
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimizing photocatalysis via electron spin control
Shaoxiong
He
 ab,
Yanxi
Chen
b,
Jingyun
Fang
*b,
Yijiang
Liu
*ac and
Zhiqun
Lin
*a
ab,
Yanxi
Chen
b,
Jingyun
Fang
*b,
Yijiang
Liu
*ac and
Zhiqun
Lin
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: z.lin@nus.edu.sg
bSchool of Environmental Science and Engineering, Guangdong Provincial Key Laboratory of Environmental Pollution Control and Remediation Technology, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: fangjy3@mail.sysu.edu.cn
cCollege of Chemistry, Key Lab of Environment-Friendly Chemistry and Application in Ministry of Education, Xiangtan University, Xiangtan 411105, Hunan Province, P. R. China. E-mail: liuyijiang84@xtu.edu.cn
First published on 22nd January 2025
Abstract
Solar-driven photocatalytic technology holds significant potential for addressing energy crisis and mitigating global warming, yet is limited by light absorption, charge separation, and surface reaction kinetics. The past several years has witnessed remarkable progress in optimizing photocatalysis via electron spin control. This approach enhances light absorption through energy band tuning, promotes charge separation by spin polarization, and improves surface reaction kinetics via strengthening surface interaction and increasing product selectivity. Nevertheless, the lack of a comprehensive and critical review on this topic is noteworthy. Herein, we provide a summary of the fundamentals of electron spin control and the techniques employed to scrutinize the electron spin state of active sites in photocatalysts. Subsequently, we highlight advanced strategies for manipulating electron spin, including doping design, defect engineering, magnetic field regulation, metal coordination modulation, chiral-induced spin selectivity, and combined strategies. Additionally, we review electron spin control-optimized photocatalytic processes, including photocatalytic water splitting, CO2 reduction, pollutant degradation, and N2 fixation, providing specific examples and detailed discussion on underlying mechanisms. Finally, we outline perspectives on further enhancing photocatalytic activity through electron spin manipulation. This review seeks to offer valuable insights to guide future research on electron spin control for improving photocatalytic applications.
 Shaoxiong He | Shaoxiong He is a PhD candidate in Professor Jingyun Fang's group in the School of Environmental Science and Engineering at Sun Yat-sen University, China. He is also a visiting student in Professor Zhiqun Lin's group in the Department of Chemical and Biomolecular Engineering at the National University of Singapore. His research focuses on photocatalyst design for energy conversion and environmental remediation. |
 Yanxi Chen | Yanxi Chen is a PhD candidate in the School of Environmental Science and Engineering at Sun Yat-sen University, China. Her research focuses on photocatalyst design and microbiome engineering for environmental remediation. |
 Jingyun Fang | Dr Jingyun Fang is a Professor in the School of Environmental Science and Engineering at Sun Yat-sen University, China. She received her bachelor and doctoral degrees from Harbin Institute of Technology in 2003 and 2010, respectively. She was a postdoctoral fellow at the Hong Kong University of Science & Technology between 2010 and 2012. Her research focuses on water chemistry, environmental functional materials, and physicochemical water treatment technologies including oxidation/reduction processes, and formation & control of disinfection byproducts. |
 Yijiang Liu | Dr Yijiang Liu is a Professor in the College of Chemistry at Xiangtan University, China. She received her MS degree from Xiangtan University in 2009 and PhD degree from the Institute of Chemistry, Chinese Academy of Sciences in 2015. She was a visiting scholar at Georgia Institute of Technology during 2017–2018. Her research work focuses on design and preparation of advanced porous carbon materials as efficient and durable electrocatalysts for energy conversion and storage; crafting of highly stable perovskite nanocrystals via functional polymer nanoreactors; and Janus materials for concurrent catalysis and emulsification. |
 Zhiqun Lin | Dr Zhiqun Lin is a Professor in the Department of Chemical and Biomolecular Engineering at the National University of Singapore. His research interests include photocatalysis, solar energy conversion, electrocatalysis, batteries, semiconductor organic–inorganic nanocomposites, multifunctional nanocrystals, conjugated polymers, block copolymers, hierarchical structure formation and assembly, and surface and interfacial properties. |
1. Introduction
The rising consumption of fossil fuels and the escalating threat of global warming underscore the urgent need for renewable, clean energy sources to address the energy crisis and associated environmental challenges. Solar energy, in particular, is as an inexhaustible and eco-friendly resource. Remarkably, the solar energy received by the Earth in just one hour (4.3 × 1020 J) is equivalent to the entire global energy consumption for an entire year (4.1 × 1020 J).1 Forecasts predict that by 2035, over 50% of global power generation will be derived from renewable sources, predominantly solar energy, highlighting the crucial role of solar energy conversion technology in future sustainable development.2 Solar-driven photocatalytic technology has been extensively researched to facilitate water splitting for green hydrogen production and convert CO2 into solar fuels (e.g., CO, CH4, CH3OH, and C2H5OH), offering an effective solution to both energy and environmental crises.3–6 Despite significant achievements in photocatalysis over recent decades – such as doping design, co-catalyst introduction, defect engineering, heterojunction construction, and external field modulation – its efficiency remains constrained by challenges including limited light absorption, inefficient charge separation, and insufficient surface reaction kinetics.7–9 Thus, developing a universal strategy to overcome these limitations and achieve highly efficient solar energy conversion is of paramount importance.Recently, electron spin control has emerged as a promising strategy for enhancing photocatalytic performance.10–13 Electron spin control involves the manipulation of both electron spin and spin states. Electron spin refers to the intrinsic angular momentum of an electron, characterized by spin-up or spin-down configurations. In contrast, the electron spin state describes the overall spin arrangement of electrons within an atomic orbital and can be classified into singlet, doublet, triplet, and other multiplet states.14,15 These two concepts are fundamental intrinsic characteristics that predominantly govern the physical properties and chemical behaviors of photocatalysts, offering innovative approaches to enhance photocatalytic efficiency.12–16 For example, electron spin control can be used to tune the energy band structures of photocatalysts, extending their light absorption capabilities across a broader range of solar spectra, including visible and near-infrared (NIR) light.17–19 Moreover, spin polarization can promote the separation efficiency of photogenerated electrons and holes, accelerating charge migration to the photocatalyst surface.13,20–23 Additionally, the interaction between photocatalysts and reactants is strengthened by modulating the electron spin state of active sites, improving surface reaction kinetics.24–26 Notably, aligning electron spin in specific directions has been proven to enhance the selectivity for desired products, offering a novel strategy for regulating surface reaction pathways. For example, this approach favors the production of triplet O2 over singlet hydrogen peroxide (H2O2) in photocatalytic water splitting processes.12,13,26–29 Therefore, electron spin control holds great potential for broadening light absorption range, mitigating charge recombination, and boosting the efficiency and selectivity of surface reactions in photocatalysis. Despite the growing interest in electron spin manipulation strategies for boosting photocatalytic activity over the past decade, a comprehensive and critical review summarizing the mechanisms, strategies, and applications of electron spin control in photocatalysis remains notably absent.
In this review, we aim at providing a systematic overview of how electron spin control can optimize photocatalysis, the strategies and materials involved, and their applications in various photocatalytic processes (Fig. 1). We begin with the fundamentals of photocatalysis and electron spin control, followed by exploration of the role of electron spin control in photocatalysis. Subsequently, we highlight advanced strategies for manipulating electron spin, including doping design, defect engineering, magnetic field regulation, metal coordination modulation, chiral-induced spin selectivity, and combined approaches. We also discuss recent developments in these strategies as applied to photocatalytic water splitting, CO2 reduction, pollutant degradation, and N2 fixation. Finally, we outline current challenges and future research directions in this rapidly evolving field.
 | ||
| Fig. 1 Overview of the mechanisms, strategies, and photocatalytic applications of electron spin control. | ||
2. Fundamentals and challenges of photocatalysis
2.1 Fundamentals of photocatalysis
Fig. 2 depicts the detailed steps of photocatalysis in a photocatalyst, from light excitation to surface redox reaction. First, when a photocatalyst is exposed to light with energy equal to or greater than its band gap (step 1), photogenerated electrons are excited from the valence band (VB) to the conduction band (CB), leaving holes in the VB (step 2). Some electron–hole pairs may return to the ground state via radiative or non-radiative relaxation, typically occurring on a nanosecond to picosecond time scale, which hinders the formation of free charge carriers. Some electrons and holes can overcome the exciton binding energy and separate into free charge carriers (step 3). However, these charge carriers may recombine within the bulk of photocatalyst (step 4), but if their diffusion length is sufficient, they can reach the surface of the photocatalyst (step 5). The diffusion of charge carriers typically happens on a picosecond time scale and requires a concentration gradient to drive their movement. Unfortunately, some charge carriers may recombine before they can participate in surface reactions (step 6). Ultimately, the photocatalytic process concludes as the remaining charge carriers are consumed in surface reactions (steps 7 and 8), which generally occur on a longer time scale. In the surface reactions, electron-driven reduction enables H2 production and the conversion of CO2 into solar fuels, N2 into NH3 or NH4+, and O2 into superoxide radical (O2˙−) or H2O2. Meanwhile, hole-driven oxidation can convert H2O into hydroxyl radicals (HO˙), H2O2, or O2.9,30–32 | ||
| Fig. 2 Schematic diagram of photocatalytic mechanism. | ||
Notably, the surface redox reactions can only be effectively driven by photogenerated electrons and holes when the redox potentials lie between the CB and VB potentials of a photocatalyst. For instance, to achieve overall water splitting, the CB must be more negative than the reduction potential for H2 production, while the VB must be more positive than the oxidation potential required for O2 generation. The redox potentials of different species in photocatalysis are shown in Fig. 3. It is accepted that a more negative CB position of a photocatalyst enhances its ability to drive reduction reactions, while a more positive VB position favors oxidation reactions. Interestingly, multi-electron reduction of O2 and CO2 are thermodynamically more favorable than their single-electron counterparts due to their lower reduction potentials. Similarly, the four-electron oxidation of H2O is more readily achieved than the two-election and single-electron oxidations, as the former has a higher oxidation potential. Therefore, whether a photocatalytic reaction can occur and its efficiency largely depend on the CB and VB positions of photocatalysts in relation to the redox potentials of target reactions. This provides guidance for selecting appropriate photocatalysts for specific target reactions. For example, TiO2, with a CB potential of −0.5 V and a VB potential of 2.7 V, is a commonly used photocatalyst for water splitting due to its suitable band potentials, stable chemical property, and low cost.33 Typically, photocatalysts with a larger band gap have VB and CB potentials that are more suitable for overall water splitting, but this often results in reduced light absorption capability. The absorption spectrum of TiO2 is confined to the ultraviolet (UV) range (about 4% of sunlight), limiting its visible light utilization efficiency. Studies have shown that doping TiO2 with metal or non-metal elements (e.g., N, S, Fe, and Co), can extend its light absorption into the visible region.33 In contrast, g-C3N4 and CdS, which have smaller band gaps, can absorb visible light and possess CB potentials of approximately −1.1 V and −0.8 V, respectively.34,35 This makes them more effective for H2 generation compared to TiO2. However, their VB potentials are inadequate for water oxidation, so a co-catalyst or a heterojunction with other suitable semiconductors is needed to achieve overall water splitting. Thus, it is essential to carefully balance and optimize the trade-off between redox potential (determined by CB and VB potentials) and light absorption capability (governed by band gap). Additionally, for certain specific reactions, the selection of photocatalyst can be based primarily on its VB or CB potential. For instance, in CO2 reduction, a photocatalyst with a relatively negative CB is ideal, as it enhances the reduction process irrespective of the VB potential.9 On the contrary, for the degradation of organic pollutants, photocatalysts with a relatively positive VB potential is preferable. This is because they possess stronger hole oxidation capability, allowing them to generate highly reactive HO˙, which significantly improves the efficiency of organic pollutant degradation.
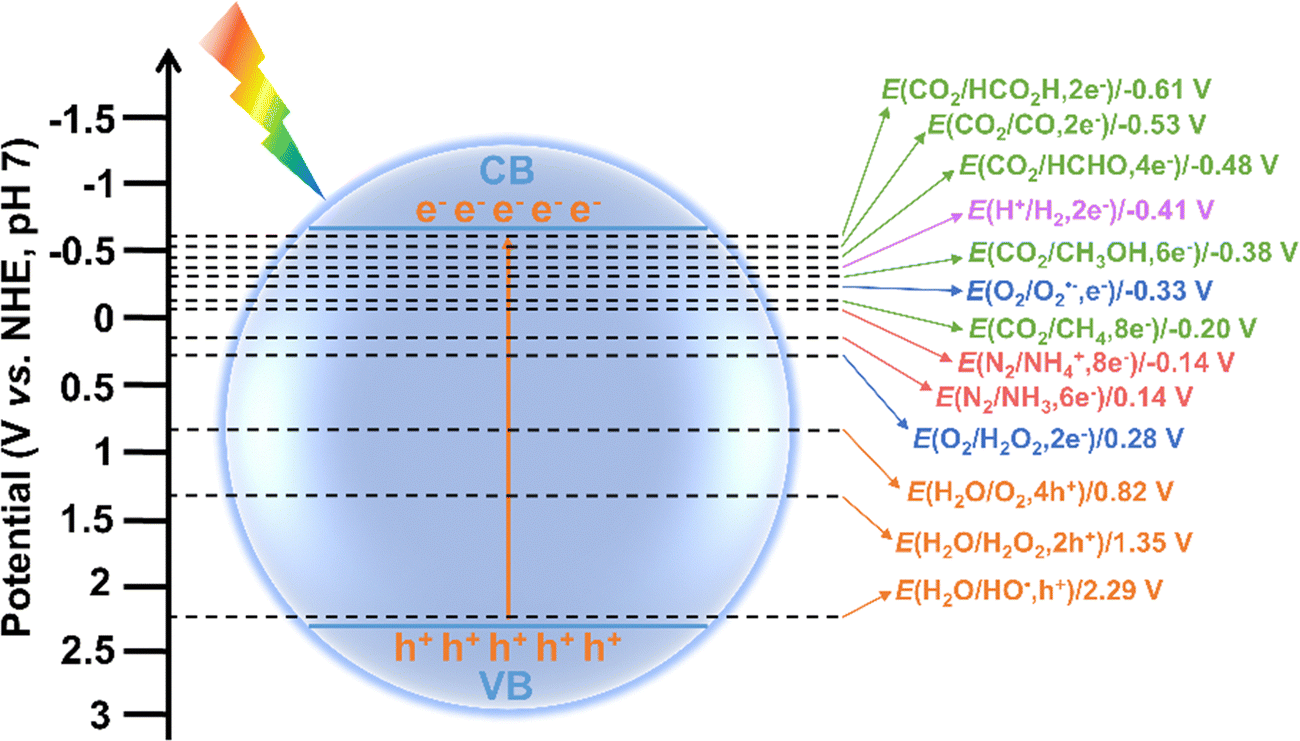 | ||
| Fig. 3 Redox potentials of different species in photocatalysis. | ||
The overall efficiency of photocatalysis is primarily influenced by the combined effects of three key steps: light absorption, charge separation, and surface reaction kinetics. Consequently, the solar energy conversion efficiency can be determined by eqn (1).
