, Germany, and obtained a doctoral degree at the same university in 2000. From 2000 to 2003 he worked as a postdoc at the Institute of Physics and Materials Science of BOKU, the University of Natural Resources and Life Sciences in Vienna, Austria. From 2003 to 2011, Ingo was a research group leader of the “Plant Biomechanics and Biomimetics” group at the Max Planck Institute of Colloids and Interfaces in the Department of Biomaterials, Potsdam, Germany. |
 Yong Ding | Dr Yong Ding obtained her doctoral degree in Wood Materials Science from ETH Zurich in 2023, where her doctoral research focused on developing functionalized wood materials. Currently, she is a postdoctoral researcher at Empa and a guest scientist at ETH Zurich. Her research is dedicated to advancing functional wood composites toward sustainable, energy-efficient smart building applications. Dr Ding's expertise lies in sustainable and carbon-negative building materials, focusing on enhancing the functionality and life cycle efficiency of wood-based composites. |
 Yi Lu | Dr Yi Lu is currently a postdoctoral research fellow at The University of British Columbia. He obtained his PhD degree in chemical engineering at the University of Alberta in 2020. He then joined Prof. Orlando J. Rojas's group in 2021, as a group leader for micro/nanomaterials and multiphase studies. His research interests are colloidal chemistry, surface and interface science, and bio-based nanoparticles. His current work focuses on understanding hierarchical and heterogeneous biobased materials via colloidal sciences to accelerate sustainable development. |
 Orlando J. Rojas | Orlando Rojas is a Canada Excellence Research Chair and Director of the Bioproducts Institute, where he synergizes a distinguished group of researchers conducting multi-disciplinary research to create fundamental knowledge and applications of biobased products. He is a highly cited researcher (top 1% by citations, Clarivate) and recognized as a worldwide leader in the area of renewable nanomaterials. This is a result of the efforts of the talented members of his Biobased Colloids and Materials group. Prof. Rojas received the Anselme Payen Award, and is an elected Fellow of the American Chemical Society and the Finnish Academy of Science & Letters. |
1 Introduction
The mitigation of climate change is a pressing contemporary issue that motivates research across diverse scientific disciplines and influences evolving government policies worldwide. The consequences of climate change are realized in their impacts on marine and freshwater ecosystems, water and food security, economy and human migration, health and well-being.1 Climate change is driven by a combination of natural variations and other contributing factors, including anthropogenic activities, such as atmospheric pollution. Among the main greenhouse gases (GHG)—carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), and water vapor (H2Ovap)—the atmospheric concentration of CO2 shows a particularly strong correlation with rising surface temperatures.2–5Since pre-industrial times, atmospheric CO2 has risen from 280 ppm to over 400 ppm.6 This trend is expected to continue unless drastic changes take place following increased public awareness, and the adoption of new regulations and policies along with remediation via sustainable and cost-effective technologies. A dominant source of anthropogenic GHG emissions has been CO2 emissions from the combustion of fossil fuels, accounting for 88% of the total emissions over the last decade.7 In the long term, a significant amount of fossil energy is anticipated to be replaced with ‘green’ sources (e.g., solar, wind, nuclear, and hydro). However, it will take time to phase out fossil fuels and products due to their ingrained relationship with modern civilization. Beyond petroleum, many other industrial activities also produce CO2, for instance, as reaction by-products (e.g., cement and steel). Therefore, in the short term, emission reduction and mitigation via CO2 capture are necessary.8
Generally, CO2 capture works by separating and concentrating it from gas streams, whereupon the CO2 is compressed and transported for storage or utilization. In the broad context, carbon capture approaches can be categorized as carbon capture and storage (CCS), carbon capture and utilization (CCU), and reactive carbon capture (RCC).9 In CCS, the captured carbon is typically stored in reservoirs such as geological formations for permanent storage. Alternatively, CCU approaches seek to utilize CO2 after capture and storage. In contrast to CCS and CCU, RCC immediately converts CO2 to another product, potentially significantly improving energy efficiency by eliminating the thermal energy requirement for CO2 release.
Current carbon capture technologies include post-combustion (CO2 and N2 from flue gas), pre-combustion (CO2 and H2 from syngas), or oxy-fuel combustion (O2 from air) as classified by the composition of the stream (i.e., the compounds to be separated).10 The implications of these distinctions are realized in the different processes and conditions required for separation. Today, with currently available carbon capture technologies, power plants could reduce their carbon emissions to close-to-zero, however, at the cost of considerable energy and financial penalties.11 For example, within the framework of post-combustion carbon capture, amine-based liquids are among the most mature technologies applied to scrub CO2 from flue gas based on the formation of water-soluble carbamates or bicarbonate species. Yet, major challenges exist in the form of the regeneration of aqueous solvents, which requires CO2 stripping at high temperatures.12 Such demands, associated with major capital and operation costs, energy, and solvent intensity, limit or prevent the adoption of technologies that have already been demonstrated for their technical feasibility.
To fulfill the ever-increasing pursuit of high-performance carbon capture materials, porous solid adsorbents have garnered attention due to their high surface area and CO2 selectivity. Compared to liquid adsorbents that rely on chemisorption, porous solids mainly work by physisorption, leading to efficient gas separation, thereby reducing energy requirements during regeneration, especially under the conditions of a low adsorption enthalpy. Porous solids can be sourced at low costs and can be tailored for morphology and stability under harsh operating conditions (high temperatures, pressures, and humidity).10,13 Of critical importance, solid adsorbents can be regenerated more efficiently than liquid adsorbents, for example, by pressure or temperature swings. Notably, these regeneration approaches imply reduced space requirements and system complexity, although, in practice, their implementation is highly dependent on the type of solid adsorbent used and the possibility of use at scale.14 Regeneration of solid adsorbents often drives the techno-economic feasibility of the technology.
Typical solid adsorbents include zeolites, metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and activated carbons, all of which display high porosity, available specific surface area and physisorption capability. However, zeolites, MOFs, and COFs are often moisture-sensitive, expensive, and difficult to regenerate.14 Various alternative technologies have also been developed, including renewable feedstocks such as amine-functionalized fibrillated cellulose, or nanochitin aerogels.15–18 Among these materials, porous carbons are promising solid adsorbents for CO2. Porous carbons can be produced from renewable, carbon-rich agricultural or industrial by-products and residual streams, offering an opportunity to add value to these resources. Current efforts highlight material conversion from a variety of bioresources such as wood sawdust,19 fruit seeds,19 oil palm,20 gelatin and starch,21 date seeds,22 nutshells,23 and food wastes,24,25 to name just a few. While these materials present significant opportunities for conversion into activated carbons, a key challenge persists: their lower CO2 adsorption capacity and selectivity compared to zeolites or MOFs. This has prompted new strategies for controlling the material pore structure and surface interactions of porous carbon, critical to adsorption performance.
In this context, lignin has emerged as a promising precursor of solid porous adsorbents. Lignin is the most abundant aromatic biopolymer found in nature, accounting for up to 10–35% by weight and 40% by energy of plant biomass.26 Lignin-containing by-products from industry represent a viable source of this biopolymer, which can be isolated downstream through fractionation processes. In such conditions, industrial lignin is produced in large quantities (∼50–70 million tons annually),27 and used primarily as a low-value biofuel through combustion that enables chemical recovery after wood pulping.
Lignin's aromatic structure endows it with a high carbon yield (∼40%, the highest of all biopolymers), while its functionalities offer various pathways for physical and chemical modification. In fact, many of the alternative resources selected for the production of activated carbon (e.g., coconut shells, wood) are actually lignin-rich, endowing them with higher carbon yields. Indeed, using lignin alone may be a more efficient approach. Further, there are pathways for exploiting the polymeric structure of lignin for strategic upgrading strategies that are not possible for other feedstocks that are complex mixtures of several biopolymers and biomolecules. Therefore, lignin has great potential as a major feedstock for producing renewable activated carbons. The use of lignin in material applications, particularly in carbon capture technologies, sequesters biogenic carbon, providing industries with a viable path toward achieving net-zero—and potentially even net-negative—CO2 emissions.
Despite decades of efforts from various stakeholders, the full potential of lignin has yet to be fully realized on the industrial scale, primarily due to the inherent composition variability and chemical complexity. However, owing to its inherent advantages, there are major incentives to find cost-effective and scalable methods that adhere to green chemistry principles for the commercialization of lignin-based carbon capture technologies. In this review, we offer a perspective on lignin-based solid gas adsorbents for CO2 capture. We discuss lignin sources and availability, its structure and chemistry, as well as the strategies employed for lignin deployment in the field. A review of the key metrics for the properties and performance of lignin-derived carbon materials is included, which is critical for developing structure–property–performance relationships. We also describe the traditional and emerging methods for processing lignin into porous materials designed for CO2 capture. We provide critical viewpoints and identify key gaps in innovation. Finally, we discuss the prospects of lignin as a technological alternative for CO2 capture and outline future directions for research and development.
2. Lignin chemistry and properties
The term “lignin” encompasses a broad class of biomacromolecules exhibiting significant variability in their chemistry, structure, bonding patterns, and properties.28 Special consideration of lignin's origins and structure is essential for processing it into value-added materials; lignin-based carbon adsorbents are no exception. In this section, we review the origins of lignin, its chemical structure, and relevant definitions. The discussion then covers lignin's thermal properties and current processing strategies. Finally, we address the sustainability aspects associated with the utilization of technical lignins in the context of the main processing routes.2.1 Lignin origins and resources
Originating roughly 400 million years ago as land plants adapted to terrestrial ecosystems – lignin is a class of polyaromatic compounds mainly derived from three canonical monolignols, p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol, which differentiate by the number of ortho methoxy groups on the phenolic ring.29 These fundamental building blocks are synthesized by plant cells in the cytoplasm from phenylalanine and transported to the cell walls, where they undergo chemically controlled free radical coupling reactions to form polymeric constructs.30 The monolignols are therefore monomeric units of the lignin polymer and are referred to as p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S), respectively. Due to the absence of biochemical control dictating polymer formation and the high functionality of its monomer precursors, lignin is a highly complex, three-dimensional random polymer with a variety of linkages and structures (Fig. 1) including covalent linkages to the polysaccharide framework of the cell wall. Further, lignin variation according to the plant source and the plasticity of the lignin biosynthetic pathways result in high compositional and macromolecular variability.31 Hence, there is no strict definition of lignin, and there are still ongoing critical discussions on key elements of its structure and definition.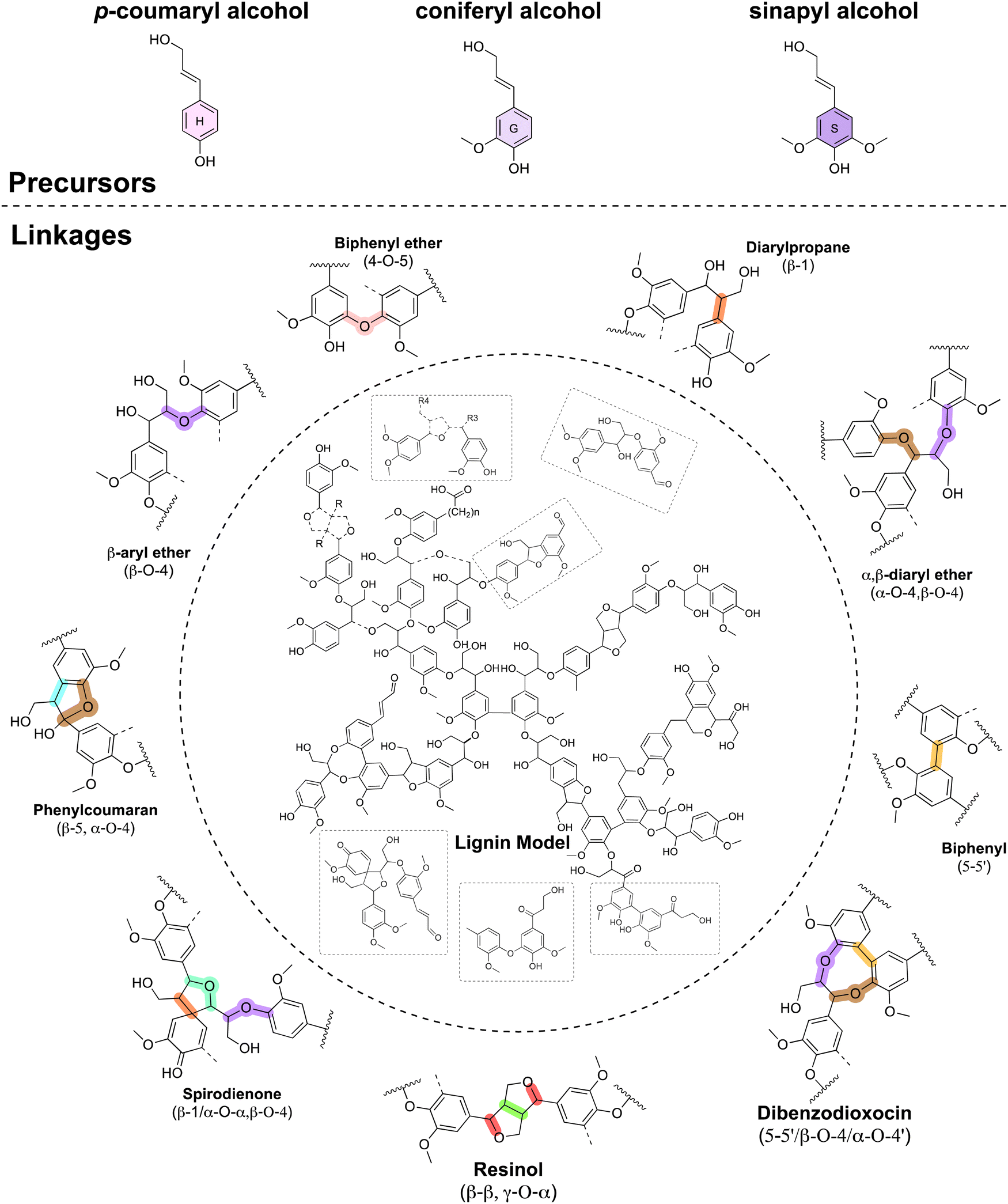 | ||
| Fig. 1 Main lignin precursors (top), typical inter-unit linkages and proposed structural model macromolecule corresponding to spruce milled-wood lignin. Some of the elements shown in this illustration are inspired by ref. 32. Adapted under terms of the CC-BY 4.0 license.32 Copyright 2020, Royal Society of Chemistry. | ||
A distinction should be made between native lignins, and lignins that have been extracted from biomass, termed technical lignins or those that remain attached to other polymeric structures, namely, residual lignins. Technical lignins are of practical relevance and have a significantly altered structure compared to their native counterparts. The former distinguish from the latter as far as its (i) reduced primary and secondary aliphatic hydroxy groups, (ii) reduced oxygenated aliphatic moieties, such as the β-O-4, and (iii) higher phenolic hydroxy and ester groups, saturated aliphatic moieties, and degree of condensation.33 The extent of these differences can vary significantly depending on processing history. Technical lignins can be bound to carbohydrate units, the so-called lignin-carbohydrate complexes (LCCs), especially lignosulfonates and hydrolysis lignins. These complexes include both covalent and non-covalent bonding between lignin and carbohydrates, and therefore, their separation and/or characterization introduce further challenges. Depending on the dissolution and fractionation processes used, technical lignins can be modified to include heteroatoms. For example, kraft lignin contains small amounts of organically bound sulfur (thiol compounds).34 Further, a significant number of native linkages are cleaved and the propyl side chain on the lignin subunits is modified, resulting in a change of chemical functionality, new linkages, and a new polymer chain architecture.
According to the plant source, three main categories of lignin exist: softwood lignins, hardwood lignins, and herbaceous lignins. Softwood lignins are primarily composed of G-type subunits and possess less compositional variability. In comparison, hardwood lignins have varying proportions of S and G units (i.e., a diverse S/G ratio), whereas herbaceous lignins are considered to be a complex mixture of H-, S-, and G-types.
In terms of processing differences, technical lignins are primarily produced by the pulp and paper industry during the chemical digestion of biomass, where lignin is dissolved to liberate the cellulosic fibers. Kraft lignins and lignosulfonates, which originate from the kraft and sulfite pulping processes, respectively, are commercially available. Lignosulfonates are a special case of technical lignins with different behavior relative to other technical lignins. The chemistry of the sulfite pulping process results in the production of highly sulfonated and amphiphilic lignin, which is soluble in water at neutral pH and possesses enhanced surface activity. Due to the significant differences of lignosulfonates with other technical lignins, in this review, it will be specified clearly when discussing them. Despite sulfite pulping being much less common than kraft, lignosulfonates have been the main source of lignin-derived commercial products.35 However, the kraft pulping process can afford partial lignin removal without offsetting the chemical and energy recovery cycle. This can be done by implementing separation processes from the so-called black liquor by the LignoBoost and the LignoForce process.36–38 As a result, the Kraft lignin market has the potential to surpass that of lignosulfonates in the near future.39
Beyond traditional pulping processes, there have been significant developments in the area of biorefining processes that incorporate organic solvent extraction, ionic liquids, acid-hydrolysis, and steam explosion.40–43 The obtained lignins may comprise new categories since these alternative processes differ significantly from established pulping systems. Hence, the term “lignin” actually describes a broad category of macromolecules and special attention should be dedicated to clearly identifying their nature or origins and processing conditions (e.g., botanical source and industrial process). Even lignins that originate from the same botanical source and industrial process possess high variability, which is not easily quantified due to their complexity.
