
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical valorization of captured CO2: recent advances and future perspectives†
Xin-Ming
Hu
 *a,
Hong-Qing
Liang
*b,
Alonso
Rosas-Hernández
*c and
Kim
Daasbjerg
*d
*a,
Hong-Qing
Liang
*b,
Alonso
Rosas-Hernández
*c and
Kim
Daasbjerg
*d
aEnvironment Research Institute, Shandong University, Qingdao, 266237, China. E-mail: huxm@sdu.edu.cn
bKey Lab of Adsorption and Separation Materials & Technologies of Zhejiang Province, MOE Engineering Research Center of Membrane and Water Treatment, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310058, China. E-mail: liang.hongqing@zju.edu.cn
cCarbon Dioxide Activation Center (CADIAC), Interdisciplinary Nanoscience Center (iNANO), Department of Chemistry, Novo Nordisk Foundation (NNF) CO2 Research Center, Aarhus University, Gustav Wieds Vej 10C, 8000 Aarhus C, Denmark. E-mail: arosas@chem.au.dk
dNovo Nordisk Foundation (NNF) CO2 Research Center, Interdisciplinary Nanoscience Center, Department of Chemistry, Aarhus University, Gustav Wieds Vej 10C, 8000 Aarhus C, Denmark. E-mail: kdaa@chem.au.dk
First published on 10th December 2024
Abstract
The excessive emission of CO2 has led to severe climate change, prompting global concern. Capturing CO2 and converting it through electrochemistry into value-added products represent promising approaches to mitigating CO2 emissions and closing the carbon cycle. Traditionally, these two processes have been performed independently, involving multiple steps, high energy consumption, and low efficiency. Recently, the electrochemical conversion of captured CO2, which integrates the capture and conversion processes (also referred to as electrochemically reactive CO2 capture), has garnered increasing attention. This integrated approach bypasses the energy-intensive steps involved in the traditional independent process, including CO2 release, purification, compression, transportation, and storage. In this review, we discuss recent advances in the electrochemical conversion of captured CO2, focusing on four key aspects. First, we introduce various capture media, emphasizing the thermodynamic aspects of carbon capture and their implications for integration with electrochemical conversion. Second, we discuss product control mediated by the selection of different catalysts, highlighting the connections between the conversion of captured CO2 and gas-fed CO2. Third, we examine the effect of reactor systems and operational conditions on the electrochemical conversion of captured CO2, shedding light on performance optimization. Finally, we explore real integration systems for CO2 capture and electrochemical conversion, revealing the potential of this new technology for practical applications. Overall, we provide insights into the existing challenges, potential solutions, and thoughts on opportunities and future directions in the emerging field of electrochemical conversion of captured CO2.
 Xin-Ming Hu | Xin-Ming Hu received his B.Sc. and M.Sc. degrees in China from Nankai University in 2008 and Graduate University of Chinese Academy of Sciences in 2011, respectively. He obtained the Ph.D. degree in 2014 from University of Copenhagen, and afterwards worked as a Postdoctoral Fellow and Assistant Professor at Aarhus University, Denmark. In 2020, he moved back to China and joined Shandong University as Professor. His research focus is on the development of new materials and technologies for CO2 capture and utilization. |
 Hong-Qing Liang | Hong-Qing Liang is a ZJU100 Young Professor at Zhejiang University. He received his B.Sc. (2011) and Ph.D. degrees (2016) from Zhejiang University, under supervision of Prof. Zhi-Kang Xu. He then carried out postdoctoral research at University of Texas at San Antonio (2016–2018, Prof. Banglin Chen), Aarhus University (2018–2019, Prof. Kim Daasbjerg), University of Padova (2020–2021, Prof. Stefano Agnoli), and Leibniz Institute for Catalysis (Alexander von Humboldt fellow, 2021–2023, Prof. Matthias Beller and Prof. Robert Francke). His current research focuses on electrochemical CO2 reduction with interfacial engineering. |
 Alonso Rosas-Hernández | Alonso Rosas-Hernández pursued his education at the National Autonomous University of Mexico (UNAM), where he obtained his BS and MS degrees in Chemistry. He then completed his doctorate in 2017 at the Leibniz Institute for Catalysis (LIKAT) under the supervision of Prof. Matthias Beller. Following this, he joined Prof. Jonas Peters’ group at the California Institute of Technology (Caltech) as a postdoctoral scholar. In 2021, he began his independent career at Aarhus University. His research interests focus on the fundamental understanding of bond-breaking and bond-forming reactions at electrified surfaces and the development of novel catalytic materials to advance a circular carbon economy. |
 Kim Daasbjerg | Kim Daasbjerg obtained his PhD in 1993, conducting research in organic electrochemistry under the supervision of Prof. Henning Lund at Aarhus University. After completing his doctoral studies, he undertook postdoctoral research at the Royal Institute of Technology in Stockholm in 1994, working with Prof. Gábor Mérenyi. He then began his academic career at Aarhus University, where he earned a Doctor of Science degree in 2006 and was appointed Professor in 2010. His research expertise spans electrochemistry, polymer brushes, responsive polymers, smart hybrid materials, and composites incorporating carbon substrates such as graphene. In recent years, he has leveraged his combined expertise in these fields to tackle the scientifically complex and socially important challenge of converting carbon dioxide, a key greenhouse gas, into valuable building blocks for the chemical industry and energy sector. |
1. Introduction
The excessive emission of CO2, the primary anthropogenic greenhouse gas, has caused severe climate change, presenting one of the most urgent global challenges.1,2 The amount of CO2 released into the atmosphere due to human-related activities is massive, with emissions of ∼35 Gt CO2 just in 2021.3 For this reason, atmospheric CO2 levels have surged from 280 ppm before the Industrial Revolution to 420 ppm.4 Consequently, global temperatures have risen by ∼1 °C compared to pre-industrial times, with projections indicating a potential increase exceeding 2 °C by the end of this century if CO2 emissions remain unchecked.4,5 Climate change has already resulted in severe events on earth, including glacier retreats, sea level elevation, extreme weather events, and ecosystem degradation.Carbon capture, utilization, and storage (CCUS) technologies have been developed rapidly over the past decades to mitigate CO2 emissions and reduce atmospheric CO2 concentration.6,7 CCUS typically involves multiple steps, including CO2 capture from flue gas by amine or other alkaline solutions/materials, release of the captured CO2 by temperature or pressure swings, followed by CO2 purification, compression, and transportation before it is finally stored or utilized.8 The long-term storage of CO2 is mainly implemented by mineral carbonation, along with oceanic and underground geological storage.9 However, CO2 can also be utilized directly in food preservation and production, as well as in industrial processes,10 or indirectly by conversion into various value-added products and fuels through thermochemical,11,12 photochemical,13–15 biochemical,16 and electrochemical techniques.17 Among these techniques, electrochemical conversion of CO2 stands out due to several advantages such as mild reaction conditions, environmental friendliness, use of renewable electricity, and controllable reaction rate and product selectivity.18 In particular, gas diffusion electrodes and flow cell configurations have been effectively utilized to boost the productivity of electrochemical CO2 reduction at industrial-relevant current densities (>200 mA cm−2).19,20
To date, the CO2 capture from diluted sources (0.04–15%) and the follow-up electrochemical conversion have been operated in three distinct routes, i.e., (1) independent, (2) coupled, and (3) integrated (Fig. 1).21,22 Route 1 involves multiple decoupled processes, where CO2 capture occurs independently of electrochemical CO2 conversion. Route 2 entails CO2 capture and release stages, immediately followed by the electrochemical valorization of the released CO2. Route 3 exhibits complete integration between the capture and electrochemical conversion of CO2, bypassing the CO2 stripping step required in Routes 1 and 2. For all three routes, producing value-added chemicals and fuels helps offset capture expenses, rendering commercial deployment economically more feasible.
 | ||
| Fig. 1 Three routes for CO2 capture and electrochemical conversion: (1) independent, (2) coupled, and (3) integrated. | ||
1.1. Route 1 (independent)
The independent systems inherently incur energy penalties and costs associated with CO2 release, purification, compression, and transportation. In particular, the CO2 desorption step can be energy-intensive if thermal regeneration at high temperatures is required. Nevertheless, Route 1 configuration can emerge as a viable scenario, considering the potential incompatibility in operating conditions between capture technologies that utilize temperature or pressure swings and the electrochemical conversion of CO2 under ambient conditions. This configuration offers flexibility for independently optimizing the capture and conversion stages. However, it is important to note that standalone electrochemical systems using gas-fed CO2 often face a significant challenge due to the typically low single-pass CO2 conversion efficiency (<20%).23–25 Consequently, this leads to low product concentration and significantly escalates downstream separation/purification costs.26,271.2. Route 2 (coupled)
An advantage of the coupled CO2 capture and conversion system is that it bypasses the CO2 purification, compression, and transportation steps, thereby reducing the complexity and energy consumption of the overall process. Here, the CO2 for the downstream conversion is directly supplied by the upstream CO2 capture unit, which usually releases CO2 by electrochemically driven redox couples or pH swings.28,29 Note that the coupled cascade system still involves independent CO2 capture and conversion, as well as the energy-intensive CO2 release step. Accordingly, the problem of low single-pass conversion efficiency for CO2 exists. In addition, by enhancing the overlap between the capture and conversion steps, fewer degrees of freedom are available for optimizing the electrocatalysts toward higher activity and selectivity for a targeted CO2 reduction product.1.3. Route 3 (integrated/reactive capture)
Compared with Route 2, these systems offer the advantage of reducing the total energy requirements for capture and conversion by eliminating CO2 release and many other follow-up steps, which typically incur a significant energy penalty. By directly converting the captured CO2 at the electrode surface, the regeneration of the capture media and the production of valuable products occur simultaneously. In this sense, Route 3 integrates the two processes of CO2 capture and follow-up electrochemical conversion and has also been coined electrochemically reactive capture of CO2.30–32As a result, Route 3 represents a more straightforward and sustainable approach for CCUS than the other two routes, at least at first glance. This unique advantage has stimulated increasing activities in the study of direct electrochemical valorization of captured CO2 in the past several years. For instance, several reaction media, such as amine/carbamate solution, hydroxide/carbonate/bicarbonate solution, and molten oxide/carbonate, have proven effective in mediating CO2 capture and subsequent electrochemical conversion. This offers the opportunity to directly transform CO2 from flue gas or the air into valuable products such as carbon monoxide, formate, methane, and ethylene. Despite the significant advances and great prospects, several critical challenges still need to be addressed to advance such an emerging technology for large-scale applications.
These challenges lie in the unclear mechanism and low efficiency of the conversion of captured CO2, poor integration of CO2 capture and conversion processes, and others. In this context, a timely and thorough review article summarizing the progress and discussing the challenges in this emerging field would benefit the community.
In this review, we summarize the recent advances in the strategy of reactive CO2 capture, with a specific focus on the electrochemical conversion of captured CO2. Techno-economic analyses comparing the three different routes for CO2 capture and conversion can be found in recent reviews and are thus not covered in this paper.30,33 Moreover, readers with interests in related topics such as electrochemical CO2 capture34,35 and electrochemical CO2 conversion36 are referred to the respective reviews.
This review is structured around four aspects of the technology: capture media, electrocatalysts, system optimization, and practical considerations for continuous operation (Fig. 2). We start by introducing various types of capture media, emphasizing the thermodynamic aspects of carbon capture and their implications for integrated Route 3 processes. We move on to examine different electrocatalyst compositions and morphologies, discussing their influence on product distribution from the conversion of captured CO2 and its relationship with the conversion of gas-fed CO2. Next, we explore the effects of reactor systems and operational conditions on the electrochemical conversion of captured CO2, aiming to dissect the key factors determining the conversion performance. Finally, we present examples of integrated CO2 capture and conversion processes. Furthermore, we offer insights into the challenges and potential solutions and present our perspectives on the opportunities and future directions for integrating CO2 capture and electrochemical conversion. This exploration uncovers promising avenues for practical applications of this emerging field in achieving more sustainable CO2 capture and conversion, potentially contributing to mitigating global climate change.
 | ||
| Fig. 2 Themes covered in this review on the electrochemical conversion of captured CO2. | ||
2. Properties of capture media
The electrochemical conversion of captured CO2 from diluted sources marks a significant advancement in the field of CCUS. Historically, CO2 capture and subsequent electrochemical valorization were treated as separate research domains. The electrochemical valorization has primarily focused on novel electrocatalyst development, electrolyte selection, cell configuration engineering, and using gas-fed concentrated CO2 without considering the carbon source (Routes 1 and 2 in Fig. 1). However, the emergence of highly efficient electrochemical systems for CO2 reduction over the past decade has reshaped the field.37,38 Consequently, the CO2 capture step and the conversion of resulting captured CO2 have begun to be integrated to optimize the system (Route 3 in Fig. 1). This section explores the various capture media wherein the captured CO2 can be utilized for electrochemical conversion, encompassing aqueous amines, alkaline solutions, and molten oxides. In this section, our focus is directed toward the underlying mechanisms of carbon capture and the thermodynamic aspects of chemical CO2 sorption.2.1. Amine solutions
The fundamental process of CO2 removal from a gas stream leveraging its reversible binding with amines was discovered in 1930.39 Initially utilized to separate CO2 from natural gas and hydrogen, such a chemical reactivity has since been employed in amine scrubbing for post-combustion capture of CO2 on small-scale gas and coal-fired plants.40 Currently, amine scrubbing is the most cost-effective and widely used technology for CO2 capture from industrial flue gases in power plants, oil refineries, cement production, and other applications. This is due to the high absorption rate and large CO2 capacity of amines. For example, NRG Energy Inc. and JX Nippon Oil & Gas Exploration Corp. have launched a post-combustion carbon capture project called Petra Nova, which aims to remove over 90% of the CO2 from the flue gas of a coal-fired power plant in Texas, USA using an amine solution.41 However, it is important to note that the high temperatures in flue gas environments can lead to amine losses due to thermal degradation and evaporation. Furthermore, the presence of poisoning gases such as SOx and NOx can decrease the yield of captured CO2. While amine scrubbing is often hailed as the most cost-effective post-combustion CO2 capture technology, it remains relatively expensive in terms of scalability. Therefore, ongoing research endeavors aim to enhance amine scrubbing processes, address their limitations, and optimize efficiency.The capture of CO2 using amine-based technologies involves two primary steps: absorption of CO2 and CO2-stripping for amine regeneration. The process is initiated by directing a CO2-diluted gas stream at or near ambient temperature through the amine solution. During this phase, CO2 is captured via a thermodynamically favorable reaction with the amine, yielding carbamate and ammonium ions (eqn (1)). Amines generally display high values of absorption enthalpies (ΔHabs), ranging from −90 to −60 kJ mol−1 when flue gas (12–15% CO2 concentration) is employed.42 Subsequently, the amine undergoes regeneration through stripping with water vapor at temperatures ranging from 100–120 °C, which shifts the equilibrium in eqn (1) toward the reactants. Following this, water is condensed from the stripper vapor, leaving behind pure CO2. The main energy demand in the process comes from stripping CO2 from the CO2-rich amine stream, which requires 115–140 kJ mol-CO2−1. This step consumes more energy than is released during the absorption phase because it produces a higher-purity CO2 stream than the original flue gas. Thus, the critical thermodynamic parameters governing the amine scrubbing process revolve around the chemical interaction between the amine and CO2.
| 2RNH2(aq.) + CO2(g) ⇌ RNHCOO−(aq.) + RNH3+(aq.) | (1) |
Different strategies have been employed to balance the efficiency of the chemical absorption step and the total energy consumption for the amine regeneration. Predictably, enhancing the chemical interaction between CO2 and the amine to improve absorption rates leads to increased energy input required to break the C–N bond of the carbamate group during amine regeneration, which is translated to the use of higher temperatures during the stripping step. For instance, primary and secondary amines display a very high heat of absorption since they can quickly generate chemically stable carbamates during their reaction with CO2.43 However, their regeneration energy requirement is high. In contrast, tertiary amines have a low heat of absorption and need less regeneration energy for solvent recovery, as bicarbonate, instead of carbamate, is formed for CO2 capture with tertiary amines.
The most commonly used amine-based capture medium is a 30 wt% aqueous monoethanolamine solution, valued for its high CO2 absorption rate, large absorption volume, and low cost.44 Other frequently employed amines include diethanolamine and methyldiethanolamine, which are examples of secondary and tertiary amines, respectively. Although these aqueous amine solutions are widely used for CO2 absorption, they have drawbacks, such as amine loss and thermal decomposition. A reported strategy to overcome such issues is mixing monoethanolamine with other secondary or tertiary amines with low energy consumption for regeneration.45 This results in a mixture in the absorber unit that simultaneously displays high absorption capacity, high absorption rate, and low regeneration energy consumption. For instance, a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of monoethanolamine and diethylenetriamine has been reported to increase CO2 absorption capacity by 53% and removal efficiency by 31% with respect to the commonly used 30 wt% monoethanolamine aqueous solution.46
:1 mixture of monoethanolamine and diethylenetriamine has been reported to increase CO2 absorption capacity by 53% and removal efficiency by 31% with respect to the commonly used 30 wt% monoethanolamine aqueous solution.46
Since 2010, amine scrubbing has been successfully applied to capture CO2 from natural gas, hydrogen, and other gases with low oxygen content.40 For instance, aqueous monoethanolamine solution can capture CO2 from flue gas of coal-fired plants.47 Unfortunately, it has displayed oxidative and thermal degradation under these conditions. When the oxygen content in the gas stream is in the range of 15%, a more concentrated amine solution (30 wt%) is required to increase the rate of CO2 capture, which is inhibited by the oxidation of the amine in the presence of oxygen. Additionally, thermal degradation has been minimized by operating at lower temperatures (100 °C).48
Stern et al. proposed in 2013 an alternative process that uses an electrochemical swing based on Cu2+/Cu to strip CO2 from the carbamate adduct and regenerate the amine.49 The electrochemically mediate amine regeneration (EMAR) cycle leverages the high stability of the Cu2+–amine complexes compared to the carbamate, thus favoring the dissociation of CO2 from the latter. Formation constants of cupric amine complexes exhibit values of 1018 for polyamines such as ethylenediamine, while binding constants between amines and CO2 typically fall in the order of ∼103.50
Currently, the EMAR technology has potential for large-scale applications in systems following Route 2 in Fig. 1. One of its most notable advantages is its ability to achieve high amine utilization, reducing the capital costs for auxiliary equipment due to lower circulation rates.51 Additionally, the EMAR technology requires less energy input since the capture and strip of CO2 are performed at room temperature. Since the process requires only electrical energy, decentralized devices can be developed at point sources to capture and convert CO2 as needed.
2.2. Aqueous alkaline solutions
CO2 capture in many industrial processes has been successfully performed using inorganic salt solutions of various weak acids.52 The most commonly employed salts are sodium and potassium carbonate, which, due to their alkaline pH (typically 9–11), absorb acid gases such as CO2. For instance, the traditional Benfield process, which has been in use since the 1950s, was developed as an alternative to amine scrubbing and uses hot K2CO3 solutions to absorb CO2 from a synthesis gas under relatively high CO2 partial pressures and high temperatures.53The capture of CO2 using aqueous K2CO3 solutions depends on the established equilibria when CO2 dissolves in water and how these equilibria are affected by pH changes. In open systems, increasing the pH raises the total concentration of dissolved carbon, while lowering the pH causes CO2 to be released, thereby decreasing CO2 absorption. When CO2 passes through aqueous K2CO3, it can react via two pathways. First, bicarbonate ions can be directly formed from the reaction of CO2 with hydroxide ions (eqn (2)). Alternatively, CO2 can be hydrated by water to produce carbonic acid (eqn (3)), which then deprotonates to form bicarbonate (eqn (4)). In either case, the resulting decrease in pH is buffered by the equilibrium between the carbonate and bicarbonate species (eqn (5)). Overall, K2CO3 in water captures CO2 as potassium bicarbonate (eqn (6)), with a ΔHabs value of approximately −40 kJ mol−1.35
| CO2(g) + OH−(aq.) ⇌ HCO3−(aq.) | (2) |
| CO2(g) + H2O(l) ⇌ H2CO3(aq.) | (3) |
| H2CO3(aq.) + H2O(l) ⇌ HCO3−(aq.) + H3O+(aq.) | (4) |
| HCO3−(aq.) + H2O(l) ⇌ CO32−(aq.) + H3O+(aq.) | (5) |
| CO2(g) + K2CO3(aq.) + H2O(l) ⇌ 2KHCO3(aq.) | (6) |
Either eqn (2) or (3) represents the process's rate-limiting step, depending on pH, as the deprotonation/protonation reactions in aqueous systems (eqn (4) and (5)) are considered barrierless. Accordingly, in solutions with pH > 9, the generally faster reaction between CO2 and hydroxide ions dominates. In contrast, in solutions with pH < 7, the slower CO2 hydration reaction becomes more prominent. For K2CO3 solutions, where the pH is usually >9, the reaction with hydroxide ions predominates and serves as the rate-determining step.