| η = ηabs × ηcs × ηsr | (1) |
Quantum efficiency (QE) is another key metric for evaluating photocatalytic reactions. It quantifies the rate at which a specific event occurs per absorbed photon and is used to assess the efficiency of reactant consumption, product formation, and light emission in photophysical and photochemical reactions.36 In photocatalysis, a higher QE indicates that more photons are effectively utilized in the reaction, suggesting a more efficient photocatalyst. Optimizing QE can lead to improved photocatalyst design and enhanced performance in photocatalytic reactions. The actual QE, often referred to as internal quantum efficiency (IQE), is defined as the number of reacted electrons participating in the reaction to the total number of photons absorbed over a given period.2 However, due to challenges such as light scattering and reflection during photocatalysis, directly measuring the number of photons absorbed is difficult. As a result, apparent quantum efficiency (AQE) is typically used as an alternative. AQE is determined by the number of reacted electrons to the number of incident photons. Common detectors used to measure incident photons include thermopiles and silicon photodiodes, which can accurately gauge the flux of incident photons. Since light intensity within the irradiated area is often uneven, it is advisable to calculate the photon number by integrating the photon flux across the entire illuminated area. This involves dividing the illuminated area into smaller segments, measuring the photon flux in each segment, and then integrating these measurements to obtain an average photon number. The IQE and AQE of a photocatalyst can be determined by eqn (2) and (3).
 | (2) |
 | (3) |
2.2 Photocatalytic applications
Photocatalytic technology holds tremendous promise in the fields of modern energy and environmental science, particularly in key processes such as H2 production via water splitting, CO2 reduction, pollutant degradation, and N2 fixation. By harnessing light energy to drive chemical reactions, photocatalysis not only enables the efficient generation of renewable energy but also offers a low-energy, environmentally friendly solution for reducing greenhouse gas emissions and addressing environmental pollution. In water splitting, photocatalysts can directly decompose water into H2 and O2, providing a clean energy source; in CO2 reduction, they transform greenhouse gases into valuable fuels and chemicals, contributing to the development of a carbon-recycling economy. Additionally, in pollutant degradation and N2 fixation, photocatalytic technology plays a crucial role in environmental remediation and agricultural production. Due to its sustainability and versatility, photocatalysis has emerged as a promising solution to global energy and environmental challenges. As photocatalysts and reaction mechanisms continue to improve, the technology is poised to make an even greater impact in large-scale industrial applications.In the photocatalytic H2 production half-reaction, research focuses on optimizing photocatalysts and reaction conditions to improve the H2 production rate and quantum efficiency. Studies have shown that introducing precious metal nanoparticles with surface plasmon resonance effects, such as Pt and Au, can enhance light absorption and improve the separation efficiency of photogenerated charge carriers.2 Additionally, regulating the crystal face structure of the photocatalyst, such as exposing specific crystal facets, can increase the selectivity and efficiency of the H2 production reaction.37
The main challenge in the photocatalytic O2 production half-reaction lies in the high overpotential and slow reaction kinetics of the water oxidation reaction. To address this, researchers have introduced highly active oxidation catalysts, such as Co3O4 and IrO2, to accelerate the water oxidation process.38 Moreover, constructing 3D nanostructured materials, such as BiVO4 photoanodes, has increased the contact area between light absorption sites and the reaction interface, thereby enhancing water oxidation efficiency.39
Despite advancements, photocatalytic overall water splitting still faces significant challenges, particularly in simultaneously improving the efficiency of both H2 and O2 production reactions. Strategies like constructing Z-scheme heterojunctions and photo-alloy catalysts have been shown to promote the overall water splitting.30 Additionally, utilizing two-photon excitation and multi-electron transfer mechanisms has improved the quantum efficiency of overall water splitting.2 As research deepens our understanding of photocatalytic mechanisms and photocatalysts continue to be optimized, photocatalytic water splitting holds great potential for practical applications in the future.
A crucial aspect of optimizing the efficiency and selectivity is adjusting both the reaction conditions and the characteristics of the photocatalyst. Research has demonstrated that the reaction efficiency and the selectivity for different products can be influenced by adjusting reaction conditions, such as temperature, pressure, CO2 concentration, and light intensity.5 Moreover, the fabrication of heterostructured photocatalysts with well-aligned energy bands is an effective strategy for achieving a broad spectral response and efficient charge separation.30 Additionally, photocatalytic CO2 reduction can be promoted using co-catalysts such as Au, Ag, Cu, and Bi, primarily due to the localized surface plasmon resonance effect.40 Notably, by altering the compositions, morphologies, and crystal structures of photocatalysts, their electronic structure can be modified, which in turn enhances its selectivity for specific products.5 With a deeper understanding of the reaction mechanisms and the development of novel photocatalysts, it is anticipated that more precise control over the products of photocatalytic CO2 reduction will be achieved in the future.
Currently, many researchers utilize photocatalysts in combination with common oxidants, such as H2O2, O3, persulfate, and hypochlorous acid.44–47 The reactions between electrons or holes and these oxidants generate additional free radicals, significantly enhancing pollutant degradation efficiency. Therefore, photocatalytic pollutant degradation technology offers a promising solution for environmental remediation.
Recent advancements in photocatalytic N2 fixation focus on the development of new photocatalysts, the optimization of surface active sites, and the enhancement of electron transfer efficiency. To address challenges related to N2 adsorption and high activation energy barriers, researchers have employed strategies such as morphology control, element doping, defect engineering, and heterojunction design.48–50 These approaches improve N2 adsorption and activation while reducing the recombination rate of photogenerated charge carriers. These innovations lay the groundwork for achieving efficient NH3 production at room temperature and pressure, paving the way for its large-scale application.
2.3 Current challenges in photocatalysis
Despite considerable progress in photocatalysis over the past few decades, its efficiency remains hindered by challenges including limited light absorption, inefficient charge separation, and insufficient surface reaction kinetics.Researchers have explored various strategies for adjusting the band gap of photocatalysts to enhance light absorption. One common approach is doping, where transition metals (e.g., Fe, Co, and Mn) and non-metals (e.g., N, S, and C) are introduced into photocatalysts.30 This can reduce the band gap and extend the absorption range to visible and NIR regions. However, doping may introduce impurity states that increase the recombination rate of photogenerated charge carriers, potentially reducing quantum efficiency. Another method to extend the absorption range is sensitization with organic dyes and quantum dots.51,52 However, the long-term stability of these sensitizers and their susceptibility to photodegradation under extended illumination remain challenges. Additionally, some narrow bandgap materials, such as black phosphorus,53 Ag2S,54 and ZnIn2S4,55 have been developed to absorb visible and even NIR light. Despite this, these materials are often vulnerable to photocorrosion and rapid recombination of electrons and holes, leading to reduced activity and poor long-term stability. Continued optimization is needed to achieve both efficient and stable visible-to-NIR solar energy conversion.
To address this issue, researchers have developed several strategies with notable progress. One effective approach is constructing semiconductor heterojunctions with appropriately matched energy band structures.30 This creates an interface that forms an electric field, which facilitates the migration of electrons and holes to different semiconductors, thereby reducing their recombination and enhancing photocatalytic efficiency. For example, the TiO2/g-C3N4 heterojunction combines TiO2's high photocatalytic activity with g-C3N4's visible light absorption capability, utilizing the built-in electric field to achieve improved charge separation.56,57 However, designing heterojunctions requires precise band structure alignment between the semiconductors, which limits the range of materials that can be used. Moreover, many materials face challenges in band structure matching and lattice compatibility, reducing the diversity and flexibility of heterojunction photocatalysts.
Introducing suitable co-catalysts (e.g., Pt, Pd, RuO2, and Co3O4) is another widely studied approach to promote charge separation.2,30 These co-catalysts facilitate the transport of electrons or holes, thereby boosting the overall photocatalytic performance. For instance, Pt and RuO2 are commonly used in photocatalytic water splitting to enhance H2 and O2 production, respectively, by serving as sinks for electrons and holes.58 However, efficient co-catalysts, particularly precious metals such as Pt, Au, and Pd, are costly, limiting their large-scale use. Additionally, the chemical stability of these co-catalysts may diminish over time, posing challenges for long-term photocatalytic applications. Therefore, there remains a need to develop efficient, cost-effective, and versatile strategies to facilitate charge separation.
Current approaches for improving surface reaction kinetics focus on increasing the number of active sites on photocatalyst surface and enhancing the adsorption of reactants and intermediates. For example, compared to bulk materials, the design of nanomaterials (e.g., nanosheets, nanowires, and nanospheres) increases the specific surface area and provides more active sites.59 Additionally, the introduction of co-catalysts can increase active sites and lower reaction energy barriers, facilitating better reactant adsorption and more efficient reaction pathways with reduced energy barriers.60 Creating defects within photocatalyst structure can also offer extra reaction sites and enhance the adsorption of reactants.61 Furthermore, functionalizing photocatalyst surface with suitable ligands (e.g., amino acids, alcohols, and organic acids) strengthens the interaction between the photocatalysts and target molecules, thereby boosting surface reaction kinetics.62
Notably, selective product generation, where catalysts preferentially produce desired products while minimizing the formation of byproducts, is crucial for maximizing surface reaction efficiency. However, common photocatalysts may lack high selectivity, leading to the formation of various byproducts, which reduces the yields of target products and complicates purification. For instance, the selectivity for O2 production from H2O is often unsatisfied, as it tends to produce H2O2 as a byproduct, making this the rate-limiting step in photocatalytic water splitting.12 Similarly, in the photocatalytic degradation of organic pollutants, photocatalysts may generate toxic intermediates and byproducts such as phenols and aldehydes.45,63 These byproducts not only reduce the overall effectiveness but also pose additional environmental risks. Therefore, it is essential to develop highly selective photocatalysts to optimize surface reactions.
While photocatalysis is hindered by limited light absorption, inefficient charge separation, and insufficient surface reaction kinetics, recent research has proposed innovative solutions to overcome these challenges. Among these, electron spin control has emerged as a particularly promising method. By manipulating the spin state of electrons within photocatalysts, electron spin control can optimize photocatalysis through enhancing light absorption, promoting charge separation, and improving surface reaction kinetics. In the following sections, we will summarize the fundamentals of electron spin control and its role in photocatalysis.
3. Fundamentals of electron spin control and its role in photocatalysis
3.1 Theories and principles of electron spin and spin state
In this review, electron spin control encompasses the manipulation of both electron spin and spin states. Electron spin refers to the intrinsic angular momentum of an electron, characterized by the spin magnetic quantum number ms, which can be either spin up (ms = ½) or spin down (ms = −½) (Fig. 4a); while electron spin state describes the overall spin configuration of electrons in an atomic orbital and can be classified as singlet, doublet, triplet, etc.14,15 These fundamental concepts significantly impacts the physical properties and chemical behavior of atoms, molecules, and materials. | ||
| Fig. 4 Schematic diagram of electron spin and spin state. (a) Spin-up and spin-down electrons. (b) Spatial illustration of the five d-orbitals. (c) Crystal field splitting of d-orbitals in a metal cation. | ||
The theories and principles of electron spin and spin state have been developed over the past century, establishing a solid foundation for research related to electron spin control. In 1925, Wolfgang Pauli proposed the famous Pauli exclusion principle, stating that each spatial orbital can accommodate a maximum of two electrons with opposite spin directions, characterized by ms of ½ or −½.64 In electron-pair bonds, electrons can exist in a singlet state, where spins are antiparallel, the spin wavefunction is antisymmetric, and the spatial part of the wavefunction is symmetric. Alternatively, they can occupy a triplet state, where spins are parallel, the spin wavefunction is symmetric, and the spatial part of the wavefunction is antisymmetric. This principle serves as the cornerstone for comprehending the electronic structure of atoms and the organization of elements in Mendeleev periodic table. At the end of 1925, George Uhlenbeck and Samuel Goudsmit introduced the concept of electron spin, which refers to the intrinsic angular momentum of an electron, independent of its orbital angular momentum.65 After that, Friedrich Hund stated that electrons preferentially occupy different orbitals with parallel spins because of the minimum energy principle.66 Hund's rules predict and explain the spin configuration of electrons, laying the foundation for the development of electron spin state. However, electron spin and spin state was not incorporated into quantum theory until 1928, when Paul A. M. Dirac published the Dirac equation to further explain the spin and magnetic moment of electrons.67 In the decades that followed, research on electron spin and spin states spurred the advancement of spin-related fields, such as spintronics and quantum computing.
3.2 Electron spin states in photocatalysis
In recent years, the electron spin states in photocatalysis have garnered significant attention due to their notable impacts on the photocatalytic performance.12,13,21,68 Typically, the central metal atom in metallic photocatalysts forms an octahedral coordination structure with six surrounding ligand atoms. In this arrangement, the electrons in the metal's d orbitals repel the electrons of the ligands, causing those electrons closer to the ligands to have higher energy than those further away. As a result, the d orbitals split into two groups with distinct energy levels: the dxy, dxz, and dyz orbitals, known as the t2g orbitals, have lower energy; while the dz2 and dx2−y2 orbitals, referred to as the eg orbitals, have higher energy (Fig. 4b).The splitting of metal d orbitals can alter the balance between crystal field splitting energy (the energy required to split the metal d orbitals) and spin pairing energy (the energy needed for two electrons to occupy the same orbital), leading to changes in the electron spin state. The spin states are categorized as low, intermediate, and high spin states (Fig. 4c). Generally, when the crystal field splitting energy exceeds the spin pairing energy, electrons first fill the t2g orbitals with two electrons of opposite spins. Then, electrons occupy the eg orbitals singly until the energy needed to fill an additional orbital surpasses the spin pairing energy. Conversely, if the crystal field splitting energy is lower than the spin pairing energy, electrons occupy all orbitals with similar energy levels singly before pairing up. As systems tend to minimize their total energy, those with crystal field splitting energy greater than spin pairing energy typically exhibit low spin configurations, while those with lower crystal field splitting energy favor high spin configurations.
3.3 Techniques for investigating electron spin state
The study of electron spin state is crucial for understanding spin-dependent chemistry. Commonly used techniques include X-ray absorption spectroscopy (XAS), Mössbauer spectroscopy, electron-energy-loss spectroscopy (EELS), electron spin resonance (ESR) spectroscopy, temperature-dependent magnetization (M–T) measurement, and theoretical modeling and calculations (Fig. 5). | ||
| Fig. 5 Techniques for studying electron spin state. | ||
In general, XAS offers high-resolution energy distribution that aid in determining the electron density of states and local spin state, particularly at the K-edge and L-edge absorption peaks. However, for metal centers with multiple coordination types, quantifying their coordination is challenging, as XAS provides average values for all absorbed atoms instead of a specific single site.14
Mössbauer spectroscopy provides advantages including strong resistance to interference and applicability to various sample types such as powders, thin films, and bulk materials. However, it is limited to the analysis of 57Fe and 119Sn and is primarily used to study the spin states in Fe-based catalysts.
EELS offers high energy resolution, enabling precise differentiation of spin states within the local band structure. When paired with transmission electron microscopy (TEM), it provides nanometer-scale spatial resolution for detailed spin state analysis in localized regions of a material. However, EELS requires samples to be sufficiently thin and stable.