2.2 Lignin thermal properties
To date, there is still no systematic understanding of the direct relationship between lignin's chemical and structural features and the corresponding thermal properties. Yet, thermal processing is critical for producing solid-porous adsorbents from lignin. Pyrolysis, the thermal decomposition of a material in the absence of oxygen, is a common lignin treatment method. Under inert conditions, lignin is decomposed at high temperatures, with no combustion, leading to a high carbon yield. This process and others have been discussed in a comprehensive review,44 which proposes lignin pyrolysis occurs in three main stages: dehydration (∼30–200 °C), active pyrolysis (∼200–450 °C), and passive pyrolysis (>450 °C) (Fig. 2). In the dehydration stage, moisture and volatiles are released, although it should be noted that significant rearrangement of the lignin structure will also occur below 200 °C. Above 200 °C, decomposition proceeds more rapidly, first cleaving weak ether bonds and eventually condensed C–C bonds, producing depolymerized primary products, including monomeric fragments. In this stage, roughly 40 wt% of the material is lost in gaseous forms. Above ca. 450 °C, the decomposition mechanism transitions to a gradual passive stage dominated by secondary degradation and transformation, including the cleavage or rearrangement of functional groups and depolymerization of carbon species. The pyrolysis degradation mechanism is very complex and includes various reactions and physiochemical phenomena – dependent on both the nature of the lignin structure, processing parameters (such as heating rate and final temperature), and their interactions. Unless otherwise stated, in this review, we consider carbon production from lignin by pyrolysis, which is most often performed under a nitrogen (N2) atmosphere.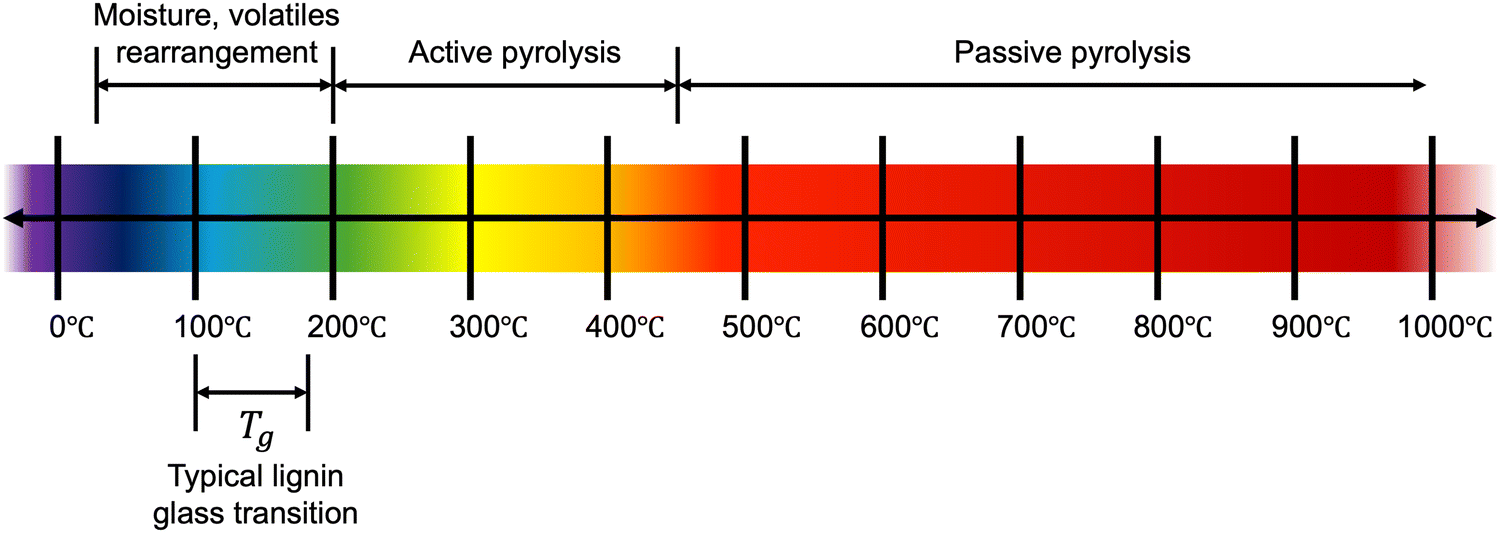 | ||
| Fig. 2 Key regions for the thermal decomposition and glass transition of typical lignin streams. | ||
Various thermal analyses can be used to investigate the thermal properties of lignin. Typical approaches include thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Along with a decomposition profile, TGA provides information regarding moisture content, volatile matter, fixed carbon, and ash content, thus serving as a valuable tool to guide thermal processing regimes and estimate carbon yield. DSC provides thermodynamic information such as phase transitions, reaction temperatures, and, importantly, the glass transition temperature (Tg) – a critical property for stabilizing the given structures during carbonization.
Kraft lignins are mostly degraded in the range of 250–450 °C (roughly 40 wt%), after which degradation begins to plateau. Most kraft lignins have a relatively low glass transition temperature, between 100 and 170 °C.45 Often, a goal prior to/during thermal processing is to increase the glass transition temperature of lignin, to facilitate direct decomposition instead of melting. The former mechanism, referred to as thermal stabilization (or oxidative pretreatment), preserves structures such as pores or hierarchical assemblies. Heating lignin at low to moderate temperatures in the presence of oxygen is an effective means to improve the thermal stability of lignin. Not all lignins are equal; in particular, thermostability strongly depends on lignin type. Lignosulfonates have higher molecular weights than most other technical lignins (Mw 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–40000 Da) and therefore may preserve the inherent morphologies and pore structures at small scales.46 Also, the fractionation of lignin impacts its glass transition, and this can be exploited for thermal stabilization.47,48
000–40000 Da) and therefore may preserve the inherent morphologies and pore structures at small scales.46 Also, the fractionation of lignin impacts its glass transition, and this can be exploited for thermal stabilization.47,48
2.3 Processing of technical lignins
In contrast to tailorable synthetic polymers with uniform structure, the heterogeneity of lignin is a major challenge in processing. Therefore, different strategies have been developed to produce lignin streams with less heterogeneity and selected features, which can be classified mainly as fractionation and assembly (Fig. 3). | ||
| Fig. 3 Typical lignin processing strategies include (a) fractionation, (b) assembly, (c) hierarchical assembly, and (d) chemical modification. | ||
Lignins are separated into more chemically uniform streams by using solvent fractionation, ultrafiltration, and/or pH shifting. Increasing lignin uniformity through fractionation may be necessary for developing solid-porous adsorbents from lignin, with tailorable and ordered pore structures. Fractionation is effective in producing fractions of (relatively) narrow molecular weight distribution. Secondary effects of fractionation lead to lignin sub-streams with chemical gradients between each other – a consequence of the inextricably linked molecular weight and functionality-type distributions inherent to lignins.49 Larger molecular weight fractions typically contain more aliphatic and less phenolic and carboxylic hydroxy groups; differences in the inter-unit linkages among fractions are also typical. Collectively, the solubility of lignin is greatly impacted, together with properties such as thermal stability, reactivity and surface energy/wettability.50
Alternatively, assembly has been applied to transform polymeric lignin into spherical colloidal particles of tailorable size (1 nm–10 μm) and surface charge. They are readily produced in wet or dry forms through solvent exchange, spray-drying, or aerosol flow processes.51 Despite the broad molecular weight distribution and functionality of the starting lignin profile, remarkably narrow particle size distribution is attainable within assembled lignin structures. The degree of uniformity can be further improved with particle size fractionation, offering avenues for highly ordered structures.52,53
Hierarchical assembly and chemical modification can be applied to tailor both the molecular- and macro-scale properties of lignin-based materials. Lignin supraparticles, which display hierarchical porous structures, have been shown for their potential for CO2 capture.54 Controlled porosity at multiple length scales facilitates molecular and gas transport, producing materials with macrostructures that meet the technical requirements for scaled processes.
The abundant functional groups of lignin, including aliphatic, phenolic, carboxyl hydroxy, and methoxy groups, as well as ether bonds, offer convenient possibilities for chemical modification and to impart properties matching given applications (Fig. 4).55 Chemical modification can take the form of depolymerization, modification of its functional groups, or graft copolymerization.56 Depolymerization of lignin macromolecules to platform chemicals enables bottom-up approaches for materials synthesis, yet is not a trivial task due to its structural complexity and recalcitrant C–C and C–O bonds.57 Functional group modification can include the synthesis of new active sites or the modification of the present hydroxy groups.58 Graft copolymerization can be performed to attach new polymer chains to the lignin structure via its reactive hydroxy groups.
 | ||
| Fig. 4 Lignin derivatization pathways: the central panel depicts a segment of a lignin macromolecule, surrounded by various derivatives corresponding to specific reactions, as indicated. | ||
While the processing strategies covered in this section have been discussed individually, more sophisticated efforts involve the combination of two or more of these strategies.59 The compositional profiles (and associated properties) of the derived lignins must be considered for its impact on the final product. This can be addressed through early-stage techno-economic analyses, which are recommended to identify viable pathways for valorization.
2.4 Sustainability aspects relevant to technical lignin valorization
Emerging carbon capture technologies must be critically evaluated in terms of their sustainability. Ideally, sustainable solutions should consider the entire environmental impact over their full life cycle, from raw material sourcing to recycling or disposal. While bio-based materials provide renewable carbon sources, they must also meet these sustainability standards. In this context, standardized life cycle assessments (LCAs) are essential.60Generally, lignin valorization approaches have the potential to reduce the environmental impact of kraft pulp mills by decreasing stack emissions from the recovery boiler.61 The utilization of lignin for carbon capture applications further expands the potential for reducing net CO2 emissions and the associated environmental impacts of lignin valorization. A critical assessment of 42 peer-reviewed LCAs regarding lignin and lignin-derived products found that they often offer better environmental performance compared to their fossil-based counterparts, particularly concerning their impact on climate change.62 Moreover, when considering the displacement (substitution) of petroleum-derived products by lignin, this impact can be even more significant. Life cycle assessments involve assumptions, boundary conditions, and standardized methods that must be taken into account when interpreting their results, as these factors can significantly influence decision-making.62
In Section 2.3, we introduced various techniques typical for technical lignin processing aimed at reducing heterogeneity and tailoring its properties. While these approaches have gained popularity in the literature, they should be approached with caution. Excessive or redundant processing steps can significantly increase the complexity of the system, as well as capital and operational costs, and may also heighten environmental impacts.
Chemical modification of lignin must adhere to the principles of green chemistry and sustainability.63,64 Molecular fractionation of lignin can be employed to reveal structure–property relationships. However, if not properly integrated, these schemes can become complex and expensive, often providing only incremental improvement in lignin uniformity and properties. A practical approach is assembling lignin into spherical particles, which addresses multiple technical challenges and produces relatively homogeneous and structured materials. However, in the most common methods for obtaining colloidal lignin particles—such as pH shift and the use of anti-solvents—considerable expenses arise due to the large volumes of organic and aqueous solvents (used for solvent exchange) and the energy-intensive drying processes.
In Section 4 to follow, we will discuss the processing of lignin to porous carbon adsorbent materials. These operations are often energy and chemical-intensive. Thermal processing incurs a significant energy penalty during the carbonization and activation steps. Chemical activation, which is a common approach because of its simplicity and efficacy, requires the use of an activation reagent typically at amounts of at least twice that of the carbon source by mass. Further, a washing step is typically necessary for post-treatment to remove the remaining activation agent and expose the formed pore structure. Assembling porous structures prior to carbonization are promising approaches to bypass or reduce the requirement for chemical activation and associated washing steps, but this is often at the cost of increased processing time, complexity, and energy for procedures such as thermal stabilization – often required to maintain those structures at the high temperatures necessary for carbonization.
In lignin valorization efforts, processing steps often drive costs and environmental impact, ultimately affecting sustainability. However, in the context of carbon capture technologies, the sustainability of lignin-based adsorbents will critically depend on their adsorption performance. In the following section, we discuss the key performance indicators for lignin-based carbon adsorbents, which ultimately determine the success of these emerging technologies.
3. Properties and performance of lignin-based carbon adsorbents
The performance indicators of lignin-based carbon adsorbents are essential for advancing their commercial-scale implementation in adsorption systems. These indicators encompass both material properties and adsorption performance, which are interrelated. Regarding material properties, standard characterization of carbon materials typically involves assessing the specific surface area, pore structure, surface chemistry, purity, and degree of graphitization. Adsorption capacity, selectivity, and kinetics are key factors that influence adsorption performance.One of the key challenges for the scaleup adoption of solid adsorbents is the lack of standardization in the characterization and evaluation. To adequately compare various lignin-based adsorbents and benchmark them against competing technologies, it is essential to align experimental methods and subsequent analyses. Therefore, this section will first introduce the properties of lignin-based carbons, which dictate adsorption performance, with a focus on methods for their determination. Subsequently, we will discuss adsorption performance in terms of the most critical performance indicators.
3.1 Carbon properties
The specific surface area of a solid material can be determined using gas physisorption experiments based on equilibrium van der Waals interactions between the gas molecules and the solid surface.65 The BET (Brunauer–Emmett–Teller) theory is the most common approach to obtain the specific surface area (SSA) of carbonized lignin.66 Adsorption isotherms can also be used to calculate pore size distributions. Extreme caution is necessary for applying the BET theory to materials with a high degree of microporosity.67 Further, the choice of adsorptive is also of critical importance, adsorption of CO2 at 273 K (0 °C) has become the accepted method for carbonaceous materials with very narrow micropores.67 The influence of porosity on CO2 uptake will be discussed in Section 5.1.1.Activated carbons typically contain disordered arrangements of aromatic carbon structures that stack to create local order.68 Their degree of graphitization is therefore dependent on many factors of the precursor material and thermal processing. Carbonizing lignin produces hard carbon, which is a disordered material that is not easily graphitized, even at very high temperatures (>1500 °C).69 The degree of graphitization or order can provide insights into the structure and performance of carbon materials. Powder X-ray diffraction (XRD) is a typical approach to gain insights into the order of the carbon and can be used to estimate the degree of graphitization by comparing peak intensities of the (002) and (100) reflections. Raman spectroscopy can also be utilized to provide information on order and crystallite or domain size. A stretching vibration in aromatic layers is known as the G (graphite) band ∼1580 cm−1, which can be compared to the D (disorder or defect) band ∼1350 cm−1 attributed to the breakdown of translational and local lattice symmetries.70
Carbon materials derived from technical lignins can also exhibit diverse surface chemistries, yet they all share in common considerable sp2-hybridized carbons with two-dimensional order in addition to many heteroatoms (mainly oxygen).71 Minor differences in surface chemistry can play an important role in dictating the adsorbent–adsorbate interactions, and therefore, characterization of these groups is critical. Common approaches to identify and attempt to quantify these groups include spectroscopy/spectrometry techniques such as Infrared spectroscopy (IR), Raman, X-ray photoelectron spectroscopy (XPS), and temperature-programmed desorption (TPD). Boehm titration can also be employed, with caution, to measure the concentration of functional groups on the carbon surface.72
Other important material properties include their structural topology, thermal stability, and mechanical stability. The structural topology can be evaluated with microscopy, in particular, electron microscopy is typical for evaluating the structural features at smaller length scales. Thermal stability, of which can be decisive under the various operating conditions necessary considering feed-gas temperatures and regeneration, is typically evaluated with TGA. Mechanical stability of structured adsorbents is essential for determining their suitability for withstanding compression forces in packed bed systems and can be evaluated with compression tests, for example with dynamic mechanical analysis.54
3.2 Adsorption performance
Adsorption capacity (equilibrium capacity and working capacity) is a primary performance indicator for solid gas adsorbents. Equilibrium capacity refers to the amount of CO2 adsorbed at equilibrium under specified conditions, while working capacity represents the amount of CO2 captured during a complete adsorption/desorption cycle, calculated as the difference in capacity between adsorption and desorption conditions.73 Working capacity is more relevant at the industrial scale; however, equilibrium adsorption capacity is often the only metric reported in the literature, particularly for adsorbents associated with low technology readiness levels (TRLs). Equilibrium adsorption capacity is typically determined using volumetric methods (gas adsorption isotherms) or gravimetric methods (TGA).Dynamic column breakthrough experiments are important indicators of adsorbent performance. The breakthrough response of an adsorption column refers to the signal observed at the column's outlet stream resulting from a step change in the concentration of one or more adsorbate components.74 Notably, breakthrough experiments provide both single- and multi-component equilibrium information, in addition to enabling the characterization of adsorption kinetics. Dynamic column breakthrough experiments are not widely reported, particularly in material development labs that do not specialize in adsorption systems, where sample production is often limited to the milligram scale. Advances in microscale dynamic column breakthrough measurements may accelerate the screening of solid adsorbents, especially from materials that exhibit high variability, such as lignin.75
Additional indicators of the performance of solid adsorbents include selectivity, heat of adsorption, and their recyclability/regeneration. In real processes, feed gas streams consist of multicomponent mixtures that compete for adsorption on the surfaces of the adsorbent. Therefore, evaluating CO2 selectivity is essential.76 Of the various models proposed for predicting multi-component gas adsorption equilibria from pure-component isotherms, the Ideal Adsorbed Solution Theory (IAST) has remained a preferred choice due to its simplicity and reliability.77,78 The isosteric enthalpy of adsorption—defined as the heat released upon binding to a surface—governs the local adsorbent temperature and, consequently, the local adsorption equilibria, kinetics, and resulting separation efficiency.79 In particular, a large isosteric enthalpy of adsorption can delay equilibrium and indicate stronger adsorbent–adsorbate interactions, which can increase the costs associated with adsorbent regeneration.80–82 The isosteric enthalpy of adsorption is typically determined indirectly by applying the Clausius–Clapeyron equation to adsorption isotherms obtained from volumetric gas adsorption measurements.83 To assess the regeneration stability (mechanical, chemical, and thermal stability) of the adsorbent, cycling experiments under the relevant operating conditions are performed.
4. Processing of lignin-based porous materials
There are several methods for producing solid porous adsorbents from lignin. Traditional approaches involve two main processes: carbonization and activation, which can be carried out either simultaneously in a one-step process or sequentially in a two-step process. In carbonization, thermal treatment is applied to induce a complex reaction that results in a material with a higher carbon content. Thermal treatment is most often performed via pyrolysis, while hydrothermal carbonization and microwave treatment are also promising alternatives. In contrast to carbonization, activation aims to introduce or augment the porosity of the material, creating cavities, which facilitate gas transport or create sites for molecular adsorption. This is typically achieved via chemical or physical activation. Chemical activation creates pores by reacting the material with an acid, base, or salt at elevated temperatures. In contrast, physical activation uses a reactive gas environment at high temperatures to etch the material's surface.The above-mentioned approaches are effective in producing highly porous lignin-based materials but lack control over pore sizes, leading to disordered morphologies and non-uniform pore-size distributions. However, ordered porosity is known to promote molecular transport and adsorption.84 Thus, recent approaches have advanced morphology control at multiple scales, reducing or eliminating the severity of the activation steps. Hierarchically structured materials are those with well-defined and reliably controlled combinations of structural features at various scales and typically exhibit multiple advantages compared to their more disordered counterparts. In particular, control over macroscopic dimensions has technical advantages for industrial adoption, for example, in the case of packed columns typical for adsorption processes, where powdery material can induce undesired pressure drop due to insufficient void volume for gas transport. In terms of pore structure, mesopores (2–50 nm) facilitate gas transport and molecular diffusion, forming scaffolds for and access to micropores (<2 nm), which in turn greatly enhance the surface area, offering adsorption sites and molecular sieving capacity. Ultramicropores (<0.7 nm) are critical in determining the dimensions of the binding sites and provide molecular sieving function.