The release of CO2 from KHCO3, along with the regeneration of K2CO3 (reverse of eqn (6)), takes place through a pressure swing between the absorption and desorption units, eliminating the need for an additional temperature swing. This engineering approach enables absorption and regeneration at the same elevated temperature (∼100 °C), improving absorption kinetics and reducing energy consumption by nearly half.53
Aqueous K2CO3 solutions offer several advantages over traditional amine-based absorption liquids, including lower toxicity and nonvolatility, which minimize absorbent losses and decrease solvent replacement costs. Most importantly, carbonates exhibit resistance to absorbent degradation and are relatively inexpensive compared to many amines. A practical challenge associated with the carbonate absorption medium is that the precipitation of bicarbonate salts limits the use of highly concentrated solutions. However, recent research has shown that purposeful precipitation of bicarbonate salts can facilitate high-pressure stripping processes, reducing the regeneration energy requirement and liquid circulation rates.54 For example, an Australian company, KC8 Capture Technologies, uses a K2CO3 solution to capture 90–95% of CO2 emissions from flue gas, forming KHCO3, which can then precipitate and be subjected to CO2 release and K2CO3 regeneration.55 The regeneration energy requirement of the technology is 88–110 kJ mol-CO2−1, which is lower than that typically needed for amine-based processes. Another challenge for the carbonate absorption medium is its slow kinetics for CO2 absorption, which can be improved by adding activators or promoters such as amines53,56 and enzymes.57,58
Analogous to carbonate solutions, aqueous hydroxide solutions can also be used to capture CO2, but in this case, carbonate salts are produced as described in eqn (7). The produced carbonate can continue to capture CO2 to form bicarbonate.
| CO2(g) + 2KOH(aq.) ⇌ K2CO3(aq.) + H2O(l) | (7) |
Initially proposed in 1999 as a solution for direct air capture (DAC), the high absorption rate of this process results from the large thermodynamic driving force of the reaction between dissolved CO2 and hydroxide ions (−ΔHabs = 90–100 kJ mol−1).59 Although the original process was limited by reduced capture capability due to the low solubility of the used Ca(OH)2, most recent implementations of this technology use KOH, which has a solubility in water over two orders of magnitude greater than that of Ca(OH)2.60 Once K2CO3 is formed in the absorption chamber (eqn (7)), the next step is the precipitation of calcium carbonate and the regeneration of reusable KOH in solution (eqn (8)). The calcium carbonate is then subjected to a calcination process at ∼900 °C to liberate CO2 and generate CaO (eqn (9)), which generates the calcium hydroxide required in the precipitation chamber by reaction with water. Industrial applications of this technology for DAC have shown promising results by achieving a capture rate of 1 Mt CO2 year−1 (Carbon Engineering ULC.).61 However, this plant needs high-grade heat (230 kJ mol-CO2−1), which is supplied by burning natural gas.
| K2CO3(aq.) + Ca(OH)2(aq.) ⇌ 2KOH(aq.) + CaCO3(s) | (8) |
 | (9) |
2.3. Molten metal oxides
Capture of CO2 using alkali and alkaline-earth metal oxides has been employed in large-scale processes for over 150 years. Its first application involved purifying hydrogen by removing CO2 with CaO in a gasification process. However, it was not until 1999 that Shimizu et al. introduced the carbonate looping process, which paved the way for CO2 capture by metal oxides at medium to high temperatures (>200 °C).62 In this process, CaO reacts with post-combustion CO2 to form CaCO3 (eqn (10)), followed by its thermal decomposition to CaO and the release of pure CO2. Compared to liquid amine scrubbing, the higher operating temperatures of metal oxides provide the advantage of incurring lower energy penalties when the carbon capture process is implemented into power plants or other high-temperature processes.63The most used metal oxide sorbents utilize alkaline-earth metals such as calcium and magnesium to capture CO2 by forming the corresponding carbonates (eqn (10) and (11)). Although the carbonation reactions for CaO and MgO are exergonic, relatively high operating temperatures are required for the CO2 capture step to achieve adequate CO2 capture rates. This ensures that the carbonation reaction is primarily controlled by thermodynamics, leading to high process efficiencies. For example, continuous CO2 capture from ambient air has been demonstrated using CaO particles at temperatures ranging from 365 to 400 °C, achieving a CO2 removal efficiency of 99.9% after five consecutive cycles.64
| CaO + CO2 → CaCO3 | (10) |
| MgO + CO2 → MgCO3 | (11) |
Using oxides of alkali metals such as Li, Na, and K as sorbents is not often considered for large-scale CO2 capture applications. The formed carbonates (eqn (12)–(14)) typically require temperatures exceeding 1200 °C to release the absorbed CO2 and regenerate the metal oxides. However, these monovalent oxides have played an important role in altering the CO2 sorption mechanisms of CaO and MgO.65 When used as promoters, the alkali metal oxides readily transform into their carbonate forms in the presence of CO2 with ΔHabs of approximately −300 kJ mol−1.65 The melting points of Li2CO3, Na2CO3, and K2CO3 are relatively low, ranging from 730–900 °C, and eutectic mixtures of the three components possess melting points as low as ∼400 °C. Taking advantage of these low melting points, a strategy has been devised to enhance the rate and extent of CO2 uptake by CaO and MgO-based sorbents. This involves coating sorbents with binary or ternary molten alkali carbonates, which enhances CO2 transport within the sorbent.
| Li2O + CO2 → Li2CO3 | (12) |
| Na2O + CO2 → Na2CO3 | (13) |
| K2O + CO2 → K2CO3 | (14) |
Huang et al. reported the remarkable effects of alkali carbonate molten salts on the performance of CaO particles for CO2 capture.66 The (Li–K)2CO3 molten salt coating was found not only to promote CO2 uptake but also to facilitate CO2 desorption from CaO. Particularly at low temperatures of 500 and 600 °C, the CO2 capture capacity increased significantly from 1.19 and 3.26 mmol g−1 to 6.93 and 10.38 mmol g−1, respectively, using 7.5 mol% (Li–K)2CO3. The coating of alkali carbonate molten salts is believed to prevent the formation of a rigid CaCO3 layer on the surface of the CaO particles. This facilitates the ongoing reaction of dissolved CO2 with O2−, originating from CaO, in the liquid molten salts, resulting in the formation of CO32− (eqn (15)). The (Li–K)2CO3 molten salt is particularly effective in enhancing CO2 uptake due to the high O2− migration, which ensures the continuous counter diffusion of CO32− and O2−.67 Furthermore, CO2 can be captured by the reaction with the continuously delivered CO32− to form dicarbonate ions (eqn (16)).
| CO2 + O2− → CO32− | (15) |
| CO2 + CO32− → C2O52− | (16) |
In 2015, Harada et al. disclosed that coating MgO particles with molten alkali metal nitrates could significantly improve the CO2 uptake,68 which increased from <1 mmol g−1 for uncoated MgO to 10.2 mmol g−1 at 300 °C with the coating. They also demonstrated the molten alkali metal nitrates prevented the formation of a rigid carbonate layer on the surface of MgO, which would otherwise be impermeable to CO2. The high concentration of oxide ions in the molten salt restricted the generation of this rigid layer and facilitated the generation of carbonate ions (eqn (11)).69 This resulted in the rapid formation of MgCO3 and eased the regeneration of particles at moderate temperatures.
2.4 Electrochemical reactions of captured CO2
The physicochemical properties of each capture medium will affect the thermodynamics of the electrochemical conversion of the corresponding CO2-adduct to any of the possible products. As depicted in Fig. 3, the electrocatalytic conversion of the captured CO2 is an energetically uphill pathway, with the initial energy level determined by the CO2 absorption enthalpy of the capture medium employed. For instance, the electrochemical conversion of bicarbonate, derived from the CO2 capture with aqueous carbonate solutions (eqn (6)), is less endothermic than the direct conversion of the carbamate obtained from the reaction between CO2 and aqueous amines (eqn (1)). This is translated to a higher energy input for the electrochemical conversion of the carbamate as the result of the stronger interaction between CO2 and the capture medium. | ||
| Fig. 3 Schematic representation of the energetics for the electrochemical conversion of captured CO2. Enthalpy changes in CO2 absorption are shown for each capture medium. For ease of representation, energy levels are not to scale. | ||
The electronic and steric properties of the CO2-adduct are also crucial in determining the kinetics of the electrochemical conversion of captured CO2. The energies of intermediates involved in the pathways leading to the reduced products are influenced by the chemical structure of the captured CO2 species, thereby affecting the reaction kinetics. Moreover, the chemical composition and morphology of the catalyst can alter the binding energies of such reaction intermediates, consequently impacting reaction rates and favoring specific products. Thus, there is a complex interplay between the nature of the captured CO2 species and the electrocatalyst, collectively determining the kinetics for generating the products.
For instance, carbamates can be directly converted at the cathode, producing CO or other carbon-based products while regenerating the amine for another capture cycle (eqn (17)). The efficiency of the electrochemical conversion of CO2 captured as carbamate can be affected by several factors. First, the energy required to break the C–N bond in the carbamate group increases with stronger binding interactions between CO2 and the amine, which raises the overpotential of the electrocatalytic reaction. Moreover, the selectivity toward carbonaceous products can be adjusted by changing the concentration of the amine solutions to improve the mass transport of the amine–CO2 adduct to the electrode, thereby suppressing the undesired hydrogen evolution reaction (HER). Also, considering that the electrochemical conversion of carbamate follows an inner-sphere electron transfer mechanism,70 the distance between the carbamate and the electrode surface is crucial in determining the kinetics of the electrocatalytic reaction. Consequently, the structure of the amine will significantly influence the efficiency of carbamate conversion.
| RNHCOO−⋯RNH3+ + 2e− + H2O → CO + 2RNH2 + 2OH− | (17) |
In the case of CO2 capture using hydroxide or carbonate solutions, bicarbonate is the final product (eqn (2) and (6)). The bicarbonate solution can undergo electrochemical reduction to obtain CO or other products while regenerating the initial carbonate ions (eqn (18)). A challenging aspect of bicarbonate reduction systems is resolving the electrochemically active species, considering the dynamic acid–base equilibria that define the relative concentrations of CO2, bicarbonate, and carbonate at the electrode surface (eqn (2)–(5)).
| 2HCO3− + 2e− → CO + CO32− + 2OH− | (18) |
The CO2 capture by molten oxide generates molten carbonate (eqn (15)), which can be reduced at elevated temperatures (400–900 °C) at the cathode to form CO or other products while regenerating the oxide (eqn (19)). Similarly, dicarbonate ions can be directly reduced at the cathode, regenerating the carbonate and oxide ions (eqn (20)). These electrochemical transformations are unique since they are carried out at much higher temperatures than the conversion of carbamate or bicarbonate solutions. It has been shown that adjusting the temperature can effectively control the selectivity of the electrocatalytic reactions.71
| CO32− + 2e− → CO + 2O2− | (19) |
| C2O52− + 2e− → CO + CO32− + O2− | (20) |
The electrocatalytic conversion of CO2 captured by various capture media, as described above, facilitates the conversion of CO2 from diluted sources into value-added products. Producing carbonaceous products other than carbon monoxide, such as formate, ethylene, and carbon materials, entails more complex reaction pathways. The next section will explore these topics in detail.
3. Catalyst-mediated product control for converting captured CO2
The electrocatalyst is crucial in converting captured CO2, affecting the type and distribution of products, and the conversion efficiency. Recent progress in the electrochemical valorization of captured CO2 has made it possible to produce various products, including CO, HCOO−, CH4, C2H4, C2H5OH, and carbon materials, as summarized in Table 1.| Cathode materials | Captured CO2 | Electrolyzer | Pathway | Products | FE (%) | |j| (mA cm−2)a | T (°C)b | Ref. |
|---|---|---|---|---|---|---|---|---|
| a |j|: current density. b T: temperature. c DTAB: dodecyltrimethylammonium bromide. d Calculated based on data from references. e N.A.: not available. | ||||||||
| Ag nanoparticles (NPs) on Ag film | K2CO3 | MEA with BPM | Indirect | CO | 28 | 100 | R.T. | 72 |
| Ag NPs | K2CO3 | MEA with cation exchange membrane (CEM) | Indirect | CO | 46 | 200 | R.T. | 73 |
| Ag NPs | K2CO3 | MEA with CEM | Indirect | CO | 42 | 200 | R.T. | 74 |
| Ag NPs-coated porous carbon | KHCO3 | MEA with BPM | Indirect | CO | 37 | 100 | R.T. | 75 |
| Ag NPs | KHCO3 | MEA with BPM | Indirect | CO | 82 | 100 | R.T. | 76 |
| Ag NPs | KHCO3 | MEA with BPM | Indirect | CO | ∼65 | 200 | R.T. | 77 |
| Ag NPs | KHCO3 | MEA with CEM | Indirect | CO | 99 | 51.5 | R.T. | 78 |
| Ag (free-standing porous electrode) | KHCO3 | MEA with BPM | Indirect | CO | 59 | 100 | R.T. | 79 |
| Ag foam | KHCO3 | MEA with CEM | Indirect | CO | 40 | 100 | R.T. | 80 |
| Ag foam | KHCO3 + DTABc | MEA with BPM | Indirect | CO | 80 | 100 | R.T. | 81 |
| Ag (electrodeposited) | KHCO3 + DTABc | MEA with BPM | Indirect | CO | 85 | 100 | 50 | 82 |
| Ag (Br-modified) | NH4HCO3 | H cell | Indirect | CO | 77.8 | 18d | R.T. | 83 |
| Ag/C | Captured CO2 from the air using KOH solution | MEA with BPM | Indirect | CO | 13 | 50 | R.T. | 84 |
| Ag NPs on a Ag film | CO2-captured monoethanolamine | Flow cell | Direct | CO | 72 | 50 | 60 | 85 |
| Ag (Cl-modified) | CO2-captured 2-amino-2-methyl-1-propanol (AMP) | H cell | Indirect | CO | 91 | 6 | R.T. | 86 |
| Au (foil) | CO2-captured AMP | Flow cell | Indirect | CO | 45 | 10 | 75 | 87 |
| Ni SAC | CO2-captured monoethanolamine | MEA with BPM | Indirect | CO | 64.9 | 50 | R.T. | 88 |
| Ni SAC | KHCO3 | MEA with BPM | Indirect | CO | 67.2 | 100 | R.T. | 89 |
| Ni SAC | KHCO3 | MEA with BPM | Indirect | CO | 93 | 200 | R.T. | 90 |
| CoPc@CNT | KHCO3 | MEA with CEM | Indirect | CO | 96.2 | 300 | R.T. | 91 |
| FeNi alloy (foam) | Na2CO3–K2CO3 | Undivided cell | Direct | CO | 90.5 | 100 | 800 | 92 |
| Bi-coated carbon | KHCO3 | MEA with BPM | Indirect | HCOO− | 64 | 100 | R.T. | 93 |
| Bi electrodeposited on carbon | NH4HCO3 | MEA with AEM | Indirect | HCOO− | 75 | 100 | 40 | 94 |
| SnO2/C | Captured CO2 from air using KOH solution | MEA with BPM | Indirect | HCOO− | 16 | 50 | R.T. | 84 |
| Sn (rotating disc electrode) | KHCO3 | H cell | Indirect | HCOO− | 66 | ∼3d | R.T. | 95 |
| Sn (foil) | KHCO3 | H cell | Indirect | HCOO− | 18 | 6 | R.T. | 96 |
| Sn (foil) | KHCO3 | Undivided cell | Direct | HCOO− | 83 | 146d | 100 | 97 |
| Pb (foil) | CO2-captured AMP | Flow cell | Indirect | HCOO− | 50 | 25 | 75 | 87 |
| Cu (foam) | KHCO3 | MEA with BPM | Indirect | CH4 | 27 | 400 | R.T. | 98 |
| Cu (mesh) | KHCO3 | MEA with BPM | Indirect | CH4 | 27 | 500 | R.T. | 99 |
| Fe | Li2CO3–Na2CO3–K2CO3–LiOH | Undivided cell | Direct | CH4 | 63.3 | 15 | 575 | 100 |
| Fe sheet | Li1.427Na0.359K0.214CO3 ∼ 0.15LiOH | Undivided cell | Direct | CH4 | 42.7 | 400 | 650 | 101 |
| Cu/Ag (bilayer) | KHCO3 | MEA with BPM | Indirect | C2+ (CH3COO-, C2H4, C2H5OH, C3H7OH) | 41.6 | 100 | R.T. | 102 |
| Cu/CoPc-carbon nanotube (CNT) | K2CO3 | MEA with CEM and interposer | Indirect | C2H4 | 34 | 300 | R.T. | 103 |
| Cu/Ag (NPs) | K2CO3 | MEA with BPM | Indirect | C2H4 | 9 | 150 | R.T. | 104 |
| Ag–Cu(OH)2 nanowire on Cu foam | K2CO3/KHCO3 | MEA with BPM | Indirect | C2H4 | 12 | 100 | R.T. | 105 |
| Cu/Zn (Muntz brass sheet; 3:2 alloy) |
Li2CO3–Na2CO3 | Undivided cell | Direct | carbon nano-scaffold | 85 | 200 | 670 | 106 |
| Sn (liquid cathode) | Li2CO3–Na2CO3–K2CO3 | Undivided cell | Direct | Sn–C spheres (Sn@C) | 84 | N.A.e | 550 | 107 |
| SnO2 | CaCO3-CaCl2-NaCl | Undivided cell | Direct | Sn@CNT | 85.1 | N.A. | 800 | 108 |
| Steel (galvanized) | CO2-captured Li1.5Na1.5BO3 | Undivided cell | Direct | CNT | 55 | 120 | 600 | 109 |
| Steel (galvanized) | Li2CO3 | Undivided cell | Direct | Carbon platelets | N.A. | 200 | 730 | 110 |
| Ni | Li2CO3–K2CO3–Na2CO3 | Undivided cell | Direct | Graphite | N.A. | 5–100 | 650–750 | 111 |
| Ni | LiCl–Li2CO3–LiBO2 | Undivided cell | Direct | Carbon nanofibers | 93.8 | 35 | 550 | 112 |
| Ni | Li2CO3 | Undivided cell | Direct | Carbon nano-onion | N.A. | 150 | 750 | 113 |
These products are important chemicals, fuels, or materials with many industrial applications. Carbon monoxide is a key component of syngas, which is extensively used to synthesize long-chain hydrocarbons through the Fischer–Tropsch process.114 In addition, CO is widely utilized in synthesizing various carbonyl compounds via carbonylation reactions.18 Formate and formic acid are used in leather and textile manufacturing, serve as preservatives in livestock feed, and act as efficient H2 carriers in fuel cells.115 Methane, the primary component of natural gas, serves as a gas fuel for electricity generation, heating buildings, transportation, and industrial processes. Ethylene is an important building block for industrial chemical production, particularly in the manufacture of polyethylene, the most widely used plastic worldwide.116 Ethanol is commonly used as a solvent, disinfector, precursor, liqueur, and liquid fuel. Carbon materials, with varying morphology and physiochemical properties, are extensively utilized in applications such as adsorption, separation, and catalysis.117
3.1. Mechanism of conversion of captured CO2
There are two possible pathways for the electrochemical valorization of captured CO2, each influencing catalyst selection. (a) An indirect pathway, where the captured CO2 is first converted back to CO2 at the electrode–electrolyte interface and then undergoes electrochemical reduction (Fig. 4a); (b) a direct pathway, where the captured CO2 is directly reduced at the electrode (Fig. 4b).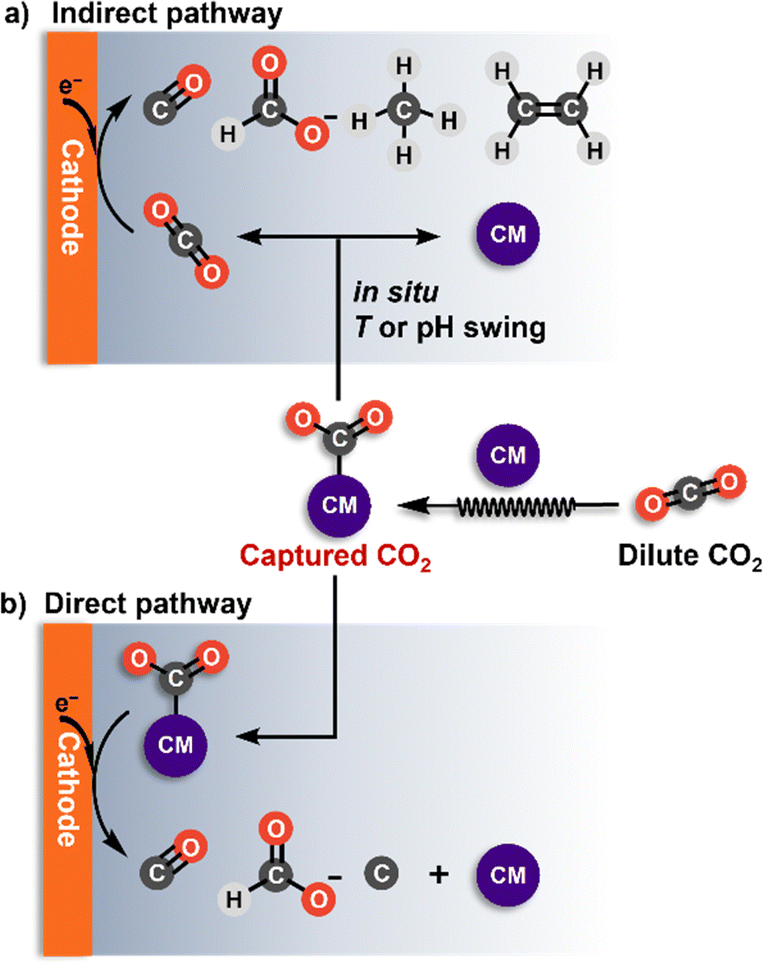 | ||
| Fig. 4 Illustration of the two pathways of electrochemically reactive CO2 capture: (a) Indirect with the captured CO2 converted back to CO2 at the electrode–electrolyte interface before its electrochemical reduction; (b) direct with the captured CO2 as a reactant in the electrochemical step. CM: capture medium. | ||
In the indirect conversion, the captured CO2 (such as bicarbonate or carbamate) dissociates, producing CO2in situ at the electrode–electrolyte interface (Fig. 4a). This dissociation process is typically triggered by either temperature or proton flux changes within the electrolyzer. Currently, most reported instances of electrochemical conversion of captured CO2 in aqueous solutions follow this indirect pathway (Table 1).72–74 Because of the in situ CO2 generation, the catalyst selection closely aligns with conventional electrochemical CO2 conversion, i.e., Ag and single-atom catalysts (SACs) facilitate CO production, Bi and Sn catalyze formate formation, while Cu-based catalysts are suitable for producing hydrocarbons and alcohols. The key aspect of the indirect pathway hinges on balancing the rate of in situ CO2 release with the rate of CO2 reduction at the electrode surface to achieve the conversion of the captured CO2 with high activity and selectivity. This requires precise design of the catalyst structures and properties, as well as optimization of the electrolyzer, electrolyte, and other operating conditions.
The second pathway is the direct conversion, which bypasses the formation of CO2 before the reduction process (Fig. 4b). Here, the captured CO2 (such as bicarbonate or carbamate) serves as the primary reactant, setting this approach apart from conventional electrochemical CO2 conversion. One significant advantage of focusing on (bi)carbonate and carbamate instead of CO2 is achieving much higher reactant concentrations. So far, only a few documented examples have utilized the direct pathway in aqueous bicarbonate and carbamate, primarily under high-temperature conditions.85,97,118 Similarly, in molten salts, the captured CO2 (as CO32−) can be directly converted, using the high temperature to overcome the energy barrier for reduction.
The direct conversion of bicarbonate or carbamate, particularly to C2+ products, presents a formidable challenge, with few studies reported. It has been observed that the direct pathway for carbonate conversion does not involve CO intermediates.118 As a result, this pathway cannot generate C2+ products, as these are primarily formed through CO–CO coupling. To effectively catalyze the conversion of bicarbonate or carbamate, the catalysts need to have a strong affinity for the reactant. Since heterogeneous electron transfer is an inner-sphere reaction, the proximity between the reactant and the electrode surface is crucial for the electrolysis reaction. Unlike charge-neutral CO2 molecules, bicarbonates and carbamates are anions that tend to migrate toward the anode, resulting in low concentrations at the cathode interface. Therefore, a positively charged catalyst surface may aid in attracting the anionic reactant through electrostatic interactions. Thus, careful tuning of the electronic configuration and the morphology of electrocatalysts, along with surface modification, is essential to promote efficient reactant adsorption. Additionally, a more comprehensive understanding of the mechanistic pathway for the direct conversion of bicarbonate or carbamate is needed to investigate the potential for C–C coupling in this system.