ESR spectroscopy is highly sensitive to paramagnetic substances, including free radicals, transition metal ions, and certain metal oxides, even at extremely low concentrations. It generally does not damage the sample and can be used across various states (e.g., solids, liquids, and gases) and conditions (e.g., room and low temperatures). However, ESR is limited to samples with unpaired electrons and cannot provide information about systems with fully paired electrons.
 where μB is the Bohr magneton with a value of 9.274 × 10−24 J T−1. After getting μeff, the effective spin quantum number Seff can be calculated by Seff = μeff/(gμB), where g is the Landé factor.14,15Seff determines the arrangement of electrons in the atomic orbits and the spin state transitions. For example, the spin states of CoIII were determined using M–T measurements at a 1000 Oe magnetic field under field-cooling procedures, demonstrating a spin state transition from low spin to high spin in CoIII.79
where μB is the Bohr magneton with a value of 9.274 × 10−24 J T−1. After getting μeff, the effective spin quantum number Seff can be calculated by Seff = μeff/(gμB), where g is the Landé factor.14,15Seff determines the arrangement of electrons in the atomic orbits and the spin state transitions. For example, the spin states of CoIII were determined using M–T measurements at a 1000 Oe magnetic field under field-cooling procedures, demonstrating a spin state transition from low spin to high spin in CoIII.79
M–T measurements reveal the magnetic phase transition energy of a material across different temperatures and identify temperature-dependent spin state transitions, such as the shift from paramagnetism to ferromagnetism. However, M–T measurements are primarily applicable to samples exhibiting paramagnetism or ferromagnetism and have limited use for nonmagnetic materials. Additionally, for samples with TC exceeding 800 K, the Curie constant may not be readily accessible.
Theoretical modeling and calculations can predict the electronic structures and chemical properties of materials prior to experiments, thereby guiding experimental design and optimizing materials. Additionally, they can forecast the chemical properties of materials under extreme conditions that may be challenging to achieve experimentally. However, the models used in theoretical calculations may not perfectly align with actual experimental conditions, potentially limiting the credibility of the results.
3.4 The role of electron spin control in photocatalysis
In recent years, electron spin control has driven notable advances in photocatalysis by: (i) enhancing light absorption of photocatalysts through tuning their energy band structures, enabling effective utilization of visible to NIR light; (ii) promoting charge separation by spin polarization effect, which accelerates the migration of photogenerated electrons and holes to the photocatalyst surface; and (iii) improving surface reaction kinetics through strengthening the interaction between photocatalysts and reactants and increasing the selectivity of target products (Fig. 6).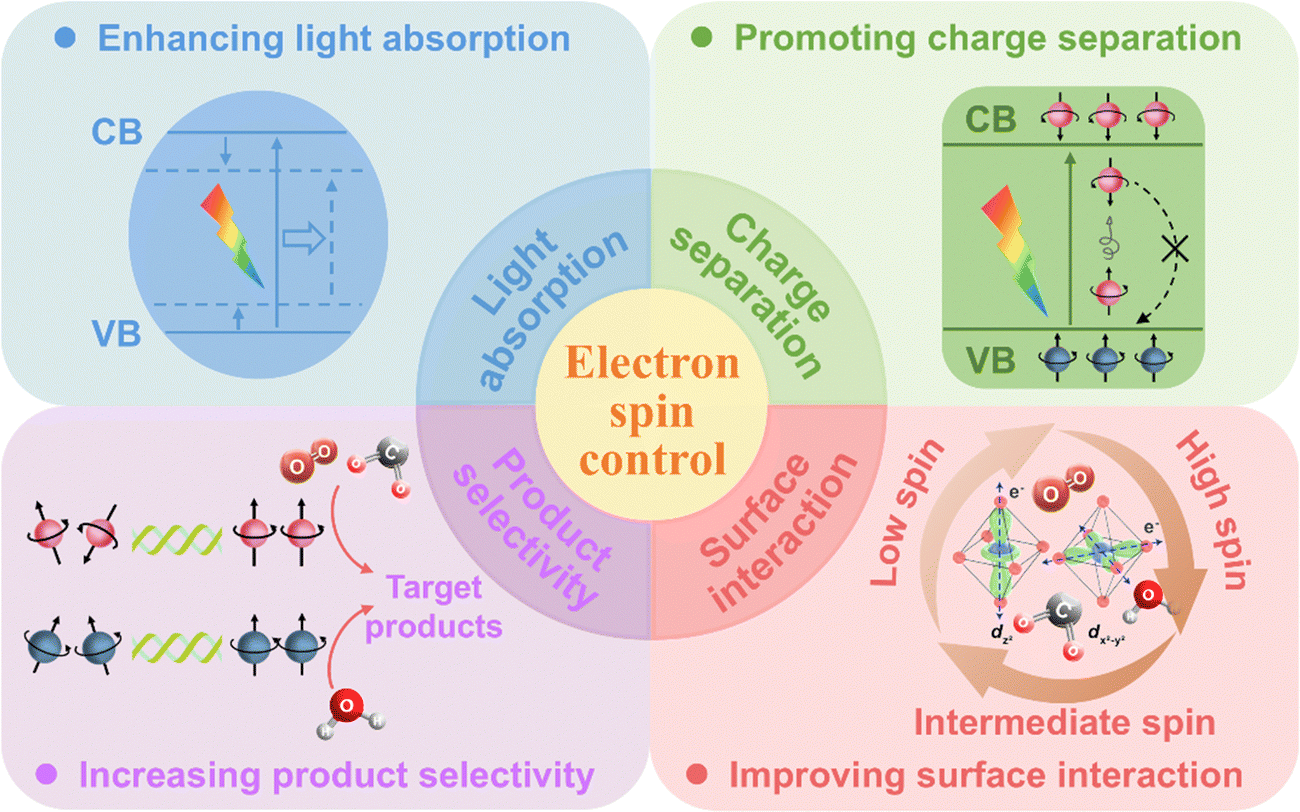 | ||
| Fig. 6 The role of electron spin control in photocatalysis. | ||
 | ||
| Fig. 7 Enhanced light absorption by the regulation of energy band structure. Schematic illustration of (a) spin-allowed excitation and (b) spin-forbidden excitation. (c) Comparison of light penetration in spin-allowed excitation and spin-forbidden excitation. Reproduced with permission from ref. 18. Copyright 2020 American Chemical Society. (d) Energy splitting introduced by Zeeman effect. Reproduced with permission from ref. 19. Copyright 2022 Cell Press. | ||
Traditional methods to enhance the light absorption of photocatalysts typically involve: (i) reducing the band gap energy through metal and non-metal doping (e.g., N, S, Fe, and Co) to enable absorption of longer-wavelength light; (ii) introducing lattice defects, such as oxygen and metal vacancies, to create new energy levels or band states, thereby increasing the absorption efficiency of visible-to-NIR light; and (iii) modifying the photocatalyst surface with photosensitizers (e.g., dyes and quantum dots) to boost light absorption via energy or electron transfer mechanisms. In contrast, electron spin control offers a novel approach by directly tuning the energy band structure through the SOC effect, without the need for complex material modifications. This presents a robust yet simple strategy for enhancing light absorption in visible and NIR regions and optimizing photocatalytic performance.
 | ||
| Fig. 8 Promoted charge separation by the application of an external magnetic field. (a) Schematic illustration of the spin polarization induced-longer photoexcited carrier lifetime in Mn–CsPbBr3 under an external magnetic field. The normalized photoinduced transient reflectivity changes (ΔR/R) of (b) CsPbBr3 and (c) Mn–CsPbBr3 with and without an external magnetic field. Reproduced with permission from ref. 20. Copyright 2022 American Chemical Society. (d) Schematic illustration of the magnetic field promoted photocatalysis system. Reproduced with permission from ref. 21. Copyright 2022 Royal Society of Chemistry. | ||
Another study revealed that Au-loaded Fe3O4/N–TiO2 superparamagnetic photocatalyst significantly enhanced the efficiency of photocatalytic water splitting under an external magnetic field because of the promoted charge separation.21 This promotion was attributed to the combined effects of the Lorentz force and spin polarization effect (Fig. 8d). In this system, the local magnetic field generated by Fe3O4 nanoparticles aligns the magnetic moments in N–TiO2 in parallel, creating a highly spin-polarized environment. Under an external magnetic field, electrons excited to the CB undergo spin relaxation, changing from their initial spin state (spin down) to an alternative state (spin up). Due to the scarcity of spin-up holes, these spin-up electrons encounter more difficulty returning to the VB, thereby reducing charge recombination and boosting photocatalytic performance.
In addition to the application of external magnetic fields, creating material defects can also regulate electron spin, thereby promoting charge separation.13,23 For example, cationic defect-rich Bi4Ti3O12 induced spin polarization, resulting in the generation of spin-parallel photogenerated electrons, which inhibited electron–hole recombination and led to efficient O2 activation (Fig. 9a).23 The reduced photoluminescence (PL) intensity in steady-state PL spectra suggest that the defects in Bi4Ti3O12 suppressed charge recombination (Fig. 9b). Meanwhile, time-resolved photoluminescence (TRPL) decay spectra reveal that defect-rich Bi4Ti3O12 facilitated a higher participation of photogenerated electrons and holes in the reaction, confirming the more effective charge separation (Fig. 9c). Moreover, steady-state surface photovoltage (SPV) spectroscopy indicates that the local electric field intensity of defect-rich Bi4Ti3O12 was approximately three times higher than that of Bi4Ti3O12, demonstrating the improved charge separation in the defect-rich Bi4Ti3O12 (Fig. 9d). In another study, intrinsic Ti vacancies in TiO2 were created by an oxygen-rich environment during the thermal assembly of Ti–O–Ti parallel lattice chains (Fig. 9e).13 These Ti vacancies resulted in the parallel alignment of electron spin orientations. Interestingly, varying the concentration of Ti vacancies allowed for tuning the degree of spin polarization. Compared to the pristine TiO2, Ti-vacancy-modified samples showed extended average TRPL lifetimes (Fig. 9f), suggesting the promoted charge separation by the spin polarization effect.
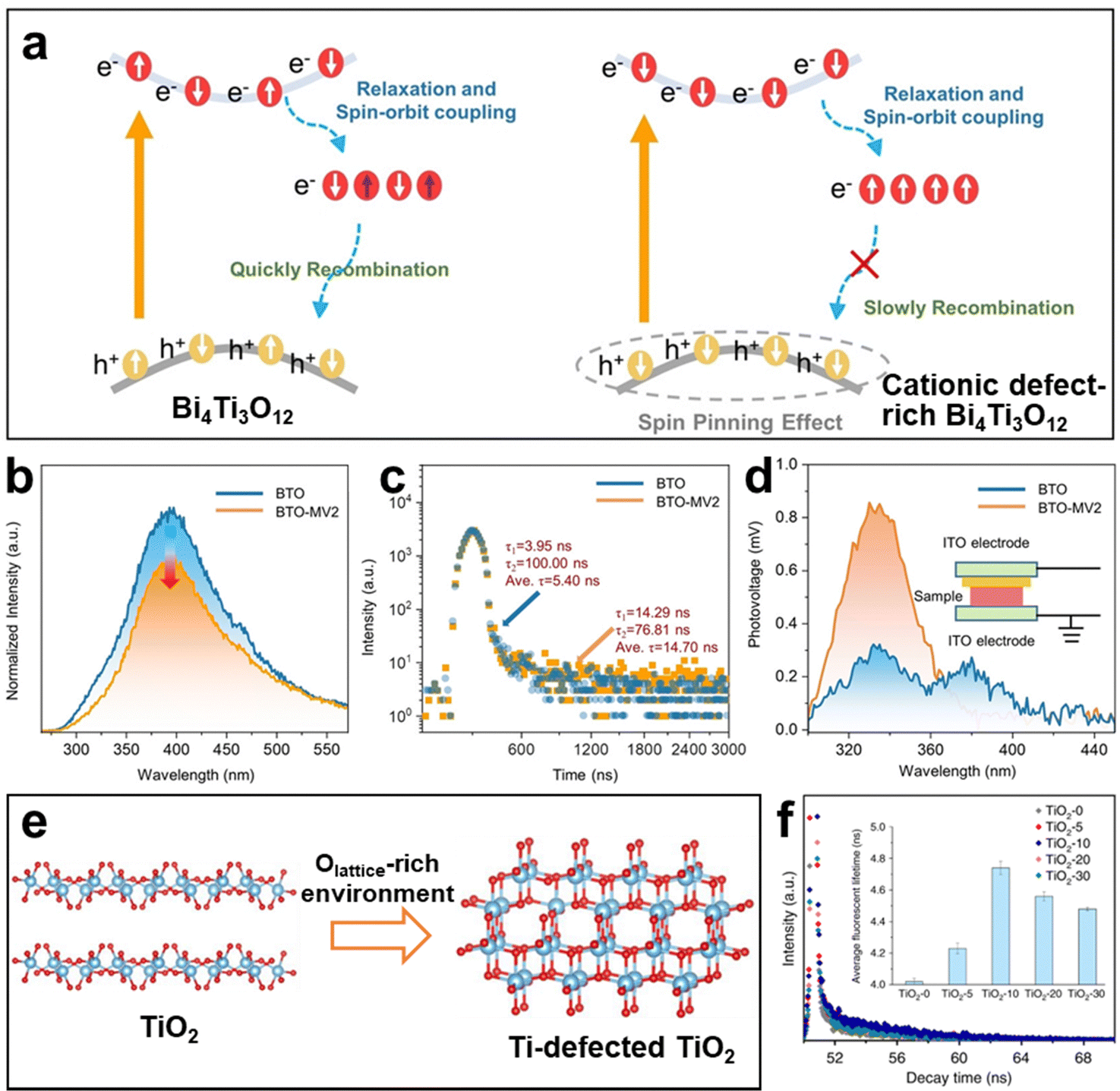 | ||
| Fig. 9 Promoted charge separation by defect engineering. (a) Schematic illustration of the excited carrier dynamics in Bi4Ti3O12 and cationic defect-rich Bi4Ti3O12. (b) Steady-state PL spectra, (c) TRPL decay spectra, and (d) SPV spectra of Bi4Ti3O12 and cationic defect-rich Bi4Ti3O12. Reproduced with permission from ref. 23. Copyright 2023 John Wiley & Sons. (e) Schematic illustration of the synthesis method of Ti-defected TiO2. (f) TRPL decay spectra of TiO2 and Ti-defected TiO2. Reproduced with permission from ref. 13. Copyright 2020 Springer Nature. | ||
Traditional methods for promoting the separation of photogenerated charge carriers generally focus on manipulating the structures, surface properties, and heterojunctions of semiconductors. Common strategies include: (i) constructing heterojunctions between different semiconductors (e.g., p–n junctions, type-II and Z-scheme heterojunctions) to utilize energy band alignment for efficient separation of photogenerated electrons and holes; (ii) depositing noble metal nanoparticles (e.g., Pt, Au, and Ag) and conductive carbon materials (e.g., graphene, carbon dots, and carbon nanotubes) on photocatalyst surface to form “electron capture” layers that enhance electron separation efficiency; (iii) optimizing charge separation efficiency by controlling the exposure of specific crystal facets of photocatalysts; and (iv) introducing lattice defects to create “electron traps”, which facilitate charge separation. In contrast to these approaches, electron spin control reduces the recombination probability of electrons and holes through the spin polarization effect, thereby extending carrier lifetimes and enhancing photocatalytic performance. This method controls charge separation by adjusting the spin state and spin-related interactions within photocatalysts, simplifying the design and fabrication process while offering a more precise and controllable way to enhance photocatalytic efficiency.