In the following section, we examine lignin processing into solid porous CO2 adsorbents and the various strategies used to increase the apparent surface area and optimize porous structure at multiple length scales. We begin with traditional processing to produce irregular porous carbon, simple one-step carbonization, and multi-step or hybrid techniques with various carbonization/activation/doping schemes. Subsequently, we cover 3D networked structures produced by crosslinking chemistries. Finally, we examine the approaches for hierarchically structured materials with more sophisticated designs and greater control over the multi-scale pore structure and macroscopic dimensions of the material, through the formation of superstructures or by hard or soft templating.
4.1 Traditional approaches to CO2 adsorbents
Pan et al. investigated the effect of pressure during pyrolysis of high ash-content alkali lignin.86 They carbonized lignin at 10 °C min−1 to 800 °C for 3 hours and under three different pressures: negative pressure (−0.1 MPa), atmospheric pressure (0 MPa), and elevated pressure (0.1 MPa). It was found that CO2 adsorption capacity, specific surface area, and micropore volume positively correlated with lower pressure during carbonization, with a trade-off in carbon yield. The optimized carbon material obtained under negative pressure had a specific surface area of 1577 m2 g−1 and micropore volume of 0.695 cm3 g−1, leading to a CO2 adsorption capacity of ∼3.6 mmol g−1 (at 0 °C). The vacuum reduced the boiling point of volatiles and reduced vapor residence time, facilitating their removal and minimizing undesired secondary reactions during pyrolysis.
The impurities of high ash-content lignin are resource-dependent and unpredictable, limiting the applicability of one-step carbonization protocols and compromising quality control over the activation process. More consistent carbon production can be achieved by adding activation agents to purified lignin. Sun et al. used a one-step procedure by soaking corn straw lignin in 2:1 w/w phosphoric acid (H3PO4), followed by heating at 30 °C min−1 to 300–600 °C for 120 min.87 Mesopores were formed below 500 °C with the presence of H3PO4, after which the pore volume decreased sharply, potentially due to the decomposition of phosphorous esters. Their approach could produce specific surface areas and pore volumes of 820 m2 g−1 and 0.8 cm3 g−1, respectively. However, the dynamic adsorption performance in a packed bed reactor indicated a relatively poor separation performance of the as-produced carbon pellet, despite a promising selectivity between CO2 and methane (CH4) from equilibrium adsorption results. The authors suggest that further modification of the functional groups on the carbon surface is necessary to improve the selectivity towards CO2.
Li et al. utilized the one-step method by using wet impregnation with KOH solutions at a 1:1 or 2:1 w/w activation ratio to process kraft lignin.88 After drying, the mixtures were carbonized at a rate of 5 °C min−1 to 600, 700, or 800 °C and holding for one hour. Regardless of the activation ratio or final temperature, the resulting carbons primarily possessed microporous structures (84–93%). Mild carbonization/activation conditions produced lignin-based adsorbents that favor adsorption at lower CO2 partial pressures, while those from high temperatures and high activation ratio conditions performed better at high CO2 partial pressures. In general, CO2 uptake depends on ultra-microporosity at low partial pressures, while total micropore volume is more important at high CO2 partial pressures. For this work, the uptake capacities can go up to 2 mmol g−1 (at 25 °C) at low CO2 partial pressures, whereas the CO2/N2 selectivity can reach 38 at 15 kPa CO2. Importantly, Li et al. provide strong evidence that KOH activation can leave intercalated potassium (K+) ions under the carbon surface that are not leached by washing in water. These ions can significantly increase favorable interactions with CO2, improving uptake by up to 50%.
In the above cases, technical lignins (such as kraft lignins) already have abundant oxygen-containing functional groups that play a role in the surface interactions with CO2, although the impact on CO2 adsorption is still a topic that remains unclear.89 Aside from harnessing the heteroatoms within the lignin, doping is the most common strategy to produce carbon materials from lignin with favorable surface chemistry for CO2 adsorption. Heteroatoms, such as nitrogen or sulfur, can increase the affinity to CO2. In the discussion that follows, it is also shown that the surface chemistry is shown to be adjustable according to the technical lignin type, i.e., its functional groups (for example in the case of lignosulfonate or enzymatic hydrolysis lignins), which contain varying quantities of sulfur or nitrogen, respectively.
Saha et al. extended the one-step approach to include S-doping by combining lignin, KOH, and sodium thiosulfate (Na2S2O3), followed by pyrolysis at 10 °C min−1 to 800 °C and holding time of 2 minutes.90 A high surface area was generated through activation, while simultaneously introducing sulfur content (1–12.6%) to the carbon surface to improve the surface interactions with CO2. The highest surface area (surface area and pore volume of 3626 m2 g−1 and 1.7 cm3 g−1, respectively) was achieved from the largest ratio of KOH and the lowest ratio of Na2S2O3, attributed to a large percentage of microporosity and, consequently, the best CO2 adsorption capacity (11 mmol g−1 at 25 °C).
N-doping has become popular in the literature, since it can significantly promote the Lewis acid–base interactions. Tkachenko et al. combined nitric acid (as the chemical activator) with urea (as the N-doping agent) to produce N-doped activated carbon from softwood kraft lignin (Fig. 5).91 In their synthesis, lignin was combined with urea and nitric acid (300 mg urea/2.5 mL 40% Nitric acid/1 g lignin) and heated at 6 °C min−1 to 800 °C. The resulting carbon material was significantly microporous with 3.5% N content and achieved a specific surface area of up to 1000 m2 g−1. The CO2 adsorption capacity boosted to 1.4 mmol g−1 at 15 kPa and 20 °C.
 | ||
| Fig. 5 Schematic illustration of the transformation of lignin to N-doped carbon. Reproduced under terms of the CC-BY 4.0 license.91 Copyright 2024, American Chemical Society. | ||
Chen et al. used microwave treatment and combined physical/chemical activation with humidified nitrogen and KOH to prepare carbon from enzymatic hydrolysis lignin (Fig. 6).92 Microwave heating can be advantageous because of its low energy consumption, short heating duration, and uniform heating gradient. Lignin powder was mixed with KOH at a 1:3 ratio and dried, then placed in a microwave under a humidified nitrogen atmosphere. The humidified nitrogen could act not only as a microwave absorber to increase temperature, but also, with steam, as a physical activator. In an optimum processing condition (30-minute microwaving), the porous carbon showed a specific surface area of 2870 m2 g−1 and 2.02 cm3 g−1 pore volume, associated with high oxygen content. Collectively, these properties enable CO2 uptake of 1.31 mmol g−1 at 30 °C. Most remarkably, the rapid carbonization time during microwave heating is the major advantage of this approach.
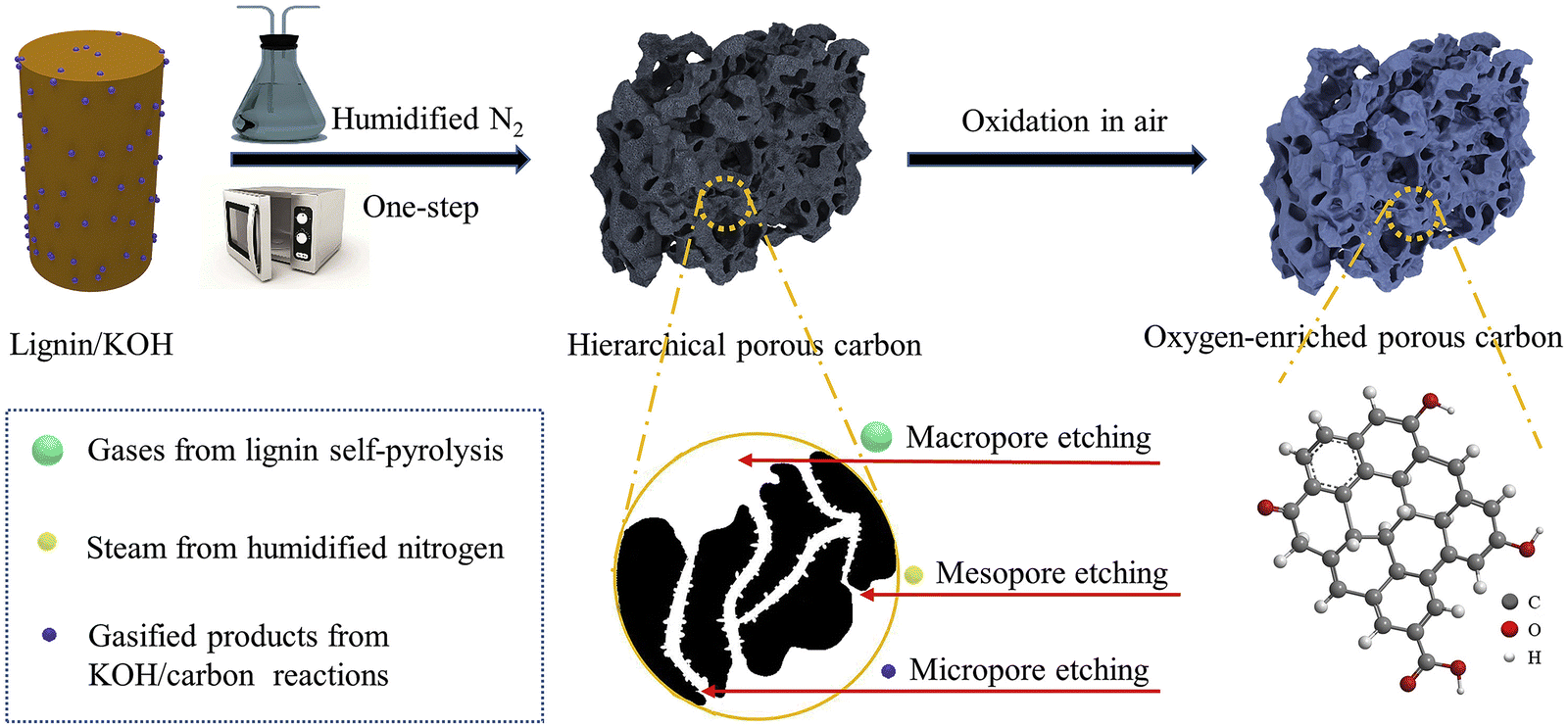 | ||
| Fig. 6 Schematic illustration of the one-step microwave synthesis in the presence of humidified nitrogen and oxygen-enriched porous carbon. Reproduced with permission.92 Copyright 2019, Elsevier. | ||
In another approach, Saha et al. synthesized a carbon with hierarchical porosity from lignin via a hybrid one-step chemical activation, followed by physical activation/nitrogen doping with ammonia (NH3).93 Lignin was first mixed at a 1:1 ratio with KOH, pyrolyzed at 10 °C min−1 to 800 °C, and held for two minutes before washing and drying. The dried carbon product was later physically activated with NH3 at 800 °C. The NH3-induced physical activation increases the surface area and pore volume, as well as grafted a variety of nitrogen groups on the carbon scaffold, such as pyridinic, amino (combined primary, secondary, tertiary), pyrrolic (in combination with pyridone), graphitic (or quaternary amine), nitro and nitroso. It is noteworthy that the physical activation process would reduce the carbon yield and cause undesired over-activation effects if not optimized. Nevertheless, CO2 selectivity increased monotonically with nitrogen content, indicating that N-doping can improve the CO2 selectivity of carbon materials. Surface area was not directly correlated with selectivity towards CO2 over N2. Their method achieved specific surface areas of up to 2922 m2 g−1 and CO2 adsorption capacities of 5.5 and 8.6 mmol g−1 at 25 °C and 0 °C, respectively.
Slow pyrolysis is the typical initial step in the two-step approach. Gong and Bao investigated a two-step carbonization and activation process to produce porous carbon from lignin.94 They first pre-carbonized the lignin at a heating rate of 5 °C min−1 to three different temperatures (350, 450, 550 °C) for 2 hours. The resulting char was ground with KOH at ratios 1:1, 1:2, and 1:3, followed by activations at 5 °C min−1 to 600 °C or 800 °C under N2. The authors found that a lower final activation temperature favored the development of pores. Their approach could achieve gas adsorption performance of 3.98 and 5.82 mmol g−1 at 100 kPa, 25 °C and 0 °C, respectively.
Hydrothermal carbonization is a promising alternative for the thermal conversion of wet organic streams such as industrial pulping residues, especially since the vast majority of technical lignins are extracted as a soluble residue in the form of black or brown liquor. The hydrothermal process can effectively avoid the energy-intensive drying and demineralization steps. Interestingly, the concept of hydrothermal processing was first conceived as a simulation of coal formation.95,96 In such a process, biomass is subjected to an aqueous treatment at low to moderate temperatures (∼180–250 °C), whereby various chemical reactions (e.g., hydrolysis, dehydration, decarboxylation, polymerization, aromatization, condensation) result in a solid carbonaceous material.97 While the carbon product of biomass pyrolysis is commonly known as biochar, the product of hydrothermal carbonization is known as “hydrochar”, as will be denoted in the following paragraphs.
Sangchoom et al. synthesized porous carbon from lignin without demineralization via a two-step process.98 Their approach involved hydrothermal carbonization at 300–390 °C, followed by chemical activation with KOH at ratios 4:1 or 2:1 via heating at a rate of 3 °C min−1 to 600–900 °C and holding for 1 hour. While the original lignin contained a 20 wt% non-combustible ash content, the hydrochar contained only organic material without minerals. This result indicates that the hydrothermal carbonization successfully subverted the demineralization step. Remarkably, the as-produced carbon products achieved high surface area and pore volumes of 1157–3235 m2 g−1 and 0.59–1.77 cm3 g−1, respectively, allowing excellent CO2 adsorption capacity of 4.6 mmol g−1 at 25 °C and 100 kPa, 17.3 mmol g−1 at 25 °C and 2000 kPa, and 7.4 mmol g−1 at 0 °C and 100 kPa. The authors denoted that pore size, particularly micropores ∼7 Å, is key for CO2 uptake at ambient conditions.
Hydrothermal carbonization can be used to produce hydrochar but can also be modified for other objectives, such as the co-production of bio-oils.99 Hao et al. used the solid by-products from the conversion of lignin to bio-oil, and incidentally, they produced a magnetic-activated carbon from eucalyptus enzymatic hydrolysis lignin and softwood kraft lignin.100 These hydrochars were considered as a side product of a lignin-to-liquid process when performing hydrothermal treatment together with formic acid. Briefly, 200 g of lignin, 200 mL of formic acid, 500 g of water, and the catalyst were added to a 5 L stainless-steel reactor. A temperature of 380 °C was used to produce hydrochars from the kraft lignin from Norway spruce and 365 °C when using the lignin derived by enzymatic hydrolysis of eucalyptus biomass. The as-produced biochars were mixed with KOH solutions and heated for 5 hours under 200 °C, followed by activation at 700 °C or 800 °C for 4 hours. The magnetic properties of the porous carbon were likely a consequence of corrosion of the stainless-steel reactor during hydrothermal treatment with formic acid, resulting in hydrochars of up to 12.3% iron by weight. These lignin-based magnetic carbons exhibited high surface area and adsorption capacities (up to 2975 m2 g−1 and 6 mmol g−1 at 101 kPa and 0 °C), but it is perhaps also a good example of the potential challenges for hydrothermal carbonization treatment, being affected by corrosion. It is further not clear the advantage of such a magnetic material.
Although the most attractive feature of hydrothermal carbonization is the energy savings (from bypassing the drying step compared to traditional pyrolysis), it has also been employed to treat dry feedstocks. Demir et al. produced N-doped porous carbon from organosolv lignin via a two-step hydrothermal carbonization and chemical activation scheme.101 Hydrothermal carbonization was performed in water at 300 °C and 10 MPa, together with probe ultrasonication. Later, the hydrochar was heated to 700–1000 °C for activation with the co-addition of KOH and adenine (N-doping source). With their approach, a high surface area was achieved (1788–2957 m2 g−1), together with up to 4.8 mmol g−1 in CO2 adsorption capacity (100 kPa, 25 °C). Heteroatom doping increased the CO2 capture performance slightly, compared to the pristine carbon. CO2 adsorption selectivity was dependent on the synergism of heteroatom doping and microporosity, rather than microporosity alone.
Atta-Obeng et al. used a two-step hydrothermal carbonization and activation with additional polyethyleneimine (PEI) functionalization.102 Hydrothermal treatment was performed at 350 °C, activation at 4:1 KOH to char ratio 10 °C min−1 to 800 °C for 1 hour. The activated samples were further functionalized with PEI at 5–25 wt%. Activation increased the surface area from 2.8 to 1341 m2 g−1, and PEI functionalization increased CO2 adsorption capacity from 1.53 mmol g−1 to 2.0 mmol g−1 at 30 °C. However, the authors also found that increasing PEI impregnation beyond 5% leads to severe pore blockage.
While typical two-step approaches separate carbonization and activation, alternative methods maintain simultaneous carbonization/activation treatments followed by a separate doping step. Dong et al. produced porous carbon from lignin using a KOH carbonization/activation step, followed by N-doping through hydrothermal treatment or pyrolysis.103 Pyrolysis was performed at 5 °C min−1 to 700 °C for 2 h at KOH:lignin mass ratios of 0.25, 0.5, 1, and 2. The resulting porous carbon was N-doped by mixing with 5 g of urea and subjected to pyrolysis under the same conditions used for carbonization/activation or by hydrothermal treatment in a reactor at 220 °C for 2 hours. The authors found that pore volumes in the range of 0.6–0.8 nm played a critical role in determining the adsorption capacity at 25 °C and 101 kPa, consistent with some density functional theory (DFT) simulation results that indicated an optimal pore size for CO2 adsorption of 0.62–0.72 nm, based on the optimal adsorption distance for CO2 molecules (0.31–0.36 nm).104 However, at lower adsorption pressures or higher adsorption temperatures, pore size became less significant, and nitrogen-containing functional groups gained importance for enhancing chemical and physical interactions with CO2, thereby increasing adsorption capacity. The chemisorption of CO2 by surface nitrogen groups at 50 °C was indicated by in situ diffuse reflectance infrared Fourier transform (DRIFT) spectra. Furthermore, the N-doped samples exhibited higher selectivity at lower adsorption pressures, suggesting that under those conditions, nitrogen-containing functional groups had a stronger affinity for CO2 than narrow micropores. Overall, their approach achieved high surface areas of up to 2126 m2 g−1 and high adsorption capacities of up to 4.79 mmol g−1 at 25 °C and 101 kPa.