3.2. Production of CO
In the electrochemical conversion of captured CO2 into CO, silver nanomaterials are extensively utilized as catalysts, similar to their prominent role in the electrochemical reduction of CO2.72,75,119 Li et al. employed Ag as the electrocatalysts for the electroreduction of a carbonate electrolyte, achieving a CO faradaic efficiency (FECO) that decreased from 28% to 12% as the current density increased from 100 to 300 mA cm−2.72 In this process, the carbonate reacts with protons from the bipolar membrane (BPM) to generate CO2in situ near the electrode surface (eqn (21)), following the indirect pathway depicted in Fig. 4a. In another work, Li et al. discovered that pure Ag foil was ineffective in catalyzing the direct reduction of NH4HCO3, while a Br-modified Ag electrode exhibited higher efficiency with a significant FECO of 77.8% and a current density of 13.8 mA cm−2 at −0.6 V vs. RHE.83,120 They hypothesized that the Br modification could increase the number of active sites on the Ag surface, promoting the release of CO2 from NH4HCO3 and its subsequent reduction.Mezza et al. conducted a systematic study on the effect of Ag mass loading on the performance of bicarbonate electrolysis to CO (eqn (18)) through the indirect pathway. Their findings showed that as the Ag loading rose from 116 to 565 μg cm−2, FECO improved from 55% to 77%, while the partial current density of CO rose from 6 to 13 mA cm−2. Further increases in the Ag loading did not lead to additional improvement because a balance was reached between the availability of active sites and the electrode permeability.121 It is important to note that there is a trade-off between selectivity and activity of CO production under these conditions. The decrease in FECO at higher current densities is due to an imbalance between the utilization rate of the in situ generated CO2 and the local CO2 concentration on the catalyst surfaces.
| CO32− + 2H3O+ → CO2 + 3H2O | (21) |
Lee et al. proposed a direct pathway for using carbamate electrolyte as the reactant in electrochemical conversion (Fig. 4b). They achieved an impressive FECO of 72% at 50 mA cm−2 in a flow cell using a Ag electrocatalyst as the cathode in an electrolyte consisting of 30 wt% monoethanolamine with 2 M KCl at 60 °C (eqn (17)).85
SACs have emerged as promising candidates for the electrolytic conversion of captured CO2, due to their exceptional selectivity for CO, high activity, and maximum atom efficiency. Kim et al. demonstrated that a Ni–N/C SAC exhibited more selective production of CO (78.3%) compared to commercially available Ag (cAg) catalysts (38.3%) in CO2-captured monoethanolamine (Fig. 5a).88 They ascribed this enhanced performance to the much higher potential of zero charge (PZC) of Ni–N/C compared to cAg, as a positively shifted PZC can increase the surface cation charge density at the same applied potential in the reduction reaction. This, in turn, stabilizes the CO2− intermediate and enhances the CO2 reduction activity. This relationship was further confirmed by studying the cation sensitivity using additional catalysts (AuAg alloy, cPd, and cAu catalysts; Fig. 5b and c). Ni–N/C could maintain a FECO of up to 50% as the cation size increased from K+ to diethanolammonium (Fig. 5d–i), showing selective CO production in different CO2-captured media. In contrast, the FECO of cAg dropped to almost zero as the bulkiness of the amine increased.
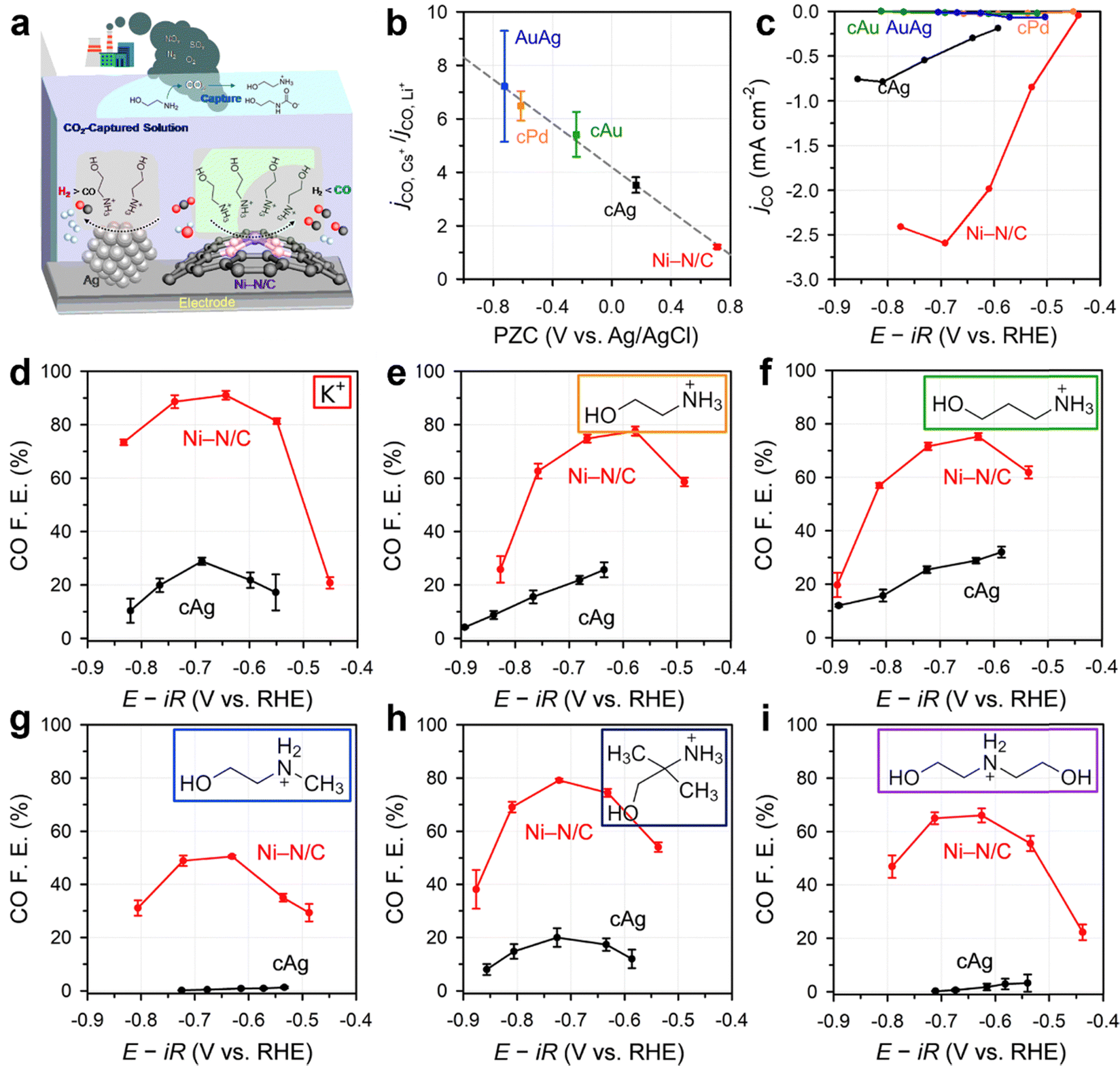 | ||
| Fig. 5 (a) Schematic illustration of the superior performance of Ni–N/C catalysts compared with Ag in electrochemical conversion of captured CO2 to CO. (b) and (c) Relationship between cation sensitivity of the catalysts and CO production performance: (b) normalized partial current density, jCO, at −1.7 V vs. NHE with respect to the PZC of the catalysts in 0.05 M carbonate electrolytes; (c) jCO of the catalysts for the electrolysis of CO2-absorbing 5 M monoethanolamine solution. FECO of Ni–N/C and cAg for electrolysis in CO2-absorbing (d) 1 M KHCO3, (e) 1 M monoethanolamine, (f) 1 M 3-amino-1-propanol, (g) 1 M 2-(methylamino)ethanol, (h) 1 M 2-amino-2-methyl-1-propanol, and (i) 1 M diethanolamine Ar-saturated solutions. Reproduced with permission from ref. 88. | ||
In addition, Yue et al. used a Ni SAC derived from a metal–organic framework (MOF) to boost the electrolytic bicarbonate conversion to CO, achieving a FECO of 67.2% at 100 mA cm−2.89 The abundant mesopores in the Ni SAC promoted the transport of the in situ released CO2 reactant. Cu electrodes have also demonstrated potential in the electrocatalytic conversion of carbamate into CO. Zhang and Kraatz showed that Cu electrodes significantly enhanced FECO to 58% compared to glassy carbon electrodes (2.3%) when using ethylenediamine as the capture media at −0.76 V vs. RHE.122 This differs substantially from numerous reports that typically show Cu-based materials catalyzing the conversion of gas-fed CO2 to C2+ products.123,124 Furthermore, the current density rose notably from 0.63 to 18.4 mA cm−2. While the morphology and crystalline properties of the Cu electrodes used were not specified, these factors are likely crucial in CO production.
3.3. Production of HCOO−
Hori and Suzuki were the first to report the electrocatalytic reduction of bicarbonate into formate at a mercury electrode at relatively low current densities (<1 mA cm−2).125 They suggested that the electrolytic reduction of HCO3− is a very slow process due to the slow dissociation of HCO3− to CO2 (reverse of eqn (2)). Min and Kanan employed a Pd electrocatalyst that achieved a formate faradaic efficiency (FEformate) of ∼54% at 6.0 mA cm−2 in aqueous bicarbonate solutions without requiring any CO2 input.126 They hypothesized that bicarbonate first decomposed into CO2, followed by the subsequent reduction of CO2 to formate (Fig. 4a, indirect pathway). In a flow cell equipped with a BPM, Bi catalysts demonstrated high efficiency in converting HCO3− to formate, achieving a FEformate of 64% at 100 mA cm−2 in a 3 M KHCO3 electrolyte.93 In this process, the proton flux facilitated by the BPM converted HCO3− to CO2 (eqn (22)), which was subsequently reduced to formate on the Bi surface. Similarly, electrodeposited Bi on a carbon paper substrate exhibited excellent performance in an ammonium bicarbonate (NH4HCO3)-fed electrolyzer, using in situ CO2 as the primary reactant at slightly elevated temperature (∼40 °C).94 Because NH4HCO3 (36 °C) has a much lower decomposition temperature than ethanolamine–CO2 (120 °C) and KHCO3 (150 °C), it can provide larger amounts of CO2 to the Bi electrode, resulting in higher current densities at a smaller energy input.| HCO3− + H3O+ → CO2 + 2H2O | (22) |
Pei et al. demonstrated that a dynamic Snδ+/Sn interface significantly enhanced the direct reduction of bicarbonate to formate, achieving an optimal partial current density of 121 mA cm−2 for formate with a FE of 83% achieved in a 3 M KHCO3 solution at 100 °C.97 Their study indicated that formate was generated directly from bicarbonate, rather than from CO2 produced by the dissociation of bicarbonate at high temperatures (Fig. 4b, direct pathway).
Ma et al. revealed that carbonate intermediates adsorbed onto a Cu electrode during the electrochemical CO2 reduction in a KHCO3 electrolyte over a potential range from −1.0 to 0.2 V vs. RHE (Fig. 6a).118 At potentials of −0.4 V vs. RHE and more negative, these intermediates were reduced to formate. Although the FEformate was quite low (0.61%), this finding demonstrated the potential for directly reducing carbonates using a modified Cu electrode. Pulse electrolysis experiments conducted under a N2 atmosphere with 0.05 M K2CO3 aqueous solution as the electrolyte corroborated the direct electroreduction of carbonate at the Cu surface (Fig. 6b).118In situ Raman spectroscopy at the applied potential of −0.05 V vs. RHE, confirmed the formation of a *CO32− intermediate (where * indicates it is bound to the surface). Upon switching the potential to −0.50 V vs. RHE, the surface-bound carbonate was reduced to formate, as detected by high-performance liquid chromatography. Isotope labeling confirmed that the carbonate was the carbon source of the produced formate. In contrast, constant potential electrolysis at −0.50 V vs. RHE under the same reaction conditions yielded no formate.
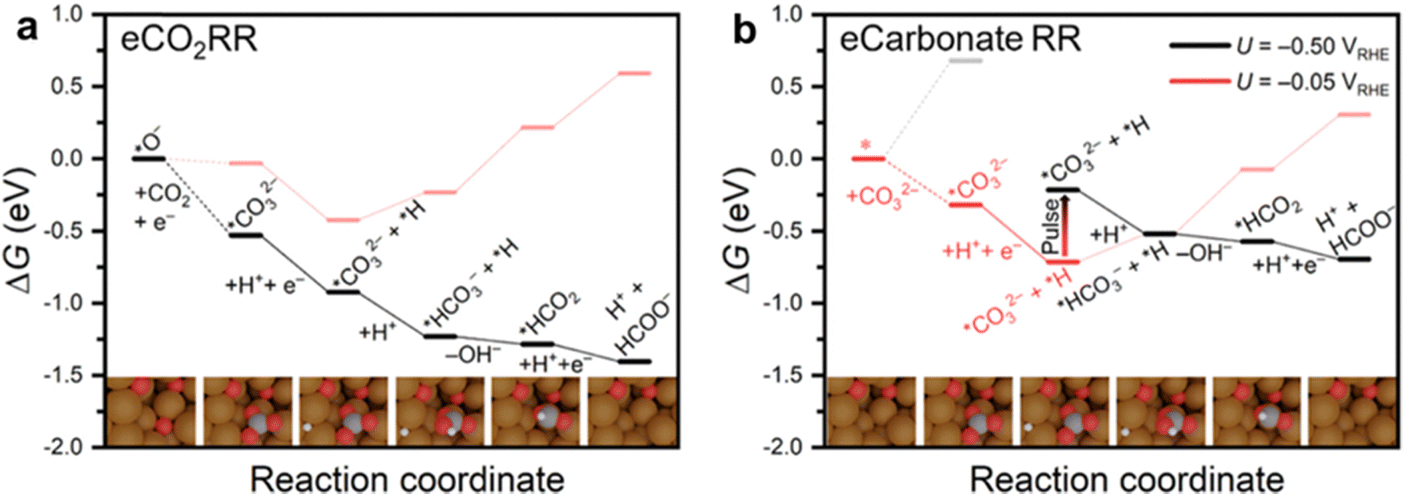 | ||
| Fig. 6 Energy profiles of (a) CO2 reduction by CO2 adsorption on an *O site at Cu and further reduction to formate at an applied potential of −0.5 V vs. RHE, and (b) CO32− reduction to formate at applied potentials of −0.05 V vs. RHE (red), −0.50 V vs. RHE (black), and with the potential pulsed from −0.05 to −0.50 V vs. RHE. Reproduced with permission from ref. 118. | ||
The carbonate reduction mechanism was modeled using density functional theory (DFT) calculations according to the two pathways shown in Fig. 6. The results indicated for the direct pathway that carbonate absorption at Cu sites is favorable at −0.05 V vs. RHE (Fig. 6b; first step of red lines). However, at this potential, the protonation of *CO32− to form *HCO3− is unfavorable (Fig. 6b; red lines). Upon switching the potential to −0.50 V vs. RHE, the formation of *HCO3− becomes feasible, along with the subsequent reaction steps leading to formate production (Fig. 6b; black lines). Notably, carbonate with ΔGads = 0.68 eV cannot be adsorbed at −0.50 V vs. RHE (Fig. 6b; gray lines) due to increased repulsion between CO32− and the negatively charged electrode. This explains the lack of formate production during constant potential electrolysis experiments.
For the indirect pathway, which involves the release of CO2 before electrochemical reduction (Fig. 6a), the calculations show that a *CO32− intermediate forms through the reaction of CO2 with a residual *O atom on the Cu surface. As expected, this reaction pathway toward formate production becomes energetically favorable at −0.50 V vs. RHE (Fig. 6a; black lines). Note that this pathway is stoichiometric in reactants and cannot continue once the *O atoms are consumed. Overall, the experimental and computational findings demonstrate that carbonate can be directly reduced at electrode surfaces, providing a possible method to activate the otherwise inert CO2-adduct.
3.4. Production of CH4
Methane is a highly desirable product due to its superior heat value of 55.5 MJ kg−1, significantly greater than that of CO (10.2 MJ kg−1) and methanol (22.7 MJ kg−1).127 Lees et al. reported that a Cu catalyst modified with cetyltrimethylammonium bromide (CTAB) facilitated CH4 formation at a partial current density of 120 ± 10 mA cm−2 and with a yield of 34 ± 7% during bicarbonate electrolysis, where the in situ generated CO2 served as the actual reactant.98 A one-dimensional continuum model of the cathode compartment suggested that H+ from the BPM in the bicarbonate electrochemical reactor would neutralize hydroxide generated at the cathode surface, favoring CH4 over multicarbon products.In the electrochemical conversion of captured CO2, CH4 is predominantly generated at elevated temperatures, with molten salts as the capture medium.100 When molten carbonate salts are used, the type and distribution of products are largely determined by the properties of these salts, as well as the electrolysis conditions, including temperature and applied potential. In this setup, cathode materials are essential as they act as electron-transfer agents, which can greatly influence the overpotential and efficiency of the reactions. For effective electrolysis in molten salts, the cathode material must fulfill two criteria: (1) resistant to corrosion by the highly alkaline molten salts during high-temperature electrolysis. (2) Unable to alloy with Li, Na, or K at high temperatures in molten salts, as this would compromise the electrode structure.100
Wu et al. reported on the electrolysis of CO2 and H2O employing Fe and Ni electrodes as the cathode and anode, respectively, in an alkali carbonate/LiOH electrolyte at 600 °C.128 The electrolysis products comprised 64.9% CH4, 34.8% H2, and 0.3% C2+. Bai et al. systematically compared the overpotential and CH4 yield of different cathode materials, including Fe, stainless steel (SS304), Ti, and cupronickel sheets, in molten Li1.427Na0.359K0.214CO3 ∼ 0.15LiOH at 650 °C.101 Their study found that Fe, cupronickel, and SS304 exhibited similar cathodic overpotentials, all higher than that of Ti. Notably, the highest CH4 yield (33.3%) was achieved with the cupronickel cathode, exceeding the yield from the standard Fe cathode (25.4%) despite its higher activity. This enhanced CH4 production from the cupronickel cathode is likely due to the synergistic electronic effects of Cu and Ni atoms, which improve the adsorption of reaction intermediates for CH4 production during electrolysis.
3.5. Production of C2+
The electrochemical conversion of captured CO2 into C2+ products offers greater potential in terms of market size and value compared to C1 products. However, this area of research has not been as extensively explored. Lee et al. applied Cu/Ag bilayer electrodes with engineered bilayer cation- and anion-conducting ionomers to convert bicarbonate into C2+ products such as acetate, ethylene, ethanol, and propanol.102 The in situ generated CO2 was first converted to CO on the Ag layer (Fig. 4a, indirect pathway), which was then efficiently transformed into C2+ products on the Cu layer, due to the locally concentrated CO. They achieved a maximum FE of 41.6 ± 0.4% for C2+ products at a current density of 100 mA cm−2. Prajapati et al. developed a fully integrated system for CO2 capture and reduction using Cu mesh catalysts, achieving a steady-state CO2 reduction FE of 57% and a high FE of 40% for C2H4 at a current density of 200 mA cm−2 (see Section 5.2).129Recently, Song et al. demonstrated the direct conversion of carbonate solutions to C2H4 using a customized Cu–Ag catalyst in a BPM-MEA (membrane electrode assembly) setup. Although the carbonate electrolysis resulted in a relatively low FE of ∼10% for C2H4 production, the Cu–Ag electrode configuration effectively achieved nearly zero CO2 concentration in the outlet stream. Increasing the operating temperature to 50 °C further enhanced C2H4 production, with a higher partial current density of 18–20 mA cm−2 compared with 10 mA cm−2 at room temperature.104
3.6. Production of carbon materials
In addition to the common products from CO2 reduction, valuable carbon materials, such as graphene and nanotubes, can be obtained in the electrochemical conversion of molten carbonate salt. The cathode's function as a substrate for carbon deposition affects the nucleation and growth of these carbon materials.130 For example, a Zn galvanized steel cathode surface was shown to yield a high proportion of carbon nanofibers (>80%), a result not achievable with a 316 stainless steel cathode.131Hu et al. successfully produced graphene using stainless steel as the cathode material in molten salt electrolysis.132 They proposed that active Fe atoms on the cathode surface catalyze carbon growth, leading to the formation of Fe3C as a key intermediate product. Subsequently, active Fe atoms are inserted into the interlayer of the graphene sheets, enabling continuous layer-by-layer growth through micro-explosion reactions in the interlayer space caused by the intense generation of CO. Moreover, different cathode materials were found to influence the structure of the carbon products, with Cu rods resulting in ball-structured graphene and Ni cathodes catalyzing the formation of flat graphene sheets.
Pint et al. demonstrated the electrochemical synthesis of carbon nanotubes from the capture and conversion of ambient CO2 using Fe as the cathode and further revealed a correlation between the thickness of the Fe metal layer and the diameter of the carbon nanotubes.133 This highlights the importance of active metal atoms on the cathode as nucleation sites that catalyze the growth of carbon films.
3.7. Designing catalysts with CO2 capture capability
Recently, a growing body of research has focused on using catalysts capable of capturing CO2. In these studies, the capture and conversion of CO2 are integrated within the catalyst itself, eliminating the need to rely on the electrolyte. For example, Liu et al. demonstrated this by depositing amine-linked covalent organic frameworks (COFs) onto a flat Ag electrode.134 They created a molecularly defined interface for the electrochemical reduction of CO2 to CO, achieving significantly higher efficiency and selectivity than a bare Ag electrode. This improved performance was attributed to the synergistic interaction between the COFs and the Ag electrode interface. The porous nature of the COFs facilitated CO2 diffusion to the electrode surface, while the amine functional groups near the electrode surface boosted the CO2 conversion efficiency by promoting the formation of a carbamate intermediate. Similar effects were observed in another study, where Liu et al. coated Ag nanoparticles with a thin 2 nm layer of 4-aminobutylphosphonic acid confined in poly(vinylpyrrolidone).135Yan et al. used redox-active 2-amino-5-mercapto-1,3,4-thiadiazole (AMT) functionalized gold nanoparticles to achieve electrochemical carbon capture and conversion from flue gas.136 The AMT ligand not only acts as a selective capture agent to concentrate CO2 near the active site but also serves as a protective layer to inhibit O2 reduction. As a result, a maximum FECO of 80.2% at −0.45 V vs. RHE in an H-type cell and 66.0% at a voltage of 2.7 V in a full cell was achieved in simulated flue gas (15% CO2, 4% O2, balanced with N2). Notably, a microporous conductive Bi-based MOF (BiHHTP, HHTP = 2,3,6,7,10,11-hexahydroxytriphenylene) was recently reported to efficiently capture CO2 from dilute sources under high humidity and catalyze the electroreduction of the adsorbed CO2 into formic acid with a high current density of 80 mA cm−2 and a FE of 90% at a low cell voltage of 2.6 V.137 This high performance was attributed to the CO2 capture capability of the micropores on BiHHTP and the lower Gibbs free energy of formation for the key intermediate *OCHO on the exposed Bi sites.