Modulating the electron spin states of metal centers enables precise control over the adsorption energy, thereby improving the surface reaction kinetics.14 For example, the spin state of Co in the covalent organic framework (COF)-367-Co can be controlled by adjusting the oxidation state of Co at the porphyrin center.68 This alteration affected the electron distribution and the orientation of the Co-3d orbitals, thereby influencing the interaction between Co and CO2 molecules. COF-367-CoIII, compared to COF-367-CoII, exhibited superior photocatalytic CO2 reduction activity and markedly enhanced selectivity for formic acid (HCOOH) (Fig. 10a). DFT calculations reveal that in COF-367-CoII, Co-3dxz or Co-3dyz orbitals couple with the O-2p orbitals of CO2, whereas in COF-367-CoIII, Co-3dz2 orbitals interact with the O-2p orbitals, resulting in the adsorption energy on COF-367-CoIII more than double that of COF-367-CoII (Fig. 10b). Moreover, COF-367-CoIII exhibits a stronger interaction with HCOOH than COF-367-CoII, favoring the formation of HCOOH while inhibiting its further conversion, thereby increasing the selectivity for HCOOH. In another study, CoII with low spin state has an empty eg orbital, making it difficult to deprotonate oxyhydroxides to form peroxide ions, which severely inhibited the reaction efficiency of oxygen evolution.86,87 However, CoIII with intermediate spin state achieved optimal adsorption of intermediates (e.g., *OOH, *O, and *OH), resulting in excellent activity. Also, tuning the spin state of the Fe center can alter its adsorption energy for reactants. For instance, when FeIII transitioned from low spin state to high spin state, the increased electrons in the eg orbitals weakened the adsorption of O2, leading to poor oxygen reduction performance.88 Conversely, when transitioned to intermediate spin state, the dz2 orbitals of FeIII easily bound to the antibonding π orbital of O2, thereby exhibiting enhanced oxygen reduction efficiency.
 | ||
| Fig. 10 Strengthened interaction between photocatalysts and reactants by the regulation of metal spin state. (a) Photocatalytic CO2 reduction activities by COF-367-Co with different Co spin states. (b) Different coupling modes of CO2 and HCOOH interacting with Co site at different spin states. Reproduced with permission from ref. 68. Copyright 2020 American Chemical Society. (c) Partial density of states of Mn obtained by DFT calculation. (d) Correlation of eg occupancy and oxygen evolution values. (e) Proposed four-electron mechanism for oxygen evolution over Mn sites. Reproduced with permission from ref. 24. Copyright 2019 John Wiley & Sons. | ||
Additionally, regulating the spin state of the Mn active center in Mn–C3N4 can modulate the interaction between the Mn–C3N4 and the reaction intermediates (e.g., *OOH, *O, and *OH), thereby enhancing the photocatalytic oxygen evolution performance.24 DFT calculations indicate that with a single Mn atom, Mn is in a high-spin state, whereas with two Mn atoms, the spin state of Mn is reduced (Fig. 10c). Consequently, the spin state of Mn can be tuned by adjusting the Mn concentration in Mn–C3N4. The binding strength between Mn and the intermediates is influenced by the antibonding eg occupancy, with the optimal eg occupancy around 0.95 (Fig. 10d). For photocatalysts with high eg occupancy, the Mn–O bond is too weak, which impedes the formation of the O–O bond. In contrast, the Mn–O bond of photocatalysts with low eg occupancy is too strong, hindering proton removal from Mn–OOH (Fig. 9e). Therefore, by precisely adjusting the spin state of Mn active center to achieve moderate eg filling, the balance between these competing processes can be optimized to enhance photocatalytic activity.
The interaction between photocatalysts and reactants is also crucial in determining the efficiency of charge transfer between photocatalysts and reactants. When reactant molecules coordinate with metal catalysts, the metal center's spin state can influence the energy distribution of the reactants’ highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).89 The HOMO represents the molecule's ability to donate electrons, while the LUMO reflects its ability to accept electrons. Generally, a smaller difference between the HOMO and LUMO energy levels facilitates the reaction. Consequently, regulating the surface interaction can directly affect the electron transfer between reactants and catalysts, thereby accelerating or slowing down the reaction rate. For instance, improving the adsorption of surface reactants (e.g., O2 and peroxide) on catalysts can regulate the electronic structures of these reactants, thereby enhancing electron transfer between the reactants and the metal center.90 Moreover, in organic synthesis, adjusting the adsorption capability of metal catalysts enables the design of reactants with optimized HOMO and LUMO energy levels, which lowers the reaction energy barrier.91
 | ||
| Fig. 11 Increased O2 selectivity by CISS effect. (a) The calculated electron transfer number as a function of applied potentials via photoelectrochemical rotating ring-disk electrode measurement. (b) The amount of hydrogen peroxide with o-tolidine indicator detected by UV-vis absorption spectra. (c) EPR spectra of DMPO–HO˙ adducts in photocatalysis. (d) Schematic representation of spin-polarized surface reaction. Reproduced with permission from ref. 12. Copyright 2023 Springer Nature. | ||
Currently, there are limited approaches for enhancing the surface reaction kinetics in photocatalysis, and these primarily focus on optimizing surface properties, active sites, and the adsorption of reactants. Common strategies include: (i) introducing additional active sites on photocatalyst surface (e.g., metal nanoparticles, acidic/basic sites, and oxygen/metal vacancies) to improve reactant adsorption; (ii) loading co-catalysts (e.g., Ni(OH)2 and RuO2) onto photocatalyst surface, which act as electron or hole capture centers to lower reaction energy barriers; and (iii) modifying surface functional groups (e.g., carboxyl, amino, and hydroxyl) to adjust surface properties, thereby optimizing the adsorption and desorption behaviors of reactants. Compared with these strategies, electron spin control offers a robust yet simple approach to not only improve the adsorption energy but also increase the product selectivity during photocatalytic processes by optimizing the electron spin states of active sites on photocatalyst surface.
4. Strategies for optimizing photocatalysis via electron spin control
In photocatalysis, the electron spin control strategy focuses on precisely manipulating the spin and spin state of electrons to optimize the separation of photogenerated electron–hole pairs and their subsequent reactions on the photocatalyst surface, which can significantly enhance photocatalytic activity. These strategies typically involve doping design, defect engineering, magnetic field regulation, metal coordination modulation, chiral induced spin selectivity, and combined strategies (the combination of these strategies). Table 1 summarizes recent advancements in electron spin control strategies for various photocatalytic applications (e.g., water splitting, CO2 reduction, pollutant degradation, and N2 fixation). We will provide specific examples illustrating the impact of electron spin control on photocatalytic light absorption (evaluated by band gap alteration), charge separation (characterized by TRPL average lifetime), and overall improvements in photocatalytic efficiency across various photocatalysts, along with detailed discussions of the underlying mechanisms.| Photocatalyst | Strategy | Photocatalytic application | Bandgap adjustment (eV) | TRPL lifetime regulation (ns) | Efficiency improvement | Ref. |
|---|---|---|---|---|---|---|
| Fe–Bi4O5Br2 | Fe doping | Pollutant degradation | — | — | 60 → 92% (25 min) | 93 |
| Fe–CN | Fe doping | Aniline hydrogenation | — | — | 7.6 → 95.0% PhNH2 | 94 |
| Co–BiVO4 | Co doping | CO2 reduction | 2.40 → 1.93 | — | 1.6 → 109.2 μmol per g per h CO | 95 |
| Co–BiOCl | Co doping | CO2 reduction | 3.32 → 3.26 | 3.6 → 6.8 | — | 96 |
| Cu–LaFeO3 | Cu doping | Pollutant degradation | 2.26 → 2.25 | — | 0.5 → 4.7 s−1 | 97 |
| Cu–TiO2 | Cu doping | Water splitting | 3.20 → 3.06 | — | — | 98 |
| Ti–CTF | Ti doping | Pollutant degradation | 2.80 → 2.68 | — | 33.5 → 97.8 (100 min) | 99 |
| Mn–Co3O4 | Mn doping | CO2 reduction | — | — | 0.8 → 23.4 μmol per g per h CH4 | 100 |
| Mn–Fe2O3 | Mn doping | Pollutant degradation | — | — | 78 → 92% (2 h) | 101 |
| Mn–CN | Mn doping | Water splitting | — | 0.6 → 1.9 | 60.0 → 695.1 μmol per g per h H2 | 24 |
| Mn–CN | Mn doping | Water splitting | 2.79 → 2.55 | 17.5 → 4.1 | 0.4 → 13.5 mmol per g per h H2 | 102 |
| Ni–BiOBr | Ni doping | CO2 reduction | — | 2.6 → 3.6 | 86.8 → 378.7 μmol per g per h CO | 103 |
| O–ZnIn2S4/Ni12P5 | O doping | Water splitting | — | 1.0 → 2.6 | 1.9 → 15.8 mmol per g per h H2 | 104 |
| S-CTF | S doping | Pollutant degradation | — | — | 9 → 100% (1 h) | 105 |
| B–CN | B doping | H2O2 production | 2.61 → 2.48 | — | 59 → 1710 μM h−1 | 106 |
| I–GO | I doping | Water splitting | — | 1.2 → 2.2 | 38.6 → 96.2 μmol per g per h H2 | 107 |
| Se–CN | Se doping | Water splitting | 3.06 → 2.73 | 20.0 → 16.3 | 0.2 → 5.4 μmol per g per h H2 | 108 |
| P/N–CN | P and N co-doping | Water splitting | — | 21.6 → 1.8 | 0.1 → 22.2 mmol per g per h H2 | 109 |
| C/B–TiO2 | C and B co-doping | — | 2.90 → 2.10 | — | — | 110 |
| Bi4Ti3O12 | Ti defect | Pollutant degradation | — | 5.4 → 14.7 | — | 23 |
| TiO2 | Ti defect | Water splitting | — | 4.1 → 4.8 | 1.8 → 18.5 mmol per g per h H2 | 13 |
| ZnO | Zn defect | Water splitting | — | 2.7 → 3.7 | 0.1 → 5.8 mmol per g per h H2 | 111 |
| WO3 | O defect | U(VI) reduction | 2.67 → 2.54 | 99.1 → 78.4 | 20.5 → 287.1 mg g−1 | 112 |
| CdS | S defect | Water splitting | 2.39 → 2.13 | 6.4 → 17.3 | 0.8 → 41.7 mmol per g per h H2 | 113 |
| SnS2 | S defect | Water disinfection | 2.29 → 2.20 | 0.1 → 1.7 | — | 114 |
| Zinc porphyrin | N defect | CO2 reduction | 1.83 → 1.80 | — | 4.6 → 12.5 μmol per g per h CO | 115 |
| CdS | Cd and S defects | Water splitting | — | 0.86 → 0.95 | 1.2 → 10.6 mmol per g per h H2 | 116 |
| Ti-based MOF | Ti and organic linker defects | Water splitting | — | — | 1.4 → 3746.7 μmol per g per h H2 | 117 |
| Fe2O3/rGO | Magnetic field | Pollutant degradation | — | — | 59 → 84% (40 min) | 118 |
| Co-MOF | Coordination regulation | CO2 reduction | — | — | — | 119 |
| Fe(Pz)[PtII/IV(CN)4]I1.2·2H2O | Coordination regulation | H2O2 production | 2.35 → 1.86 | — | 0 → 66 mM g−1 h−1 | 120 |
| Au3Fe1/Mo | Coordination regulation | N2 fixation | — | — | 60 → 484 μmol per g per h NH3 | 49 |
| Chiral ZnO | CISS effect | Water splitting | — | 1.8 → 4.2 | 0.8 → 1.3 mmol per g per h H2 | 12 |
| Fe–CsPbBr3 | Fe doping and magnetic field | CO2 reduction | — | 13.3 → 2.9 | 19.8 → 33.3 μmol per g per h CO | 121 |
| Fe–BiVO4 | Fe doping and magnetic field | Water splitting | — | 3.9 → 5.5 | 0 → 61.5 μmol per g per h H2 | 122 |
| Co–CsPbBr3 | Co doping and magnetic field | Water splitting | 2.54 → 2.13 | — | 581.7 → 1042.1 μmol per g per h H2 | 123 |
| Co–CN | Co doping and magnetic field | Water splitting | — | — | 12 → 3979 μmol per g per h H2 | 124 |
| Mn–CsPbBr3 | Mn doping and magnetic field | CO2 reduction | — | — | 0.6 → 17.6 μmol per g per h CO | 20 |
| Ni–CdS/MoS2 | Ni doping and magnetic field | Water splitting | — | 5.9 → 11.0 | 339 → 1322 μmol per g per h H2 | 125 |
| Co3O4 | Co defect and magnetic field | CO2 reduction | — | 0.14 → 3.51 | 9.4 → 70.8 μmol per g per h CO | 126 |
| Bi2WO6/CuS | W, Cu defects and magnetic field | Pollutant degradation | — | 3.27 → 3.60 | 74.5 → 90.5% | 127 |
| BaTiO3 | O defect and magnetic field | N2 fixation | — | — | 0.1 → 1.9 mg L−1 h−1 | 50 |
| CuInP2S6 | S defect and magnetic field | CO2 reduction | — | — | 4.5 → 12.5 μmol per g per h CH4 | 128 |
| Ni–CdS | Ni doping and S defect | Water splitting | 2.37 → 2.26 | 3.6 → 20.3 mmol per g per h H2 | 129 | |
| K-CN/AgCl/ferrihydrite | K doping and Ag regulation | Pollutant degradation | 2.87 → 2.44 | — | 75 → 100% (30 min) | 130 |
| MoS2/graphene | Heterostructure construction | Water splitting | — | 6.0 → 27.7 | 0 → 240 μmol per g per h H2 | 131 |
| CN/graphene | Heterostructure construction | Water splitting | 2.51 → 2.42 | 6.8 → 4.7 | 0.9 → 14.2 μmol per g per h H2 | 132 |
| Co–COF | Metal valence regulation | CO2 reduction | 1.10 → 1.03 | — | 48.6 → 93.0 μmol per g per h HCOOH | 68 |
| Fe2O3/CN | Crystal size regulation | Pollutant degradation | 2.75 → 2.47 | 4.2 → 3.8 | 0.04 → 0.21 min−1 | 133 |
| FeS2/CuCo2O4 | Pyroelectric field | Water splitting | — | 5.7 → 15.0 | 1.9 → 19.5 mmol per g per h H2 | 134 |
4.1 Doping design
Doping design is a widely used modification technique for photocatalysts. It introduces metal and non-metal elements to alter the electronic energy levels and band structure of the photocatalyst. This can broaden the light absorption range and improve carrier separation efficiency, ultimately leading to increased photocatalytic performance.2 In recent years, doping design has been employed to manipulate electron spin in photocatalysts. In certain metals and non-metals, unpaired electrons carry spin angular momentum. When elements with unpaired electrons are doped into a photocatalyst, they create local magnetic moments within the material. These local magnetic moments can interact with the surrounding electron spins, influencing the overall electron arrangement and potentially leading to an ordered spin arrangement.93,100,135 This, in turn, can affect the electron transport properties during photocatalytic reactions. Furthermore, doping elements into the photocatalyst lattice can introduce new electronic energy levels or alter the existing energy level structure,136 which may cause a rearrangement of electrons and result in the splitting of energy levels. The split energy levels will be preferentially occupied by electrons with different spin states, leading to a spin selection effect. As lower-energy spin states are more readily occupied by electrons, the energy level splitting induced by doping can cause electrons in the system to preferentially adopt specific spin states, resulting in overall spin polarization.137Doping magnetic elements (e.g., Fe, Co, and Ni) into photocatalysts is an effective strategy for manipulating electron spin state. For example, Li et al. employed Fe-doped Bi4O5Br2 (Fe–Bi4O5Br2) to photoactivate H2O2 for the degradation of organic pollutants (e.g., ciprofloxacin (CIP) and Cu–ethylenediaminetetraacetic acid (Cu–EDTA)) in water.93 They optimized the spin interactions between Fe sites and coordinated O atoms (derived from H2O2) and increased high-spin Fe sites to generate O2˙− with high pH tolerance, thus enhancing Fenton-like degradation under neutral or alkaline environment (Fig. 12a). It was observed that pH had minimal impact O2˙− production (Fig. 12b), and Fe–Bi4O5Br2 exhibited longer carrier lifetimes and superior CIP degradation compared to Bi4O5Br2 (Fig. 12c). This is because the O–O bonds of H2O2 adsorbed on Fe–Bi4O5Br2 showed increased resistance to scission, facilitating the generation of *OOH in the subsequent reaction and thereby enhancing the formation of O2˙− (Fig. 12d). This work demonstrates that doping with high-spin Fe can enhance charge separation and promote the formation of surface *OOH intermediates, ultimately boosting the generation of O2˙− for more efficient pollutant degradation.
 | ||
| Fig. 12 Generation of O2˙− mediated by high-spin Fe doping. (a) Schematic of the challenge in H2O2 activation and this work, M represents surface transition-metal atoms. (b) The production of O2˙− at various pH levels. (c) Transient absorption spectroscopy (TAS) kinetics probed at 450 nm. (d) Reaction path of H2O2 activation and free energy profile of O2˙− production process. Reproduced with permission from ref. 93. Copyright 2024 National Academy of Sciences. | ||
Another study introduced spin-polarized electrons into BiOCl nanosheets by doping with magnetic Co, which significantly improved the efficiency of photocatalytic CO2 reduction and achieved a high hydrocarbon selectivity of 76.9% for converting CO2 into CH4 and C2+ products.96 Compared to BiOCl, Co–BiOCl exhibited a narrower band gap (3.32 → 3.26 eV) and longer carrier lifetime (3.6 → 6.8 ns). Mechanistic analysis reveals that the spin-polarized electrons enhanced CO adsorption and reduced the kinetic barrier for *CH2 formation, ultimately leading to highly selective hydrocarbon generation.