Bai et al. employed a two-step method involving simultaneous carbonization and N-doping treatment, followed by chemical activation.105 Lignin was mixed with melamine at a 1:1 mass ratio and pyrolyzed at a rate of 10 °C min−1 to 500 °C for 1 h under an Argon atmosphere. The resulting carbon powder was then mixed with CuCl2·2H2O at a weight ratio of 1:2 for activation and heated at 10 °C min−1 to 800, 850, or 900 °C for 2 h. The authors found that melamine not only served its N-doping function but also introduced mesoporosity and enhanced micropore development during CuCl2 activation. Notably, the selected lignin contained approximately 2% sulfur, which contributed oxidized sulfur functionalities to the carbon surface, increasing its basic character. The analysis revealed that narrow micropores (<1 nm) exhibited a strong linear correlation with adsorption capacity; meanwhile, nitrogen and sulfur content only showed a weak correlation. However, when comparing the IAST selectivity of the prepared adsorbents, it was found that heteroatom content (nitrogen and sulfur) was critical for enhancing selectivity towards CO2 over N2. Overall, the optimized method achieved high surface areas (1678 m2 g−1) and adsorption capacities (6.78 and 3.57 mmol g−1 at 100 kPa and 0 °C and 30 °C, respectively), along with remarkable CO2/N2 IAST selectivity (132 and 81 at 100 kPa and 0 °C and 30 °C, respectively).
:1 mass ratio, and dispersed in water for thermal treatment (150 °C, 20 hours). In the second stage, the dried material was combined with KOH at a 4:1 ratio and compacted into 1.5 cm pellets at different pressures (10, 20, 30 MPa) and residence times (10, 20, 40 min). Finally, the resulting pellets (∼3 mm thick) were pyrolyzed in a tube furnace at a heating rate of 3 °C min−1 to 500–800 °C and held for 1 hour. The advantage of introducing mechanochemical processing (step 2) is its operational simplicity and scale-up capability. Mechanical treatments also bypass more intensive chemical reaction schemes, such as high temperatures. After the multi-step treatment process, both the microporosity and pore volume of the lignin-based carbon material increased. In particular, CO2 uptake is measured to be improved with the introduction of narrow micropores (<1 nm) and N-containing groups. The resulting material could achieve large surface area (1233.2 m2 g−1), micropore volume (V(d<1.0nm) 0.27 cm3 g−1) and CO2 adsorption capacity (5.00 mmol g−1 at 0 °C).
 | ||
| Fig. 7 Multistep process for mechanochemical processing of enzymatic hydrolysis lignin to nitrogen-doped activated carbon. (LDD: lignin-based hydrothermal carbon; LSY: lignin-based N-doped porous carbon). Reproduced with permission.106 Copyright 2024, Elsevier. | ||
Lignosulfonates represent a special case of lignin resources due to their sulfonated structure, higher average molecular weight distribution, and solubility in water, as previously introduced in Section 2.1. Yet, lignosulfonates are, to date, the most commercially relevant lignin resource. Gao et al. produced porous carbon from sodium lignosulfonate at various initial pH using a three-step approach, that sequentially involves hydrothermal carbonization, pyrolysis, and CO2 physical activation.107 Sodium lignosulfonate (7.5 g) was first added to 100 mL aqueous solutions and titrated by sulfuric acid at a 1:7 ratio. Hydrothermal carbonization was then conducted at 260 °C for 24 hours. The resulting char was further carbonized at 10 °C min−1 to 900 °C for 1.5 hours and, subsequently, physically activated using CO2 gas at 800–900 °C. The first step of acid treatment increased the degree of graphitization of the resulting carbon. Also, increasing acid dosage raised the carbon yield and decreased the oxygen content, reflected in a rise of carboxyl and a decrease of hydroxy and carbonyl groups.
The structural and chemical differentiations resulted in various CO2 adsorption capacities at different temperatures. The authors found that adsorption is primarily influenced by CO2-substrate interactions at low temperatures (e.g., electrostatic and van der Waals forces), whereas the diffusion resistance of the pore channel becomes the dominating factor at higher temperatures, when CO2 diffuses fast. With a vast pore size and functional group selection from the multi-step production of lignin-based carbon materials, Gao et al. investigated their corresponding interactions towards CO2 molecules by quantum DFT (Fig. 8). It was shown that smaller pores and oxygen-containing functional groups increased the favorable adsorption energy between CO2 and the carbon surface, with carboxylic groups displaying the strongest attraction to CO2. This is because the oxygen-containing groups act as a Lewis base to interact with the acidic CO2 molecule. Ultimately, these lignin-derived carbons could achieve an adsorption capacity of up to 5.10 mmol g−1 at 0 °C and 100 kPa.
 | ||
| Fig. 8 Carbon sheets with varying pore sizes represented by ball-and-stick model and electron cloud density with bond lengths between the oxygen atoms of CO2, as well as calculated adsorption energy specified. Here, (a) depicts the comparison between interactions with different pore structures (non-porous, microporous, macroporous) and (b) shows the comparison of various functional groups on the carbon sheets (pure carbon, carbonyl, hydroxy, and carboxylic acid group). Reproduced with permission.107 Copyright 2024, Elsevier. | ||
Park et al. synthesized an N-doped porous carbon from sodium lignosulfonate via the multi-step approach, which included (i) hydrothermal carbonization, (ii) KOH activation and (iii) post-doping with urea at high temperatures and under Ar gas flow.108 Hydrothermal carbonization was performed at 200 °C for 12 hours. The resulting char was activated with KOH at a 1:3 ratio at 5 °C min−1 to 700 °C. The activated carbon was doped with urea at a 1:1–1:8 ratio, and subjected to a heating rate of 5 °C min−1 to 800 °C. The KOH activation step significantly increased the surface area from 125 to 3170 m2 g−1. However, the doping step could introduce undesired blocking of micropores. The highest CO2 adsorption capacity was characterized to be 13.6 mmol g−1 at 1 MPa, achieved with the 4:1 mass ratio of urea to activated carbon, originating from the optimal balance of the high surface area and nitrogen-containing surface functional groups.
Following the concept of RCC, Zhao et al. produced a porous carbon CO2 reduction electrocatalyst from sodium lignosulfonate. ZnCl2 activation was applied to pre-carbonized samples (pyrolysis at 500 °C for 1 hour), followed by the N-/P-doping with melamine or triphenylphosphorus.109 The activation was operated with ZnCl2 at a ratio of 2:1 at 800 °C for 2 hours. Then, the carbon materials were mixed with melamine (N) or triphenylphosphorus (P) at a 5:1 doping agent to lignin ratio, and pyrolyzed at 700–1000 °C for 2 hours under N2. Both N and P doping are beneficial for CO2 reduction and hydrogen evolution. However, P doping was more favorable towards hydrogen evolution than CO2 reduction, compared to the N-doped samples. The authors found that the enhanced CO2 reduction performance from their N-doped porous carbon was actually the result of a combination of multiple factors. For example, the textural properties, degree of graphitization, pore structure, and surface functionalities were highly dependent on the carbonization temperature, and the resulting electrocatalytic performance of the material was determined by the combined effect of these variables. Overall, their material was reported with surface areas of up to 1459 m2 g−1 and adjustable syngas H2/CO ratios from 2.3 to 8.0.
4.2 Structured porosity via crosslinking
Amorphous hyper-crosslinked polymers are a type of porous materials with high surface area and pore volume, as well as adjustable surface chemistry. They can be synthesized with lignin via Friedel–Crafts reactions, which proceed from electrophilic aromatic substitution. Hyper-crosslinking is a promising approach for constructing highly rigid crosslinked bridges between the aromatic repeat units of lignin. These hyper-crosslinked polymeric materials may not necessarily require carbonization steps, as they are designed to exhibit pore structures from crosslinking. These crosslinked samples are typically more mechanically and thermally stable than the raw material, giving them a higher tolerance to harsh environments. The major drawback of the crosslinking approach is the extra cost of chemical reagents and the complex chemistry of the reactions.Meng and Weber crosslinked organosolv lignin with formaldehyde dimethyl acetal (FDA). The resulting materials were evaluated by their CO2 capture performance, either with or without further low-temperature pyrolysis (550 °C). In their scheme, organosolv lignin, anhydrous iron chloride (FeCl3), and FDA were mixed in 1,2-dichloroethane (DCE) and added to a round bottom flask equipped with a condenser under an Ar atmosphere. The mixture was stirred for 1 hour at room temperature, followed by heating at 45 °C for 5 hours to form a crosslinked network. Subsequently heated at 80 °C for 19 hours to complete the reaction. Both materials were found to be primarily ultra-microporous. Notably, the crosslinked material was capable of maintaining its shape during carbonization, in addition to having increased CO2 uptake, despite a reduced selectivity.110
Shao et al. used six model lignin monomer units to prepare a hyper-crosslinked polymer in a one-pot Friedel–Crafts reaction, also utilizing the FDA as the crosslinker.111 These six phenols were first dissolved in DCE at 35 °C. Catalysts (FeCl3, or AlCl3) were added to the mixture and then heated to 80 °C under reflux for 24 hours. The crosslinking reaction took place in the porous materials, while some of the samples could preserve micro-scale spherical morphologies. However, the materials exhibited relatively low specific surface areas for all six monomer types (7.8–246.9 m2 g−1). Of the monomers studied, 4-ethyl phenol produced the highest surface area by far (246.9 m2 g−1). The authors hypothesized that this higher surface area is due to more highly reactive sites leading to increased crosslinking and more abundant micro/mesopores. They proposed that for the other phenol monomers, the Friedel–Crafts reaction suffer from steric hindrance. Further, they speculated that methoxy groups have an adverse impact on crosslink formation via Friedel–Crafts reaction due to catalyst deactivation by complexation with FeCl3 and the oxygen atoms. Their hyper-crosslinked polymers could achieve up to ∼1.5 mmol g−1 at 0 °C.
Liu et al. produced lignin-modified hyper-crosslinked porous resins, using extracted ethanol organosolv lignin from poplar sawdust (Fig. 9).112 A free-radical copolymerization step was first conducted within the organic phase of lignin, VBC (monomers), DVB (crosslinker), ethyl acetate, and 2,2-azobisisobutyronitrile (AIBN) (initiator) in a 1 wt% aqueous poly(vinyl alcohol) (PVA) solution at 45 °C. Continuous stirring was applied while the mixture was gradually heated to 75 °C for 3 hours, 85 °C for 3 hours, and 95 °C for 2 hours, respectively. After purifying and drying the lignin-modified polymer spheres, Friedel–Crafts reactions were performed to crosslink the resins.113 While their results showed that lignin could be incorporated in the hyper-crosslinked composites, most of the lignin-modified resins became broken particles after Friedel–Crafts reactions. It is most likely that lignin destabilizes and disrupts the network of copolymers during suspension polymerization. They also found that the control resin (without the addition of lignin) had superior CO2 adsorption capacity. Therefore, the lignin addition was predominantly not beneficial for the formation of micropores, potentially because of reduced crosslinking in the presence of lignin. The authors found that CO2 adsorption was highly dependent on both specific surface area and micropore volume. Notably, at reduced pressure (25 °C and 15 kPa), one of the lignin-modified resins could achieve better performance than the control sample, potentially the results of weaker surface chemistry without the addition of lignin. Their lignin-modified hyper-crosslinked porous resins could achieve up to ∼2 mmol g−1 at 0 °C and 100 kPa.
 | ||
| Fig. 9 Lignin-modified hyper-crosslinked porous resins using from poplar sawdust organosolv lignin. (OL: organosolv lignin; CR-0: resin without lignin; LMCR-2: resin with lignin). Reproduced with permission.112 Copyright 2024, Elsevier. | ||
Karaaslan et al. used epoxide chemistry to react kraft lignin in alkaline solutions with epichlorohydrin and then crosslinked into hydrogels using diamine hardeners (Fig. 10).114 The hydrogels were subsequently solvent exchanged into organogels and then supercritically dried with CO2 to keep the internal pore structure. Carbonization produced a high surface area structured carbon aerogels that contained both mesopores and micropores. Activation resulted in an increase in surface area (up to 1609 m2 g−1) and micropore volume and 5.4 mmol g−1 of CO2 adsorption capacity at 0 °C was achieved at 100 kPa.115
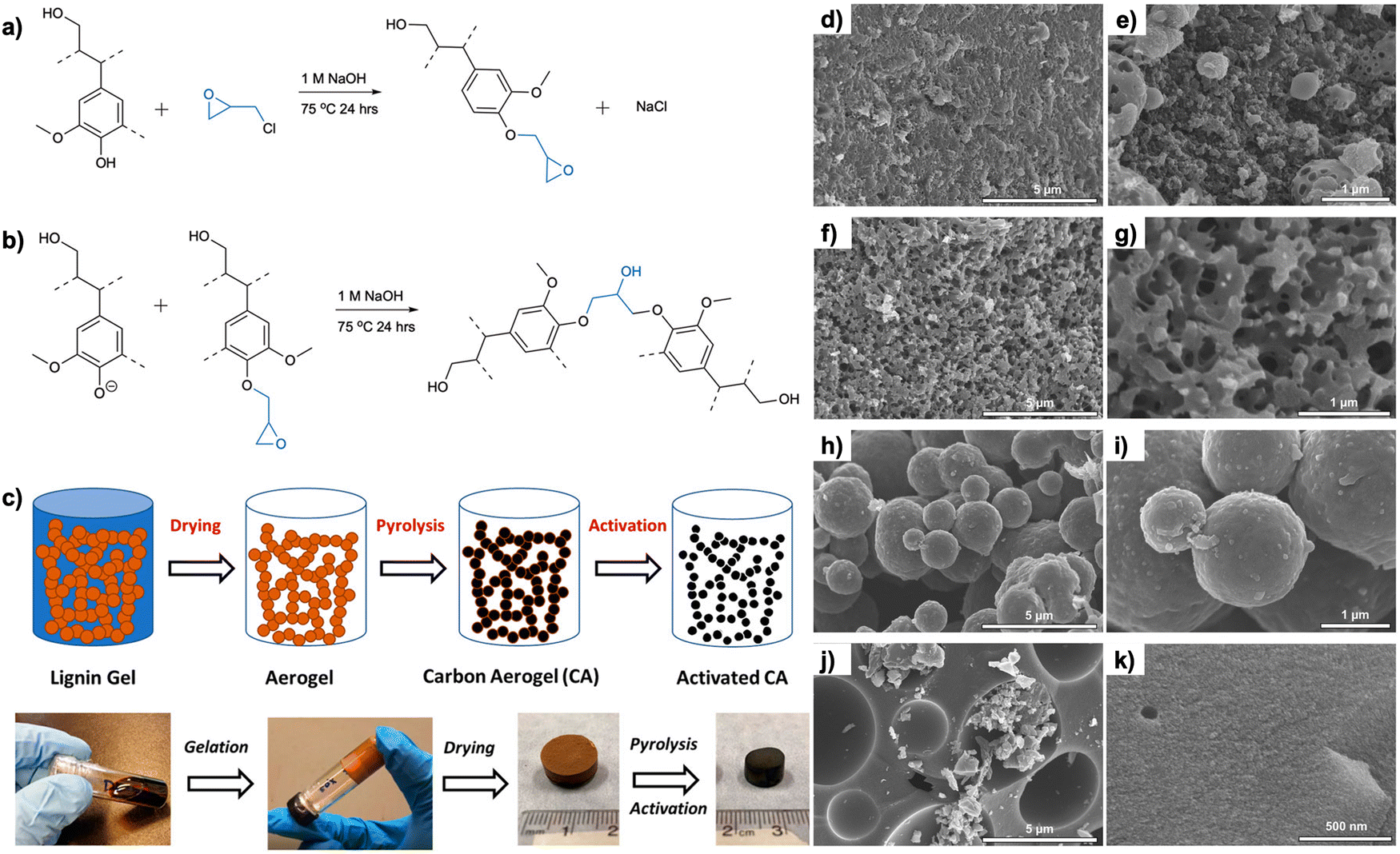 | ||
| Fig. 10 Epoxy chemistry for lignin treatment and carbonization. Reaction mechanism between lignin and epichlorohydrin: (a) Epoxidation of hydroxyl groups of lignin, followed by (b) formation of crosslinks. (c) Schematic and photography of the processing steps of lignin carbon aerogels. (d)–(k) SEM images of the lignin carbon aerogels, showing how surface morphologies were affected by the lignin-to-crosslinker ratio: (d), (e) 55% lignin, (f), (g) 68% lignin, (h), (i) 81% lignin, (j), (k) 87% lignin. Reproduced under terms of the CC-BY 4.0 license.114 Copyright 2022, Frontiers Media S.A. | ||
4.3 Hierarchical-structured adsorbents
The approaches discussed so far produce materials with multiscale porosity, but most of them do not attempt to control the hierarchical structure at multiple length scales. More recently, controlled synthesis of multiscale architectures has become increasingly desirable because it directly impacts CO2 capture applications. These approaches offer improved integration in scalable processes, improved gas phase transportation efficiency, and facilitate the accessibility of CO2 gas molecules to surface adsorption sites. In this section, we cover the approaches that attempt to control the hierarchical architecture of the material.The conversion of lignin to carbon fiber materials has been discussed for at least 55 years,118 since carbon fibers have excellent functional properties potentially useful for CO2 capture applications, such as high strength-to-weight ratio, stiffness, conductivity, and excellent thermal and chemical resilience.119 Particularly, lignin is a promising feedstock for carbon fibers because of its low cost, high carbon content, and thermoplastic properties. Lignin-based carbon fibers are typically produced via extrusion method, such as melt-spinning, wet-spinning, dry-spinning, gel-spinning, and electrospinning.