Generally, amine-based molecules135,138,139 and porous framework materials140–142 are employed as agents for capturing CO2. The immobilization of amine groups onto the catalysts will enable the catalysts to concentrate CO2 molecules near the active sites, thereby enhancing the electrochemical conversion of CO2. Furthermore, functional groups such as amines help stabilize key intermediates during CO2 reduction, improving selectivity for specific products. Using catalysts with CO2 capture capabilities is particularly valuable when dealing with flue gas, as it significantly suppresses the competing reduction of O2.
4. Reactor and system optimization
Besides the capture medium and the catalyst, several other factors influence the outcome of the electrochemical conversion of captured CO2, including the electrolyzer design, electrode configuration, ion exchange membranes, electrolyte with additives, and reaction conditions. This section will explore these aspects in detail.4.1. Batch cell versus flow reactor
The electrochemical conversion of captured CO2 can be conducted in a batch or flow process.143 The conversion of molten carbonate has typically been carried out in an undivided cell equipped with heating elements under batch conditions, as high temperature is required for this process (Fig. 7a). At elevated temperatures, the electrolysis of molten carbonate can typically achieve a current density of 100–200 mA cm−2 and a FE of 60–90% for carbonaceous products.108,144 However, for the conversion of captured CO2 in aqueous solution (such as bicarbonate and carbamate) at room temperature, both batch (e.g., H-cell) and flow (e.g., MEA) reactors have been used. Nonetheless, the reaction process and outcomes vary significantly depending on the type of reactor employed. | ||
| Fig. 7 Three types of reactors used for the electrochemical conversion of captured CO2: (a) an undivided cell for molten carbonate electrolysis at high temperatures, (b) an H-cell, and (c) an MEA for electrolysis of aqueous bicarbonate and carbamate solutions at room temperature. | ||
In an H-cell, the potential can be easily adjusted to control the product distribution, but this setup typically achieves low current density and FE (Fig. 7b). For instance, Hu et al. carried out electrolysis of a KHCO3 solution in an H-type cell using a hybrid catalyst composed of cobalt porphyrin and carbon nanotubes, achieving a current density of a few mA cm−2 and FECO of 18%.145 This performance is much lower than that of gas-fed CO2 conversion using the same H-cell with the same catalyst and is also significantly poorer than that achieved in an MEA setup. Similarly, Chen et al. conducted the electrochemical reduction of carbamate formed through CO2 capture by 30% monoethanolamine solution in an H-cell using various smooth or porous metal electrodes, such as In, Sn, Bi, Pb, Pd, Ag, Cu, and Zn.146 The highest FECO and FEHCOO− reported were 39% and 45%, respectively, with current densities below 50 mA cm−2. These studies indicate that the electrochemical conversion performance of aqueous bicarbonate and carbamate solutions in an H-cell reactor is relatively low. This is due to the very low concentration of in situ liberated CO2, produced only from the dissociation of RNHCOO− or HCO3− (reverse of eqn (1) and (2)), and the mass transport limitation of the liberated CO2 in the H-cell.125 The liberated CO2 must diffuse through the reaction layer to be reduced at the electrode surface.146
The current density and FE can be significantly enhanced when converting captured CO2 in flow reactors, such as MEA electrolyzers (Fig. 7c). This enhancement is due to the specific configuration within the MEA electrolyzers, which includes the CO2-captured electrolyte, catalyst layer, and membrane. Besides the dissociation of RNHCOO− or HCO3−, a much higher concentration of CO2 can be generated near the catalyst layer due to the reaction of RNHCOO− or HCO3− with H+ produced from the heterolytic dissociation of water at the BMP. As a result, most studies on captured CO2 conversion have been conducted using MEA electrolyzers, achieving significantly higher FE up to 90% and larger current density up to 200 mA cm−2 in some cases (Table 1).90,91,98 These performance metrics are comparable to those achieved with gas-fed CO2 conversion and are suitable for meeting the requirements of industrial applications.147,148
In summary, conducting the conversion of aqueous bicarbonate/carbamate solutions in an H-cell offers high flexibility and quick startup, but it often results in low overall efficiency. In contrast, MEA electrolyzers operate with higher efficiency, leading to increased production rates, which makes them more advantageous for large-scale applications. The next section will further explore how the key components of MEA electrolyzers impact the performance of the electrochemical conversion of bicarbonate/carbamate solutions.
4.2. Electrode configuration
In the electrochemical conversion of gas-fed CO2, gas diffusion electrodes (GDEs) are required to achieve a large current density.149 Increasing the hydrophobicity of GDEs can enhance performance, suppress the flooding of the electrodes, and thus maintain stability for CO2 electrolysis over a longer period.150,151 However, the electrodes for the electrolysis of captured CO2 differ significantly from those typically employed for gaseous CO2 electrolysis because the source of CO2 is different. In general, a certain level of hydrophilicity is needed for these electrodes to ensure adequate contact between the catalyst on the electrode and the CO2-captured solution, whether to facilitate the direct conversion of the CO2-captured solution or reduction of the in situ generated CO2.Lees et al. systematically investigated the impact of electrode structure on the electrolysis of bicarbonate solutions using a Ag catalyst.76 They found that adding hydrophobic polytetrafluoroethylene (PTFE), a microporous layer (MPL), or both, negatively affected bicarbonate electrolysis, significantly reducing FECO. The hydrophobic PTFE and MPL hinder the transport of solvated HCO3− ions through the GDE, thereby decreasing the rate of in situ CO2 generation at the electrode via the reaction of HCO3− with H+ from the bipolar membrane (eqn (22)). They also examined the effect of Nafion loading in the catalyst layer and observed that FECO initially increased to a plateau of ∼58% and then decreased as the Nafion loading increased at a current density of 100 mA cm−2. The optimal Nafion loading was found to be 4.0 wt%. This behavior is likely due to variations in the catalyst layer morphology; lower Nafion loading leads to poor adhesion of the Ag catalyst to the electrode, while higher loading leads to a densified catalyst layer that blocks the pores of the gas diffusion layer. Similar behavior has been observed in the electrochemical conversion of gas-fed CO2.152 By removing the hydrophobic components of the GDE and optimizing the catalyst coverage, optimal performance for bicarbonate electrolysis was achieved, with a FECO of 82% at 100 mA cm−2.
Based on this understanding, the same group developed a free-standing porous Ag electrode, which demonstrated higher hydrophilicity than the commonly used GDE and could be more easily integrated into commercial electrolyzers.79 As a result, the free-standing Ag electrode achieved a respectable FECO of 59% at 100 mA cm−2 under ambient pressure for converting 3 M KHCO3.
Later, Lee et al. successfully modeled the generation, diffusion, and consumption of chemical species, identifying an optimal electrode architecture for the electrochemical conversion of carbonate solution to C2+ products over a Cu catalyst.103 Their modeling results showed that the spacing distance between the cation exchange layer (CEL) and the catalyst layer (CL) influenced the concentrations of CO32−, H+, in situ generated CO2(g), and CO2(aq.) in the carbonate solution (Fig. 8). When the CEL and the CL were closely spaced, the local pH at the CEL decreased to no more than 10, as the diffusion of CO32− and OH− could easily neutralize the H+ coming from the CEL. This prevented the in situ generation of CO2(g) or products during electrolysis. However, when the spacing distance between the CEL and the CL was in the range of 100–300 μm, optimal conditions for converting carbonate to C2+ products could be achieved, with a low pH (<4) at the CEL, promoting the in situ generation of a high concentration of CO2(g) (>4 vol%) at the CL. Despite this, the pH remained >13 at the CL, as OH− was continuously produced by the conversion of the in situ generated CO2(g) from carbonate. The high local pH favored the C–C coupling to form C2+ products over C1 products. Finally, increasing the spacing distance to >540 μm decreased the concentration of CO2(g) and hindered the carbonate conversion due to the increased likelihood of CO2 being recaptured over the extended layer distance.
 | ||
| Fig. 8 (a) MEA electrolyzer for aqueous carbonate conversion, where the cathode and anode are separated by an interposer. (b) Schematic of the cathode configuration, including the cation-exchange layer (CEL), the interposer, catalyst layer, and carbon paper. (c) CO2(g) volume fraction for different spacing (LI) conditions, i.e., 0, 64, 135, and 540 μm at current densities of 200, 250, 300, and 350 mA cm−2 in 1.5 M K2CO3 electrolyte. (d) FE of C2+ products in carbonate electrolysis using a Cu electrocatalyst in 1.5 M K2CO3 electrolyte with various interposer thicknesses from 0–540 μm. Reproduced with permission from ref. 103. | ||
Based on this modeling, the authors used a porous hydrophilic mixed cellulose ester as an interposer to create a well-defined spacing between the CEL and the CL. The electrolysis of carbonate solutions at 250 mA cm−2 over a Cu catalyst using an interposer thickness of 130–270 μm increased FEC2+ to 40%. This was three times larger than with an interposer thickness of 60 μm (FEC2+ = 14%) and nearly twice as high as observed in the 540 μm case (FEC2+ = 25%). These results highlight the critical role of electrode configuration design in enhancing the selective electrochemical conversion of carbonate solution to C2+ products.
4.3. Ion exchange membrane
Ion exchange membranes serve as separators and regulate ion transport between the cathodic and anodic chambers of the electrolyzer. The choice of ion exchange membrane has a significant impact on the electrolysis process (Fig. 9). BPMs are widely used in the electrochemical conversion of bicarbonate solutions because HCO3− can react with the H+ produced by the BPM to generate in situ CO2 at the BPM–electrode interface (eqn (22)). The in situ generated CO2 subsequently receives electrons from the electrode and undergoes reduction to produce various value-added products such as CO, formate, and C2H4. Using BPMs for bicarbonate conversion offers several benefits, including rapid water dissociation, minimized crossover of generated products, and extended operational lifespan.153 However, employing BPMs requires an additional potential of 0.83 V for H+ generation via water dissociation under standard conditions, resulting in a large overpotential, increased energy consumption, and reduced energy efficiency.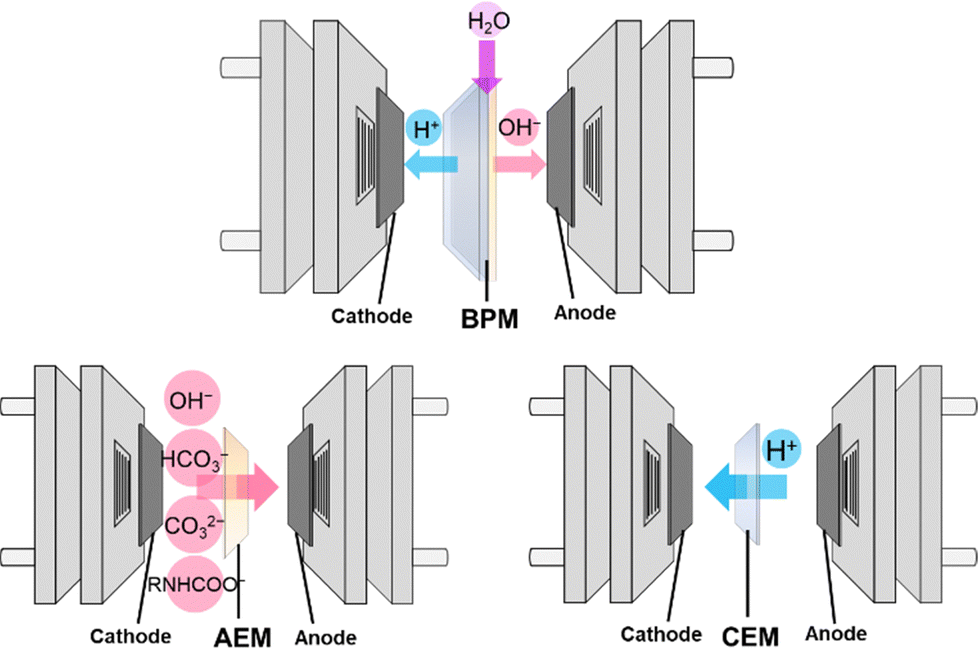 | ||
| Fig. 9 Schematic illustration of three typical types of ion exchange membranes used in electrochemical conversion of captured CO2. | ||
Replacing the BPM with an anion exchange membrane (AEM) or a cation exchange membrane (CEM) can lower the cell voltage but may negatively impact the conversion efficiency. Li et al. found that an electrolysis cell with a BPM showed a higher FEformate (+35%) for KHCO3 conversion than a corresponding AEM cell, which was attributed to the higher local CO2 concentration at the electrode in the BPM system.93 In the AEM cell, CO2 was formed only through the bicarbonate dissociation equilibrium (reverse of eqn (2)). However, the trend reversed when using a different bicarbonate (NH4HCO3) solution, where replacing the BPM with the AEM resulted in an increased FEformate.94 This was explained by the favorable microenvironment created by NH4+, which would suppress the HER at the electrode–membrane interface. It is important to note that formate crossover could be observed in the AEM cell, as AEMs are designed for anion exchange.154
Utilizing a CEM can also create an acidic environment at the cathode–membrane interface, as protons are generated from water oxidation (i.e., the oxygen evolution reaction (OER)) or the acidic electrolyte in the anodic chamber permeating through the membrane.103 These protons can react with the captured CO2 (in the form of bicarbonate or carbamate) to produce high local concentrations of free CO2 near the catalyst layer, thereby enhancing the electrolysis efficiency. To ensure effective proton transport through the CEM, it is crucial to maintain a neutral or acidic electrolyte in the anodic chamber. However, this condition makes the OER more challenging, requiring a larger overpotential.155 To address this issue, Zhang et al. conducted the hydrogen oxidation reaction at the anode instead of the conventional OER during electrolysis of 3 M KHCO3 and used a CEM in place of the BPM. This approach allowed them to produce CO at a high partial current density of 220 mA cm−2 at a cell voltage of only 2.3 V.80
4.4. Electrolyte with additives
As highlighted in the preceding section, the distance between the reactant and the electrode has a crucial impact on the reaction outcomes. Consequently, using a CO2-captured amine solution as the electrolyte presents efficiency challenges, unlike converting the bicarbonate solution. This disparity arises because the bulky carbamate (RNHCOO−) and ammonium (RNH3+) ions impede mass transport at the electrochemical double layer (EDL).To address this issue, Khurram et al. investigated the introduction of various electrolyte salts, including LiPF6, LiClO4, NaClO4, KClO4, and TBAClO4, into CO2-captured amine solutions in dimethyl sulfoxide.156 They observed that while the anions had a minor impact, the cations significantly influenced the speciation of amine–CO2 adducts. Specifically, pairing the K+ cation with RNHCOO− could enhance the rates of electrochemical reactions due to improved K+ transfer from the bulk solution to the reaction site. Therefore, the judicious selection of strong electrolyte salt that facilitates rapid cation transfer is essential for achieving high efficiency in converting amine-captured CO2.
Later, Lee et al. demonstrated that introducing alkali metal ions into the aqueous CO2-captured amine solution could significantly improve the electrochemical conversion of the amine–CO2 adduct (carbamate).85 They proposed that the alkali cations (e.g., K+) modified the EDL to facilitate the heterogeneous electron transfer from the catalyst to the carbamate (Fig. 10a). Using this approach, efficient conversion of the carbamate to CO was achieved with a Ag catalyst, resulting in a FECO = 72% at 50 mA cm−2.
 | ||
| Fig. 10 Effect of (a) strong electrolyte KCl (reproduced with permission from ref. 85), (b) surfactant (reproduced with permission from ref. 146), (c) temperature (reproduced with permission from ref. 94), and (d) pressure (reproduced with permission from ref. 79) on the conversion of aqueous carbamate or bicarbonate solution. | ||
Surfactants exert a large influence on the efficiency of gas-fed CO2 conversion,157,158 as well as the electrochemical conversion of captured CO2. Chen et al. reported that adding the cationic surfactant, CTAB, suppressed the competing HER, thereby greatly enhancing the conversion of carbamate derived from the CO2 capture by monoethanolamine (Fig. 10b).146 This enhancement was evident in the increase of FEformate from 2.4% to 45.4% over an In electrode. In contrast, other surfactants, such as sodium dodecylsulfate (anionic) and TritonX-100 (non-ionic), showed a negligible effect, indicating that the performance of the carbamate conversion depends on the type of surfactant added. Ahmad et al. found that adding CTAB to the CO2-captured 2-amino-2-methyl-1-propanol solution increased the FECO over a Cl-modified polycrystalline Ag electrode due to a more compact EDL.86 Similarly, for the conversion of a bicarbonate solution, Lees et al. observed that the presence of just 3 mM CTAB increased the CO2 reduction current density from 11 to 162 mA cm−2 and the FECH4 from 0 to 27% at an applied current density of 400 mA cm−2.98 This performance improvement was attributed to reduced oxide coverage on the Cu electrode during electrolysis when CTAB was added.
4.5. Operation parameters
The concentration of the captured CO2 significantly impacts the electrochemical performance of the system. For bicarbonate conversion, higher concentrations lead to higher FE of carbonaceous products.93 Increasing the bicarbonate concentration accelerates the rate at which HCO3− dissociates and reacts with H+ from the membrane to liberate CO2. Thus, a high concentration (3 M, near saturation) is typically used for bicarbonate conversion.Temperature can influence the performance of bicarbonate conversion in several ways. First, increasing the temperature of the electrolyte is expected to promote CO2 generation by shifting the bicarbonate equilibrium toward CO2 (reverse of eqn (2)). Moreover, a temperature rise causes an increase in pH due to the formation of OH−, which suppresses the HER. Raising the temperature also enhances mass transport, thereby accelerating the electrochemical process. This effect has been observed in numerous studies, resulting in improved FE for forming carbonaceous products (Fig. 10c).79,94 Similarly, the effect of the temperature was exploited in the conversion of CO2-captured amine (carbamate) solutions. Pérez-Gallent et al. reported that the reaction rate of the conversion of CO2-captured 2-amino-2-methyl-1-propanol in a propylene carbonate solution was significantly enhanced when the temperature was raised from 15 to 75 °C.87 The increase in reaction rate was attributed to the much faster liberation of CO2 at elevated temperatures, which was eight times higher at 75 °C than at 15 °C.
The effect of temperature becomes more complex in the electrochemical conversion of molten carbonate salts. Deng et al. reported that the electrolysis of CaCO3-containing molten LiCl–KCl over a Ni electrode produced carbon materials with various shapes and morphologies.159 At 450 °C, micron-sized hollow carbon spheres and ultrathin carbon sheets were the major products, depending on the applied cell voltage. As the temperature increased to 550 and 650 °C, a range of other carbon materials, such as quasi-spherical carbon particles, coral-like carbon, and carbon nanofibers, were formed. In a subsequent study, it was demonstrated that raising the operational temperature led to an increase in current density or a reduction in cell voltage for the conversion of CaCO3 in the LiCl–KCl melt.160 These conditions facilitated the dissolution of both the reactant CaCO3 and the product CaO in the melt, thereby preventing the accumulation of solid CaO on the electrode. As a result, the process exhibited greater durability and energy efficiency. The energy consumption for producing 1 kg of carbon was calculated to be 16.3 kW h kg−1 at 650 °C, compared with 31.4 kW h kg−1 at 450 °C.
Zhang et al. demonstrated that pressure, in addition to temperature, affects the performance of bicarbonate conversion (Fig. 10d).79 When the inlet pressure of the electrolyzer was increased from 1 to 4 atm, FECO increased from 55% to 95%, and the cathodic energy efficiency from 22% to 34% at a current density of 100 mA cm−2. At 4 atm pressure, an impressive current density of 400 mA cm−2 for bicarbonate conversion with FECO > 55% could be achieved. This performance enhancement was attributed to the kinetically improved supply of CO2 to the catalyst at elevated pressures.
The authors also explored how the electrolyte flow rate influenced the performance of bicarbonate conversion. They increased the flow rate from 30 to 100 mL min−1, which led to an increase in in situ generated CO2 and, consequently, a higher FECO. This effect was attributed to the enhanced convective mass transport of HCO3−. Building on this finding, they modified the flow plate geometry to further enhance convective mass transport. Three types of flow plates were tested, i.e. interdigitated, serpentine, and parallel (Fig. 11). Convection was the primary mode of mass transport in the interdigitated flow plate, while diffusion was dominant in the parallel and serpentine flow plates. As a result, the electrolyzer with the interdigitated flow pattern exhibited higher FECO (≈ 69% at 100 mA cm−2) compared to the other two. These results demonstrate that enhancing the convective mass transport can increase FECO significantly for converting captured CO2.
 | ||
| Fig. 11 (a) Interdigitated, serpentine, and parallel cathodic flow plate designs used in bicarbonate electrolysis. (b) FECO as a function of current density for interdigitated, serpentine, and parallel flow plates tested in a bicarbonate electrolyzer equipped with porous silver electrodes. Reproduced with permission from ref. 79. | ||
5. Practical integration of CO2 capture and conversion
In this section, we present practical examples that fully integrate the two processes of CO2 capture and electrochemical conversion (Fig. 1, Route 3). We begin by examining technologies based on different CO2 sources, including air,161,162 flue gas,163,164 and ocean water,165,166 as these sources significantly influence the outcome of the integration due to differences in CO2 concentration and purity.5.1. Conversion of captured CO2 from various sources
If conventional energy sources such as coal were used to power a DAC system with 5% efficiency, there would be no net CO2 capture, as producing the required 400 kJ to capture one mol of CO2 would generate 2.5 moles of CO2. This is also true for natural gas, the least carbon-intensive fossil fuel, which emits one mol of CO2 while generating 396 kJ of energy. Therefore, CO2-free power sources such as nuclear, wind, solar, and hydro must drive air capture systems. For example, the energy requirement of 400 kJ mol-CO2−1 is equivalent to ∼2500 kW h t-CO2−1, which could be provided by a large 3 MW wind turbine operating for 1 h.