Single-atom doping can not only alter the electron spin environment of photocatalysts by introducing unpaired electrons but also provide a strategy to maximize atomic utilization, offering an efficient and sustainable strategy for optimizing photocatalysis. For instance, Zhu et al. developed a single-atom Ti-doped covalent organic framework (Ti-CTF) for the photocatalytic degradation of 2,2,4,4′-tetrahydroxybenzophenone (BP-2), with O2˙− being the primary reactive species.99 Compared to pristine CTF, the spin-polarized Ti-CTF significantly enhanced the adsorption and degradation of BP-2, achieving a degradation rate 17 times higher than that of CTF. Experimental results and theoretical calculations indicate that single-atom Ti, bound to pyridine and triazine N, induced electron spin-down polarization near the Fermi level, which promoted electron transfer and subsequent surface reactions (Fig. 13a). Moreover, Ti doping reduced the bandgap from 2.80 to 2.68 eV, thereby improving light absorption. This demonstrates the synergistic effect of electron spin control and Ti doping in optimizing photocatalytic performance. In another study, single-atom Mn was incorporated into Co3O4 photocatalyst to replace the octahedral Co (Fig. 13b), inducing electron spin polarization and achieving a CH4 generation rate 28.8 times higher than that of Co3O4 in CO2 photoreduction.100 Notably, the doping of Mn significantly suppressed the recombination of photogenerated electrons and holes by aligning their spin directions antiparallel (Fig. 13c). Mechanistic analysis reveals that Mn sites enriched with holes initiate direct hydrogen transfer from H2O, while adjacent Co sites preferentially capture electrons to activate CO2 and convert it into *COOH intermediates.
 | ||
| Fig. 13 Photocatalytic electron spin control by metal doping. (a) Schematic illustration of application scheme on spin polarization-related Ti-CTF for micropollutants removal. Reproduced with permission from ref. 99. Copyright 2023 John Wiley & Sons. (b) The schematically illustration of substitute Co by Mn. (c) Photocatalytic CO2 methanation mechanism of Mn–Co3O4. Reproduced with permission from ref. 100. Copyright 2024 American Chemical Society. | ||
Compared with metal element doping, non-metallic element doping, such as C, N, and B, typically reduces the overall material cost and minimizes environmental pollution risks, making it a low-cost and eco-friendly strategy. Wan et al. utilized O–ZnIn2S4/Ni12P5 to create a polarization-induced internal electric field through O doping and the formation of ohmic junctions, resulting in an 8.16-fold increase in photocatalytic H2 generation compared to the pristine ZnIn2S4.104 Herein, O doping induced electron spin polarization, while the ohmic junctions enabled rapid migration of photogenerated electrons to the Ni12P5 active sites, effectively suppressing the recombination of photogenerated electron–hole pairs. TRPL decay spectra reveal that the average lifetime of photogenerated carriers in O–ZnIn2S4/Ni12P5 (2.58 ns) was significantly longer than that of ZnIn2S4 (1.01 ns), O–ZnIn2S4 (1.53 ns), and ZnIn2S4/Ni12P5 (2.21 ns). Additionally, the preferential dehydrogenation of the α-C–H bond in benzyl alcohol further facilitated hole transport and charge separation, ultimately enhancing both H2 generation and benzaldehyde synthesis.
In another work, 2D CN nanosheets were fluorinated and then thermally defluorinated in Se vapor to prepare atomically thin Se–CN nanosheets.108 Notably, Se doping expanded the light absorption edge of CN from 416 to 584 nm (bandgap reduced from 3.06 to 2.73 eV). In addition, the electron spin polarization in Se–CN significantly improved charge separation efficiency and surface catalytic reactions, which enhanced the performance of photocatalytic water splitting by 27 times compared to the pristine CN. Furthermore, Wang et al. designed 2D P/N–CN nanosheets through co-doping with P and N, leading to an optimized photocatalytic water splitting system that exhibited 200 times higher hydrogen production compared to CN, while enabling the selective oxidation of benzylamine via holes.109 The spin–orbit coupling between N 2p and P 2p orbitals significantly increased the parallel spin arrangement, thereby accelerating the charge separation and surface catalysis in P/N–CN.
4.2 Defect engineering
It is commonly understood that introducing or adjusting defects (e.g., defect type, concentration, and distribution) in photocatalysts can enhance the light absorption range and charge separation efficiency.30 Notably, defect engineering also plays an important role in manipulating electron spin by modifying the local magnetic environment within the material, which can further influence the photocatalytic performance.13,23 Specifically, when defects are introduced into photocatalysts, they can create unsaturated atomic bonds or incomplete electron pairs within the material's lattice, leading to the presence of unpaired electrons. These unpaired electrons can regulate the local magnetic moments, thereby causing spin polarization.Metal defects can enhance the light absorption range, promote charge separation, and ultimately improve the catalytic activity of photocatalysts by modulating the electron spin structure around the metal atoms near the defects. For example, Zhang et al. prepared Ti-defective Bi4Ti3O12 for the activation of O2 to degrade tetracycline (TC) in water, which achieved a degradation rate constant 3.3 times higher than that of the pristine Bi4Ti3O12.23 The Ti defects on Bi4Ti3O12 induced electron spin polarization, reducing the recombination of photogenerated electron–hole pairs. Additionally, surface Ti defects formed a center for adsorbing O2 and extracting electrons, effectively generating HO˙, O2˙−, and 1O2 for TC degradation. In another work, ZnO with abundant Zn defects was synthesized to optimize the spin structure and induce spin polarization by adjusting the electron occupancy of the eg orbital, enabling simultaneous H2 production and pollutant degradation.111 Compared to the pristine ZnO, the Zn-defective ZnO achieved a 56.4-fold increase in H2 production rate and a 27.5-fold improvement in pollutant degradation efficiency.
In photocatalytic defect engineering, non-metallic defects are often preferred due to their ease of introduction through simple synthesis methods and their lower impact on the crystal structure, which preserves the photocatalyst's properties. Yang et al. employed WO3−x nanowires rich in O defects to convert highly mobile and toxic UVI into low-solubility and less toxic UIV, achieving 79.9% reduction and immobilization of UVI (initial concentration of 10 mg L−1) at pH 5.112 Compared to pristine WO3, the band gap of WO3−x was reduced from 2.67 to 2.54 eV, enhancing its visible light absorption capacity (Fig. 14a). Additionally, the TRPL average lifetime decreased from 99.1 to 78.4 ns, which indicates the facilitated migration of electrons to the WO3−x surface for UVI reduction (Fig. 14b). In partial density of state (PDOS) spectra, the broader green region in WO3−x compared to WO3 suggests the presence of more O dangling bonds, promoting the spin polarization of W 5d band electrons (Fig. 14c). Moreover, density of state (DOS) spectra reveal that the CB of WO3−x shifted to a lower energy region compared to WO3, which indicates an increased electron density in WO3−x (Fig. 14d). Notably, the spin polarization in WO3−x facilitated the separation of electrons and holes, while the O defects created abundant active sites for photocatalytic reduction reactions (Fig. 14e). Together, these factors worked synergistically to enhance photocatalytic performance.
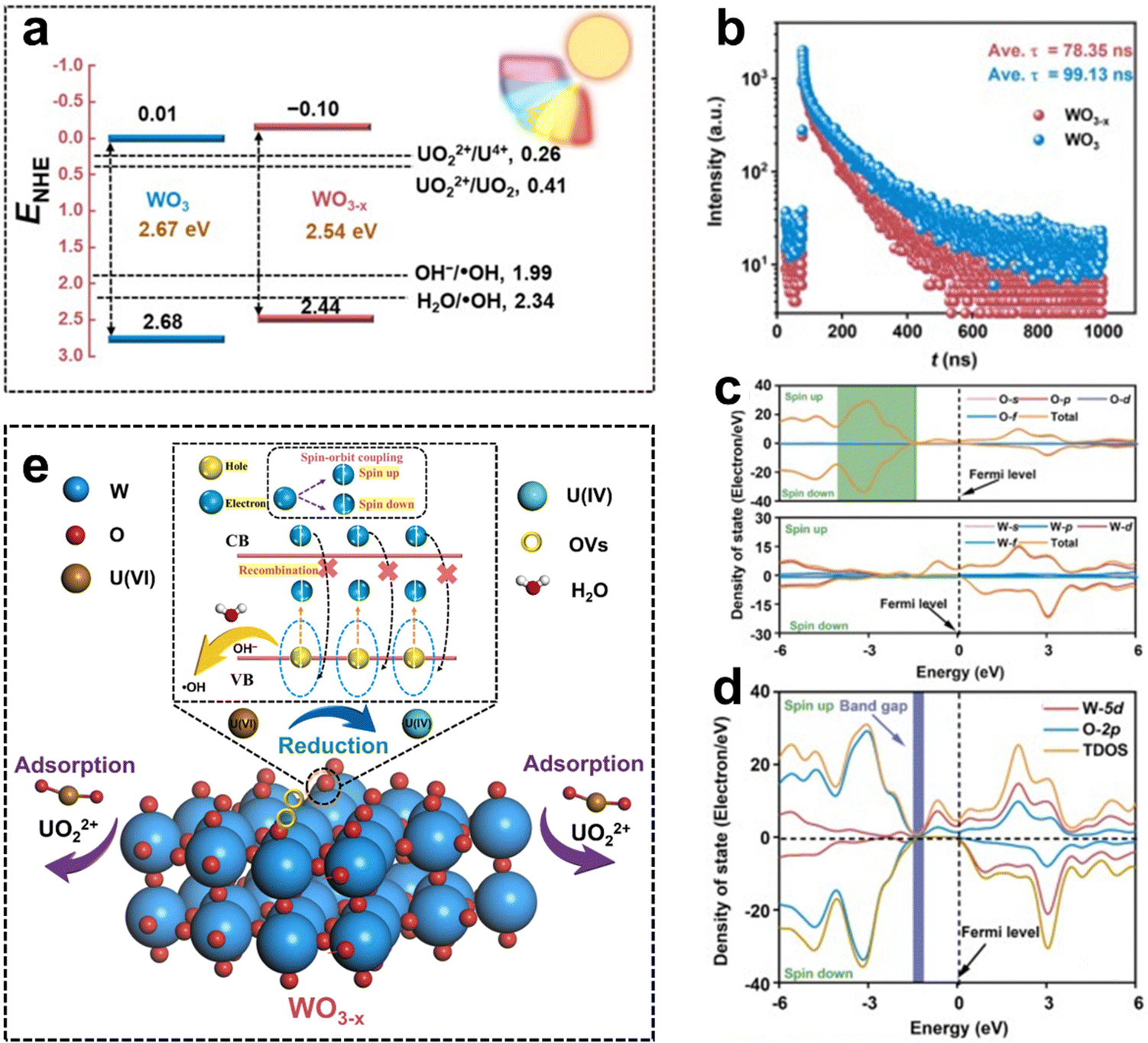 | ||
| Fig. 14 O defect-induced spin polarization in WO3−x for photocatalytic reduction and immobilization of U(VI). (a) Band structures and (b) TRPL spectra of WO3−x and WO3. (c) PDOS and (d) DOS spectra of WO3−x. (e) The schematic diagram on removal of U(VI) by WO3−x through adsorption and photocatalytic reduction. Reproduced with permission from ref. 112. Copyright 2023 Elsevier. | ||
In another study, S defects were introduced into CdS, inducing a spin polarization effect that accelerated carrier transfer dynamics from the bulk phase to surface redox sites for photocatalytic water splitting.113 Following the introduction of S defects, the band gap of CdS decreased from 2.39 to 2.13 eV, while the TRPL average lifetime extended from 6.4 to 17.3 ns. This enhancement in light absorption and carrier separation efficiency resulted in a 50-fold increase in H2 production compared to pristine CdS. Theoretical calculations reveal that H2O molecules tend to adsorb onto Cd atoms adjacent to S defects. Subsequently, the –H group in H2O is reduced to H2, along with another –H group from an adjacent adsorbed H2O molecule. Simultaneously, O atoms occupy the S defects, forming Cd–O bonds, after which the two adjacent O atoms are oxidized to O2. Therefore, the spin polarization induced by S defects accelerated the separation of photogenerated charge carriers and enhanced the kinetics of surface photocatalytic reactions, effectively facilitating the overall H2O splitting.
Recently, the introduction of dual-element defects in photocatalysts has been found to be effective in modulating the material's electronic structure and enhancing the surface reaction activity.116,117 Additionally, the interactions between dual-element defects may create a synergistic effect, further improving the photocatalytic performance. For instance, Qi et al. introduced Cd and S defects on CdS nanorods by heat treatment, enhancing the photocatalytic H2 production performance.116 The interfacial and polarization electric fields generated by the defects improved TRPL average lifetime from 0.86 to 0.95 ns and promoted faster interfacial charge migration, thereby improving the photocatalytic hydrogen production rate, with an AQE 8.5 times higher than that of pristine CdS. Moreover, Xu et al. developed Ti-based metal–organic frameworks (MOF) with Ti and organic linker defects to simultaneously achieve photocatalytic water splitting and pollutant degradation.117 Mechanistic analysis reveals that defect-induced electron spin polarization can control the CB position, resulting in an upward shift of the d-band center. This enhanced the adsorption of *H and facilitated the H2 production. Additionally, spin polarization mitigated the recombination of photogenerated electrons and holes within Ti-MOF, leading to obvious improvements in photocatalytic activities.