Calvo-Muñoz et al. produced lignin-based carbon fibers from organosolv lignin with almost exclusively ultra-micropores (<0.7 nm) and reported promising breakthrough adsorption capacities of 1.3 mmol g−1 or 5.7 wt% (in a column system, 25 °C, 15 kPa).120 The electrospun lignin/ethanol fibers were treated with an intensive oxidative thermostabilization under an air atmosphere with a ramp of 0.08 °C min−1 to 200 °C, followed by a 48-hour hold. The materials were then carbonized at 900 °C. Carbon fibers from this protocol feature narrow micropores, which are likely to be responsible for the superior CO2 uptake. A general and major challenge of this approach to utilize lignin-based carbon fibers is the requirement for a time- and energy-intensive thermostabilization step. Further, the density of the material, specifically of the carbon fibers, is much less than that of regular granulated lignin-based activated carbon, which has implications for their translation to scalable processes.
4.3.2.1 Hard templating. Sani et al. produced ordered mesoporous amine-functionalized lignin-based carbons, using a siliceous mesostructured cellular foam as a hard template in a solvothermal process. In their approach, 2.4 g of lignin was mixed with 1.2 g of template in 15 mL tetrahydrofuran (THF).123,124 After drying, the composite was carbonized (700–900 °C) for 1 hour, and the carbon product was subsequently stirred in 1 M NaOH for 24 hours to remove the hard silica template. The resulting carbons were demonstrated to be mainly mesoporous high BET surface areas (up to 960 m2 g−1) and mesopore volumes of 1.50 cm3 g−1. Further, the carbon preserved a quasi-spherical porous morphology after carbonization, inherited from the mother template. While the pristine mesoporous carbon demonstrated low CO2 uptake, the mesoporous carbon could be used as a support for polyethylenimine (PEI) to enhance its CO2 adsorption properties. Given the mesoporous structure, the material could accommodate high PEI loadings of up to 60 wt%, at which point the CO2 adsorption was highest at 2.95 mmol g−1 at 75 °C and 15 kPa. Due to this combination, the PEI-modified carbons exhibited enhanced adsorption kinetics and good cycle stability.
Zhao et al. developed a templating method with basic magnesium carbonate (BMC) or magnesium oxide (MgO) for the production of activated carbon from acid-precipitated alkali lignin, and compared the obtained results with those of samples chemically activated with ZnCl2 or KOH.125 In the templating approach, lignin was dispersed in water with BMC or MgO at a 1:3 or 2:1 ratio, respectively. The dispersion was later heated to 80 °C, followed by drying and calcination at 5 °C min−1 to 600 °C for 2 hours. The templated carbons appeared rough with slit-like pores, whereas relatively flat with etched pores were found with the chemically activated samples (Fig. 11). However, the templated samples exhibited weaker performance than the chemical activation counterparts, in terms of specific surface area, total pore volume, and CO2 adsorption capacity. The latter reached specific surface areas of 1336.5 m2 g−1 and adsorption capacity of 3.60 mmol g−1 CO2 at 25 °C. Apparently, chemically activated samples had a more pronounced microporosity (89.1–93.2%) compared to the templated samples (58.7–44.6%). This is probably because hard templating methods mainly influence macroscale morphologies. Besides, CO2 interaction relies more on microporosity, whereas macroporosity is more critical for transport phenomenon.
 | ||
| Fig. 11 Porous carbon materials prepared from lignin by using four different activation strategies: (a) chemical activation with ZnCl2 and (b) KOH. Templating method with (c) BMC and (d) MgO. (C-BLL: carbonized black liquor lignin; BMC: basic magnesium carbonate) Reproduced with permission.125 Copyright 2023, Elsevier. | ||
4.3.3.2 Soft templating. The amorphous 3D nature of the lignin macromolecular structure limits its potential for producing well-defined structures such as fibers. In efforts to fully unleash the potential of multiscale design, new lignin morphologies have garnered interest in combination with available bio-based additives. Such lignified hierarchical assemblies of plants found in nature can inspire the construction of new hierarchically structured materials with engineering porosity.
Geng et al. used an ice-templating approach to produce carbon aerogels from kraft lignin.126 Lignin was pre-mixed with TEMPO-oxidized cellulose nanofibers (TOCNF) of ∼8–12 wt%. The high aspect ratio of TOCNF can establish a robust fibrous network that forms the aerogels during ice-templating and freeze-drying. The as-produced aerogel was carbonized with a 2-hour hold at 500 °C to stabilize the material, followed by heating to 1000 °C. This ice-templating approach could produce an anisotropic macroporous structure, while the carbonization process could introduce meso- and micropores, demonstrating hierarchical porosity (Fig. 12). The carbon aerogels at 12 wt% TOCNF exhibited a high surface area (1101 m2 g−1) and adsorption capacity of 5.23 mmol g−1 at 0 °C and 100 kPa.
 | ||
| Fig. 12 (a) Synthesis of lignin/TOCNF carbon aerogels, and SEM images of (b) cross section and (c) longitudinal section of the lignin/TOCNF precursors with different TOCNF contents. Reproduced under terms of the CC-BY 4.0 license.126 Copyright 2022, American Chemical Society. | ||
In most literature, a structural polymer would be required to interconnect spherical lignin particles during soft templating since lignin itself is difficult to form a mechanical-robust interconnected network. Under such scenarios, the most common choice is cellulose. Cellulose naturally assembles in the form of fibrils and can be liberated from the cell wall matrix, in the form of cellulose nanofibrils (CNF) or nanocrystals (CNC).127 Both CNFs and CNCs have garnered significant attention as functional agents in nanocomposite materials due to their attractive physical and chemical properties, not least their universal availability. For example, it has been demonstrated that the topology of nanonetworks formed from CNFs induces interparticle cohesion and enables universal and robust superstructure of nanoscale particles for the lay-in of spherical lignin particles.54,128,129 Further, the cellulosic network acts as a sacrificial template during carbonization due to its lesser relative thermal stability and carbon content. These features are particularly attractive given the increasing momentum for spherical lignin particles as promising and versatile assemblies.
The rate of CO2 adsorption/desorption in lignin-based carbon materials is often limited by the diffusion rate of CO2 gases in amorphous carbon networks. To address such bottlenecks, the superstructuring of spherical lignin particles can be employed to tailor the molecular transport. In this context, Zhao et al. produced multiscale carbon supraparticles, utilizing the drying cohesion of cellulose nanofibrils to bind spherical lignin particles (Fig. 13).54 Softwood kraft lignin was applied to produce spherical particles at 200 and 420 nm via solvent exchange. Supraparticles were formed via evaporation-induced self-assembly of aqueous lignin particles and CNF over various ratios (CNF solid fraction in the dried supraparticles ranging from 0.5–15 wt%) by casting on a superhydrophobic Teflon-coated surface at 60 °C. The dry supraparticles required oxidative thermostabilization (0.01–5 °C min−1) at 250 °C for 2 hours under an air atmosphere. After thermo-stabilization, the supraparticles could be carbonized at 10 °C min−1 to 600–900 °C for 1 hour. Physical activation was also performed with N2 gas saturated with water (steam activation) or N2 gas saturated with aqueous ammonia vapor (ammonia-steam activation).
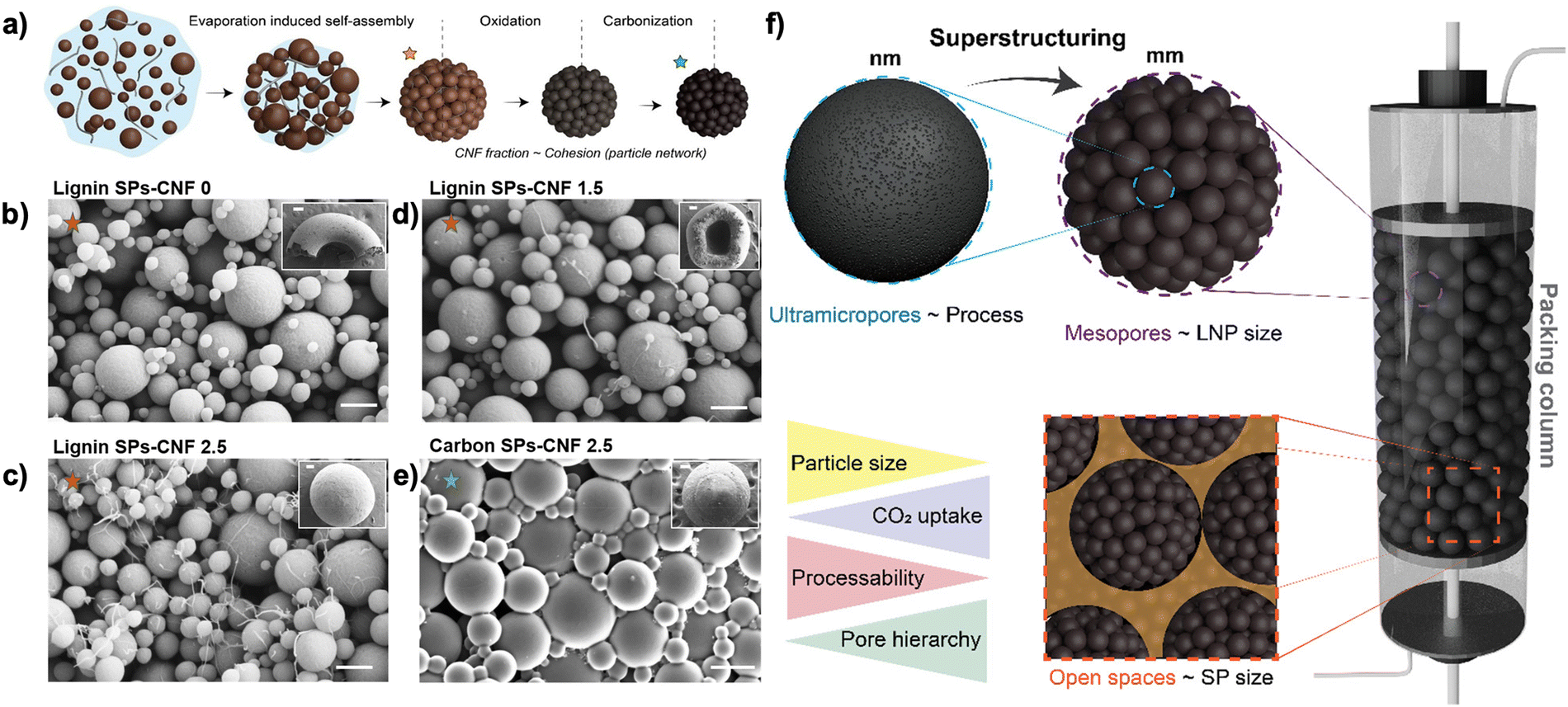 | ||
| Fig. 13 (a) Process of carbon supraparticle formation via evaporation-induced self-assembly followed by oxidation and carbonization. (b)–(e) Electron microscope images of the lignin-based supraparticles with different contents of CNF (insets are at lower magnification). (f) Super structuring of lignin particles (nanometer-scale) into millimeter-scaled carbon supraparticles (SPs) for use in CO2 capture in a packed column. Reproduced under terms of the CC-BY 4.0 license.54 Copyright 2021, American Chemical Society. | ||
The properties of lignin supraparticles are largely dependent on their specific preparation protocol. The thermal stability of lignin supraparticles could be improved by a slow oxidative thermo-stabilization, transforming lignin from a fusible thermoplastic to a thermosetting material. The rate of pre-oxidation is also critical to preserve the particles’ hierarchical properties. In general, smaller particles require slower heating rates, which requires more time and energy investment. Since the preparation of supraparticles relies on drop casting, the droplet size is critical to ensure the roundness of the as-produced supraparticles. Too large droplets would be deformed by gravity into more elliptic shapes. CNF is also critical for providing mechanical robustness and avoiding structural failure; without CNF, a ring-shaped superstructure was obtained. CNF loadings > 2.5 wt%, and particle size in the range of 200–500 nm turned out to be the optimal processing conditions for the coassembly with the fibers.128 The incorporation of CNF was also critical for preserving the suprastructure during thermal treatment. After oxidative thermo-stabilization and carbonization, the resulting carbon supraparticles with CNF were preserved, while the CNF-free particles were reduced to a powder.
The excellent mechanical strength and controlled macrostructure of the spherical supraparticles are well-suited for packed bed adsorption systems, since they are able to withstand the load of an entire packed column and reduce pressure drop (i.e., enabling sufficient gas transport during operation), especially compared to smaller materials. The specific surface area and CO2 uptake of the carbon supraparticles ranged from 348 to 405 m2 g−1 and ∼1.6 to 1.3 mmol g−1 (40 °C and 101 kPa), respectively, depending on the final carbonization temperature. In an attempt to further improve the CO2 capacity of the supraparticles and also to investigate the potential synergism of ultramicropores and N-doping on CO2 uptake, the effect of N-doping on CO2 content was analyzed with the same structure but different nitrogen content with steam and ammonia-steam activation. It was found that steam could enhance the surface area to up to 967 m2 g−1 and ammonia 1152 m2 g−1 with N-content of 4.78 wt%. Further, the maximum CO2 uptake for steam activation and ammonia-steam activation was 1.8 and 1.7 mmol g−1, respectively. Comparing the samples with steam vs ammonia-steam physical activation, showed similar porosity, surface area, graphitic structure, and CO2 uptake, suggesting incorporation of nitrogen heteroatoms may not have a significant influence on CO2 uptake. In summary, it was found that the carbon supraparticles with larger surface area or doped with nitrogen actually did not show higher CO2 uptake, highlighting the complexity and multivariate nature of CO2 uptake of porous carbon materials.
Shao et al. produced a naturally N-doped porous carbon adsorbent through the self-assembly of sodium lignosulfonate and chitosan to gel microbeads in the presence of KOH for chemical activation (Fig. 14).130 In their protocol, sodium lignosulfonate and chitosan were mixed at different mass ratios (3:1, 2:2, 1:3) in water followed by the addition of 2 M hydrochloric acid (HCl) to dissolve the chitosan with dispersed lignosulfonate. The dispersion was subsequently dropped into an 8 wt% KOH solution to form the gel beads. After drying, the beads were carbonized at 5 °C min−1 to 600 °C for 2 hours. If the beads were washed to remove KOH prior to carbonization, the beads could maintain their structure to produce an aerogel, compared to those reduced into powder forms if no washing. Nonetheless, the powdered material (i.e., no washing treatment) exhibited a high surface (∼892–1308 m2 g−1) area, whereas the washed sample had a very low surface area (13.4 m2 g−1). XPS measurements showed that the N-content of the carbon materials ranged from ∼3–5% according to chitosan content. The porosity and N content of N-doped microporous carbon could be effectively regulated by adjusting the ratio of lignin to chitosan in the precursor gel microbeads. Surface area, total pore volume, micropore volume, and N-content increased as the lignin:chitosan ratio decreased.