Integrating the CO2 capture and electrochemical conversion processes is challenging because the rate of capture is usually much lower than the achievable rate of conversion. A study exemplifying this issue involved absorbing atmospheric CO2 over 8.5 h using an alkaline solution (1 M KOH), resulting in the formation of a bicarbonate/carbonate mixture (Fig. 12).84 This solution was then used as the catholyte in a zero-gap flow electrolyzer to produce industrially significant carbon products. At a current density of 50 mA cm−2, a FEformate of 16% (using SnO2/C as the electrocatalyst) and FECO of 13% (using Ag/C) were achieved. To improve electrochemical performance, it would be necessary to capture higher concentrations of CO2 by increasing the base concentration and/or extending the absorption period. From an economic perspective, this would reduce the attractiveness of integrating the two technologies.
 | ||
| Fig. 12 Integration of atmospheric CO2 capture and the subsequent electrochemical conversion of the captured CO2: (a) diagram of the CO2 capture setup and (b) the electrolytic cell for converting captured CO2. Reproduced with permission from ref. 84. | ||
Recently, Almajed et al. evaluated the feasibility of directly integrating DAC with (bi)carbonate electrolysis using KOH and K2CO3 solutions as the capture media (Fig. 13a).168 They found that the presence of CO32− alongside HCO3− in the effluent from the air contactor significantly reduced electrolysis performance due to incomplete CO2 capture, ultimately decreasing the CO2 capture fraction from 78% to ≤1%. As a result, they estimated that air contractors would need to be 5–14 times larger than those typically required for DAC to produce suitable effluents for (bi)carbonate electrolysis, leading to unfavorable process economics. Furthermore, they demonstrated that regenerating the capture media after the electrolysis was insufficient for effective CO2 recapture. This incompatibility between DAC and (bi)carbonate electrolysis was attributed to the mismatched pH values between the air contactor outlet/electrolyzer inlet and the air contactor inlet/electrolyzer outlet. To resolve this issue, the authors suggested acidifying the air contactor effluent and basifying the electrolyzer effluent by incorporating an electrodialysis unit or directly feeding acidic/basic streams to the respective effluents (Fig. 13b). However, this approach would inevitably increase both capital and operational costs.
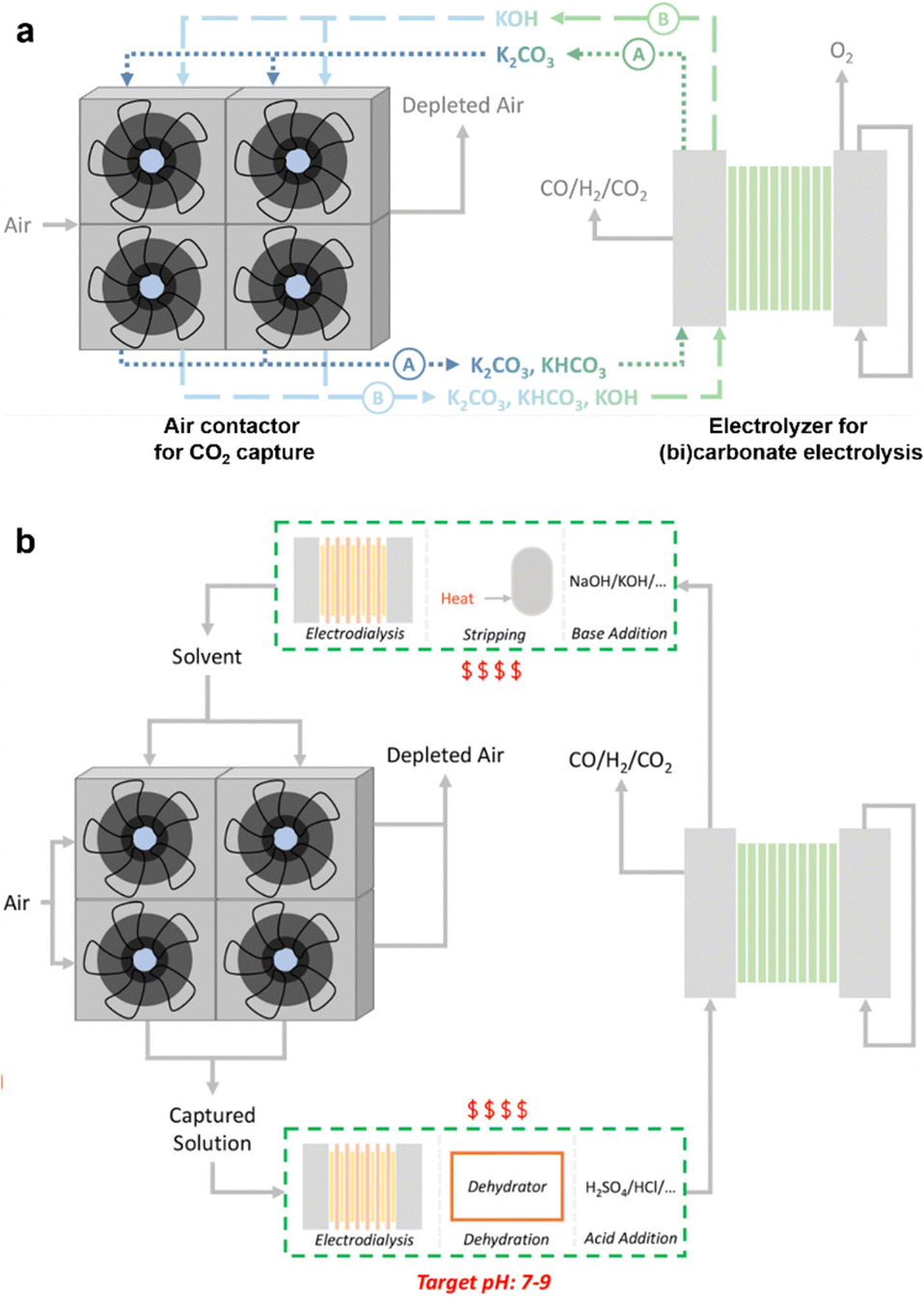 | ||
| Fig. 13 Schematic of the integration route proposed in the literature, with the air contactors on the left and the electrolyzer stacks on the right. (a) The two pathways for CO2 capture from the air are (A) K2CO3-based and (B) KOH-based. (b) Potential solutions to address the different pH requirements of the capture and conversion processes are shown inside the green dashed boxes at the top and bottom. Reproduced with permission from ref. 168. | ||
The third example concerns molten metal oxides, such as Li2O, which have shown potential for directly sequestering CO2 from the air at elevated temperatures, resulting in the formation of carbonate salts.131 These carbonates can then undergo electrochemical reduction, producing solid carbon on and attached to the cathode and oxygen at the anode. This process has been demonstrated on a laboratory scale to successfully convert ambient air into carbon and oxygen, producing, e.g., 10 g h−1 of carbon nanofibers at 100 A.
These examples highlight the feasibility of integrating electrochemical conversion with CO2 capture from the air. However, the efficiency of the integration is currently not satisfactory due to the slow CO2 capture. One approach to overcome this limitation is to use a significantly larger amount of capture agent while vigorously feeding air into the solution under stirring. Alternatively, existing methods could be suitable for small, decentralized units with minimal time constraints. Integrating electrochemical conversion with DAC technology provides the flexibility to be deployed anywhere, making it ideal for reducing emissions from dispersed sources when combined with renewable energy for power generation. Nevertheless, the widespread implementation of DAC infrastructure presents challenges, including substantial investment and land use. Furthermore, DAC technology is relatively new and less mature, which may pose hurdles regarding scalability and cost-effectiveness, at least in the short term.
In most cases, the capture agents and methodologies developed for DAC can also be applied to flue gas and vice versa. This is similarly true for the integrated CO2 capture and conversion approach. However, significant amounts of gas impurities in flue gas, such as NOx and SOx, can cause considerable inefficiencies and increase cost, as they may interfere with both the capture and the conversion processes. For example, removing SO2 from flue gas by converting it into CaSO3 requires a substantial energy input of 380 kJ mol−1.169 Removing NOx involves its selective reduction to N2 over a supported vanadia catalyst using ammonia as a reductant. Although this chemical reaction is exergonic, ∼500 kJ of energy is required per mol of NOx removed in commercial systems.169
Most studies on the electrochemical conversion of flue gas neglect these impurities by using simulated flue gas, which consists of a variable mixture of CO2 and usually N2. In general, the risk of interference from acidic impurities like SO2 and NO2 is minimal when CO2 is captured using alkaline (bi)carbonate solutions, as these can neutralize such impurities.36,170,171 Among the NOx gases, the relatively inert NO is usually the predominant component, requiring oxidation to NO2 using, e.g., hydrogen peroxide for effective removal.170 Unfortunately, even small amounts of impurities can negatively impact catalyst performance and, consequently, the conversion process. Therefore, several studies on gas-fed CO2 have thoroughly investigated this issue, with findings highly relevant for integrated processes, especially those involving in situ generated CO2.
Ko et al. examined the influence of NOx on Cu, Ag, and Sn catalysts by introducing 8300 ppm NO into a CO2 stream.172 For Ag and Sn catalysts, NO reduction reactions reduced FE by 35%. Similarly, introducing 10000 ppm SOx led to a decrease in FE by 25–40% when targeting CO or formate.173 In both cases, the catalytic efficiency was restored upon reintroducing a pure CO2 stream into the reactor. For the Cu catalyst, a significant shift in selectivity toward formate was observed, along with a decrease in the production of C2+ products, highlighting Cu's high sensitivity to SO2 impurities.173
Removing these impurities beforehand may be necessary, although it often requires a combination of specialized and expensive technologies for each impurity. A more effective approach would be to develop catalysts that can tolerate these impurities. This was demonstrated in a study,174 where stability experiments were conducted with the presence of ∼200 ppm SO2 or NO in the feed gas stream using Bi2O3 (producing formate) and Ag (producing CO) as catalysts. Encouragingly, the results showed consistent performance and high FE (>90%) for both catalysts toward the target products over 20 h.
Zhang et al. evaluated the effects of nitrogen and sulfur contaminants (NH4+, NO3−, SO42−, and SO32−) on the catalytic performance of a porous silver electrode for the conversion of 3 M bicarbonate.79 At concentrations of 100 ppm for these ions, there was no significant effect on FECO or FEH2, except for NO3−, which reduced FECO from 56% to 28%. At 500 ppm NO3−, no CO or H2 was produced due to the competitive reduction of NO3−. Later, Pimlott et al. conducted a detailed study on the impact of nitrogen- and sulfur-containing impurities at varying concentrations.81 They observed that adding anions such as SO42− and SO32− (originating from SOx impurities) in 3 M KHCO3, had no impact on product formation (Fig. 14a). In contrast, dissolved NO2− and NO3− ions (originating from NOx impurities) at a concentration of 2000 ppm in the same electrolyte reduced FECO by up to 55% at a current density of 100 mA cm−2. This decrease in FECO was attributed to the relatively more positive electrochemical potentials at which NOx anions are reduced compared to CO2, rather than catalyst degradation. Switching to a fresh KHCO3 solution restored performance, indicating minimal long-term impact from NO3−. This demonstrates that direct bicarbonate conversion is more tolerant to impurities than gaseous CO2 conversion.
 | ||
| Fig. 14 FECO for electrolysis of 3 M KHCO3 solution at 100 mA cm−2 using a porous silver foam cathode with (a) 0–2000 ppm of flue gas impurities and (b) as a function of DTAB concentration. Reproduced with permission from ref. 81. | ||
An alternative strategy to minimize the impact of NOx impurities is to remove NO2− and NO3− from the bicarbonate solutions before they reach the electrolyzer, using methods such as reverse osmosis, ion exchange, distillation, or biodenitrification.175 However, these processes can be cumbersome. To address this, Pimlott et al. demonstrated that adding surfactants like 10 mM dodecyltrimethylammonium bromide (DTAB) to the bicarbonate solution suppressed the reduction of aqueous NOx impurities and increased the FECO from 56% to 80% at 100 mA cm−2, even slightly higher than without impurities (Fig. 14b).81 This improvement occurs because the surfactant's alkyl chains suppress the HER and increase the local CO2 concentration by displacing ions near the catalyst surface.157,176 From this perspective, adding surfactants to the electrolyte solution could be a cost-effective way to mitigate the poisonous effects of anionic contaminants during the conversion of captured CO2.
Understanding the effect of oxygen on the electrochemical conversion process is crucial, given that it is often present in flue gas streams at concentrations of several percent. A recent study examined an electrolyzer supplied with either CO2 or bicarbonate.177 When 10% O2 was introduced into the CO2 stream entering a gas-fed electrolyzer, CO selectivity decreased by over 90%. In contrast, the presence of O2 in the bicarbonate solution did not affect the performance of a liquid-fed electrolyzer. Regardless of the O2 concentration in the gas streams (ranging from 0–100%), the conversion of bicarbonate solutions consistently produced CO at a Ag/C electrode with a FECO of ∼65% at a current density of 100 mA cm−2. This indifference to O2 is a very encouraging result for integrated CO2 capture and electrochemical conversion. Nonetheless, a potential strategy to mitigate the adverse effects of O2, even for the feed-stream approach, is to operate the electrolyzer at a high total current density to ensure all O2 at the electrode is reduced. Although this approach entails higher electricity usage, the associated costs appear negligible for low O2-containing feed streams (≤3% O2) at 300 mA cm−2.174
Many industrial factories already have infrastructure for capturing and processing flue gas, which facilitates the integration of CO2 capture with conversion. However, this does not change the fact that implementing CO2 conversion technologies in industrial settings requires significant initial investments in infrastructure and equipment. Another limitation of flue gas capture is that it is confined to areas with substantial industrial activity, making it ineffective for addressing CO2 emissions from dispersed sources such as transportation, buildings, or agriculture.
The potential for integrating ocean capture with electrochemical conversion is immense. Although studies to date have focused on the release/capture of CO2 or the coupled capture/conversion approach (Fig. 1, Route 2), we will describe one such study as a source of inspiration for future integration efforts. Digdaya et al. reported a reactor operated using ocean water with a bipolar membrane electrodialysis (BPMED) cell followed by vapor-fed CO2 reduction.165 The BPMED cell utilized a one-electron reversible redox couple (Fe(CN)63−/Fe(CN)64−) at the electrodes instead of the conventional water-splitting reaction. This demonstrated efficient capture of CO2 (released upon acidifying the bicarbonate-containing ocean water) at an electrochemical energy consumption of 155.4 kJ mol−1 or 0.98 kWh kg−1 of CO2, with a CO2 capture efficiency of 71%.
Once CO2 was released and led to the vapor-fed electrochemical reduction, various fuels and chemicals, such as CO, CH4, ethylene, and ethanol, could be produced with a total FE of up to 73% at current densities of 58 mA cm−2 when using a Cu electrocatalyst. Similarly, CO could be selectively obtained with a FE of up to 95% at a current density of 11.2 mA cm−2 using a Ag electrocatalyst. It is important to note that the energy required for CO2 capture constitutes only a small fraction of the total capture and conversion energy. For example, converting CO2 to CH4 would require 13.9kWh kg−1 of CO2. To date, this process has been executed coupled, with the CO2 being released from the bicarbonate-containing ocean water and directed to the vapor-fed electrode. However, there is nothing preventing the integration of these steps, as bicarbonate can be electrochemically converted to products directly or indirectly (Table 1).
5.2. Continuous operation of CO2 capture and conversion
With a comprehensive understanding of CO2 capture processes and the conversion of captured CO2, the natural progression is to integrate these processes to streamline operations and enhance overall efficiency. Below, we highlight three studies that have carefully considered aligning the two processes for continuous operation. While the first two examples utilize simulated flue gas, the third employs pure CO2.Wang and Luo discussed both a coupled and an integrated system designed for CO2 capture, electrocatalytic reduction, and purification of gaseous products.23 An alkaline solution was generated via a BPMED cell to capture CO2 from the simulated flue gas (N2:CO2 = 5:1). The absorbed CO2 was then released by acidifying the absorbent solution with the acidic solution generated from the BPMED cell. The liberated CO2 was electrocatalytically reduced into CO at a Ag GDE using a flow-cell setup. However, due to low single-pass efficiency, the CO concentration reached only ∼10% at 200 mA cm−2 and ∼15% at 300 mA cm−2. Nevertheless, any unreacted CO2 could be absorbed by the alkaline solution produced by the BPMED cell, resulting in a pure stream of syngas. The authors also explored an integrated process, bypassing the CO2 release process, by examining the performance of a Ag electrode directly in 1 M KHCO3 (Fig. 1, Route 3). In this scenario, a FECO of 40% was achieved at a current density of 50 mA cm−2. Unfortunately, the current density could not be further increased, as this led to the continuous decline in CO production, accompanied by a gradual increase in H2 from the HER.88,94 Thus, the integrated approach will require significant optimization before it can be scaled up and operated stably.
Prajapati et al. integrated CO2 capture and electrochemical conversion using a continuous flow system (Fig. 15).129 To capture CO2, they employed a migration-assisted moisture-gradient (MAMG) process, where gaseous CO2 was absorbed as HCO3− in a CO2-binding organic liquid. The captured CO2 was then led through an anion exchange membrane under an electric field to an aqueous solution, where it was converted to dissolved CO2. For CO2 reduction, they devised an electrochemical cell setup using a Cu mesh electrode, enabling the extraction of CO2-free products such as CO, CH4, and C2H4. In practice, they operated the MAMG CO2 capture unit at a current of 600 mA and the CO2 reduction unit at a current density of 200 mA cm−2, which achieved a 40% FE for C2H4 from a simulated flue gas feed and could be operated stably for 24 h. To effectively integrate the CO2 capture and CO2 reduction processes without diminishing the overall efficiency or necessitating downstream purification or recycling of CO2, the rate of CO2 capture must match the rate of CO2 reduction.
 | ||
| Fig. 15 Illustration of the integrated system featuring MAMG CO2 capture and electrochemical conversion. Reproduced with permission from ref. 129. | ||
Bai et al. developed a molten-salt-based electrolyzer capable of continuously capturing and converting CO2 with 100% efficiency into CH4 and CO, along with H2 from the HER (Fig. 16).101 Notably, the CO2 fed into the reactor was completely captured by the molten salt electrolyte by carefully optimizing the feeding rate. The highest CH4 selectivity (33%) was achieved at 400 mA cm−2 using a commercial CuNi18 sheet as the cathode and a corrosion-resistant Ni sheet as the anode in a molten Li1.427Na0.359K0.214CO3 ∼ 0.15LiOH electrolyte at 650 °C (reactor dimensions: diameter = 2 cm, length = 4 cm). Pure CO2 was supplied to the reactor at a flow rate of 20 mL min−1. Unlike previous molten-salt single cells based on the one-pot method, this reactor operated without the need for membranes and with a conversion efficiency of 100% for a single pass, eliminating the need to separate unreacted CO2 from the gaseous products. Unfortunately, the operation of this system lasted for only 3.6 h, and a gradual increase in the electrolysis voltage was observed, showing its unsatisfactory stability. In general, molten salt systems require substantial energy input to maintain operations at high temperatures. In addition, for molten salt systems that generate carbonaceous products,131,132 regular pauses in production are necessary to remove carbon deposits accumulated on the cathode.
 | ||
| Fig. 16 Schematic illustration of a reactor for reactive CO2 capture, featuring a commercial CuNi18 sheet as the cathode and a corrosion-resistant Ni sheet as the anode in molten Li1.427Na0.359K0.214CO3 ∼ 0.15LiOH. Reproduced with permission from ref. 101. | ||
Although examples of the practical integration of CO2 capture with subsequent conversion are limited, they have demonstrated the feasibility and potential of this technology. One approach involves running the two processes sequentially, with the conversion starting only after the capture is complete. The cycle can then restart after the conversion and regeneration of the capture agent. Alternatively, the conversion of captured CO2 can be conducted concurrently with the capture process, which appears promising if the capture can be performed efficiently, as in the case of CO2 from flue gas. However, if this is not the case, this approach may result in overall low conversion efficiency and selectivity issues due to the low steady-state concentration of the captured CO2.
5.3. Energy cost analysis
An important aspect of the practical integration of CO2 capture and conversion is the analysis of the energy costs of the entire process, from capturing CO2 from dilute sources to generating the desired product. This analysis is crucial for assessing the feasibility of the technology from both technical and economic perspectives. Since CO is the most reported product from the electrolysis of both gas-fed CO2 and captured CO2 systems, we first compare the energy consumption for producing CO between independent and integrated CO2 capture and electrochemical conversion (Table 2). In the analysis, we considered three key steps that incur the main energy costs in the entire chain of CO2 capture and conversion, i.e., CO2 capture/release, electrolysis, and product separation. The energy consumption analysis used experimentally reported data, including faradaic efficiencies, full-cell potentials, and CO2 utilization efficiencies. As a result, the calculated energy values inherently carry some degree of uncertainty due to the variability and limitations of the experimental conditions and measurements (see the ESI† for details).| System | Product | CO2 utilization (%) | CO2 regenerationb | FE (%) | Electrolysis EE (%)c | Electrolysisb | Outlet CO2 (%) | Product purity (vol%) | Product separationbd | Total energyb | CCU EE (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a See the ESI for details on the calculation procedure.
b Unit: kJ mol-product−1.
c EE: Energy efficiency.
d Separation energy of unreacted CO2 from reaction products.
e Electrolysis energy without thermal considerations is 4881 kJ mol-product−1; an extra 32108 kJ mol−1 has been added due to the process being conducted at 650 °C (see the ESI).
|
|||||||||||||
| Route 1 | CO2 | CO | 20 | 892 | 77 | 41 | 633 | 50 | 26 | 1250 | 2775 | 9 | 179 |
| CO2 | CO | 17 | 1049 | 99 | 54 | 480 | 36 | 32 | 1050 | 2579 | 10 | 180 | |
| Route 3 | HCO3− | CO | 60 | 0 | 70 | 25 | 1014 | 25 | 54 | 200 | 1214 | 21 | 90 |
| CO32− | CO | 100 | 0 | 25 | 35 | 734 | 0 | 25 | 0 | 734 | 35 | 72 | |
| Carbamate | CO | 90 | 0 | 72 | 40 | 634 | 0 | 72 | 0 | 634 | 41 | 85 | |
| Route 1 | CO2 | HCOO− | 9 | 1981 | 96 | 29 | 917 | N.A. | N.A. | 265 | 3163 | 9 | 181 |
| Route 3 | HCO3− | HCOO− | 70 | 0 | 85 | 40 | 499 | 0 | 0.8 M | 265 | 764 | 26 | 94 |
| Route 1 | CO2 | CH4 | 1 | 13715 | 62 | 16 | 5495 | 99 | 1 | 38000 |
57210 | 1 | 182 |
| Route 3 | HCO3− | CH4 | 25 | 0 | 27 | 4 | 20584 |
75 | 7 | 1500 | 22084 | 4 | 98 |
| Molten salt | CH4 | 100 | 0 | 43 | 21 | 36989e |
0 | 16 | 0 | 36989 | 3 | 101 | |
| Route 1 | CO2 | C2H4 | 5 | 3566 | 70 | 34 | 3974 | 77 | 3 | 15400 |
22940 | 6 | 19 |
| Route 3 | CO32− | C2H4 | 100 | 0 | 34 | 10 | 13971 |
0 | 8 | 0 | 13971 | 10 | 103 |
The independent CO2 capture and electrochemical conversion strategy (Fig. 1, Route 1) requires a substantial energy investment to release CO2 from the capture media. For DAC technologies using potassium hydroxide solutions, the energy required to release CO2 from the subsequently formed CaCO3 is 178.3 kJ mol−1.183 Furthermore, in alkaline CO2-fed electrolyzers, CO2 gas can be lost due to its reaction with OH− ions generated during the reduction reaction. Therefore, the CO2 utilization efficiency, defined as the percentage of input carbon converted to a targeted product, is typically no greater than 20% for electrolyzers that generate CO.179,180 In other words, to obtain one mol of CO product, 5 moles of CO2 need to be captured, thus leading to an energy consumption of 892 kJ mol-CO−1 for this CO2 regeneration step. As expected, the energy required for the CO2 capture and release steps with CO2-fed electrolyzers increases as the CO2 utilization efficiency decreases. For the electrolysis step, gas-fed CO2 electrolyzers, whether in flow cell or MEA configurations, require relatively low energy costs, ranging from 480 to 633 kJ mol-CO−1. This is due to the high level of optimization of the electrocatalysts for the CO2-to-CO conversion, resulting in systems displaying high FEs, large current densities, and low cell voltages.