4.3 Magnetic field regulation
Compared to traditional methods for enhancing photocatalytic performance, applying an external magnetic field during photocatalytic reactions is regarded as an efficient, safe, convenient, and contactless technique that significantly improves photocatalytic efficiency. The core mechanism behind this approach involves the spin–orbit and magnetic interactions of electrons. Electrons possess spin angular momentum, meaning that each electron has an intrinsic magnetic moment, akin to a tiny magnet. When exposed to an external magnetic field, electrons in the photocatalyst undergo spin–orbit and magnetic interactions, leading to adjustments in their spin states based on the field's direction and intensity.10 The spin–orbit interaction couples the electrons’ spin with their orbital motion, while the magnetic interaction aligns the electrons’ magnetic moments with the external field. This combined effect not only reconfigures the electrons’ arrangement but also breaks the degeneracy of energy levels, causing them to split, which is known as the Zeeman effect. Specifically, the spin states divide into two levels: “parallel” (with spin aligned to the magnetic field) and “antiparallel” (with spin opposite to the magnetic field). Electrons preferentially occupy the lower-energy parallel state, resulting in spin polarization, where more electrons adopt the same spin orientation.138Additionally, the external magnetic field influences photogenerated charge carriers via the Lorentz force. The Lorentz force acts on moving charges in a magnetic field, effectively aiding in the separation and transfer of electron–hole pairs. This is particularly useful in magnetic materials, where the Lorentz force prevents their recombination and enhances photocatalytic efficiency. Moreover, the Lorentz force helps reduce catalyst particle agglomeration and increases the exposure of active sites, further enhancing the catalytic reaction.19
Li et al. found that applying a magnetic field enhanced the pollutant degradation efficiency of α-Fe2O3/reduced graphene oxide (rGO).118 This improvement is attributed to the magnetic field facilitating carrier transfer from α-Fe2O3 to rGO, thereby greatly increasing carrier transfer efficiency. Moreover, it is reported that the application of an external magnetic field can control electron spin polarization in Mn–CsPbBr3, which enhanced the efficiency of photocatalytic CO2 reduction reactions.20 When exposed to a 300 mT magnetic field or a permanent magnet, the photocatalytic performance of Mn–CsPbBr3 improved by 5.7 times compared to pristine CsPbBr3. This is because of the synergistic effect of doping with magnetic elements and applying a magnetic field, which increased the number of spin-polarized carriers and extended the carrier lifetimes.
In another study, applying an external magnetic field was found to manipulate spin polarization in Co3−xO4, achieving 100% selectivity for CO production during the photocatalytic reduction of CO2.126 Notably, the intrinsic spin polarization of Co3−xO4 (Fig. 15a), combined with the antipodal Lorentz forces (Fig. 15b), enhanced the TRPL average lifetime from 0.14 to 3.51 ns, significantly boosting the charge carrier mobility. Additionally, spin polarization induced by Co defects and the external magnetic field provided a thermodynamic advantage for CO2 reduction by facilitating the conversion of CO2 to *COOH and subsequently to *CO, while lowering the free energy required for the reduction reaction (Fig. 15c and d).
 | ||
| Fig. 15 Spin polarization-enhanced photocatalytic CO2 reduction by Co3−xO4 under a magnetic field. (a) Crystal structure and electron spin distribution of Co3O4 and Co3−xO4. (b) Schematic diagram of the Lorentz force effect on photogenerated carrier transfer. (c) Proposed pathway for CO2 photoreduction to CO. (d) Calculated free energy diagrams of photoreduction of CO2 to CO on Co3O4 and Co3−xO4 with and without consideration of spin polarization in the calculation. Reproduced with permission from ref. 126. Copyright 2024 American Chemical Society. | ||
In addition, controlling charge separation by the combination of ferroelectric polarization and spin polarization has recently gained attention as a promising approach to boost the efficiency of photocatalytic reactions. For instance, Chiang et al. demonstrated the modulation of charge separation in 2D ferroelectric CuInP2S6 crystals through the ferroelectric field and an applied magnetic field, which enhanced the photocatalytic reduction of CO2.128 Specifically, the ferroelectric polarization of CuInP2S6 can be regulated through ferroelectric phase transitions and electric polarization, while the electron spin polarization can be tuned by introducing S defects and applying an external magnetic field, enabling the simultaneous manipulation of both ferroelectric and spin polarizations, which facilitated the conversion of CO2 to CO and CH4. The in situ diffuse reflectance infrared Fourier transformed (DRIFT) spectra exhibit the formation of various surface adsorbed species during the photocatalytic CO2 reduction, including *COOH, *CH3O, and *CHO, which are all key intermediates for the conversion of CO2 to CH4. Notably, compared to pristine CuInP2S6, the increased DRIFT intensity in CuInP2S6 with S defects indicates enhanced CH4 production, which is attributed to the promotion of charge separation through both ferroelectric and spin polarizations.
4.4 Metal coordination modulation
Modulating the metal coordination environment in a photocatalyst can typically alter the energy level distribution of the metal center and the local electron cloud around the metal, thereby manipulating the spin state of the metal and inducing spin polarization.119 Current methods to adjust the metal coordination environment include controlling the number of ligands around the metal, altering the ligand types at the metal center, and modifying the geometric symmetry of the coordination environment (e.g., octahedral and tetrahedral).139 For instance, Sun et al. synthesized three different Co-doped Zn-based MOFs, with Co centers coordinated to –CH3COO (Co–OAc), –Br (Co–Br), and –CN (Co–CN), by varying the Co precursor (Fig. 16a).119 It was proposed that during the photocatalytic CO2 reduction process, spin-up electrons are excited from the HOMO to the LUMO under visible light irradiation, while spin-up holes remain in the HOMO. Due to spin–orbit coupling (SOC) and hyperfine interactions (HFI), the excited spin-up electrons transition to a spin-down, while the spin-up holes retain their spin orientation. This spin mismatch between electrons and holes effectively suppresses their recombination (Fig. 16b). Both experimental results and DFT calculations reveal that modulating the coordination environment of the Co site adjusted its spin state, resulting in varying levels of charge separation efficiency and CO2 reduction capacity in Co–OAc, Co–Br, and Co–CN. Notably, Co–OAc, with its highest spin state, exhibited exceptional charge separation and optimized CO2 adsorption and activation energy barrier (Fig. 16c), leading to a remarkable photocatalytic CO2 reduction rate of 2.3 mmol g−1 h−1 and a 99.1% selectivity for CO production. | ||
| Fig. 16 Manipulating the coordination environment of Co sites for boosting CO2 photoreduction. (a) Synthesis of Co–OAc, Co–Br, and Co–CN featuring different spin states of CoII and CoIII species via a postsynthetic exchange strategy. (b) Mechanism of spin polarization for improved charge separation. (c) Energy variations of CO2 photoreduction along the reaction path of the three Co-based MOFs. Reproduced with permission from ref. 119. Copyright 2024 American Chemical Society. | ||
Additionally, Huang et al. proposed to regulate the spin state of the Fe center in the Fe–Pt Hofmann clathrates through facilitating the valence state conversion of Pt coordinated with Fe from PtIV to PtII by iodine treatment.120 Notably, when the spin state of FeII in the Fe–Pt Hofmann clathrates is high spin, only the photocatalytic oxygen reduction reaction (ORR) occurs, leading to the synthesis of H2O2. Conversely, when FeII is in a low spin state, both ORR and water oxidation reactions (WOR) can take place. This spin state transition enables the switching between ORR and WOR in H2O2 photosynthesis (Fig. 17a). This is because the low spin state of FeII reduced the energy barriers for both ORR and WOR processes. Moreover, compared to the high spin state, the low spin state of FeII exhibited enhanced light harvesting ability, improved photogenerated carrier separation efficiency, and superior charge transfer capability.
 | ||
| Fig. 17 Modulation of metal coordination environment for electron spin control. (a) Schematic illustrations of the dynamic spin-state transition for FeII in the Hofmann clathrates and the spin-related photocatalytic H2O2 synthesis overall reaction. Reproduced with permission from ref. 120. Copyright 2023 American Chemical Society. (b) Schematic of orbital interaction between the metal and N2 in medium-spin Au3Fe1/Mo. Reproduced with permission from ref. 49. Copyright 2024 Elsevier. | ||
In another study, the spin state of FeIII in a Au3Fe1/Mo alloy photocatalyst can be finely tuned to enhance photocatalytic N2 fixation by altering the metal coordination environment.49 Theoretical calculations indicate that the strong electronic interactions among Mo, Fe, and Au enable the Fe site to serve as a crucial center for N2 adsorption and activation. Specifically, the unpaired electrons of N2 first occupy the empty d orbital of FeIII, after which the unpaired electrons from FeIII fill the antibonding orbital of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, resulting in its dissociation. In high spin Fe sites, the lack of an empty orbital hinders N2 activation. In contrast, medium spin Fe sites have an empty orbital that can accept these unpaired electrons from N2, which then feed back into the NN antibonding orbital, weakening the NN bond and facilitating the N2 fixation process (Fig. 17b).
N, resulting in its dissociation. In high spin Fe sites, the lack of an empty orbital hinders N2 activation. In contrast, medium spin Fe sites have an empty orbital that can accept these unpaired electrons from N2, which then feed back into the NN antibonding orbital, weakening the NN bond and facilitating the N2 fixation process (Fig. 17b).
4.5 Chiral induced spin selectivity
The chiral induced spin selectivity (CISS) effect refers to the preferential alignment of electron spins in a specific direction within a chiral molecule (whose structure cannot be superimposed on its mirror images) after charge polarization (Fig. 18a), which plays a crucial role in controlling chemical reactions, enantiomer separation, and biorecognition processes.16,140 When an electron moves through a chiral molecule, the curvature of the potential energy within the chiral structure generates a centripetal force that acts perpendicular to the electron's velocity (Fig. 18b).16 The direction of this force depends on the handedness (left- or right-handed) of the chiral molecule. Moreover, the centripetal force can create an effective magnetic field (B) along the electron's path, which interacts with the electron's magnetic moment, stabilizing one spin orientation while destabilizing the other. This preferred spin orientation is determined by the chiral axis of the chiral molecule, which is influenced by its handedness and linear momentum. Additionally, when a chiral molecule is subjected to an electric field along its axis, charge polarization occurs, accompanied by spin polarization (δ(+) and δ(−) at the ends of the helix) (Fig. 18c).16 Notably, the spin polarization associated with each pole is influenced by the handedness of the chiral molecule. Thus, when a chiral molecule participates in an electron transfer process, it can selectively facilitate or inhibit electron transfer based on the spin direction of the electrons. In other words, the CISS effect enables chiral molecules to act as spin filters, allowing only electrons with a specific spin orientation to pass while blocking others (Fig. 18d). In addition, the spin–orbit coupling (SOC) model posits that the spin of electrons within a chiral molecule is intertwined with its orbital motion, linking the CISS effect to the intricate interaction between electron spin and molecular chirality (Fig. 18e).140 As a result, the electron spin is closely tied to the molecular chirality.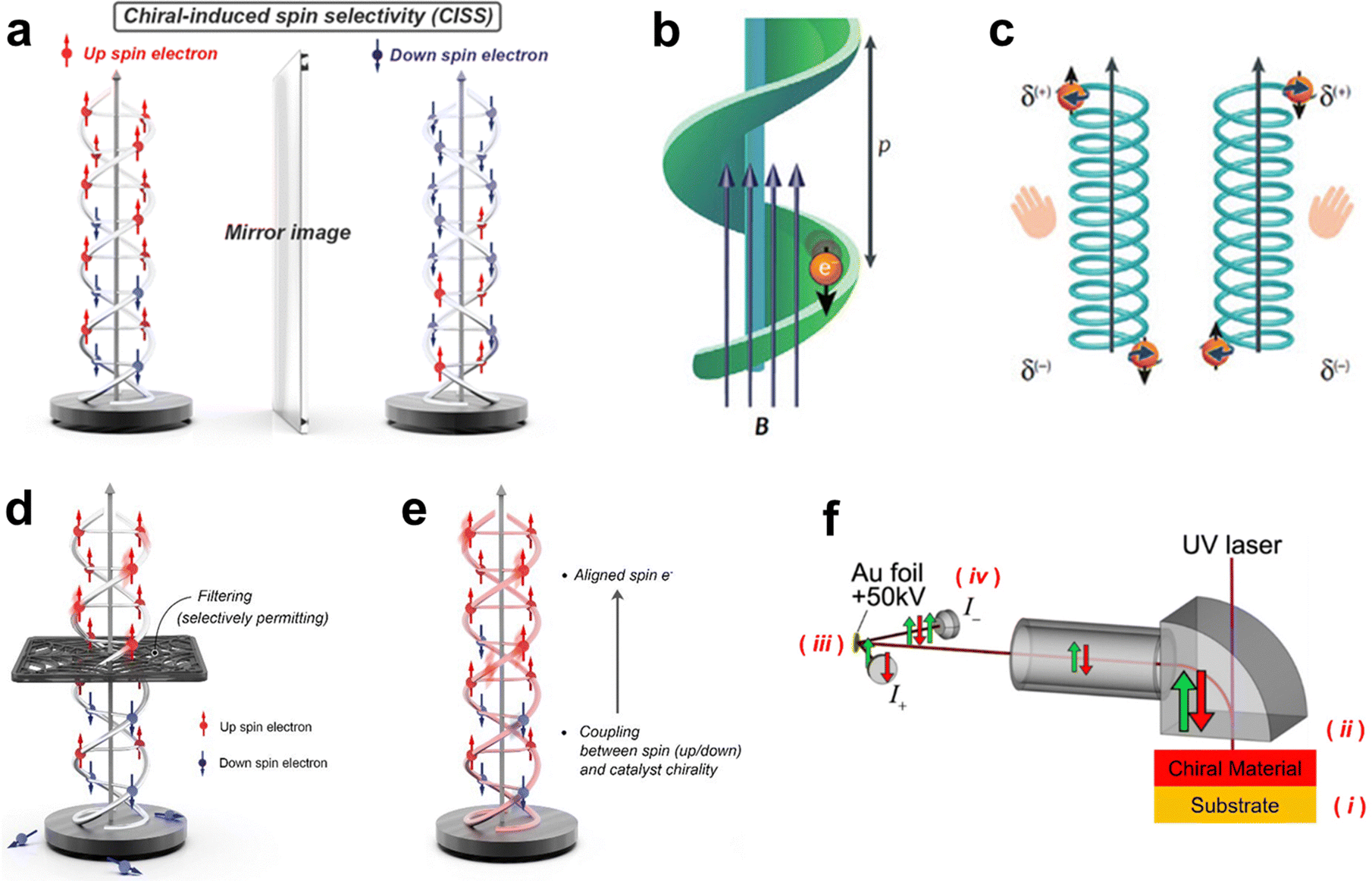 | ||
| Fig. 18 Fundamentals of CISS effect. (a) Schematic of CISS effect in the transport of electrons with opposite spin through the chiral structure. Reproduced with permission from ref. 140. Copyright 2024 Royal Society of Chemistry. (b) Schematic of the electron transmission through a chiral potential of pitch p. (c) Schematic of the charge and spin polarization in chiral molecules, when the molecules are exposed to an electric field acting along their axes. Reproduced with permission from ref. 16. Copyright 2019 Springer Nature. (d) The spin-filter model demonstrating how electrons with specific spin states are selectively transmitted. (e) The SOC model highlighting the interaction between an electron's spin and its orbital motion within the chiral structure. Reproduced with permission from ref. 140. Copyright 2024 Royal Society of Chemistry. (f) Schematic of CISS determination using Mott polarimetry measurements. Reproduced with permission from ref. 141. Copyright 2024 American Chemical Society. | ||
Mott polarimetry is a powerful technique for investigating the magnetic properties of thin films and characterizing the CISS effect. In this approach, electrons striking an Au foil with significant spin–orbit coupling are scattered at varying angles based on their spin orientation, with the angle-dependent detection providing quantitative data about the electron spin population (Fig. 18f).141 Specifically, the typical procedure involves first exciting photoelectrons in the substrate (i), which then pass through a chiral spin filter (ii), resulting in spin polarization. These photoelectrons are subsequently scattered onto the Au foil according to their spin orientation (iii) and quantified using two independent detectors (iv). Additionally, the CISS effect is often demonstrated using magnetic conductive probe atomic force microscopy (mCP-AFM), which combines AFM with magnetic conductivity measurements.142 This technique assesses a material's conductivity and magnetic properties by applying an external magnetic field during the scanning process. The structure of chiral molecules modulates the electron spin during migration, resulting in spin-selective charge transfer. Using mCP-AFM, researchers can quantify the strength of the CISS effect and elucidate how chiral molecules influence electron transfer.