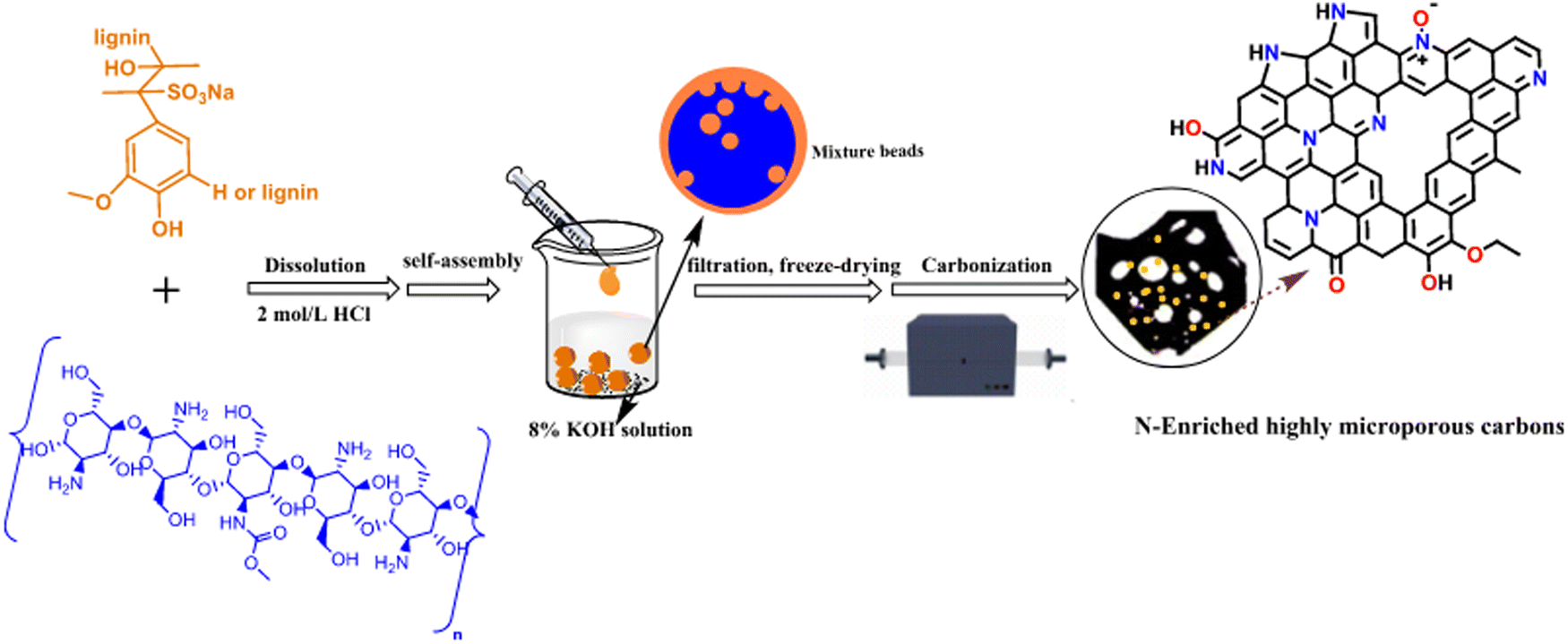 | ||
| Fig. 14 Preparation process of N-doped highly microporous carbon derived from lignin/chitosan composites beads. Reproduced with permission.130 Copyright 2023, Elsevier. | ||
5. Perspectives on the rational design of lignin-based adsorbents
In the previous section, we reviewed the state-of-the-art of lignin-to-carbon processing and its implications in CO2 capture. Table 1 provides a summary of the lignin-based porous adsorbents covered in this review, from traditional approaches to structured porosity via crosslinking and hierarchically structured materials. However, one has to admit that the development of lignin-based materials is mostly in the early stages, and rational design will become essential to improve CO2 capture performance. In this section, we provide several perspectives on the future development of lignin-based carbon materials, that are design-driven following (i) a fundamental understanding, and (ii) computational simulations. Lastly, we introduce recent developments that use artificial intelligence (AI).| Feedstock | Other chemicals | Processing technique | BET SSAb | Adsorption capacityc | Selectivityd | Ref. |
|---|---|---|---|---|---|---|
| a Performance metrics reported are maximums achieved and do not necessarily correspond to the same sample. b SSA reported in units of m2 g−1. c CO2 adsorption capacity in mmol g−1, adsorption capacity can be assumed to be measured at 101 kPa unless otherwise specified. d Selectivity measurements assumed to be conducted at 101 kPa and 15% CO2/N2 unless otherwise specified. | ||||||
| Traditional approaches | ||||||
| High-ash content alkali lignin | — | Pyrolysis, negative/atm./elevated pressure | 1577 | 3.6 (0 °C) | — | 86 |
| Corn straw lignin | Chemical activation (H3PO4) | Pyrolysis | 820 | 0.4 (0 °C) | IS: ∼15 (20 °C) | 87 |
| Kraft lignin | Chemical activation (KOH) | Pyrolysis | 2121 | 3.3, 2.0 (0, 25 °C) | IAST: 38 (25 °C & 15 kPa) | 88 |
| Dealkaline lignin (TCI America) | Chemical activation (KOH), S-doping (Na2S2O3) | Pyrolysis | 3626 | 11 (25 °C) | IAST: ∼60 (25 °C) | 90 |
| Softwood kraft lignin (UPM BioPiva) | Chemical activation (HNO3), N-doping (Urea) | Pyrolysis | 1000 | 1.4 (20 °C & 15 kPa) | IAST: ∼18 (25 °C) | 91 |
| Enzymatic hydrolysis lignin | Chemical activation (KOH), physical activation (humidified N2) | Microwave | 2870 | 1.3 (30 °C) | — | 92 |
| Dealkaline lignin (TCI America) | Chemical activation (KOH), physical activation/N-doping (NH3) | Pyrolysis | 2922 | 8.6, 5.5 (0, 25 °C) | IAST: ∼100 (25 °C) | 93 |
| Dealkalized lignin | Chemical activation (KOH) | Pyrolysis | 2604 | 5.8, 4.0 (0, 25 °C) | IAST: ∼16 (25 °C, CO2/N2 10:90) |
94 |
| High ash content lignin (Borregaard LignoTech) | Chemical activation (KOH) | Hydrothermal carbonization, pyrolysis | 3235 | 7.4, 4.6 (0, 25 °C) | — | 98 |
| Eucalyptus enzymatic hydrolysis lignin, Norway spruce kraft lignin | Bio-oil production (formic acid, catalyst), chemical activation (KOH) | Hydrothermal carbonization, pyrolysis | 2875 | 6.0 (0 °C) | IAST: ∼4 (0 °C) | 79 |
| Organosolv lignin (lignol innovation company) | Chemical activation (KOH), N-doping (adenine) | Hydrothermal carbonization, pyrolysis | 2957 | 4.8 (25 °C) | IS: 18 (0 °C, CO2/N2 10:90) |
101 |
| IAST: 22 (0 °C, CO2/N2 10:90) |
||||||
| Not specified | Chemical activation (KOH), functionalization (PEI) | Hydrothermal carbonization, pyrolysis | 1341 | 2.0 (30 °C) | — | 102 |
| Dealkalized lignin (TCI Shanghai) | Chemical activation (KOH), N-doping (Urea) | Pyrolysis, hydrothermal carbonization | 2126 | 4.79, 1.44 (25 °C at 101, 15 kPa) | IAST: 36 (25 °C) | 103 |
| 2.97, 0.63 (50 °C at 101, 15 kPa) | ||||||
| Lignin (Rizhao Huatai Paper Industry) | N-Doping (melamine), chemical activation (CuCl2·2H2O) | Pyrolysis | 2703 | 6.78, 3.57 (0, 30 °C at 100 kPa) | IAST: 132, 81 (0, 30 °C) | 105 |
| Model lignin (methylene bridged mesitylene) | Polymer synthesis (mesityylene, HCl, HCHO, NaCl, ZnCl2, CH2Cl2, FeCl3, CH3OH), chemical activation (KOH) | Oxidative thermostabilization, pyrolysis | 1899 | 6.16 (0 °C) | IAST: 24, 28 (0, 25 °C) | 131 |
| Enzymatic hydrolysis lignin | N-Doping (melamine), chemical activation (KOH) | Hydrothermal carbonization, mechanochemical processing, pyrolysis | 2030 | 5.0 (0 °C) | IAST: 525 (0 °C) | 106 |
| Henry's Law: 29 (0 °C) | ||||||
| Sodium lignosulfonate | pH adjusting (H2SO4), physical activation (CO2) | Hydrothermal carbonization, pyrolysis | 1336 | 5.1 (0 °C) | IAST: 27 (25 °C, CO2/N2 10:90) |
107 |
| Sodium lignosulfonate | Chemical activation (KOH), N-doping (Urea) | Hydrothermal carbonization, pyrolysis | 3064 | 2.7, 13.0 (25 °C & 101, 1013 kPa) | — | 108 |
| Sodium lignosulfonate | Chemical activation (ZnCl2), N-/P-doping (melamine/triphenylphosphorus) | Pyrolysis | 1459 | CO2RR performance: H2/CO ratios 2.3–8.0 | 109 | |
| Structured porosity via crosslinking | ||||||
| Poplar organosolv lignin | Crosslinking (FDA, FeCl3, DCE) | Pyrolysis | 140 | 2.2–2.5 (25 °C) | 60 (30 °C) | 110 |
| Model lignin monomers | Crosslinking (FDA, FeCl3, AlCl3, DCE) | — | 247 | 1.5 (0 °C) | IAST: 257 (0 °C) | 111 |
| Henry's Law: 35 (0 °C) | ||||||
| Poplar sawdust ethanol organosolv lignin | Crosslinking (VBC, DVB, ethyl acetate, AIBN, PVA) | — | 969 | 2.0 (0 °C) | — | 112 |
| Southern pine softwood kraft lignin (BioChoiceTM) | Crosslinking (NaOH, epichlorohydrin) | Supercritical-drying, pyrolysis | 1609 | 5.4 (0 °C) | — | 114 |
| Hierarchically structured-adsorbents | ||||||
| Not specified | Formalin, NaOH, ferric nitrate | Pyrolysis | 193 | 0.7 (25 °C) | — | 117 |
| Alcell® lignin | Electrospinning (ethanol) | Oxidative thermostabilization, pyrolysis | 850 | 1.3 (25 °C & 15 kPa) | — | 120 |
| Lignin (Sigma-Aldrich) | Hard template (siliceous mesostructured foam, THF), functionalization (polyethyleneimine) | Pyrolysis | 960 | 3.0 (75 °C & 15 kPa) | — | 123,124 |
| Acid precipitated lignin (alkali pulping company, Hunan, China) | Templating (BMC, MgO), chemical activation (ZnCl2, KOH) | Pyrolysis/calcination | 1337 | 5.2, 3.6, 2.2 (0, 25, 50 °C) | IAST: 14.2, 12.7 (25 °C, CO2/N2 10:90, 15:85) |
125 |
| Kraft lignin (Sigma-Aldridge) | TEMPO-oxidized cellulose nanofibers | Ice-templating, thermal stabilization, pyrolysis | 1101 | 5.2 (0 °C) | — | 126 |
| Softwood kraft (BioPiva 100), pine kraft lignin (indulin AT) | Particle formation (THF/DMF), superstructuring (CNF), physical activation (ammonia-steam) | EISA, oxidative thermostabilization, pyrolysis | 1152 | 1.75 (40 °C) | — | 54 |
| Sodium lignosulfonate | Bead formation & N-doping (chitosan), chemical activation (KOH) | Pyrolysis | 1308 | 4.9, 3.2 (0, 25 °C) | IAST: 146 (0 °C) | 132 |
| Henry's Law: 24 (0 °C) | ||||||
5.1 Fundamental design-driven carbon capture with lignin
The pore size distribution requires specific attention, when aiming at improving the carbon capture efficiency of biomass-derived porous materials. The significance of pore size and size distributions lies in their impact on the accessibility of reaction sites and the accommodation of CO2 molecules within the material matrix. For well-defined synthetic nanomaterials (e.g., MOFs, zeolites, porous organic polymers), their CO2 capture capability can be precisely controlled by tuning the microporosity and mesoporosity during material synthesis.136,137 For heterogeneous bio-based materials, pore sizes could exhibit a broad distribution, ranging from a few nanometers to micrometers, depending on the pretreatment and activations. Such a hierarchical porosity is generally considered to be beneficial for CO2 capture efficiency with a sacrifice in selectivity.138 Macroporosity facilitates the gas flow and transportation within a torturous porous medium. Mesopores could promote local mass transports and act as local constraints to enhance gas–solid interactions, being suitable for high-pressure CO2 adsorption, while improving microporosity is the key to generating active adsorption sites in both low- and high-pressure conditions.
Accordingly, tailoring pore sizes within lignin-based materials affects the kinetics of CO2 molecule adsorption. In general, faster CO2 uptake kinetics is favorable. The early-stage diffusion of CO2 gas into microporous regions is the rate-limiting step for adsorption kinetics. Increasing the pressure could facilitate the accessibility of CO2 molecules to micropores that are smaller than 0.33 nm to allow strict physical adsorption.139,140 In real practice, functional groups are introduced to improve the CO2 affinity with adsorbent substrates, accelerate their early-stage diffusion, and enhance low-pressure adsorption performance. Templating and controlled activation processes are the most common methodologies for producing consistent and well-dispersed pore structures.17,54,126,141 These endeavors seek to strike a balance between pore size and distribution, aiming for materials that offer enhanced adsorption kinetics and selectivity in CO2 capture applications. Recently, Keffer et al.142,143 utilized computational approaches to investigate the effect of N-doping in lignin-derived carbon quantum dots (CQDs) for carbon capture. They found that both N-doping within the aromatic structure of the CQDs and the introduction of amine groups positively influenced CO2 capture efficiency and selectivity over N2, O2, and H2O.
| 2R1R2NH ⇌ R1R2NH2+ + R1R2N− | (1) |
| R1R2N− + CO2 → R1R2NCOO− | (2) |
Additionally, the nitrogen atoms from tertiary amines (R1R2R3N) tend to undergo protonation to generate hydroxide, which serves as the Lewis base to the capture the acidic CO2 gas:149,150
| CO2 + H2O → H2CO3 → HCO3− + H+ | (3) |
| HCO3− → CO32− + H+ | (4) |
| R1R2R3N + H+ → R1R2R3NH+ | (5) |
Alkoxides are another highly promising group of substances for adsorbing CO2. They are less sensitive to O2 and have the ability to directly interact with CO2 to create a carbonate compound.
| RO− + CO2 → ROCO2− | (6) |
Even though the above-mentioned functional groups are likely to provide chemical reaction sites for CO2 capture, the steric effects of the applied macromolecules cannot be ignored. The exchange-repulsion from the non-polar groups could cancel the attractive effects between CO2 and reaction sites.151 Due to the propensity of lignin to induce coil-globule transitions at high temperatures or in a cosolvent environment,152–155 it is more probable that it would augment the exposed surface area, hence improving the accessibility of lignin for CO2 chemisorption.
Cui et al. grafted the active amine group on a lignin-derived compound vanillin and alkaline lignin by Mannich reaction.156 The ratio of material n(vanillin):n(formaldehyde):n(amine reagent) is 1:1:1 in the protocol, all of which are 0.06 mol. The vanillin modified by acrylamide obtained a CO2 uptake capacity of 0.114 g CO2 per gram (2.6 mmol g−1) of the material under 25 °C and 100 kPa. In-depth analysis identified the multi-site synergistic adsorption process, in which CO2 double interacts with the amide group and single interacts (hydroxy oxygen on the adsorbent's benzene ring) due to a large local difference in electronegativity and a small steric hindrance.
CO2 adsorption mechanisms can involve a complex interplay of both physisorption and chemisorption. It is crucial to approach fundamental interpretations carefully, as an oversimplified acid–base interaction model may not adequately account for all interactions. Sun et al.157 synthesized four conjugated azacyclo-copolymers that self-assembled into two-dimensional layered structures with well-defined and uniform amino and azacyclo groups. Experimental and computational results revealed that the negative electrostatic potential around the azacyclo-N-doping sites could induce strong van der Waals interactions, facilitating selective CO2 adsorption. Although the reported CO2 uptake efficiency was below the theoretical limit (0.255 mmol g−1 at 0 °C and 1 bar, compared to the 1 mmol g−1 predicted for N-doping), this advancement represents a significant shift in the design of highly effective porous carbon adsorbents through chemical and surface modification.158,159
PEI impregnation is another common practice for lignin chemisorption requires amine modification/grafting, in which PEI is commonly applied with alkaline combined high-temperature treatment. An example has been given in previous sections,124 where the derived mesoporous carbon material demonstrates a high CO2 adsorption capacity, above 2 mmol g−1. Some mechanistic analysis was also performed to identify the CO2 interactions with amide and hydroxy groups.
5.2 Computational design-driven carbon capture with lignin
Computational chemistry and molecular modeling approaches offer fundamental insights into complex physiochemical phenomena that would otherwise be inaccessible with experiments alone. These approaches have been employed in efforts to address a range of challenges in lignin chemistry, from its polymerization, linkages and supramolecular interactions in the plant cell wall,160–162 to its functional properties such as solubility and glass transition temperature.152,163,164Computational methods, such as DFT and quantum mechanics/molecular mechanics (QM/MM), can be utilized to investigate molecular-level phenomena and support the development of modeling approaches at larger length scales, such as those conducted with molecular dynamics (MD) and Monte Carlo (MC) simulations. Of particular relevance for the production of carbon from lignin include reactive force field approaches that enable the modeling of thermochemical reactions (i.e., pyrolysis).165 Yet, the variability and multiscale complexity of lignin offers a significant challenge for modeling and simulation approaches.166 To address the challenge of lignin heterogeneity and variability in molecular simulations, tools have been developed for the generation of libraries of lignin polymer topologies suitable for classical and quantum-level studies.167,168 With the introduction of these tools, new levels of topological complexity may be studied, offering an improved representation of broader lignin populations.
Insights on the molecular level provide an in-depth understanding of the interaction of CO2 and porous carbon and guidelines on further materials design and optimization. Theoretical analysis has shown that there is physisorption between CO2 and cyclo[18]carbon,169 as well as acyclic imine-based functional molecules and a range of functional molecules that contain multi-N-containing superbases and heteroaromatic ring complexes (Fig. 15).170,171 In order to gain a deeper comprehension and to distinguish the significant and insignificant factors of non-covalent interaction between CO2 and the lignin molecules, the utilization of energy decomposition analysis (EDA) is crucial. This method effectively decomposes the overall interaction energy into distinct physical components that are readily comprehensible. Briefly, EDA can be categorized into two main methods, one of which involves Kitaura–Morokuma,172 LMOEDA,5 GKS-EDA,173 NEDA,174 and ALMO-EDA.175 Another representative one is the symmetry-adapted perturbation theory (SAPT) method.176 Lu et al. employed SAPT theoretical calculations to forecast the physical adsorption of CO2 on cyclo[18]carbon, incorporating a London Dispersion interaction energy of −7.6 kcal mol−1, resulting in a total attraction ratio of 87.36%.169 Neutral compounds, like aromatic systems, can exhibit a similarly high ratio of London Dispersion interactions.171 On the other hand, this ratio will exhibit a bias towards electrostatic interaction (Ele) dominance, when the physical absorption occurs between CO2 and ionic solvents.177 Remarkably, the interaction strength between COO− and CO2 is nearly equal to that of pyrrole and indole, with a similar magnitude of higher than −10 kcal mol−1. This result could provide a theoretical insight for surface modification on lignin molecules, such as amine-functionalization and carboxylation,178,179 to enhance non-covalent interaction with CO2.
 | ||
| Fig. 15 Non-covalent interactions involved in CO2 capture mechanisms. (a) Optimized structures and non-covalent interaction maps of CO2 molecule containing cyclo[18]carbon. (b) Components of interaction energy between cyclo[18]carbon and various chemical species obtained from symmetry-adapted perturbation theory (SAPT) calculations. Reproduced with permission.169 Copyright 2021, Elsevier. | ||
5.3 Artificial intelligence (AI) design-driven carbon capture with lignin
In recent years, the development of AI has created a paradigm shift in material design strategies. Compared to the conventional bottom-up (i.e., fundamental-driven design) or top-down (i.e., performance screening) approaches, AI-driven design relies on the collection of large databases (e.g., materials’ structural, properties) through literature, experiments and/or simulations, and then using a nonlinear pathway within high-dimensional geometric-variable space180 (i.e., the machine learning algorithm) to optimize and predict performances. The most critical criterion for successful machine learning research is the access to large amounts of reliable data. For example, machine-learning or deep-learning methods have been successful in determining the reaction pathways for catalyst designs, CO2 adsorption, and particle analysis, among many others,181–184 owing to the abundant data availability.In the regime of lignin research, state-of-the-art research is mostly still in the stage of data exploration, collection, and validation. Establishing a more comprehensive lignin knowledge library would be beneficial to boost the AI-driven designs for lignin-based materials in general. In this regard, it is essential for researchers to provide a comprehensive description of the feedstock properties, including lignin supplier, biomass source, extraction process, chemical structure characteristics and purity indicators.185 Apart from the experimental results, simulations are less expensive for data collection without the need for materials gathering and consumption, despite a debatable extent of reliability. Nevertheless, current simulation means can be efficient in predicting intra-/intermolecular forces, colloidal interactions, particle assembly, etc.,186–188 all of which can be the resources for machine learning research.