After electrolysis, the outlet gas stream consists of CO, unreacted CO2, and H2 byproduct, which requires separation before obtaining pure CO. Greenblatt et al. outlined general approaches to separating product mixtures resulting from CO2 valorization.184 The unreacted CO2 and unwanted H2 gases can be separated through, e.g., pressure/temperature-based swings, adsorption/desorption, and membrane separation. Since the CO2 utilization efficiency is low, the percentage of unreacted CO2 at the outlet gas stream can be as high as 50%, and the purity of CO is estimated to be 26%.179 Such a low CO purity leads to high energy consumption for the separation step, estimated at 1250 kJ mol-CO−1. In total, the energy consumption from cradle to grave is 2775 kJ mol-CO−1. Given that the theoretical energy consumption for converting gas-fed CO2 to CO is 257 kJ mol-CO−1,38 the global carbon capture and utilization (CCU) energy efficiency for the entire chain is 9%.
In the case of the electrochemically reactive capture of CO2 (Fig. 1, Route 3), the absence of energy costs associated with the CO2 capture/release step is a significant advantage, highlighting the potential of this technology in reducing energy consumption. The primary energy cost in the integrated pathway is the electrochemical conversion of the captured CO2 solution. For instance, in the electrochemical conversion of a bicarbonate solution to CO,90 the energy consumption for this step is 1014 kJ mol-CO−1, which is higher than for the electrolysis of gas-fed CO2. However, the CO2 utilization efficiency for converting bicarbonate is 60%, significantly higher than that of gas-fed CO2 electrolyzers. Since some in situ generated CO2 exits the electrolyzer without being converted to CO, the concentration of CO in the outlet gas stream is 54%, thus requiring 200 kJ mol-CO−1 to separate the small amount of unconverted CO2. The total energy consumption for the entire process, from CO2 capture in dilute sources to CO production via a bicarbonate system, amounts to 1214 kJ mol-CO−1, which is significantly lower than for the independent CCU process. Moreover, the global CCU energy efficiency of the integrated pathway is 21%, twice as high as that of the independent CCU process.
In the case of carbonate and carbamate solutions, the in situ generated CO2 can be quantitatively converted to CO, as almost no CO2 is detected in the outlet gas stream.72,85 This suggests that the CO2 utilization efficiency for both cases is nearly 100%. In this scenario, the total energy consumption for converting carbonate or carbamate solutions can be estimated at 734 and 634 kJ mol-CO−1, respectively. These values are approximately four times lower than those for independent systems, highlighting their potential for practical applications (Table 2). The global CCU energy efficiencies are calculated to be 35% for the carbonate system and 41% for the carbamate system, significantly higher than those achieved with CO2-fed electrolyzers. Notably, if the targeted product is syngas (a combination of CO and H2), the separation energy can be considered zero since no unreacted CO2 is present in the outlet gas stream.
The estimation of the separation energy for the CO product focused exclusively on the energy required to remove unreacted CO2, without accounting for the separation of the H2 byproduct. This approach is based on several considerations. First, CO2 removal is the most energy-intensive step,185 while extracting H2 from syngas after CO2 removal requires significantly less energy. For example, the energy cost for CO2 separation can vary widely, ranging from 100–900 kJ mol−1 of CO2, depending on the separation method used. For the energy calculations, a fixed value of 500 kJ mol−1 of CO2 was assumed for separating unreacted CO2 from the gaseous output.72 In contrast, the energy required for H2 removal from syngas (with a ratio of ∼1:3 CO:H2) is about 85 kJ mol-H2−1.186 Secondly, the H2 concentration in the gas stream is typically much lower than that of unreacted CO2 or the primary product, CO. As a result, the energy cost for H2 separation generally accounts for a small fraction of the overall energy consumption for various CO2 capture and conversion methods.
For instance, in cases where FECO ≥ 70%, the estimated energy cost for H2 separation is ≤34 kJ mol-CO−1, representing <2% of the total energy consumption for both Route 1 scenarios and under 5% for the two Route 3 scenarios (e.g., bicarbonate and carbamate conversion). In the context of carbonate conversion, where FECO is only 25% (i.e., CO/H2 = 1:3), the estimated energy cost for H2 separation rises to 255 kJ mol-CO−1. Even with this increased cost, the total energy consumption for Route 3 (e.g., carbonate conversion, 989 kJ mol-CO−1) remains lower than that for the two Route 1 scenarios (2775 and 2559 kJ mol-CO−1, respectively). It is important to note that the energy cost for H2 separation heavily depends on the gas stream composition and the required purity levels of CO and H2. Consequently, the cost will vary based on the specific components of the gas stream. These factors introduce complexity and result in uncertainties that are challenging to quantify. Given the relatively minor contribution of H2 separation and its associated complexities, these energy costs are not included in Table 2.
Among the different types of captured CO2 (i.e., bicarbonate/carbonate/carbamate), the energy consumption for converting carbamate to CO/syngas is the lowest, which aligns with the weaker binding energy of amines with CO2 compared to hydroxide solutions. Therefore, from an energy consumption standpoint, integrating CO2 capture by amine with the electrolysis of the resulting carbamate to produce CO/syngas is the most advantageous option.
Similar conclusions can be drawn for the generation of HCOO− as the product. In the case of CO2-fed electrolyzers, due to the low CO2 utilization efficiency (9%), 1981 kJ mol-formate−1 of energy is required for the CO2 capture and release steps.181 Additionally, considering the electrolysis and purification steps, the total energy consumption amounts to 3163 kJ mol-formate−1, corresponding to a global CCU energy efficiency of 9%. In contrast, the direct electrochemical upgrading of a bicarbonate solution in the integrated process requires only 764 kJ mol-formate−1, leading to a global CCU energy efficiency of 26%.94 These efficiency values demonstrate the promising potential of the integrated strategy in producing an important chemical such as HCOO−.
A key difference between the systems producing CO and those generating HCOO− lies in the purification methods to separate HCOO− from the liquid electrolyte solution. For one thing, the formate after electrolysis usually has a low concentration (0.8 M) in the electrolyte, which brings difficulty for separation (Table 2). For another, since HCOO− is a charged product that can combine with electrolyte cations to form salts, an additional acidification step is required to obtain the more valuable formic acid. The resulting formic acid in water can then be separated by azeotropic distillation or extraction, which requires 265 kJ mol-formate−1.187
In producing highly reduced products such as CH4 and C2H4, CO2-fed electrolyzers typically exhibit low CO2 utilization efficiency. For example, electrolyzers converting CO2 into CH4 have demonstrated efficiencies of ∼1%,182 while those producing C2H4 achieve ∼5%.19 These low efficiencies result in high energy costs associated with the CO2 capture and release steps. Moreover, for both independent and integrated pathways, the energy costs of the electrolysis step are significantly higher compared to electrolyzers that generate CO or HCOO−. This is due to the greater number of electrons required to convert CO2 into CH4 (8 electrons) or C2H4 (12 electrons), as opposed to the 2 electrons needed for producing CO or HCOO−, substantially increasing the energy consumption of the electrolysis. For instance, in the direct electrochemical conversion of a bicarbonate solution to CH4,98 the energy required for the electrolysis step can be calculated to amount to 20584 kJ mol-CH4−1. However, when considering the CO2 capture/release and product purification steps, the direct conversion of bicarbonate into CH4 has a lower total energy consumption (22084 kJ mol-CH4−1) compared to CO2-fed electrolyzers (57210 kJ mol-CH4−1). The higher concentration of CO2 in the electrolyzer outlet makes the independent pathway more energetically expensive, as more energy is required to separate the unreacted CO2 from CH4 than in the integrated approach.
An energy analysis of CH4 production using molten salts indicates that even in an electrolysis step carried out at 650 °C, the integrated strategy is energetically more favorable.101 With molten salts, the CO2 utilization efficiency is exceedingly high, and the energy consumption for the electrolysis step is relatively low (4881 kJ mol-CH4−1). However, additional energy is required to heat the electrolyzer to 650 °C, amounting to 32108 kJ mol-CH4−1. This brings the total energy cost to 36989 kJ mol-CH4−1, which is still lower than the total energy costs for producing CH4 using a CO2-fed electrolyzer (57210 kJ mol-CH4−1).
Based on the energy consumption analysis shown in Table 2, we conclude that the integrated CO2 capture and electrochemical conversion strategy (Fig. 1, Route 3) is more energy-efficient than independent processes (Fig. 1, Route 1), regardless of the products formed. This makes the integrated approach potentially more economical. The analysis also suggests that CO or syngas would be the most feasible initial targets, followed by HCOO−, as the total energy consumption for producing CO and HCOO− from dilute CO2 sources via electrochemically reactive carbon capture is lower than for other products such as CH4 and C2H4. This conclusion is further supported by Debergh et al.,188 who examined the economics of electrochemical syngas production via DAC. The authors compared the levelized cost of syngas (LCOS) for different CO2 capture and electrochemical conversion routes from air. They found that the integrated route offers the lowest LCOS due to reduced downstream separation (DSP) and CO2 capture costs from the air (i.e., DAC), even though it incurs higher electrolysis costs (Fig. 17). Furthermore, their analysis revealed that CO is the most promising target for the integrated route among the various products.
 | ||
| Fig. 17 Comparison of the LCOS of different routes of capture and electrochemical conversion of CO2 from air. The terms “alkaline flow,” “MEA,” and “PSE” (porous solid electrolyte) refer to the independent route using these respective cell configurations. The “General OPEX” category accounts for operational and maintenance costs. Reproduced with permission from ref. 188. | ||
Highly reduced products such as CH4 and C2H4 have important applications as chemical feedstocks. To improve the economic viability of producing these valuable compounds via the integrated route, it is essential to reduce the overall energy consumption. A significant portion of the energy used in these processes is attributed to the direct electrolysis of captured CO2 solutions. Currently, the electrocatalytic materials employed in these processes suffer from high overpotentials and low selectivity for CH4 and C2H4, which drives up energy costs for electrolysis. Addressing this challenge requires developing novel electrocatalysts optimized for directly upgrading captured CO2 solutions to lower energy consumption and enhance the economic competitiveness of CH4 and C2H4 production. Methanol and ethanol, both valuable as fuels and chemical intermediates, can be produced through the electrochemical conversion of gas-fed CO2. However, there are currently only a few reports of direct electrochemical conversion of captured CO2 into these products with low selectivity (e.g., FEethanol < 20%).102 Therefore, developing efficient and selective catalysts for the integrated process in these cases is essential for accurately evaluating their energy requirements and economic viability.
It should be noted that the integrated route for CO2 capture and conversion demonstrates a similarly high CO2 utilization efficiency as thermochemical CO2 conversion by hydrogenation, typically ranging between 70% and 90%.189–191 The CO2 hydrogenation reactions are generally conducted at temperatures between 300 and 700 °C, enabling the production of valuable products such as CO, HCOOH, CH3OH, and CH4. While both CO2 valorization methods achieve comparable CO2 conversion efficiencies, thermochemical processes tend to produce higher CO2 emissions due to their energy-intensive conditions, unless powered by carbon-neutral heat sources.
5.4. Stability evaluation
The energy costs mentioned above are based on electrolysis data obtained from short-term experiments, typically lasting several hours to tens of hours. For industrial-scale applications, it is crucial to comprehensively study the long-term stability of integrated CO2 capture and electrochemical conversion systems operating at high current densities. Unfortunately, only relatively few studies have addressed this issue.Table 3 summarizes studies investigating the stability of electrochemical conversion of captured CO2 for at least 5 hours at current densities of ≥50 mA cm−2. In an early report, Li et al. demonstrated the stable operation of an integrated system for 145 h using a Ag catalyst (Fig. 18a and b).72 Here, a KOH solution was used to capture CO2, resulting in the formation of carbonate, which was then reduced to syngas while regenerating the KOH solution for another round of CO2 capture via electrolysis. Throughout the operation, the current density remained stable at ∼180 mA cm−2, and the H2/CO ratio consistently ranged between 2 and 3. However, slight fluctuations in the syngas ratio and a decrease in FECO were observed, attributed to metal contamination and deposition over time.
| Captured CO2 | |j| (mA cm−2) | Cell voltage (V) | Product FE | Duration of electrolysis (h) | Reason for performance decrease | Ref. |
|---|---|---|---|---|---|---|
| a Data selected with |j| ≥ 50 mA cm−2 and electrolysis time of ≥5 h. b Electrolyte refreshed at regular intervals. | ||||||
| Captured CO2 in 2 M KOH | ∼180 | 3.8 | CO: 33–25% | 145 | Contamination by metal deposition | 72 |
| 3 M KHCO3 | 100 | ∼3.5 | CO: 37–27%b | 5 | Consumption of KHCO3 and pH increase | 75 |
| 3 M KHCO3 | 100 | 3.5–3.7 | CO: 42–32%b | 8 | Consumption of KHCO3 and pH increase | 76 |
| 3 M KHCO3 | 65 | 3.4–3.5 | CO: 88–85%b | 80 | Consumption of KHCO3 and pH increase | 79 |
| Captured CO2 in 2 M KOH | 100 | 3.3 | CO: 40–36% | 23 | N.A. | 73 |
| 1 M K2CO3 | 200 | ∼3.7 | CO: 42–35% | 25 | N.A. | 74 |
| 3 M KHCO3 with CO2 purging | 200 | ∼3.2 | CO: 95–86% | 29 | Partial detachment of Ni SAC | 90 |
| Captured CO2 in 2 M KOH | 200 | 3.5–3.8 | C2 products: 42–32% | 22.5 | Degradation of the CEM interposer pore structure | 103 |
| Captured CO2 in 2 M KOH | 100 | 3.4–3.5 | C2 products: 22–18% | 50 | Reduction of exposed Cu(OH)2 nanowire catalyst | 105 |
| 5 M K2CO3 | 100 | ∼3.7 | C2H4:10–9% |
23 | Consumption of K2CO3, degradation of Cu–Ag catalyst | 104 |
 | ||
| Fig. 18 Stability evaluation of the integrated CO2 capture and electrochemical conversion. (a) Experimental setup, where CO2 is captured into 2 M KOH (1) to generate carbonate, which is then pumped into a new bottle (2) and reduced in a direct carbonate cell (3). The gas products are measured by a mass flow meter (4) to determine the total volume. (b) Syngas production performance during electrolysis at a constant potential of 3.8 V using a Ag catalyst with the same experimental setup. Reproduced with permission from ref. 72. (c) Stability comparison of electrolysis between gas-fed CO2 and 3 M KHCO3 with/without CO2 purging at a constant current density of 200 mA cm−2 using a Ni SAC. Reproduced with permission from ref. 90. (d) Long-term operation for CO2 capture and electrolysis in a 2 M KOH solution, performed with a Cu/CoPc-CNTs electrocatalyst and a 135 mm MEC interposer. Reproduced with permission from ref. 103. | ||
Later, Zhang et al. conducted electrolysis of a 3 M KHCO3 solution over 80 h at an applied current density of 65 mA cm−2 using a free-standing porous silver electrode.79 Throughout the experiment, FECO decreased by only 3% and the cell voltage increased by just 100 mV, demonstrating the good stability of the system. However, this stability was achieved by manually refreshing the 3 M KHCO3 electrolyte every 500 s. When the free-standing electrode was replaced with silver–carbon composite electrodes, a much larger decrease in FECO (16%) was observed over the 80 h electrolysis. Both Li et al. and Lees et al. also demonstrated that refreshing the captured CO2 electrolyte was essential to maintaining good stability in their electrolysis systems.75,76 Without refreshing the electrolyte, a gradual decrease in FECO was observed due to the consumption of (bi)carbonate, resulting in an increase in pH over electrolysis time (Table 3).
Recently, Song et al. investigated the electrolysis of a 3 M KHCO3 solution with continuous CO2 purging for 29 h using a Ni SAC. They found that the system remained very stable for continuous CO production with FECO of >90% during the first 18 h due to the regeneration of bicarbonates by the CO2 purging (Fig. 18c).90 Under such conditions, electrolyte refreshing had no impact on the electrolysis performance, and therefore, it was not required. In contrast, the electrolysis of gas-fed CO2 and 3 M KHCO3 without CO2 purging showed a rapid decline in FECO under the same conditions. In addition, a slight decrease of FECO to <90% was observed after 20 h of electrolysis, which was attributed to the partial detachment of the catalysts.
Similar stability was observed in the electrochemical conversion of (bi)carbonate solution to C2 products, such as C2H4. Ma et al. observed a decrease in FE for C2 products and increased cell voltage during the first 10 h of electrolysis of captured CO2 in a 2 M KOH solution using a Ag–Cu(OH)2 nanowire catalyst.105 The authors attributed this initial performance deterioration to the reduction of the exposed Cu(OH)2 nanowires. After this phase, the system remained stable for 40 h of operation. Song et al. evaluated the stability of converting 5 M K2CO3 solution to C2H4 using a Cu–Ag catalyst at an applied current density of 100 mA cm−2.104 The FE for C2H4 was stable for the first 9 h but decreased after 18 h, attributed to various factors, including the consumption of K2CO3 and degradation of the Cu–Ag catalyst.
Lee et al. constructed a prototype system capable of operating CO2 capture and electrochemical conversion to continuously produce C2 products, similar to a previously reported system by the group (Fig. 18a).103 The authors employed a Cu/CoPc-CNT catalyst (CoPc denotes cobalt phthalocyanine) and used a porous hydrophilic mixed cellulose ester (MCE) as the interposer to establish a well-defined spacing between the catalyst layer and the ion exchange membrane. They demonstrated continuous operation of the setup for 20 h at a current density of 200 mA cm−2. However, they observed a decline in performance after 8 h of operation (Fig. 18d), which was attributed to the degradation of the pore structure in the MCE membrane. This resulted in an increase in cell voltage and HER.
These results highlight the importance of optimizing the catalyst, electrolyte, interposer materials, and electrode configuration to enhance the stability of electrolysis of the captured CO2 solution under relevant conditions. Note that during continuous operation, the electrolyte is refreshed through the reaction of CO2 with the continuously basified electrolyte. One potential solution to address stability issues is the incorporation of additives or specifically engineered functionalities to prevent degradation of the catalyst and interposer. Until now, most stability studies have concentrated on the (bi)carbonate system, with limited research on the carbamate system. Furthermore, continuous CO2 capture and electrochemical conversion over extended periods (≥1000 h) has not yet been demonstrated experimentally for either system, a crucial milestone for ensuring long-term durability in CO2 capture and conversion.
5.5. Scalability consideration
Scalability is another important metric for integrated CO2 capture and electrochemical conversion that has not yet been thoroughly explored in the literature. The production rate, which reflects the scalability of a specific target product, is determined by the current density and FE achieved during electrolysis, as well as the geometric area of the electrode. Therefore, increasing these parameters is expected to enhance the production of target products. It should be noted, however, that there is a trade-off between current density and electrode area when aiming for a specific production rate. The current density determines the size of the electrolyzer. At lower current densities, the conversion of captured CO2 is more energy efficient and incurs lower operating costs due to reduced energy losses from internal resistance, but this results in larger electrolyzers, increasing capital costs. In addition, it should be noted that scaling up production can be achieved either by increasing the size of the electrolyzer or by stacking multiple electrolyzers. An optimal current density balances operating and capital costs, estimated to be 200–400 mA cm−2 for the electrochemical conversion of gas-fed CO2.192,193 Such current densities are achievable for the electrolysis of captured CO2 in many systems (Table 1).To determine the electrolyzer size required to produce CO at a scale of 1 ton day−1, we may use the optimal performance metrics reported by Song et al. (Table 3).90 This calculation shows that operating electrolysis at a current density of 200 mA cm−2 with FECO = 90% can achieve a CO production rate of 22.6 kg m−2 day−1. Accordingly, an electrolyzer with a total electrode area of 44.2 m2 or an electrolyzer stack reaching this total area would be needed to reach the target production of CO. Doubling the current density is expected to reduce the electrolyzer size by half. Based on this calculation, scaling up the production of other products, such as C2H4, would require significantly larger electrolyzers due to the lower FE currently achieved for this product (<40%, Table 1).