Chiral materials encompass both organic materials, such as chiral polymers, chiral MOFs, and chiral quantum dots, as well as inorganic materials, including chiral oxides, chiral metals, and chiral silicon materials. Inorganic chiral materials typically exhibit superior thermal stability, mechanical strength, and optical properties, making them suitable for a broader range of applications, including photocatalysis, optoelectronic devices, and sensors.143 Additionally, the synthesis of inorganic materials often allows for more precise control over their crystal structure and chiral characteristics, enhancing their overall performance and functionality. Chirality in inorganic nanomaterials is intricately related to their shape, crystal structure, and interactions with chiral ligands. The origins of chirality in these materials can be categorized into four main types (Fig. 19): (a) intrinsic chirality from chiral crystals;144–146 (b) chiral interactions between inorganic nanomaterials and chiral ligands;147–150 (c) chiral shapes at subwavelength scales;151–153 and (d) chiral assemblies formed by inorganic nanoparticles.154–157 Intrinsic chirality in chiral crystals originates from the asymmetric arrangement of atoms within their lattice, primarily involving Sohncke space groups (such as rotation, rotational translation, and translation), as well as lattice distortions or defects (Fig. 19a).144–146 When synthesized under appropriate conditions, nanocrystals with chiral space groups can develop chiral lattices featuring uniform rotational stacking. A robust method for introducing chirality into inorganic nanomaterials is interacting with chiral ligands or molecules, such as deoxyribonucleic acid (DNA) and amino acids, which can induce chirality in nearby achiral materials (Fig. 19b).147–150 This can be achieved either by direct synthesis in the presence of chiral ligands or through post-synthesis ligand exchange, where initial achiral ligands are replaced by chiral ones. Chiral-shaped inorganic nanomaterials, characterized by mirror-asymmetric geometries, are typically synthesized using two main methods: bottom-up and top-down (Fig. 19c).151–153 In the bottom-up approach, chiral ligands direct atoms to form chiral stacking patterns, enabling the synthesis of chiral nanostructures from various inorganic materials. Top-down methods, on the other hand, use advanced tools such as focused ion beams, electron beam lithography, and multi-photon direct laser writing to precisely shape chiral nanostructures, allowing for meticulous control over their size, geometry, and symmetry. However, the top-down methods typically come with high equipment costs and complex processes. Chiral assembly refers to the organization of inorganic nanomaterials into chiral configurations, typically facilitated by chiral ligands and polymer composites acting as scaffolds or bridges (Fig. 19d).154–157 These ligands promote interactions between nanomaterials, leading to their chiral arrangement. Furthermore, the chiral ligands on the surface of inorganic nanomaterials can mutually attract, resulting in the formation of chiral superstructures.
 | ||
| Fig. 19 Schematic diagram of chirality origin in inorganic nanomaterials. Reproduced with permission from ref. 143. Copyright 2023 American Chemical Society. (a) Intrinsic chirality from chiral crystals. Reproduced with permission from ref. 144. Copyright 2015 American Chemical Society. Reproduced with permission from ref. 145. Copyright 2020 Springer Nature. Reproduced with permission from ref. 146. Copyright 2013 Springer Nature. (b) Chiral interactions between inorganic nanomaterials and chiral ligands. Reproduced with permission from ref. 147. Copyright 2016 American Chemical Society. Reproduced with permission from ref. 148. Copyright 2018 American Chemical Society. Reproduced with permission from ref. 149. Copyright 2018 John Wiley and Sons. Reproduced with permission from ref. 150. Copyright 2019 American Chemical Society. (c) Chiral shapes at subwavelength scales. Reproduced with permission from ref. 151. Copyright 2018 Springer Nature. Reproduced with permission from ref. 152. Copyright 2020 Springer Nature. Reproduced with permission from ref. 153. Copyright 2022 John Wiley and Sons. (d) Chiral assemblies formed by inorganic nanoparticles. Reproduced with permission from ref. 154. Copyright 2012 American Chemical Society. Reproduced with permission from ref. 155. Copyright 2015 American Chemical Society. Reproduced with permission from ref. 156. Copyright 2014 Springer Nature. Reproduced with permission from ref. 157. Copyright 2012 American Chemical Society. | ||
Recent advancements in the CISS effect have demonstrated its significant potential for controlling the spin orientation of electrons, providing a novel approach to enhancing the performance of photocatalytic water splitting. In this process, the oxygen evolution reaction is usually regarded as the rate-limiting step, as the oxidation of H2O by photogenerated holes generates HO˙ with random spin orientations. These free radicals with antiparallel spin orientations tend to combine into the singlet byproduct H2O2 rather than forming triplet O2. The CISS effect can align the unpaired electrons in HO˙ in a parallel configuration, facilitating their combination along the triplet pathway to produce O2.140 For example, Pan et al. synthesized chiral ZnO to induce the CISS effect in photocatalytic water splitting, offering a robust strategy for manipulating electron spin-dependent redox reactions.12 Magnetic circular dichroism (CD) spectroscopy reveals that the chiral-structured ZnO functioned as a spin filter, inducing the CISS effect in the photoinduced carriers (Fig. 20a). The photocatalytic water splitting performance of chiral L- and D-ZnO was notably higher than that of the achiral DL-ZnO, with activities 2.0 and 1.9 times greater in photocatalytic O2 production, respectively (Fig. 20b). Moreover, the charge carrier dynamics of chiral and achiral ZnO were investigated using TAS, with the data analyzed using a triple exponential decay function to account for electron capture. The average carrier lifetimes for L- and D-ZnO were determined to be 4.2 and 2.1 ns, respectively, representing increases of 2.4 and 1.2 times compared to DL-ZnO (Fig. 20c). This extended carrier lifetime is attributed to the high spin polarization induced by the CISS effect (Fig. 20d). Mechanistic analysis indicates that polarized holes regulated the spin alignment of HO˙ intermediates, preventing the formation of H2O2 from spin-antiparallel HO˙ recombination and ultimately enhancing the efficiency of O2 production (Fig. 20e).
 | ||
| Fig. 20 CISS effect in chiral ZnO for photocatalysis. (a) CD spectra of ZnO and after infiltration with water. (b) Photocatalytic oxygen production of powdered photocatalysts. (c) Normalized carrier decay kinetics of L-, D-, and DL-ZnO. (d) Schematic diagram of spin selection of photoinduced carriers in chiral ZnO. (e) Formation of singlet or triplet products by HO˙ with different spin directions. Reproduced with permission from ref. 12. Copyright 2023 Springer Nature. | ||
4.6 Combined strategies
Generally, combining two electron spin control strategies can generate a synergistic effect, optimizing light absorption, electron–hole pair separation efficiency, and surface reaction kinetics, resulting in a comprehensive enhancement of photocatalysis. For example, Li et al. utilized an external magnetic field along with single-atom Co doping to synergistically enhance the electron spin polarization of C3N4, promoting photocatalytic water splitting and the simultaneous oxidation of benzylamine.124 With a modest external magnetic field of 24.5 mT, the H2 production rate of Co–C3N4 reached approximately 340 times that of pristine C3N4 in the absence of a magnetic field. Experimental results and theoretical calculations reveal that the interaction between Co d and N p orbitals altered the symmetry center of C3N4, leading to increases in both the dielectric constant and spin polarization.Furthermore, the magnetic field enhanced the alignment of electron spins, increasing the number of electrons with parallel spins. Thus, the combination of the magnetic field and Co doping strategy created a significant built-in electric field and active sites, which improved charge transfer and surface reactions. In another study, a dilute magnetic Ni–CdS/MoS2 photocatalyst with high ferromagnetic spin polarization was developed by doping Ni ions into CdS/MoS2 for photocatalytic water splitting.125 The electron spin polarization induced by Ni doping and the applied magnetic field reduced charge recombination in Ni–CdS/MoS2 and improved the interfacial transfer efficiency between CdS and MoS2, resulting in a 3.89-fold increase in the photocatalytic H2 production rate.
The combination of defect engineering and magnetic field regulation also exhibits synergistic electron spin control for boosting photocatalytic activities. For instance, BaTiO3 with O defects exhibited remarkable activity under an applied magnetic field, achieving an NH3 yield of over 1.93 mg L−1 h−1 in photocatalytic N2 fixation.50 The preparation of O defect-modified BaTiO3 involves mechanically grinding BaTiO3 with NaBH4 in a glove box, followed by reducing the mixture with molten NaBH4 in an Ar atmosphere to create O defects on the BaTiO3 surface. Characterization tests reveal that O defects not only induced lattice distortion and valence state changes of BaTiO3 but also increased local disorder, facilitating the spontaneous polarization of surface electrons. DFT calculations indicate that the desorption of NH3 to form *N is the rate-determining step in the photocatalytic N2 fixation process. Interestingly, the internal electric field induced by O defects and the applied magnetic fields reduced the energy barrier of this step, thereby accelerating NH3 production. Thus, the electromagnetic synergy between the internal electric field and the external magnetic field was achieved by controlling O defects, effectively suppressing the recombination of photogenerated charge carriers and promoting the reduction of N2.
Besides using magnetic fields, combining doping with defect engineering is also an effective strategy for electron spin control. For example, Ni–CdS photocatalyst was synthesized by incorporating highly dispersed Ni atoms into the CdS lattice to replace Cd atoms.129 This incorporation induced local lattice distortion and S defects, which increased the dipole moment and enhanced the spin-polarized electric field. The resulting charge redistribution, driven by the enhanced internal electric field, shifted the S p-band center downward, facilitating the desorption of the *H intermediate during photocatalytic water splitting, ultimately achieving a hydrogen yield 5.58 times higher than that of pristine CdS.
4.7 Other strategies
Recently, the development of van der Waals heterostructures has been shown to modify the electronic band structure and local electron density distribution in photocatalysts.131,132 These alterations influence the distribution of electron spin states, leading to spin polarization effects. By integrating heterostructures with spin polarization, it becomes possible to suppress the recombination of photogenerated charge carriers while enhancing the activity and selectivity of surface redox reactions. For example, spin-polarized monolayer graphene was in situ grown on 2D MoS2 using chemical vapor deposition to form van der Waals heterostructures capable of photocatalytic overall water splitting under visible light.131 The spin polarization facilitated the spatial separation of photogenerated electrons and holes in MoS2 and directed electrons to active sites on the graphene surface through heterojunctions, resulting in an extended TRPL average lifetime from 6.0 to 27.7 ns. Moreover, CN/graphene composites with van der Waals heterostructures and spin-polarized electronic properties have demonstrated high charge separation efficiency and enhanced surface catalytic reactions, achieving efficient photocatalytic water splitting.132Altering the oxidation state of a transition metal can directly change the number of d-electrons, which in turn affects the orbital occupancy. As a result, the number of unpaired electrons varies, influencing the spin state. Gong et al. regulated the spin state of Co-COF by altering the oxidation state of Co at the porphyrin center, significantly promoting photocatalytic CO2 reduction and enhancing selectivity for HCOOH production.68 Theoretical calculations reveal that in CoIII-COF, the Co-3dz2 orbitals interact with the O-2p orbitals of CO2, whereas in CoII-COF, the Co-3dxz or Co-3dyz orbitals couple with the O-2p orbitals. Notably, the CO2 adsorption energy in CoIII-COF is more than twice that in CoII-COF. Moreover, CoIII-COF exhibited stronger interactions with HCOOH, further promoting selective HCOOH generation. These findings highlight that the spin state transition induced by metal valence regulation plays a crucial role in optimizing photocatalytic performance.
In addition, regulating the size of a nanocrystal can influence its spin state, primarily due to the enhanced quantum confinement effects that arise as the nanocrystal size decreases.158 In smaller nanocrystals, discrete energy levels replace continuous ones, altering the distribution and alignment of electron spins. For instance, Fe2O3 nanoclusters with low spin Fe were synthesized through a nanocrystal size modification strategy and subsequently anchored on g-C3N4 for photocatalytic pollutant degradation.133 Notably, the low spin Fe2O3/g-C3N4 exhibited superior photocatalytic activity compared to its high spin counterpart. This improvement was attributed to the d-band center of the Fe 3d orbital in low spin Fe being closer to the Fermi energy level, which weakened the antibonding state. Consequently, the Fe–O interaction was strengthened, enhancing the production of HO˙ and O2˙− for the degradation of diclofenac.
It has been reported that the pyroelectric field can induce the reconstruction of electron spin states.134 He et al. prepared FeS2/CuCo2O4 hollow core–shell heterojunctions and utilized temperature fluctuations between hot and cold regions to drive the pyroelectric field. Specifically, FeS2 generated heat under NIR light, providing the necessary high-temperature endpoint for the pyroelectric field in CuCo2O4. This pyroelectric field enabled CuCo2O4 to release surface charges and generate spontaneous polarization. Additionally, it modified the filling of electrons on Co sites and reversed the electron spin state. This resulted in an extension of TRPL average lifetime from 5.7 to 15.0 ns, enhancing photocatalytic water splitting with an average AQE of up to 19.8%.