6. Conclusion and outlook
With growing concerns about climate change, developing efficient and sustainable CO2 capture materials has become a matter of high relevance, including the exploration of related technologies for their upstream and downstream applications (e.g., CO2 storage, utilization, and adsorbent regeneration). Despite its advancements, the aminated liquid-based carbon capture technologies face challenges due to the energy intensity required for solution regeneration, which limits the sustainability of its carbon-negative life cycle. In this context, carbon adsorbents are highly promising materials due to their cost-effective synthesis, customizable properties, and potential to lower costs associated with adsorbent regeneration.Bio-derived residues can be effectively converted into carbon materials with high surface area and multi-scale porosity. In particular, lignin stands out as a promising source due to its high carbon content, versatile functional structure, and low cost. This work reviewed recent developments in lignin-based carbon adsorbents for CO2 capture. Unlike petroleum-based chemicals, which are commercially upgraded into homogeneous compounds, the biomass processing industry, particularly in the context of lignin utilization, has not yet reached full maturity. We began this review by examining the origins of lignin feedstocks and their industrial varieties, thermal properties, processing strategies, and sustainability aspects. We follow with an overview of the key performance indicators for lignin-derived carbon adsorbents, including their carbon properties and adsorption performance. We then covered state-of-the-art processing of technical lignins to produce carbon adsorbents and offered a critical discussion on this emerging interdisciplinary field, focusing on the potential of rational designs in future developments.
The field of lignin-based materials for CO2 capture is still in its early stages. Several challenges need to be addressed for future development. The primary challenge of lignin valorization lies in its natural variability, heterogeneity, and chemical complexity. The impact of lignin's origins on the final products is not yet fully understood, and there is a lack of consistency and broad applicability across various processing techniques. Further investigation into the structure–property relationships for carbon development is necessary. Despite these challenges, the field is advancing and aligning with current trends in the broader lignin community, such as fractionation, chemical modification, and the assembly into particles.
As discussed in this review, the diversity of lignin has significant implications for processing. This serves as both a caution and a reminder of the importance of selecting the appropriate lignin type. For instance, different grades of lignin can be utilized for specific purposes, such as using pulping salts as chemical activators. Additionally, the presence of heteroatoms in various technical lignins can enable novel processing strategies or provide options for additive-free doping, such as with water-soluble sulfur-containing lignosulfonates or nitrogen-containing enzymatic hydrolysis lignins. Thermal processing also varies with lignin selection due to differences in molecular weight and thermal stability. Furthermore, there are ongoing efforts to use advanced protocols, such as assembly and supraparticle formation, to address challenges related to disordered porosity and technological scalability, crucially, these approaches are highly dependent on lignin source and grade.
Due to the variety of new competing technologies, techno-economic assessments should accompany lignin-based technologies to evaluate their feasibility at an early stage, identify key process variables, and establish benchmarks. New technologies require knowledge of various thermodynamic and kinetic parameters for design and optimization that go beyond the typical metrics such as adsorption capacity and selectivity.189 Alternative technologies, such as MOFs and COFs, are also rapidly advancing, often exhibiting high adsorption capacities relative to carbon adsorbents. Membranes of various types of COFs have achieved up to 22.6 mmol g−1 for TAPB-PDA COF aerogels.190,191 While these materials have typically faced challenges with moisture, mechanical stability and scalable production, many of these challenges are also being addressed.192 Furthermore, emerging functional materials such as photo-responsive adsorbents offer additional opportunties.193,194 In this context, a systems approach to the rational design and comprehensive assessment of lignin-based adsorbents is essential to facilitate development in this active field.
Finally, current research on lignin carbonization is predominantly driven by top-down approaches, where experimental screening is used to identify optimal performance. Future developments could shift towards bottom-up designs, utilizing fundamental principles or AI-driven methods, which involve synthesizing designed structures followed by performance validation. This shift is due to the long-standing challenges in understanding the complex carbonization process. We anticipate that in-depth mechanistic studies will enhance the rational design of lignin-based carbon materials,195,196 and advance the broader field of carbon capture. Additionally, leveraging existing experimental and simulation data, we expect that combining machine-learning techniques with materials synthesis will enable data-driven optimization of the carbonization process.
Author contributions
D. B. R.: conceptualization, visualization, writing – original draft, writing – review & editing; J. C., Z. W.: writing – original draft; S. R.: writing – review & editing; I. B.: conceptualization, writing – review & editing; Y. D.: conceptualization, writing – original draft, writing – review & editing; Y. L.: conceptualization, supervision, writing – original draft, writing – review & editing; O. J. R.: conceptualization, supervision, funding acquisition, writing – review & editing.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. B. R., J. C., Z. W., Y. L., S. R., O. J. R. would like to acknowledge financial support from Natural Resources Canada/Innovation and Clean Growth Research, Development, and Demonstration Programs (CCUS-CAP-092, award number: AWD-027574 NRCan 2023). D. B. R., J. C., Z. W., Y. L., O. J. R. also acknowledge the Canada Excellence Research Chair program (CERC-2018-00006), Canada Foundation for Innovation (Project number 38623) and Pacific Economic Development Canada (PacifiCan). S. R. acknowledges the Canada Research Chairs Program, Tier 2, in Advanced Renewable Materials (#950-232330) for financial support. I. B. and Y. D. would like to acknowledge the financial support from the ETH Joint Initiative: Sustainable Pathways of Environmental and Energy Development towards Net Zero Switzerland (SPEED2ZERO).References
- W. H. Organization, Ecosystems and human well-being: health synthesis: a report of the Millennium Ecosystem Assessment, World Health Organization, 2005.
- J. D. Shakun, P. U. Clark, F. He, S. A. Marcott, A. C. Mix, Z. Liu, B. Otto-Bliesner, A. Schmittner and E. Bard, Nature, 2012, 484, 49–54 CrossRef CAS.
- P.-S. Wei, Y.-C. Hsieh, H.-H. Chiu, D.-L. Yen, C. Lee, Y.-C. Tsai and T.-C. Ting, Heliyon, 2018, 4, e00785 CrossRef PubMed.
- H. S. Kheshgi, S. J. Smith and J. A. Edmonds, Mitigation and Adaptation Strategies for Global Change, 2005, vol. 10, pp. 213–220 Search PubMed.
- J. W. Rae, Y. G. Zhang, X. Liu, G. L. Foster, H. M. Stoll and R. D. Whiteford, Annu. Rev. Earth Planet. Sci., 2021, 49, 609–641 CrossRef CAS.
- T. M. Letcher, Future energy, Elsevier, 2020, pp. 3–17 Search PubMed.
- P. Friedlingstein, M. O’Sullivan, M. W. Jones, R. M. Andrew, D. C. Bakker, J. Hauck, P. Landschützer, C. Le Quéré, I. T. Luijkx, G. P. Peters and W. Peters, Earth System Science Data, 2023, 15, 5301–5369 CrossRef.
- P. Fennell, J. Driver, C. Bataille and S. J. Davis, Nature, 2022, 603, 574–577 CrossRef CAS PubMed.
- R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2022, 13, 766–784 CrossRef.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. W. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 RSC.
- L. Joos, J. M. Huck, V. Van Speybroeck and B. Smit, Faraday Discuss., 2016, 192, 391–414 RSC.
- L. Zheng, Oxy-fuel combustion for power generation and carbon dioxide (CO2) capture, Elsevier, 2011 Search PubMed.
- M. Pardakhti, T. Jafari, Z. Tobin, B. Dutta, E. Moharreri, N. S. Shemshaki, S. Suib and R. Srivastava, ACS Appl. Mater. Interfaces, 2019, 11, 34533–34559 CrossRef CAS.
- D. G. Boer, J. Langerak and P. P. Pescarmona, ACS Appl. Energy Mater., 2023, 6, 2634–2656 CrossRef CAS.
- M. Maluszynska-Hoffman, N. Repond, J. A. Wurzbacher and C. Gebald, WO 2017/009241 A1, 2017.
- F. Baraka and J. Labidi, Sci. Total Environ., 2024, 912, 169093 CrossRef CAS.
- Y. Lu, M. Kamkar, S. Guo, X. Niu, Z. Wan, J. Xu, X. Su, Y. Fan, L. Bai and O. J. Rojas, Small, 2023, 19, 2300686 CrossRef CAS.
- T. Guo, Z. Wan, M. Panahi-Sarmad, G. Banvillet, Y. Lu, S. Zargar, J. Tian, F. Jiang, Y. Mao, Q. Tu and O. J. Rojas, ACS Nano, 2024, 18, 14954–14967 CrossRef CAS PubMed.
- E. Gomez-Delgado, G. V. Nunell, A. L. Cukierman and P. R. Bonelli, Carbon Lett., 2020, 30, 155–164 CrossRef.
- J. Y. Lai, L. H. Ngu, S. S. Hashim, J. J. Chew and J. Sunarso, Carbon Lett., 2021, 31, 201–252 CrossRef.
- A. Alabadi, S. Razzaque, Y. W. Yang, S. Chen and B. Tan, Chem. Eng. J., 2015, 281, 606–612 CrossRef CAS.
- A. E. Ogungbenro, D. V. Quang, K. Al-Ali and M. R. M. Abu-Zahra, Activated carbon from date seeds for CO2 capture applications, 13th International Conference on Greenhouse Gas Control Technology (GHGT), Lausanne, Switzerland, 2016.
- J. Serafin, M. Ouzzine, O. F. Cruz, J. Srenscek-Nazzal, I. C. Gómez, F. Z. Azar, C. A. R. Mafull, D. Hotza and C. R. Rambo, Waste Manage., 2021, 136, 273–282 CrossRef CAS PubMed.
- M. Wu, W. Chen, J. Hu, D. Tian, F. Shen, Y. Zeng, G. Yang, Y. Zhang and S. Deng, Sci. Total Environ., 2019, 695, 133898 CrossRef CAS PubMed.
- Y. Lu, Y. Chun, X. Shi, D. Wang, F. Ahmadijokani and O. J. Rojas, Adv. Mater., 2024, 36, 2400311 CrossRef CAS.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 11 CrossRef.
- O. Musl and A. Potthast, Lignin Chemistry, 2024, pp. 61–83 Search PubMed.
- J. K. Weng and C. Chapple, New Phytol., 2010, 187, 273–285 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- Y. Mottiar, R. Vanholme, W. Boerjan, J. Ralph and S. D. Mansfield, Curr. Opin. Biotechnol, 2016, 37, 190–200 CrossRef CAS PubMed.
- M. Balakshin, E. A. Capanema, X. Zhu, I. Sulaeva, A. Potthast, T. Rosenau and O. J. Rojas, Green Chem., 2020, 22, 3985–4001 RSC.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5, 87187–87199 RSC.
- A. Berlin and M. Balakshin, in Bioenergy Research: Advances and Applications, ed. V. K. Gupta, M. G. Tuohy, C. P. Kubicek, J. Saddler and F. Xu, Elsevier, Amsterdam, 2014, pp. 315–336 Search PubMed.
- M. Y. Balakshin, E. A. Capanema, I. Sulaeva, P. Schlee, Z. E. Huang, M. Feng, M. Borghei, O. J. Rojas, A. Potthast and T. Rosenau, ChemSusChem, 2021, 14, 1016–1036 CrossRef CAS PubMed.
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53 CAS.
- L. Kouisni, P. Holt-Hindle, K. Maki and M. Paleologou, J. Sci. Technol. For. Prod. Processes, 2012, 2, 6–10 Search PubMed.
- M. Hubbe, R. Alén, M. Paleologou, M. Kannangara and J. Kihlman, BioResources, 2019, 14, 2300–2351 CAS.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. Xu, Renewable Sustainable Energy Rev., 2020, 123, 11 CrossRef.
- N. Izaguirre, E. Robles, R. Llano-Ponte, J. Labidi and X. Erdocia, Ind. Crops Prod., 2022, 175, 114251 CrossRef CAS.
- S. K. Singh, Bioresour. Technol. Rep., 2022, 17, 100958 CrossRef CAS.
- D. M. de Carvalho and J. L. Colodette, BioResources, 2017, 12, 6907–6923 CAS.
- Q. He, I. Ziegler-Devin, L. Chrusciel, S. N. Obame, L. Hong, X. Lu and N. Brosse, ACS Sustainable Chem. Eng., 2020, 8, 5380–5392 CrossRef CAS.
- E. Leng, Y. Guo, J. Chen, S. Liu, E. Jiaqiang and Y. Xue, Fuel, 2022, 309, 122102 CrossRef CAS.
- L. A. Riddell, J. P. B. Lindner, P. de Peinder, F. Meirer and P. C. Bruijnincx, ChemSusChem, 2024, 17, e202301464 CrossRef CAS.
- A. Berlin and M. Balakshin, Bioenergy Res.: Adv. Appl., 2014, 315–336 CAS.
- M. A. Karaaslan, M. Cho, L.-Y. Liu, H. Wang and S. Renneckar, ACS Sustainable Chem. Eng., 2021, 9, 458–470 CrossRef CAS.
- M. Cho, M. Karaaslan, H. Wang and S. Renneckar, J. Mater. Chem. A, 2018, 6, 20973–20981 RSC.
- O. Musl, S. Galler, G. Wurzer, M. Bacher, I. Sulaeva, I. Sumerskii, A. K. Mahler, T. Rosenau and A. Potthast, Biomacromolecules, 2022, 23, 1413–1422 CrossRef CAS.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- J. Chen, J. Tian, N. Feng, L. Ning, D. Wang, B. Zhao, T. Guo, J. Song and O. J. Rojas, Small, 2024, 2309756 CrossRef CAS.
- J. Liu, M. Nero, K. Jansson, T. Willhammar and M. H. Sipponen, Nat. Commun., 2023, 14, 3099 CrossRef CAS PubMed.
- B. Zhao, M. Borghei, T. Zou, L. Wang, L. S. Johansson, J. Majoinen, M. H. Sipponen, M. Österberg, B. D. Mattos and O. J. Rojas, ACS Nano, 2021, 15, 6774–6786 CrossRef CAS PubMed.
- J. Carrier, C.-Y. Lai and D. Radu, ACS Environ. Au, 2024, 4, 196–203 CrossRef CAS.
- P. Figueiredo, K. Lintinen, J. T. Hirvonen, M. A. Kostiainen and H. A. Santos, Prog. Mater. Sci., 2018, 93, 233–269 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- T. Zou, M. H. Sipponen, A. Henn and M. Österberg, ACS Nano, 2021, 15, 4811–4823 CrossRef CAS.
- I. Standard, Environmental management-Life cycle assessment-Requirements and guidelines, ISO, 2006.
- C. Culbertson, T. Treasure, R. Venditti, H. Jameel and R. Gonzalez, Nord. Pulp Pap. Res. J., 2016, 31, 30–40 CrossRef CAS.
- C. Moretti, B. Corona, R. Hoefnagels, I. Vural-Gürsel, R. Gosselink and M. Junginger, Sci. Total Environ., 2021, 770, 144656 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford university press, 2000 Search PubMed.
- C. Libretti, L. Santos Correa and M. A. R. Meier, Green Chem., 2024, 26, 4358–4386 RSC.
- R. Bardestani, G. S. Patience and S. Kaliaguine, Can. J. Chem. Eng., 2019, 97, 2781–2791 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- S. Zhang, M. Zheng, Y. Tang, R. Zang, X. Zhang, X. Huang, Y. Chen, Y. Yamauchi, S. Kaskel and H. Pang, Adv. Funct. Mater., 2022, 32, 2204714 CrossRef CAS.
- M. Khosravi, N. Bashirpour and F. Nematpour, Adv. Mater. Res., 2014, 829, 922–926 Search PubMed.
- M. B. Vázquez-Santos, E. Geissler, K. László, J.-N. Rouzaud, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2012, 116, 257–268 CrossRef.
- A. Carlos, D. L. y Leon and L. R. Radovic, Chemistry & Physics of Carbon, 2023, pp. 213–310 Search PubMed.
- R. B. Fidel, D. A. Laird and M. L. Thompson, J. Environ. Qual., 2013, 42, 1771–1778 CrossRef CAS.
- F. Raganati, R. Chirone and P. Ammendola, Ind. Eng. Chem. Res., 2020, 59, 3593–3605 CrossRef CAS.
- N. S. Wilkins, A. Rajendran and S. Farooq, Adsorption, 2021, 27, 397–422 CrossRef CAS.
- N. S. Wilkins, J. A. Sawada and A. Rajendran, Ind. Eng. Chem. Res., 2022, 61, 7032–7051 CrossRef CAS.
- T. Lu, J. Bai, J. Huang, Q. Yu, M. Demir, M. Kilic, B. N. Altay, L. Wang and X. Hu, Energy Fuels, 2023, 37, 3886–3893 CrossRef CAS.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- K. S. Walton and D. S. Sholl, AIChE J., 2015, 61, 2757–2762 CrossRef CAS.
- S. Sircar and D. V. Cao, Chem. Eng. Technol., 2002, 25, 945–948 CrossRef CAS.
- L. Tian, Y. Zhi, Q. Yu, Q. Xu, M. Demir, S. G. Colak, A. A. Farghaly, L. Wang and X. Hu, Energy Fuels, 2024, 38, 13186–13195 CrossRef CAS.
- D. Levesque and F. D. Lamari, Mol. Phys., 2009, 107, 591–597 CrossRef CAS.
- C. Ma, J. Bai, M. Demir, Q. Yu, X. Hu, W. Jiang and L. Wang, Sep. Purif. Technol., 2022, 303, 122299 CrossRef CAS.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- Y. Chun, Y. Zhu, C. Stubenrauch, Y. Lu and O. J. Rojas, Curr. Opin. Colloid Interface Sci., 2024, 73, 101822 CrossRef CAS.
- W. Cao, H. Xu, X. Zhang, W. Xiang, G. Qi, L. Wan and B. Gao, Chem. Eng. J., 2023, 471, 144523 CrossRef CAS.
- Z. Pan, G. Qi, X. Zhang, Q. You, Y. Zheng, W. Xiang, Y. Zhao and B. Gao, Sep. Purif. Technol., 2024, 345, 127398 CrossRef CAS.
- Y. Sun, G. Yang, J.-P. Zhang, Y. Wang and M.-S. Yao, Chem. Eng. Technol., 2012, 35, 309–316 CrossRef CAS.
- M. B. Li, X. Liu, C. G. Sun, L. Stevens and H. Liu, J. Environ. Chem. Eng., 2022, 10, 11 Search PubMed.
- G. Yin, Z. Liu, Q. Liu and W. Wu, Chem. Eng. J., 2013, 230, 133–140 CrossRef CAS.
- D. Saha, G. Orkoulas and D. Bates, Materials, 2023, 16, 12 CrossRef PubMed.
- O. Tkachenko, A. Nikolaichuk, N. Fihurka, A. Backhaus, J. B. Zimmerman, M. Strømme and T. M. Budnyak, ACS Appl. Mater. Interfaces, 2024, 16, 3427–3441 CrossRef CAS.