6. Conclusions and perspective
6.1. Summary
The electrochemical conversion of captured CO2 has garnered increasing attention in recent years due to its advantage of bypassing the traditional CO2 release, purification, compression, transport, and storage steps. In this review, we outlined the four key factors that influence the success and applicability of this emerging technology. Table 4 summarizes the three typical approaches for electrochemically reactive CO2 capture.| Systems | RNH2 (aq.) | OH−/CO32− (aq.) | Molten oxide |
|---|---|---|---|
| Captured CO2 | RNHCOO− (aq.) | CO32−/HCO3− (aq.) | Molten carbonate |
| Direct or indirect conversion | Both possible | Both possible | Direct |
| Main products | CO, HCOO− | CO, HCOO−, CH4, C2H4 | C, CO, CH4 |
| Key benefits | Room-temperature operation; fast CO2 absorption kinetics | Room-temperature operation; improved stability and durability; larger |j| and FE for product formation | More compatible with flue gas systems; larger |j| and FE for product formation |
| Key challenges | Prone to thermal and oxidative degradation; lower |j| and FE for product formation | Inefficiency of CO32− (aq.) in capturing CO2 from the air | Operation at high temperature; batch production; interruptions due to buildup of solid carbon products on the cathode |
(1) The choice of capture medium plays a crucial role in determining the thermodynamics and kinetics of the CO2 absorption process and its subsequent electrochemical conversion. Each capture medium reviewed exhibits unique chemical properties that can be exploited to minimize the energy penalties associated with both capture and electrochemical conversion. Aqueous amines are the most widely studied medium for post-combustion CO2 capture, although they are particularly vulnerable to thermal and oxidative degradation. Due to the strength of the C–N bond, the electrochemical reduction of the carbamate typically results in low current density values (<50 mA cm−2). Capturing CO2 as a (bi)carbonate solution using aqueous carbonate and hydroxide solutions has emerged as a more economical and thermally stable alternative to amine scrubbing. The reduction of (bi)carbonate has shown higher reaction rates than carbamate reduction. However, several acid–base equilibria between CO2 and (bi)carbonate complicate mechanistic investigations to identify the electrochemically active species at the electrode surface. Molten alkali carbonates are particularly beneficial for capturing CO2 from flue gas, as this medium is compatible with the high temperature of such streams. Electrochemical conversion of captured CO2 in molten carbonates typically occurs at temperatures ranging from 500–950 °C, with the added advantage of generating solid carbon in many cases, thereby eliminating the need for costly separation and purification processes.
(2) Various carbonaceous products can be produced through the electrochemical conversion of captured CO2. The product types are determined mainly by the choice of catalysts, although the capture medium can also have an impact. For the electrochemical conversion of aqueous carbamate and (bi)carbonate solutions, CO is typically generated using Ag nanoparticles or SACs, formate is produced over Bi and Sn, while CH4 and C2H4 are formed using Cu-based catalysts. This pattern mirrors that observed in the electrochemical conversion of gas-fed CO2. In molten carbonate systems, the product outcome is more temperature dependent, with CO formed at ∼900 °C and carbon materials at lower temperatures (∼500 °C). Notably, the conversion of captured CO2 can follow either an indirect pathway, where CO2 is generated in situ and converted, or a direct pathway, where the captured CO2 is directly converted at the electrode.
(3) Several other factors also significantly impact the electrochemical conversion of captured CO2. Key elements such as the electrolyzer design, electrode materials, electrolyte composition, ion exchange membranes, and operating conditions (e.g., temperature, pressure, and flow plate design) can all affect conversion efficiency. These factors influence the availability of in situ generated CO2 by shifting the dissociation equilibrium between carbonate, bicarbonate, or carbamate, and free CO2, as well as impacting proton production and transport. Thus, optimizing these operational parameters is essential for improving the overall conversion efficiency.
(4) Based on these understandings, integrating CO2 capture with subsequent conversion for practical applications becomes feasible. The outcomes of this integration will vary depending on the source of CO2 (i.e., air or flue gas), as each source contains different levels of CO2, O2, and gas impurities. These factors impact, e.g., the availability of the CO2 reactant, the charge transfer processes (e.g., CO2vs. oxygen reduction), and catalyst poisoning. For effective integration, the rates of capturing CO2 and its subsequent conversion should be aligned. A comparison of energy costs between the conventional independent route (Fig. 1, Route 1) and the integrated route (Fig. 1, Route 3) shows that the integrated route consumes less energy and, as a result, achieves higher overall efficiency for the entire process, spanning from CO2 capture from dilute sources to the production of the final products.
6.2. Challenges and potential solutions
Although previous studies have highlighted the significant potential for the electrochemical conversion of captured CO2, several key challenges impede the rapid advancement of this emerging field and its large-scale implementation.(1) The mechanism behind the conversion of captured CO2 remains a topic of debate. It is not always clear whether the CO2-bound capture agent itself or the CO2 released from it is the actual species converted at the electrode. Most reports show that (bi)carbonate and carbamate are converted via the in situ generation of CO2 at the electrode interface, although some reports indicate that they can also be directly converted. Due to the equilibrium between (bi)carbonate/carbamate and CO2 during electrochemical conversion, determining the exact reaction pathway using only ex-situ techniques can be difficult. Kinetic studies are crucial for this clarification, and in situ/operando techniques, such as infrared reflection–absorption spectroscopy and Raman spectroscopy, are valuable for identifying the adsorbed reactive species (bicarbonate/carbamate vs. CO2) at the catalytic sites during the conversion process.194,195 For example, Lu et al. recently reported an electrolysis optical coherence tomography platform to visualize the chemical reactions occurring in a CO2 electrolyzer, including reactants, intermediates, and products.77 This platform, or a similar one, may serve as an effective tool for monitoring the dynamic movement of the various species during the electrolysis of captured CO2, providing valuable insight into the mechanism. Furthermore, theoretical simulations based on DFT calculations can be employed to determine the adsorption energy of reactive species and intermediates, as well as the free energy change of each elementary step in the conversion process.196,197
(2) In general, the conversion efficiency for captured CO2 is low. Over the past decades of intensive research, the electrochemical conversion of gas-fed CO2 can now be achieved at a large current density (>1 A cm−2) and with high FE (>95%) for producing CO and formate.198,199 In comparison, the current density (usually <200 mA cm−2, although 400 mA cm−2 can be achieved under certain conditions) and the product FE (usually <80%, with 90% achievable under certain conditions) are much lower for the electrochemical conversion of captured CO2 (Table 1), making the technology less efficient. The main reason for this is the low CO2 concentration at the electrode/electrolyte interface, caused by the slow dissociation of carbonate/bicarbonate/carbamate to generate in situ CO2 in the indirect conversion or the electrostatic repulsion between the negatively charged electrode and the carbonate/bicarbonate/carbamate ions in the direct conversion. Different strategies should be adopted to overcome these obstacles. For instance, modifying the electrode configuration, ion exchange membrane, and even the anolyte can enhance proton production and transport, thereby increasing the generation of in situ CO2 and improving the conversion process. Drawing inspiration from electrochemical nitrate conversion, where negatively charged species are also involved, functionalizing the electrode surface with positively charged groups can reduce electrostatic repulsion and concentrate carbonate/bicarbonate/carbamate ions, promoting their direct conversion.200,201 Additionally, designing new catalysts specifically tailored for captured CO2 conversion is crucial to enhance efficiency.
(3) Inefficient integration of CO2 capture and conversion can hinder practical applications. To date, most studies have focused on improving the electrochemical conversion efficiency of captured CO2 (i.e., carbonate, bicarbonate, or carbamate). A few examples demonstrate that integrating the two processes is feasible at the proof-of-concept stage. However, these reports rarely address the recyclability of the resulting electrolyte for CO2 capture, nor do they explore the impact of different CO2 sources or the long-term stability of the capture-conversion cycles. Currently, the electrolysis of captured CO2 has been conducted for no longer than 150 h, with performance degradation arising mainly due to electrolyte basification and catalyst deactivation. Electrolyte basification can be mitigated by continuous and efficient CO2 capture, along with a judicious selection of the capture medium, tailored to the specific operating conditions.
While air has a low concentration of CO2, flue gas has a much higher CO2 concentration but also includes gas impurities such as NOx and SOx. In an air capture scenario, a medium with selective and fast CO2 uptake is necessary to concentrate the low levels of CO2 for subsequent conversion. Molten oxides would be a suitable option in this case. For flue gas, aqueous amine and carbonate solutions can be effective for CO2 capture, but careful attention must be given to selecting catalysts resistant to gas impurities. The decision to extract CO2 from flue gas or ambient air depends on various factors, including location, emission sources, available resources, and technological readiness. Additionally, new catalysts that demonstrate long-term stability for extended electrolysis periods are needed, or additives and specialized functionalities should be introduced to the electrode or electrolyte to prevent catalyst deactivation. Ideally, the electrochemical conversion process should regenerate the capture medium with minimal loss or degradation. Ultimately, the full integration of the capture and conversion processes must operate stably for hundreds or even thousands of hours without a significant decline in efficiency for both CO2 capture and conversion.
One promising approach is integrating the capture and conversion sites at a molecular level. This approach closely mimics the working mechanism of enzymes, creating a precise microenvironment for molecular recognition and specific catalysis.202 These studies highlight the strategy of tailoring microenvironments at a molecular level to enhance the performance of CO2 capture and conversion.
(4) The scalability and economics of integrated systems have not yet been thoroughly explored. The few existing studies on such systems remain on the laboratory scale. In contrast, other large-scale CO2 technologies have reached pre-commercial stages, including high-temperature electrochemical CO2-to-CO production,203 thermal catalytic hydrogenation of CO2 to methanol,204 and biological CO2 upgrading to methane.205 These technologies can serve as valuable reference points for evaluating the scalability of integrated CO2 capture and electrochemical conversion.
Any technology nearing scalability requires a comprehensive techno-economic analysis (TEA), which should extend beyond energy cost analysis. This means factoring in operational expenditures (OPEX) and capital expenditures (CAPEX) to fully understand the economic viability of the process.206 This assessment should encompass the entire value chain, including CO2 capture methods (post-combustion or DAC), electrolysis using various capture media, and downstream processes for product purification and recycling of unconverted CO2. While a detailed TEA is beyond the scope of this review, it would be highly beneficial for experts in the field to explore these aspects further. An important direction would be to conduct a comparative TEA of the integrated and independent routes over the entire life cycle of the CO2 capture and conversion system.
To scale up the electrochemical conversion of captured CO2 from a technological point of view, several parameters, such as current density, faradaic efficiency, and electrode area, must be optimized to increase product formation rates. However, once a target production rate is defined, a balance must be struck between current density and electrode area, as these factors heavily influence the economic feasibility of the electrolysis process. For instance, operating at lower current densities tends to be more energy efficient and reduces OPEX by minimizing energy losses from internal resistance. However, this approach requires larger electrolyzers, driving up CAPEX. Conversely, higher current densities might lower CAPEX but increase OPEX due to less efficient energy usage. In any case, highly durable electrolyzer systems are essential to reduce overall costs, as frequent replacement of electrolyzer components incurs additional expenses and should, therefore, be minimized.
6.3. Future directions
In addition to addressing the challenges mentioned, there are numerous other opportunities to advance this emerging field. Two potential future directions are outlined below:Author contributions
All authors contributed to the manuscript's writing, reviewing, and revising. K. Daasbjerg coordinated and supervised the overall project.Data availability
All data supporting the findings in this review are available from the referenced sources listed in the manuscript. The data presented in the tables are derived from the corresponding literature references. For the calculated values in Table 2, details of the calculation methodology are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
X.-M. Hu acknowledges the financial support from the National Natural Science Foundation of China (Grant No. 22376120) and Shandong Provincial Science Foundation for Excellent Young Scholars Overseas (2022HWYQ-002). H.-Q. Liang acknowledges the financial support from the National Natural Science Foundation of China (Grant No. 22302174) and the research startup package from Zhejiang University. A. Rosas-Hernández acknowledges the financial support from the Danish National Research Foundation (Grant No. DNRF118). Part of this work was supported by the Novo Nordisk Foundation CO2 Research Center (CORC) with grant numbers NNF21SA0072700 (K. Daasbjerg) and NNF22SA0072700 (A. Rosas-Hernández) and is published under the number CORC_24_33.Notes and references
- D. Jiménez-de-la-Cuesta and T. Mauritsen, Nat. Geosci., 2019, 12, 902–905 CrossRef.
- C. Le Quéré, G. P. Peters, P. Friedlingstein, R. M. Andrew, J. G. Canadell, S. J. Davis, R. B. Jackson and M. W. Jones, Nat. Clim. Change, 2021, 11, 197–199 CrossRef.
- S. J. Davis, Z. Liu, Z. Deng, B. Zhu, P. Ke, T. Sun, R. Guo, C. Hong, B. Zheng, Y. Wang, O. Boucher, P. Gentine and P. Ciais, Nat. Clim. Change, 2022, 12, 412–414 CrossRef CAS.
- A. M. Vicedo-Cabrera, N. Scovronick, F. Sera, D. Royé, R. Schneider, A. Tobias, C. Astrom, Y. Guo, Y. Honda, D. M. Hondula, R. Abrutzky, S. Tong, M. D. S. Z. S. Coelho, P. H. N. Saldiva, E. Lavigne, P. M. Correa, N. V. Ortega, H. Kan, S. Osorio, J. Kyselý, A. Urban, H. Orru, E. Indermitte, J. J. K. Jaakkola, N. Ryti, M. Pascal, A. Schneider, K. Katsouyanni, E. Samoli, F. Mayvaneh, A. Entezari, P. Goodman, A. Zeka, P. Michelozzi, F. de’Donato, M. Hashizume, B. Alahmad, M. H. Diaz, C. D. L. C. Valencia, A. Overcenco, D. Houthuijs, C. Ameling, S. Rao, F. Di Ruscio, G. Carrasco-Escobar, X. Seposo, S. Silva, J. Madureira, I. H. Holobaca, S. Fratianni, F. Acquaotta, H. Kim, W. Lee, C. Iniguez, B. Forsberg, M. S. Ragettli, Y. L. L. Guo, B. Y. Chen, S. Li, B. Armstrong, A. Aleman, A. Zanobetti, J. Schwartz, T. N. Dang, D. V. Dung, N. Gillett, A. Haines, M. Mengel, V. Huber and A. Gasparrini, Nat. Clim. Change, 2021, 11, 492–500 CrossRef CAS PubMed.
- R. Swaminathan, R. J. Parker, C. G. Jones, R. P. Allan, T. Quaife, D. I. Kelley, L. de Mora and J. Walton, J. Clim., 2022, 35, 29–48 Search PubMed.
- A. I. Osman, M. Hefny, M. I. A. Abdel Maksoud, A. M. Elgarahy and D. W. Rooney, Environ. Chem. Lett., 2021, 19, 797–849 CrossRef CAS.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- M. D. Aminu, S. A. Nabavi, C. A. Rochelle and V. Manovic, Appl. Energy, 2017, 208, 1389–1419 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef.
- B. Guene Lougou, Y. Shuai, G. Chaffa, H. Xing, H. Tan and H. Du, J. Energy Chem., 2019, 28, 61–72 CrossRef.
- A. M. Parvez, M. T. Afzal, T. G. Victor Hebb and M. Schmid, J. CO2 Util., 2020, 40, 101217 CrossRef CAS.
- J. Lan, X. Lu, B. Ren, F. Duo, X. Niu and J. Si, Org. Biomol. Chem., 2024, 22, 682–693 RSC.
- X. He, L.-Q. Qiu, W.-J. Wang, K.-H. Chen and L.-N. He, Green Chem., 2020, 22, 7301–7320 RSC.
- Z. Fan, Z. Zhang and C. Xi, ChemSusChem, 2020, 13, 6201–6218 CrossRef CAS.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754 CrossRef CAS.
- P. R. Yaashikaa, P. Senthil Kumar, S. J. Varjani and A. Saravanan, J. CO2 Util., 2019, 33, 131–147 CrossRef CAS.
- D. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- X. Wang and C. Song, Front. Energy Res., 2020, 8, 560849 CrossRef.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- M. Wang and J. Luo, eScience, 2023, 3, 100155 CrossRef.
- C.-T. Dinh, Y. C. Li and E. H. Sargent, Joule, 2019, 3, 13–15 CrossRef.
- G. O. Larrazábal, M. Ma and B. Seger, Acc. Mater. Res., 2021, 2, 220–229 CrossRef.
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- H. Seo, M. Rahimi and T. A. Hatton, J. Am. Chem. Soc., 2022, 144, 2164–2170 CrossRef CAS PubMed.
- J. C. Bui, É. Lucas, E. W. Lees, A. K. Liu, H. A. Atwater, C. Xiang, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2023, 16, 5076–5095 RSC.
- Q. Xia, K. Zhang, T. Zheng, L. An, C. Xia and X. Zhang, ACS Energy Lett., 2023, 8, 2840–2857 CrossRef CAS.
- D. J. D. Pimlott, Y. Kim and C. P. Berlinguette, Acc. Chem. Res., 2024, 57, 1007–1018 CrossRef CAS.
- R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2023, 13, 766–784 CrossRef CAS.
- S. Kumar De, D.-I. Won, J. Kim and D. H. Kim, Chem. Soc. Rev., 2023, 52, 5744–5802 RSC.
- R. Sharifian, R. M. Wagterveld, I. A. Digdaya, C. Xiang and D. A. Vermaas, Energy Environ. Sci., 2021, 14, 781–814 RSC.
- S. E. Renfrew, D. E. Starr and P. Strasser, ACS Catal., 2020, 10, 13058–13074 CrossRef CAS.
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Acc. Chem. Res., 2022, 55, 638–648 CrossRef CAS.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- F. Meng, Y. Meng, T. Ju, S. Han, L. Lin and J. Jiang, Renewable Sustainable Energy Rev., 2022, 168, 112902 CrossRef CAS.
- A. Larson, POWER, 2017, https://www.powermag.com/worlds-largest-post-combustion-carbon-capture-project-completed/.
- S. E. Jerng and B. M. Gallant, iScience, 2022, 25, 104558 CrossRef CAS.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS.
- Z. Cesaro, M. Ives, R. Nayak-Luke, M. Mason and R. Bañares-Alcántara, Appl. Energy, 2021, 282, 116009 CrossRef CAS.
- B. Aghel, S. Janati, S. Wongwises and M. S. Shadloo, Int. J. Greenhouse Gas Control, 2022, 119, 103715 CrossRef CAS.
- D. Zhu, M. Fang, Z. Lv, Z. Wang and Z. Luo, Energy Fuels, 2012, 26, 147–153 CrossRef CAS.
- F. de Meyer and S. Jouenne, Curr. Opin. Chem. Eng., 2022, 38, 100868 CrossRef.
- S. J. Vevelstad, V. Buvik, H. K. Knuutila, A. Grimstvedt and E. F. da Silva, Ind. Eng. Chem. Res., 2022, 61, 15737–15753 CAS.
- M. C. Stern, F. Simeon, H. Herzog and T. A. Hatton, Energy Environ. Sci., 2013, 6, 2505–2517 RSC.
- J. L. Hall and W. E. Dean, J. Am. Chem. Soc., 1958, 80, 4183–4188 CrossRef CAS.
- C. Alie, L. Backham, E. Croiset and P. L. Douglas, Energy Convers. Manage., 2005, 46, 475–487 CrossRef CAS.
- A. Kohl, Physical solvents for acid gas removal, Gulf Professional Publishing, Houston, USA, 1997 Search PubMed.
- K. H. Smith, N. J. Nicholas and G. W. Stevens, in Absorption-Based Post-combustion Capture of Carbon Dioxide, ed. P. H. M. Feron, Woodhead Publishing, 2016, pp. 145–166 DOI:10.1016/B978-0-08-100514-9.00007-X.
- K. Smith, G. Xiao, K. Mumford, J. Gouw, I. Indrawan, N. Thanumurthy, D. Quyn, R. Cuthbertson, A. Rayer, N. Nicholas, A. Lee, G. da Silva, S. Kentish, T. Harkin, A. Qader, C. Anderson, B. Hooper and G. Stevens, Energy Fuels, 2014, 28, 299–306 CrossRef CAS.
- Carbon Capture (KC8), https://optimalgroup.com.au/carbon-capture-kc8).
- T. N. G. Borhani, A. Azarpour, V. Akbari, S. R. Wan Alwi and Z. A. Manan, Int. J. Greenhouse Gas Control, 2015, 41, 142–162 CrossRef CAS.
- S. Zhang and Y. Lu, Chem. Eng. J., 2015, 279, 335–343 CrossRef CAS.
- S. Zhang, Z. Zhang, Y. Lu, M. Rostam-Abadi and A. Jones, Bioresour. Technol., 2011, 102, 10194–10201 CrossRef CAS.
- F. Zeman, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS.
- T. Yuan, J. Wang and Z. Li, Fluid Phase Equilib., 2010, 297, 129–137 CrossRef CAS.
- F. Bisotti, K. A. Hoff, A. Mathisen and J. Hovland, Chem. Eng. Sci., 2024, 283, 119416 CrossRef CAS.
- T. Shimizu, T. Hirama, H. Hosoda, K. Kitano, M. Inagaki and K. Tejima, Chem. Eng. Res. Des., 1999, 77, 62–68 CrossRef CAS.
- M. Haaf, R. Anantharaman, S. Roussanaly, J. Ströhle and B. Epple, Resour., Conserv. Recycl., 2020, 162, 104973 CrossRef.
- V. Nikulshina, C. Gebald and A. Steinfeld, Chem. Eng. J., 2009, 146, 244–248 CrossRef CAS.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- L. Huang, Y. Zhang, W. Gao, T. Harada, Q. Qin, Q. Zheng, T. A. Hatton and Q. Wang, Energy Technol., 2017, 5, 1328–1336 CrossRef CAS.
- L. Huang, C. Xu, R. Ren, Q. Zheng, Z. Wang, B. Louis and Q. Wang, Sustainable Energy Fuels, 2018, 2, 68–72 RSC.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
- S.-I. Jo, Y.-I. An, K.-Y. Kim, S.-Y. Choi, J.-S. Kwak, K.-R. Oh and Y.-U. Kwon, Phys. Chem. Chem. Phys., 2017, 19, 6224–6232 RSC.
- A. J. Bard, J. Am. Chem. Soc., 2010, 132, 7559–7567 CrossRef CAS PubMed.
- Q. Zhu, Y. Zeng and Y. Zheng, Ind. Chem. Mater., 2023, 1, 595–617 RSC.
- Y. C. Li, G. Lee, T. Yuan, Y. Wang, D.-H. Nam, Z. Wang, F. P. García de Arquer, Y. Lum, C.-T. Dinh, O. Voznyy and E. H. Sargent, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
- Y. C. Xiao, C. M. Gabardo, S. Liu, G. Lee, Y. Zhao, C. P. O’Brien, R. K. Miao, Y. Xu, J. P. Edwards, M. Fan, J. E. Huang, J. Li, P. Papangelakis, T. Alkayyali, A. Sedighian Rasouli, J. Zhang, E. H. Sargent and D. Sinton, EES Catal., 2023, 1, 54–61 RSC.
- S. Prakash, X. Ge, H. K. Welgama, P. Gogoi, M. Janpandit, T. R. Cook and Y. C. Li, Energy Fuels, 2024, 38, 6223–6229 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- X. Lu, C. Zhou, R. S. Delima, E. W. Lees, A. Soni, D. J. Dvorak, S. Ren, T. Ji, A. Bahi, F. Ko and C. P. Berlinguette, Nat. Chem., 2024, 16, 979–987 CrossRef CAS.
- A. Yoshizawa, M. Higashi, A. Anzai and M. Yamauchi, Energy Adv., 2024, 3, 778–783 RSC.
- Z. Zhang, E. W. Lees, F. Habibzadeh, D. A. Salvatore, S. Ren, G. L. Simpson, D. G. Wheeler, A. Liu and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 705–713 RSC.
- Z. Zhang, E. W. Lees, S. Ren, B. A. W. Mowbray, A. Huang and C. P. Berlinguette, ACS Central Sci., 2022, 8, 749–755 CrossRef CAS PubMed.
- D. J. D. Pimlott, A. Jewlal, B. A. W. Mowbray and C. P. Berlinguette, ACS Energy Lett., 2023, 8, 1779–1784 CrossRef CAS.
- C. Larrea, J. R. Avilés-Moreno and P. Ocón, Molecules, 2023, 28, 1951 CrossRef CAS PubMed.
- H. Li, J. Gao, Q. Du, J. Shan, Y. Zhang, S. Wu and Z. Wang, Energy, 2021, 216, 119250 CrossRef CAS.
- O. Gutiérrez-Sánchez, B. de Mot, N. Daems, M. Bulut, J. Vaes, D. Pant and T. Breugelmans, Energy Fuels, 2022, 36, 13115–13123 CrossRef.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- N. Ahmad, Y. Chen, X. Wang, P. Sun, Y. Bao and X. Xu, Renewable Energy, 2022, 189, 444–453 CrossRef CAS.
- E. Pérez-Gallent, C. Vankani, C. Sánchez-Martínez, A. Anastasopol and E. Goetheer, Ind. Eng. Chem. Res., 2021, 60, 4269–4278 CrossRef.
- J. H. Kim, H. Jang, G. Bak, W. Choi, H. Yun, E. Lee, D. Kim, J. Kim, S. Y. Lee and Y. J. Hwang, Energy Environ. Sci., 2022, 15, 4301–4312 RSC.
- P. Yue, K. Xiong, L. Ma, J. Li, L. Zhang, X. Zhu, Q. Fu and Q. Liao, ACS Appl. Mater. Interfaces, 2022, 14, 54840–54847 CrossRef CAS.
- H. Song, C. A. Fernández, H. Choi, P.-W. Huang, J. Oh and M. C. Hatzell, Energy Environ. Sci., 2024, 17, 3570–3579 RSC.
- M. Shen, L. Ji, D. Cheng, Z. Wang, Q. Xue, S. Feng, Y. Luo, S. Chen, J. Wang, H. Zheng, X. Wang, P. Sautet and J. Zhu, Joule, 2024, 8, 1999–2015 CrossRef CAS.
- H. Shi, M. Cai, W. Li, X. Chen, K. Du, L. Guo, P. Wang, P. Li, B. Deng, H. Yin and D. Wang, Chem. Eng. J., 2023, 462, 142240 CrossRef CAS.
- T. Li, E. W. Lees, Z. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 CrossRef CAS.
- H. Liu, Y. Chen, J. Lee, S. Gu and W. Li, ACS Energy Lett., 2022, 7, 4483–4489 CrossRef CAS.
- O. Gutiérrez-Sánchez, N. Daems, W. Offermans, Y. Y. Birdja, M. Bulut, D. Pant and T. Breugelmans, J. CO2 Util., 2021, 48, 101521 CrossRef.
- A. Bonet Navarro, A. Nogalska and R. Garcia-Valls, Electrochem, 2021, 2, 64–70 CrossRef CAS.
- Y. Pei, W. Gu, S. Cheng, S. Xiao, C. Wang, Y. Yang, H. Zhong and F. Jin, ACS Catal., 2023, 13, 12082–12091 CrossRef CAS.
- E. W. Lees, A. Liu, J. C. Bui, S. Ren, A. Z. Weber and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 1712–1718 CrossRef CAS.
- C. A. Obasanjo, G. Gao, J. Crane, V. Golovanova, F. P. García de Arquer and C.-T. Dinh, Nat. Commun., 2023, 14, 3176 CrossRef CAS PubMed.
- D. Ji, Z. Li, W. Li, D. Yuan, Y. Wang, Y. Yu and H. Wu, Int. J. Hydrogen Energy, 2019, 44, 5082–5089 CrossRef CAS.
- W. L. Bai, Y. Zhang and J. N. Wang, ACS Sustainable Chem. Eng., 2023, 11, 15364–15372 CrossRef CAS.
- J. Lee, H. Liu and W. Li, ChemSusChem, 2022, 15, e202201329 CrossRef CAS.
- G. Lee, A. S. Rasouli, B.-H. Lee, J. Zhang, D. H. Won, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi, H. Liu, I. Grigioni, J. Abed, T. Alkayyali, S. Liu, K. Xie, R. K. Miao, S. Park, R. Dorakhan, Y. Zhao, C. P. O’Brien, Z. Chen, D. Sinton and E. Sargent, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- H. Song, C. A. Fernández, A. Venkataraman, V. D. Brandão, S. S. Dhingra, S. S. Arora, S. S. Bhargava, C. M. Villa, C. Sievers, S. Nair and M. C. Hatzell, ACS Appl. Energy Mater., 2024, 7, 1224–1233 CrossRef CAS.
- S. Ma, Y. Kim, Z. Zhang, S. Ren, C. Donde, L. Melo, A. S. R. Williams, M. Stolar, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2024, 9, 2326–2332 CrossRef CAS.
- X. Wang, G. Licht, X. Liu and S. Licht, Sci. Rep., 2020, 10, 21518 CrossRef CAS.
- S. Jing, R. Sheng, X. Liang, D. Gu, Y. Peng, J. Xiao, Y. Shen, D. Hu and W. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202216315 CrossRef CAS PubMed.
- J. Cao, S. Jing, H. Wang, W. Xu, M. Zhang, J. Xiao, Y. Peng, X. Ning, Z. Wang and W. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202306877 CrossRef PubMed.
- M. P. Nitzsche, L. Bromberg and T. A. Hatton, ACS Sustainable Chem. Eng., 2023, 11, 11012–11018 CrossRef CAS.
- X. Liu, X. Wang, G. Licht and S. Licht, J. CO2 Util., 2020, 36, 288–294 CrossRef CAS.
- R. Yu, B. Deng, K. Du, D. Chen, M. Gao and D. Wang, Carbon, 2021, 184, 426–436 CrossRef CAS.
- L. Hu, B. Deng, K. Du, R. Jiang, Y. Dou and D. Wang, iScience, 2020, 23, 101607 CrossRef CAS.
- E. Laasonen, V. Ruuskanen, M. Niemelä, T. Koiranen and J. Ahola, J. Environ. Chem. Eng., 2022, 10, 106933 CrossRef CAS.
- Y. Hu, C. C. Lee, M. Grosch, J. B. Solomon, W. Weigand and M. W. Ribbe, Chem. Rev., 2023, 123, 5755–5797 CrossRef CAS PubMed.
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS.
- M. Selvin, S. Shah, H. J. Maria, S. Thomas, R. Tuladhar and M. Jacob, Ind. Eng. Chem. Res., 2024, 63, 1200–1214 CrossRef CAS.
- H. Wang, Y. Shao, S. Mei, Y. Lu, M. Zhang, J.-K. Sun, K. Matyjaszewski, M. Antonietti and J. Yuan, Chem. Rev., 2020, 120, 9363–9419 CrossRef CAS.
- H. Ma, E. Ibáñez-Alé, R. Ganganahalli, J. Pérez-Ramírez, N. López and B. S. Yeo, J. Am. Chem. Soc., 2023, 145, 24707–24716 CAS.
- N. Gao, C. Quiroz-Arita, L. A. Diaz and T. E. Lister, J. CO2 Util., 2021, 43, 101365 CrossRef CAS.
- H. Li, J. Gao, J. Shan, Q. Du, Y. Zhang, X. Guo, M. Xie, S. Wu and Z. Wang, J. Environ. Chem. Eng., 2021, 9, 106415 CrossRef CAS.
- A. Mezza, M. Bartoli, A. Chiodoni, J. Zeng, C. F. Pirri and A. Sacco, Nanomaterials, 2023, 13, 2314 CrossRef CAS PubMed.
- M. Abdinejad, Z. Mirza, X.-A. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS.
- Y. Zhou, Y. Liang, J. Fu, K. Liu, Q. Chen, X. Wang, H. Li, L. Zhu, J. Hu, H. Pan, M. Miyauchi, L. Jiang, E. Cortés and M. Liu, Nano Lett., 2022, 22, 1963–1970 CrossRef CAS PubMed.
- Y. Hori and S. Suzuki, J. Electrochem. Soc., 1983, 130, 2387 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS.
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, 2101741 CrossRef CAS.
- H. Wu, D. Ji, L. Li, D. Yuan, Y. Zhu, B. Wang, Z. Zhang and S. Licht, Adv. Mater. Technol., 2016, 1, 1600092 CrossRef.
- A. Prajapati, R. Sartape, M. T. Galante, J. Xie, S. L. Leung, I. Bessa, M. H. S. Andrade, R. T. Somich, M. V. Rebouças, G. T. Hutras, N. Diniz and M. R. Singh, Energy Environ. Sci., 2022, 15, 5105–5117 RSC.
- A. Douglas, R. Carter, N. Muralidharan, L. Oakes and C. L. Pint, Carbon, 2017, 116, 572–578 CrossRef CAS.
- J. Ren, F.-F. Li, J. Lau, L. González-Urbina and S. Licht, Nano Lett., 2015, 15, 6142–6148 CrossRef CAS PubMed.
- L. Hu, Y. Song, S. Jiao, Y. Liu, J. Ge, H. Jiao, J. Zhu, J. Wang, H. Zhu and D. J. Fray, ChemSusChem, 2016, 9, 588–594 CrossRef CAS.
- A. Douglas, R. Carter, M. Li and C. L. Pint, ACS Appl. Mater. Interfaces, 2018, 10, 19010–19018 CrossRef CAS.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS.
- Z. Liu, T. Yan, H. Shi, H. Pan and P. Kang, Appl. Catal., B, 2024, 343, 123456 CrossRef CAS.
- T. Yan, S. Liu, Z. Liu, J. Sun and P. Kang, Adv. Funct. Mater., 2024, 34, 2311733 CrossRef CAS.
- Z.-H. Zhao, J.-R. Huang, D.-S. Huang, H.-L. Zhu, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2024, 146, 14349–14356 CrossRef CAS PubMed.
- Y. Cheng, J. Hou and P. Kang, ACS Energy Lett., 2021, 6, 3352–3358 CrossRef CAS.
- J. Wang, X. Jing, Y. Yang, B. Xu, R. Jia and C. Duan, J. Am. Chem. Soc., 2024, 146, 19951–19961 CrossRef CAS.
- K. Van Daele, D. Balalta, S. Hoekx, R. Jacops, N. Daems, T. Altantzis, D. Pant and T. Breugelmans, ACS Appl. Energy Mater., 2024, 7, 5517–5527 CrossRef CAS.
- S. Mukhopadhyay, M. S. Naeem, G. Shiva Shanker, A. Ghatak, A. R. Kottaichamy, R. Shimoni, L. Avram, I. Liberman, R. Balilty, R. Ifraemov, I. Rozenberg, M. Shalom, N. López and I. Hod, Nat. Commun., 2024, 15, 3397 CrossRef CAS.
- S. Cui, C. Yu, X. Tan, W. Li, Y. Zhang and J. Qiu, Chem. Eng. J., 2023, 470, 144083 CrossRef CAS.
- C. P. O’Brien, R. K. Miao, A. Shayesteh Zeraati, G. Lee, E. H. Sargent and D. Sinton, Chem. Rev., 2024, 124, 3648–3693 CrossRef.
- L. Hu, B. Deng, K. Du, R. Jiang, Y. Dou and D. Wang, iScience, 2020, 23, 101607 CrossRef CAS.
- X.-M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2017, 10, 4109–4118 CrossRef CAS PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- L. Ge, H. Rabiee, M. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS.
- T. N. Nguyen and C.-T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS.
- M. Wang, L. Lin, Z. Zheng, Z. Jiao, W. Hua, G. Wang, X. Ke, Y. Lian, F. Lyu, J. Zhong, Z. Deng and Y. Peng, Energy Environ. Sci., 2023, 16, 4423–4431 RSC.
- Y.-J. Kong, Y.-B. Zou, T.-W. Jiang, K. Jiang and X.-M. Hu, ACS Sustainable Resour. Manage., 2024, 1, 778–786 CrossRef CAS.
- M. A. Blommaert, D. Aili, R. A. Tufa, Q. Li, W. A. Smith and D. A. Vermaas, ACS Energy Lett., 2021, 6, 2539–2548 CrossRef CAS PubMed.
- J. Lin, H. Chi, H. Liu, Q. Fan, T. Yan, S. Kuang, H. Wang, M. Li, Y. Yan, T. Zhang, S. Zhang and X. Ma, AlChE J., 2024, 70, e18382 CrossRef CAS.
- C. Liang, Y. Katayama, Y. Tao, A. Morinaga, B. Moss, V. Celorrio, M. Ryan, I. E. L. Stephens, J. R. Durrant and R. R. Rao, J. Am. Chem. Soc., 2024, 146, 8928–8938 CrossRef CAS.
- A. Khurram, L. Yan, Y. Yin, L. Zhao and B. M. Gallant, J. Phys. Chem. C, 2019, 123, 18222–18231 CrossRef CAS.
- S. Banerjee, X. Han and V. S. Thoi, ACS Catal., 2019, 9, 5631–5637 CrossRef CAS.
- Z. Tao, Z. Wu, Y. Wu and H. Wang, ACS Catal., 2020, 10, 9271–9275 CrossRef CAS.
- B. Deng, J. Tang, M. Gao, X. Mao, H. Zhu, W. Xiao and D. Wang, Electrochim. Acta, 2018, 259, 975–985 CrossRef CAS.
- B. Deng, M. Gao, R. Yu, X. Mao, R. Jiang and D. Wang, Appl. Energy, 2019, 255, 113862 CrossRef CAS.
- F. Sabatino, A. Grimm, F. Gallucci, M. van Sint Annaland, G. J. Kramer and M. Gazzani, Joule, 2021, 5, 2047–2076 CrossRef CAS.
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS.
- Z. Shi, Y. Tao, J. Wu, C. Zhang, H. He, L. Long, Y. Lee, T. Li and Y.-B. Zhang, J. Am. Chem. Soc., 2020, 142, 2750–2754 CrossRef CAS.
- H. Lyu, O. I.-F. Chen, N. Hanikel, M. I. Hossain, R. W. Flaig, X. Pei, A. Amin, M. D. Doherty, R. K. Impastato, T. G. Glover, D. R. Moore and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 2387–2396 CrossRef CAS PubMed.
- I. A. Digdaya, I. Sullivan, M. Lin, L. Han, W.-H. Cheng, H. A. Atwater and C. Xiang, Nat. Commun., 2020, 11, 4412 CrossRef CAS PubMed.
- M. D. Eisaman, K. Parajuly, A. Tuganov, C. Eldershaw, N. Chang and K. A. Littau, Energy Environ. Sci., 2012, 5, 7346–7352 RSC.
- J. K. Soeherman, A. J. Jones and P. J. Dauenhauer, ACS Eng. Au, 2023, 3, 114–127 CrossRef CAS.
- H. M. Almajed, R. Kas, P. Brimley, A. M. Crow, A. Somoza-Tornos, B.-M. Hodge, T. E. Burdyny and W. A. Smith, ACS Energy Lett., 2024, 9, 2472–2483 CrossRef CAS.
- K. Z. House, A. C. Baclig, M. Ranjan, E. A. van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428–20433 CrossRef CAS PubMed.
- S. Valluri and S. K. Kawatra, J. Environ. Sci., 2021, 103, 279–287 CrossRef CAS.
- A. Dal Pozzo, R. Moricone, A. Tugnoli and V. Cozzani, Ind. Eng. Chem. Res., 2019, 58, 6316–6324 CrossRef CAS.
- B. H. Ko, B. Hasa, H. Shin, E. Jeng, S. Overa, W. Chen and F. Jiao, Nat. Commun., 2020, 11, 5856 CrossRef CAS PubMed.
- W. Luc, B. H. Ko, S. Kattel, S. Li, D. Su, J. G. Chen and F. Jiao, J. Am. Chem. Soc., 2019, 141, 9902–9909 CrossRef CAS.
- S. Van Daele, L. Hintjens, S. Hoekx, B. Bohlen, S. Neukermans, N. Daems, J. Hereijgers and T. Breugelmans, Appl. Catal., B, 2024, 341, 123345 CrossRef CAS.
- R. D. K. I. Bogárdi and W. G. Ennenga, Nitrate Contamination: Exposure, Consequence, and Control, Springer Berlin, Heidelberg, 2013 Search PubMed.
- S. Banerjee, Z.-Q. Zhang, A. S. Hall and V. S. Thoi, ACS Catal., 2020, 10, 9907–9914 CrossRef CAS.
- D. J. D. Pimlott, A. Jewlal, Y. Kim and C. P. Berlinguette, J. Am. Chem. Soc., 2023, 145, 25933–25937 CrossRef CAS.
- S. Kim, M. P. Nitzsche, S. B. Rufer, J. R. Lake, K. K. Varanasi and T. A. Hatton, Energy Environ. Sci., 2023, 16, 2030–2044 RSC.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Energy Environ. Sci., 2019, 12, 1950–1968 RSC.
- S. S. Bhargava, F. Proietto, D. Azmoodeh, E. R. Cofell, D. A. Henckel, S. Verma, C. J. Brooks, A. A. Gewirth and P. J. A. Kenis, ChemElectroChem, 2020, 7, 2001–2011 CrossRef CAS.
- L.-P. Chi, Z.-Z. Niu, Y.-C. Zhang, X.-L. Zhang, J. Liao, Z.-Z. Wu, P.-C. Yu, M.-H. Fan, K.-B. Tang and M.-R. Gao, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312876120 CrossRef CAS PubMed.
- X. Wang, A. Xu, F. Li, S.-F. Hung, D.-H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- J. B. Greenblatt, D. J. Miller, J. W. Ager, F. A. Houle and I. D. Sharp, Joule, 2018, 2, 381–420 CrossRef CAS.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. O’Brien, A. Gaona, J. Wicks, A. Obradović, A. Sarkar, S. A. Jaffer, H. L. MacLean, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 4405–4412 CrossRef CAS.
- A. Naquash, M. A. Qyyum, Y. D. Chaniago, A. Riaz, F. Yehia, H. Lim and M. Lee, Chemosphere, 2023, 313, 137420 CrossRef CAS.
- B. Mahida, H. Benyounes and W. Shen, Chem. Pap., 2021, 75, 599–609 CrossRef CAS.
- P. Debergh, O. Gutiérrez-Sánchez, M. N. Khan, Y. Y. Birdja, D. Pant and M. Bulut, ACS Energy Lett., 2023, 8, 3398–3403 CrossRef CAS.
- S. Sun, H. Sun, P. T. Williams and C. Wu, Sustainable Energy Fuels, 2021, 5, 4546–4559 RSC.
- V. S. K. Yadav, M. J. Al-Marri, M. A. H. S. Saad and A. Kumar, Emergent Mater., 2024, 7, 1–16 CrossRef CAS.
- Y. Xie, J. Chen, J. Wen, Z. Li, F. Cao, S. Zhang, Q. Sun, P. Ning, Q. Zhang and J. Hao, ACS Catal., 2024, 14, 12214–12224 CrossRef CAS.
- S. Verma, B. Kim, H.-R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- W. Shan, R. Liu, H. Zhao and J. Liu, J. Phys. Chem. Lett., 2022, 13, 7296–7305 CrossRef CAS.
- E. P. Delmo, Y. Wang, Y. Song, S. Zhu, H. Zhang, H. Xu, T. Li, J. Jang, Y. Kwon, Y. Wang and M. Shao, J. Am. Chem. Soc., 2024, 146, 1935–1945 CrossRef CAS PubMed.
- H. Jing, J. Long, H. Li, X. Fu and J. Xiao, ACS Catal., 2023, 13, 9925–9935 CrossRef CAS.
- S. Gorthy, S. Verma, N. Sinha, S. Shetty, H. Nguyen and M. Neurock, ACS Catal., 2023, 13, 12924–12940 CrossRef CAS.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X.-M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef CAS.
- J. Bi, P. Li, J. Liu, Y. Wang, X. Song, X. Kang, X. Sun, Q. Zhu and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202307612 CrossRef CAS PubMed.
- K. Chu, W. Zong, G. Xue, H. Guo, J. Qin, H. Zhu, N. Zhang, Z. Tian, H. Dong, Y.-E. Miao, M. B. J. Roeffaers, J. Hofkens, F. Lai and T. Liu, J. Am. Chem. Soc., 2023, 145, 21387–21396 CrossRef CAS PubMed.
- C. Zhang, Y. Zhang, R. Deng, L. Yuan, Y. Zou, T. Bao, X. Zhang, G. Wei, C. Yu and C. Liu, Adv. Mater., 2024, 36, 2313844 CrossRef CAS.
- S. D. Fried and S. G. Boxer, Annu. Rev. Biochem., 2017, 86, 387–415 CrossRef CAS PubMed.
- C. Mittal, C. Hadsbjerg and P. Blennow, Chem. Eng. World, 2017, 52, 44–46 Search PubMed.
- Renewable methanol plant: first production of fuel from CO2 at industrial scale, https://carbonrecycling.com/projects/george-olah.
- D. Rusmanis, R. O’Shea, D. M. Wall and J. D. Murphy, Bioengineered, 2019, 10, 604–634 CrossRef CAS.
- M. Li, E. Irtem, H.-P. Iglesias van Montfort, M. Abdinejad and T. Burdyny, Nat. Commun., 2022, 13, 5398 CrossRef CAS.
- J. H. Rheinhardt, P. Singh, P. Tarakeshwar and D. A. Buttry, ACS Energy Lett., 2017, 2, 454–461 CrossRef CAS.
- S. Voskian and T. A. Hatton, Energy Environ. Sci., 2019, 12, 3530–3547 RSC.
- S. Dongare, O. K. Coskun, E. Cagli, K. Y. C. Lee, G. Rao, R. D. Britt, L. A. Berben and B. Gurkan, ACS Catal., 2023, 13, 7812–7821 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cs00480a |
| This journal is © The Royal Society of Chemistry 2025 |