- W. M. Chen, X. Wang, Z. Hashisho, M. Feizbakhshan, P. Shariaty, S. Niknaddaf and X. Y. Zhou, Microporous Mesoporous Mater., 2019, 280, 57–65 CrossRef CAS.
- D. Saha, S. E. Van Bramer, G. Orkoulas, H. C. Ho, J. H. Chen and D. K. Henley, Carbon, 2017, 121, 257–266 CrossRef CAS.
- L. Z. Gong and A. Bao, J. CO2 Util., 2023, 68, 11 Search PubMed.
- T. Wang, Y. Zhai, Y. Zhu, C. Li and G. Zeng, Renewable Sustainable Energy Rev., 2018, 90, 223–247 CrossRef CAS.
- F. Bergius, in Die Anwendung hoher Drucke bei chemischen Vorgängen und eine Nachbildung des Entstehungsprozesses der Steinkohle, ed. W. Knapp, 1913 Search PubMed.
- H. Wikberg, T. Ohra-aho, F. Pileidis and M.-M. Titirici, ACS Sustainable Chem. Eng., 2015, 3, 2737–2745 CrossRef CAS.
- W. Sangchoom and R. Mokaya, ACS Sustainable Chem. Eng., 2015, 3, 1658–1667 CrossRef CAS.
- M. O. Bengoechea, A. Hertzberg, N. Miletić, P. L. Arias and T. Barth, J. Anal. Appl. Pyrolysis, 2015, 113, 713–722 CrossRef.
- W. Hao, F. Björnerbäck, Y. Trushkina, M. Oregui Bengoechea, G. Salazar-Alvarez, T. Barth and N. Hedin, ACS Sustainable Chem. Eng., 2017, 5, 3087–3095 CrossRef CAS.
- M. Demir, T. D. Tessema, A. A. Farghaly, E. Nyankson, S. K. Saraswat, B. Aksoy, T. Islamoglu, M. M. Collinson, H. M. El-Kaderi and R. B. Gupta, Int. J. Energy Res., 2018, 42, 2686–2700 CrossRef CAS.
- E. Atta-Obeng, B. Dawson-Andoh, E. Felton and G. Dahle, Waste Biomass Valorization, 2019, vol. 10, pp. 2725–2731 Search PubMed.
- J. Dong, R. Wang, X. Wang, S. Tan, Z. Zhao, Q. Yin and X. Lu, Carbon, 2024, 229, 119530 CrossRef CAS.
- J. Zhang, D. Huang, J. Shao, X. Zhang, H. Yang, S. Zhang and H. Chen, J. Cleaner Prod., 2022, 355, 131642 CrossRef CAS.
- Y. Bai, Y. Li, M. Li, X. Lin, C. Cai, H. He and S. Liu, Ind. Crops Prod., 2024, 220, 119183 CrossRef CAS.
- D. Liu, L. Shao, P. Zhan, L. Zhang, Z. Wu, J. Wang, X. Ma and J. Huang, Sep. Purif. Technol., 2024, 347, 127657 CrossRef CAS.
- J. Gao, Z.-Q. Wang, B. Li, W. Zhao, Z.-R. Ba, Z.-Y. Liu, J.-J. Huang and Y.-T. Fang, Bioresour. Technol., 2024, 393, 130171 CrossRef CAS PubMed.
- S. Park, M. S. Choi and H. S. Park, Carbon Lett., 2019, 29, 289–296 CrossRef.
- J. Zhao, Q. Wang, W. Zhang, D. Shen and C. Cheng, Int. J. Hydrogen Energy, 2024, 67, 127–135 CrossRef CAS.
- Q. B. Meng and J. Weber, ChemSusChem, 2014, 7, 3312–3318 CrossRef CAS.
- L. S. Shao, N. Liu, L. Z. Wang, Y. F. Sang, H. A. Wan, P. Zhan, L. Zhang, J. H. Huang and J. N. Chen, Chemosphere, 2022, 288, 11 CrossRef PubMed.
- N. Liu, L. S. Shao, C. Wang, F. B. Sun, Z. P. Wu, P. Zhan, L. Zhang and H. A. Wan, Int. J. Biol. Macromol., 2022, 221, 25–37 CrossRef CAS.
- N. Liu, J. Chen, Z. Wu, P. Zhan, L. Zhang, Q. Wei, F. Wang and L. Shao, ACS Appl. Polym. Mater., 2021, 3, 2178–2188 CrossRef CAS.
- M. A. Karaaslan, L. T. Lin, F. Ko and S. Renneckar, Front. Mater., 2022, 9, 894061 CrossRef.
- M. A. Karaaslan, L. C. Lin, F. Ko and S. Renneckar, presented in part at the American Chemical Society, San Diego, CA, USA, March 20-24, 2022, 2022.
- Z. Li, B. Li, C. Yu, H. Wang and Q. Li, Adv. Sci., 2023, 10, 2206605 CrossRef CAS PubMed.
- H. Qin, S. Kang, Y. Huang, S. Liu, Y. Fang, X. Li and Y. Wang, Mater. Lett., 2015, 159, 463–465 CrossRef CAS.
- S. Otani, Y. Fukuoka, B. Igarashi and K. Sasaki, US3461082A, 1969.
- W. Fang, S. Yang, X.-L. Wang, T.-Q. Yuan and R.-C. Sun, Green Chem., 2017, 19, 1794–1827 RSC.
- E. M. Calvo-Muñoz, F. J. García-Mateos, J. M. Rosas, J. Rodríguez-Mirasol and T. Cordero, Front. Mater., 2016, 3, 14 Search PubMed.
- Y. Lu, M. Mehling, S. Huan, L. Bai and O. J. Rojas, Chem. Soc. Rev., 2024, 53, 7363–7391 RSC.
- L. Zhang, L. Jin, B. Liu and J. He, Front. Chem., 2019, 7, 22 CrossRef CAS.
- S. Sani, X. Liu, M. B. Li, L. Stevens and C. G. Sun, Microporous Mesoporous Mater., 2023, 347, 11 CrossRef.
- S. Sani, X. Liu, L. Stevens, H. M. Wang and C. G. Sun, Fuel, 2023, 351, 11 CrossRef.
- J. Zhao, W. J. Zhang, D. K. Shen, H. Y. Zhang and Z. H. Wang, J. Energy Inst., 2023, 107, 9 Search PubMed.
- S. Geng, J. Wei, S. Jonasson, J. Hedlund and K. Oksman, ACS Appl. Mater. Interfaces, 2020, 12, 7432–7441 CrossRef CAS PubMed.
- C. Salas, T. Nypelö, C. Rodriguez-Abreu, C. Carrillo and O. J. Rojas, Curr. Opin. Colloid Interface Sci., 2014, 19, 383–396 CrossRef CAS.
- B. D. Mattos, B. L. Tardy, L. G. Greca, T. Kämäräinen, W. Xiang, O. Cusola, W. L. E. Magalhães and O. J. Rojas, Sci. Adv., 2020, 6, eaaz7328 CrossRef CAS PubMed.
- H. Wang, C. Zeng, C. Wang, J. Fu, Y. Li, Y. Yang, Z. Du, G. Tao, Q. Sun and T. Zhai, Nat. Mater., 2024, 23, 596–603 CrossRef CAS PubMed.
- L. Shao, H. a Wan, L. Wang, J. Wang, Z. Liu, Z. Wu, P. Zhan, L. Zhang, X. Ma and J. Huang, Sep. Purif. Technol., 2023, 313, 123440 CrossRef CAS.
- S.-C. Qi, Y. Liu, A.-Z. Peng, D.-M. Xue, X. Liu, X.-Q. Liu and L.-B. Sun, Chem. Eng. J., 2019, 361, 945–952 CrossRef CAS.
- L. S. Shao, H. A. Wan, L. Z. Wang, J. J. Wang, Z. H. Liu, Z. P. Wu, P. Zhan, L. Zhang, X. C. Ma and J. H. Huang, Sep. Purif. Technol., 2023, 313, 15 CrossRef.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. B. Yi, D. Y. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- C. Goel, S. Mohan and P. Dinesha, Sci. Total Environ., 2021, 798, 21 CrossRef PubMed.
- V. K. Singh and E. A. Kumar, Appl. Therm. Eng., 2016, 97, 77–86 CrossRef CAS.
- N. Planas, A. L. Dzubak, R. Poloni, L. C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed.
- G. K. Cui, J. J. Wang and S. J. Zhang, Chem. Soc. Rev., 2016, 45, 4307–4339 RSC.
- Y. Q. Chen, X. Zhang, W. Chen, H. P. Yang and H. P. Chen, Bioresour. Technol., 2017, 246, 101–109 CrossRef CAS PubMed.
- A. E. Creamer and B. Gao, Environ. Sci. Technol., 2016, 50, 7276–7289 CrossRef CAS PubMed.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M. C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- G. Banvillet, S. Pritchard, J. J. Kaschuk, X. Shi, M. Imani, Y. Lu, A. Takagi, M. Kamkar and O. J. Rojas, Mater. Today Nano, 2023, 24, 100424 CrossRef CAS.
- M. Samandari, M. T. Broud, D. P. Harper and D. J. Keffer, J. Phys. Chem. B, 2024, 128, 8530–8545 CrossRef CAS.
- M. T. Broud, M. Samandari, L. Yu, D. P. Harper and D. J. Keffer, J. Phys. Chem. C, 2023, 127, 13639–13650 CrossRef CAS.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- X. Ma, Y. Yang, Q. Wu, B. Liu, D. Li, R. Chen, C. Wang, H. Li, Z. Zeng and L. Li, Fuel, 2020, 282, 118727 CrossRef CAS.
- Z. Zhang, A. L. Kummeth, J. Y. Yang and A. N. Alexandrova, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2123496119 CrossRef CAS.
- A. Lancheros, S. Goswami, M. R. Mian, X. Zhang, X. Zarate, E. Schott, O. K. Farha and J. T. Hupp, Dalton Trans., 2021, 50, 2880–2890 RSC.
- Y. Lu, D. Sun, J. Ralston, Q. Liu and Z. Xu, J. Colloid Interface Sci., 2019, 557, 185–195 CrossRef CAS PubMed.
- M. Zhang, Y. Nan, Y. Lu, Q. You and Z. Jin, Fuel, 2023, 331, 125773 CrossRef CAS.
- T. Lu and Q. Chen, J. Phys. Chem. A, 2023, 127, 7023–7035 CrossRef CAS PubMed.
- L. Petridis, R. Schulz and J. C. Smith, J. Am. Chem. Soc., 2011, 133, 20277–20287 CrossRef CAS.
- A. S. Patri, B. Mostofian, Y. Pu, N. Ciaffone, M. Soliman, M. D. Smith, R. Kumar, X. Cheng, C. E. Wyman, L. Tetard, A. J. Ragauskas, J. C. Smith, L. Petridis and C. M. Cai, J. Am. Chem. Soc., 2019, 141, 12545–12557 CrossRef CAS.
- S. V. Pingali, M. D. Smith, S.-H. Liu, T. B. Rawal, Y. Pu, R. Shah, B. R. Evans, V. S. Urban, B. H. Davison, C. M. Cai, A. J. Ragauskas, H. M. O’Neill, J. C. Smith and L. Petridis, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16776–16781 CrossRef CAS.
- Z. Nezafat, M. Nasrollahzadeh, S. Javanshir, T. Baran and Y. H. Dong, Green Chem., 2023, 25, 9603–9643 RSC.
- Y. D. Cui, B. He, Y. Lei, Y. Liang, W. T. Zhao, J. Sun and X. M. Liu, Chin. J. Chem. Eng., 2023, 54, 89–97 CrossRef CAS.
- S.-C. Qi, J.-K. Wu, J. Lu, G.-X. Yu, R.-R. Zhu, Y. Liu, X.-Q. Liu and L.-B. Sun, J. Mater. Chem. A, 2019, 7, 17842–17853 RSC.
- S.-C. Qi, X.-J. Lu, Y.-C. Lou, R. Zhou, D.-M. Xue, X.-Q. Liu and L.-B. Sun, Engineering, 2022, 16, 154–161 CrossRef CAS.
- A.-Z. Peng, S.-C. Qi, X. Liu, D.-M. Xue, S.-S. Peng, G.-X. Yu, X.-Q. Liu and L.-B. Sun, Chem. Eng. J., 2019, 372, 656–664 CrossRef CAS.
- S. Beck, P. Choi and S. H. Mushrif, Phys. Chem. Chem. Phys., 2022, 24, 20480–20490 RSC.
- S. Beck and S. H. Mushrif, Org. Biomol. Chem., 2023, 21, 4596–4600 RSC.
- R. L. Silveira, S. R. Stoyanov, S. Gusarov, M. S. Skaf and A. Kovalenko, J. Phys. Chem. Lett., 2015, 6, 206–211 CrossRef CAS PubMed.
- J. V. Vermaas, M. F. Crowley and G. T. Beckham, ACS Sustainable Chem. Eng., 2020, 8, 17839–17850 CrossRef CAS.
- D. Vural, J. C. Smith and L. Petridis, Phys. Chem. Chem. Phys., 2018, 20, 20504–20512 RSC.
- T. Zhang, X. Li, L. Guo and X. Guo, Energy Fuels, 2019, 33, 11210–11225 CrossRef CAS.
- P. N. Ciesielski, M. B. Pecha, A. M. Lattanzi, V. S. Bharadwaj, M. F. Crowley, L. Bu, J. V. Vermaas, K. X. Steirer and M. F. Crowley, ACS Sustainable Chem. Eng., 2020, 8, 3512–3531 CrossRef CAS.
- M. J. Orella, T. Z. H. Gani, J. V. Vermaas, M. L. Stone, E. M. Anderson, G. T. Beckham, F. R. Brushett and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2019, 7, 18313–18322 CrossRef CAS.
- J. V. Vermaas, L. D. Dellon, L. J. Broadbelt, G. T. Beckham and M. F. Crowley, ACS Sustainable Chem. Eng., 2019, 7, 3443–3453 CrossRef CAS.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2021, 171, 514–523 CrossRef CAS.
- S. Anila, C. H. Suresh and H. F. Schaefer, III, J. Phys. Chem. A, 2022, 126, 4952–4961 CrossRef CAS PubMed.
- H. M. Lee, I. S. Youn, M. Saleh, J. W. Lee and K. S. Kim, Phys. Chem. Chem. Phys., 2015, 17, 10925–10933 RSC.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- P. Su, Z. Jiang, Z. Chen and W. Wu, J. Phys. Chem. A, 2014, 118, 2531–2542 CrossRef CAS.
- E. D. Glendening, J. Am. Chem. Soc., 1996, 118, 2473–2482 CrossRef CAS.
- P. R. Horn, Y. Mao and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067–23079 RSC.
- K. Szalewicz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 254–272 CAS.
- E. I. Izgorodina, J. L. Hodgson, D. C. Weis, S. J. Pas and D. R. MacFarlane, J. Phys. Chem. B, 2015, 119, 11748–11759 CrossRef CAS PubMed.
- J.-Y. Chung, U. Hwang, J. Kim, N.-Y. Kim, J. Nam, J. Jung, S.-H. Kim, J. K. Cho, B. Lee, I.-K. Park, J. Suhr and J.-D. Nam, ACS Sustainable Chem. Eng., 2023, 11, 2303–2313 CrossRef CAS.
- J. Sun, C. Wang, L. P. Stubbs and C. He, Macromol. Mater. Eng., 2017, 302, 1700341 CrossRef.
- H. Sidky, W. Chen and A. L. Ferguson, Mol. Phys., 2020, 118, e1737742 CrossRef.
- Y. Wang, J. M. Lamim Ribeiro and P. Tiwary, Curr. Opin. Struct. Biol., 2020, 61, 139–145 CrossRef CAS PubMed.
- F. Noé, A. Tkatchenko, K.-R. Müller and C. Clementi, Annu. Rev. Phys. Chem., 2020, 71, 361–390 CrossRef PubMed.
- S. Bhakat, RSC Adv., 2022, 12, 25010–25024 RSC.
- R. Jones, M. Khalkhali, Y. Lu, Q. Liu, A. Ghosh and R. Lespiat, US Pat., US20240025807A1, 2024.
- G. Tofani, E. Jasiukaitytė-Grojzdek and B. Likozar, BioResources, 2024, 19, 6967–6969 CAS.
- J. Zhang, Y.-K. Lei, Z. Zhang, J. Chang, M. Li, X. Han, L. Yang, Y. I. Yang and Y. Q. Gao, J. Phys. Chem. A, 2020, 124, 6745–6763 CrossRef CAS.
- L. Bonati, G. Piccini and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113533118 CrossRef CAS PubMed.
- D. Ray, E. Trizio and M. Parrinello, J. Chem. Phys., 2023, 158, 204102 CrossRef CAS PubMed.
- A. Rajendran, G. K. H. Shimizu and T. K. Woo, Adv. Mater., 2024, 36, 2301730 CrossRef CAS PubMed.
- G. Zhao, Z. Li, B. Cheng, X. Zhuang and T. Lin, Sep. Purif. Technol., 2023, 315, 123754 CrossRef CAS.
- L.-G. Ding, B.-J. Yao, F. Li, S.-C. Shi, N. Huang, H.-B. Yin, Q. Guan and Y.-B. Dong, J. Mater. Chem. A, 2019, 7, 4689–4698 RSC.
- T. Nguyen, G. Shimizu and A. Rajendran, Chemrxiv, 2024 DOI:10.26434/chemrxiv-2024-nx3vg.
- P. Tan, Y. Jiang, Q. Wu, C. Gu, S. Qi, Q. Zhang, X. Liu and L. Sun, Chin. J. Chem. Eng., 2022, 42, 104–111 CrossRef CAS.
- Y. Qiao, J. J. Bailey, Q. Huang, X. Ke and C. Wu, Renewable Sustainable Energy Rev., 2022, 158, 112079 CrossRef CAS.
- Z. Pan, A. Bodi, J. A. van Bokhoven and P. Hemberger, Phys. Chem. Chem. Phys., 2022, 24, 21786–21793 RSC.
- Z. Pan, X. Wu, A. Bodi, J. A. van Bokhoven and P. Hemberger, Green Chem., 2024, 26, 9899–9910 RSC.
| This journal is © The Royal Society of Chemistry 2025 |