5. Conclusions and outlook
Electron spin control of photocatalysts has emerged as a promising strategy for precisely manipulating the spin and spin state of electrons to optimize the separation of photogenerated electron–hole pairs and their subsequent reactions on the photocatalyst surface, offering an innovative approach to improve photocatalytic efficiency. By modulating the electron spin of active sites to enhance interactions with reactants and aligning the electron spin in a specific direction to improve product selectivity, this approach also offers a novel strategy for regulating surface reaction pathways. Summarizing recent progress in electron spin control within the field of photocatalysis is therefore of particular importance. This review provides a comprehensive overview of advanced strategies for manipulating electron spin, including doping design, defect engineering, magnetic field regulation, metal coordination modulation, CISS effect, and combined strategies, aiming at enhancing photocatalytic processes such as water splitting, CO2 reduction, pollutant degradation, and N2 fixation.Among these strategies, doping design, defect engineering, magnetic field regulation, metal coordination modulation, and CISS effect are recognized as key approaches. We compared the number of published academic papers utilizing these strategies to promote photocatalytic applications, with the results presented in a pie chart in Fig. 21a. The findings indicate that doping design is the most widely employed strategy, while defect engineering and magnetic field regulation have also received considerable attention. In contrast, research on electron spin manipulation through metal coordination modulation and the CISS effect remains relatively limited.
 | ||
| Fig. 21 Comparative analysis of doping design, defect engineering, magnetic field regulation, metal coordination modulation, and the CISS effect. (a) Percentage of publications employing the five strategies. (b) Comparison of the five strategies from multiple metrics. (c) Comparison of the advantages and limitations of the five strategies. | ||
A comparison of the five strategies based on multiple metrics, including light absorption, charge separation, surface reaction efficiency, selectivity, stability, and practicality, is presented in Fig. 21b, with a detailed overview of the advantages and limitations provided in Fig. 21c. Notably, each strategy has unique strengths and limitations that must be carefully considered when designing spin control approaches. For instance, doping design remains the most popular strategy due to its ability to enhance light absorption, increase active sites, provide high stability, and offer a straightforward synthesis process. However, it also presents several challenges, including the potential formation of recombination centers that reduce the separation efficiency of photogenerated carriers. Additionally, structural defects introduced during doping can adversely impact the optical and electronic properties of photocatalysts. Furthermore, controlling the doping level is often difficult, ultimately affecting the reliability of photocatalytic performance. In contrast, defect engineering significantly enhances charge separation but introduces limitations, such as uncontrolled defect formation, structural instability, and reduced efficiency over time, making instability a key challenge to address. The application of an external magnetic field during photocatalytic reactions is an efficient and contactless technique that not only promotes charge separation but also improves reaction selectivity. Nevertheless, this approach requires specialized magnetic field generation equipment and precise control, inevitably increasing the complexity and cost of operation. Furthermore, not all photocatalysts are compatible with magnetic field, particularly flexible or non-magnetic materials. Additionally, the effectiveness of magnetic field can be sensitive to environmental factors such as temperature and humidity, potentially affecting the stability and repeatability of the photocatalytic reaction.
Currently, research on metal coordination modulation and the CISS effect remains relatively limited. This may be due to unclear mechanisms of spin control and the underdeveloped state of material preparation techniques, which complicate the implementation of these strategies. The challenges typically associated with metal coordination modulation and the CISS effect include complex photocatalyst synthesis, stability issues, and limited scalability. However, these two strategies also offer significant advantages. Metal coordination modulation allows for precise active site tuning and design flexibility, while the CISS effect promotes charge separation and improves reaction selectivity. These benefits are likely to attract increased research attention, driving the development of new photocatalysts that enable robust electron spin control. Thus, when designing photocatalysts and spin control strategies, it is essential to carefully consider both the advantages and limitations to select the most appropriate approach.
Despite the continuous progress made in recent years in electron spin control for optimizing photocatalysis, several areas still require further development and improvement:
1. Further improving strategies for electron spin control. Developing more robust manipulation techniques is essential to fully realize the potential of electron spin control and enhancing photocatalytic efficiency. For instance, advanced doping techniques such as co-doping can boost light absorption and improve spin polarization, while employing suitable non-metallic dopants reduces reliance on rare metals and minimizes ecological impact.109,110 Moreover, optimizing the concentration and distribution of dopants can lead to more precise electron spin control, ultimately raising photocatalytic performance.
Employing defect engineering to create and control defects within the photocatalyst lattice can introduce local states that facilitate spin manipulation. Exploring the relationship between different defect types (e.g., metallic and non-metallic defects) and their impacts on electron spin control is essential for enhancing photocatalytic performance. Additionally, developing defect engineering techniques that precisely regulate the location and concentration of defects, while also improving the stability of the synthesized materials, is essential to ensure their long-term efficacy in photocatalytic applications.
Currently, most studies on magnetic fields in photocatalysis focus on static magnetic fields, where both the direction and intensity remain constant throughout the process. However, varying the frequency of magnetic field can trigger different mechanisms, such as natural resonances and exchange resonances, both of which may influence electron spin alignment.19 Unfortunately, research providing specific parameters for field intensity and frequency settings is lacking. Further investigation into the effects of external magnetic fields on spin polarization, particularly the impact of magnetic field direction and intensity, could lead to breakthroughs in charge separation and reaction kinetics, offering valuable insights for optimizing electron spins. Additionally, designing electrically, magnetically, or dielectrically sensitive materials as supports for photocatalysts, or utilizing these supports directly as composite catalysts, could enhance response to external fields without compromising the optimal performance of the photocatalyst components.19
Modulating the metal coordination environment to enhance electron spin polarization has opened new avenues of research in photocatalysis. By meticulously designing the geometric structure and electronic properties of metal ligands, it is possible to improve electron spin polarization in photocatalysts, thereby optimizing the electronic environment of active sites and enabling selective modulation of reaction pathways. Future research should integrate theoretical calculations with experimental investigations to develop a comprehensive understanding of how metal coordination affects electron spin polarization, ultimately guiding photocatalyst optimization. Additionally, crucial to advance synthetic methods that enable precise design of the metal coordination environment.
Integrating chiral materials in photocatalysis has shown promise in inducing spin selectivity, leading to improved charge separation and selective product generation. Future research should concentrate on synthesizing innovative chiral nanostructures and exploring their influence on electron spin. By merging insights from theoretical studies with practical applications, researchers can develop new chiral photocatalysts, such as chiral MOFs, chiral COFs, and chiral halide perovskites.159–161 Additionally, a deeper understanding of the mechanisms underlying the CISS effect is essential for fully harnessing its potential. Notably, combining different materials or strategies can create hybrid systems that leverage the advantages of each component, thereby enhancing spin control and photocatalytic efficiency.
2. Developing spin-regulated photocatalysts that fulfill the requirements for practical photocatalytic applications. In practical photocatalytic applications, complex environmental conditions and the presence of multiple components can weaken the spin polarization effect, posing a significant challenge to its practical implementation. To address practical needs, it is imperative to develop stable and efficient spin-regulated photocatalysts for photocatalytic water splitting, CO2 reduction, pollutant degradation, and N2 fixation. This will facilitate their integration into industrial and environmental settings.
Certain photocatalysts have the potential for practical applications due to their stability, efficiency, and scalability. For instance, TiO2 is one of the most widely employed photocatalysts, known for its excellent photostability and non-toxic characteristics. Researchers are actively exploring methods to control its electron spin through techniques such as morphology control, defect engineering, and the integration of chiral materials.13,162,163 COFs also offer a promising avenue for development, as their tunable structures facilitate the incorporation of various functional groups and metals, allowing for precise regulation of electron spin.161 Chiral halide perovskites are potential candidates due to their unique optical properties and ability to induce spin selectivity.159 Their compositional flexibility imparts the design of photocatalysts that efficiently utilize light energy while maintaining favorable spin characteristics. Overall, future research on spin-regulated photocatalysts should focus on integrating these advanced materials into practical systems, ensuring effective operation under diverse environmental conditions while achieving high efficiency and stability.
3. Innovating synthesis methods to achieve precise control of electron spin. The development of novel synthetic techniques that enable precise tuning of spin-selective sites in photocatalysts will pave the way for materials with optimized spin states. Future research should prioritize integrating molecular and atomic-level spin control mechanisms during synthesis, including the precise introduction of dopants, defects, ligands, and chiral components to promote spin polarization without compromising material stability or scalability. Advanced techniques such as atomic layer deposition and template-assisted methods allow for fine-tuning of morphology, lattice structure, and coordination environment of materials, thereby facilitating precise control over electron spin. Furthermore, combining theoretical modeling with experimental synthesis is crucial for understanding how these modifications impact electron spin. As the field progresses, developing environmentally sustainable and cost-effective synthesis methods will be key to translating these advances into practical applications, driving the next generation of efficient and spin-regulated photocatalysts.
4. Deepening understanding of mechanisms underlying electron spin control. Acquiring a deeper understanding of the mechanisms governing electron spin control is crucial for advancing the development of efficient spin-regulated photocatalysts. Future research should elucidate these mechanisms through a synergistic approach that combines experimental techniques with theoretical calculations. Advanced techniques include XAS, Mössbauer spectroscopy, EELS, ESR spectroscopy, and M–T measurement render the scrutiny of local electronic environments and spin dynamics within photocatalysts. Additionally, TRPL and TAS can be employed to investigate the ultrafast processes related to charge carrier dynamics influenced by electron spin control. Furthermore, the generation and transformation of intermediates at active sites of photocatalysts during reaction can be examined using in situ techniques such as in situ EPR, FTIR, and Raman spectroscopy, offering insights into spin-affected surface reactions.
Theoretical calculations can be utilized to model how various factors such as doping, defects, and coordination environmental affect electron spin behavior. This approach elucidates the underlying mechanisms from a theoretical perspective while offering supplementary analysis of experimental results. Additionally, these calculations enable the prediction and optimization of electronic structure and spin characteristics of photocatalysts. By integrating these theoretical insights with experimental data, we could deepen our understanding of spin control mechanisms and guide the design of new materials and strategies aimed at optimizing photocatalytic performance.
One of the key challenges in mechanism studies is separating the effects of electron spin control from the inherent impacts of modification strategies on photocatalytic performance. Accurately making this distinction is critical, as any ambiguity could result in misinterpretation of the results and lead to flawed conclusions. When investigating the effects of electron spin induced by different strategies, it is essential to first consider how each strategy influences photocatalysis. For instance, defect-induced electron spin polarization can enhance the separation efficiency of photogenerated carriers, while the defects may also act as reaction sites, working synergistically to improve photocatalytic performance. Furthermore, it is crucial to differentiate whether the enhancement in photocatalytic performance stems from the intrinsic effects of strategies like doping or defect engineering, or from the spin polarization induced by these strategies. A combination of experimental and theoretical approaches could clarify this distinction. Comparative experiments using non-spin-active materials or adjusting the strength of spin polarization (e.g., through application of magnetic field) could isolate the spin-related contributions. Advanced characterization techniques, such as EPR and spin-resolved photoelectron spectroscopy (PES), could directly probe spin polarization. Coupled with time-resolved spectroscopy and surface reaction kinetics measurements, these techniques enable the assessment of how spin polarization influences carrier dynamics and reaction pathways. Additionally, theoretical calculations and dynamic simulations could provide insights into the independent effects of spin polarization and structural or electronic modifications.
However, the characterization techniques and theoretical calculations for evaluating electron spin behavior are still in development, posing challenges for research in this area. Future efforts should emphasize interdisciplinary collaboration, integrating expertise from chemistry, physics, materials science, and engineering. This collaborative approach will facilitate the design and optimization of next-generation spin-regulated photocatalysts.
In summary, both opportunities and challenges coexist in the field of spin-regulated photocatalysis. Researchers are encouraged to undertake persistent efforts to advance electron spin control strategies aimed to optimize photocatalysis. This involves transitioning fundamental research into practical applications, including photocatalytic water splitting, CO2 reduction, pollutant degradation, and N2 fixation. We hope this review provides valuable insights and inspiration for those interested in leveraging electron spin control to enhance photocatalytic activity.
Data availability
This review does not include any original research results, software, or code, nor were any new data generated or analyzed. Additional information about the review is available upon request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. Lin acknowledges the financial support by the Ministry of Education (MOE), Singapore, under its Academic Research Fund Tier 2 program (T2EP50123-0032) and Agency for Science, Technology and Research (A*STAR) under its RIE2025 MTC IRG program (M23M6c0106). J. Fang acknowledges the financial support by the National Natural Science Foundation of China (22325607 and 22176228). Y. Liu acknowledges the financial support by the National Natural Science Foundation of China (52173207), the Science Fund for Distinguished Young Scholars of Hunan Province of China (2023JJ10040), and the Science and Technology Innovation Program of Hunan Province (2022RC1077). S. He acknowledges the financial support by the China Scholarship Council.References
- N. S. Lewis, Science, 2016, 351, aad1920 Search PubMed.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 Search PubMed.
- B. Dai, G. M. Biesold, M. Zhang, H. Zou, Y. Ding, Z. L. Wang and Z. Lin, Chem. Soc. Rev., 2021, 50, 13646–13691 Search PubMed.
- B. Samanta, Á. Morales-García, F. Illas, N. Goga, J. A. Anta, S. Calero, A. Bieberle-Hütter, F. Libisch, A. B. Muñoz-García and M. Pavone, Chem. Soc. Rev., 2022, 51, 3794–3818 Search PubMed.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 Search PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 Search PubMed.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 Search PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 Search PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 Search PubMed.
- Z. Zhao, T. Zhang, S. Yue, P. Wang, Y. Bao and S. Zhan, ChemPhysChem, 2024, 25, e202300726 Search PubMed.
- T. Wu, X. Ren, Y. Sun, S. Sun, G. Xian, G. G. Scherer, A. C. Fisher, D. Mandler, J. W. Ager and A. Grimaud, Nat. Commun., 2021, 12, 3634 Search PubMed.
- M. Ai, L. Pan, C. Shi, Z.-F. Huang, X. Zhang, W. Mi and J.-J. Zou, Nat. Commun., 2023, 14, 4562 Search PubMed.
- L. Pan, M. Ai, C. Huang, L. Yin, X. Liu, R. Zhang, S. Wang, Z. Jiang, X. Zhang and J.-J. Zou, Nat. Commun., 2020, 11, 418 Search PubMed.
- Y. Zhang, Q. Wu, J. Z. Y. Seow, Y. Jia, X. Ren and Z. J. Xu, Chem. Soc. Rev., 2024, 53, 8123–8136 Search PubMed.
- Z. Zhang, P. Ma, L. Luo, X. Ding, S. Zhou and J. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202216837 Search PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 Search PubMed.
- A. Manchon, H. C. Koo, J. Nitta, S. M. Frolov and R. A. Duine, Nat. Mater., 2015, 14, 871–882 Search PubMed.
- B. D. Ravetz, N. E. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053–2059 Search PubMed.
- Z. Wang, Y. Li, C. Wu and S. C. E. Tsang, Joule, 2022, 6, 1798–1825 Search PubMed.
- C.-C. Lin, T.-R. Liu, S.-R. Lin, K. M. Boopathi, C.-H. Chiang, W.-Y. Tzeng, W.-H. C. Chien, H.-S. Hsu, C.-W. Luo and H.-Y. Tsai, J. Am. Chem. Soc., 2022, 144, 15718–15726 Search PubMed.
- Y. Li, Z. Wang, Y. Wang, A. Kovács, C. Foo, R. E. Dunin-Borkowski, Y. Lu, R. A. Taylor, C. Wu and S. C. E. Tsang, Energy Environ. Sci., 2022, 15, 265–277 Search PubMed.
- D. Zhang, P. Wang, J. Wang, Y. Li, Y. Xia and S. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2114729118 Search PubMed.
- D. Zhang, Y. Li, P. Wang, J. Qu, S. Zhan and Y. Li, Angew. Chem., Int. Ed., 2023, 135, e202303807 Search PubMed.
- S. Sun, G. Shen, J. Jiang, W. Mi, X. Liu, L. Pan, X. Zhang and J. J. Zou, Adv. Energy Mater., 2019, 9, 1901505 Search PubMed.
- A. Vadakkayil, C. Clever, K. N. Kunzler, S. Tan, B. P. Bloom and D. H. Waldeck, Nat. Commun., 2023, 14, 1067 Search PubMed.
- X. Ren, T. Wu, Y. Sun, Y. Li, G. Xian, X. Liu, C. Shen, J. Gracia, H.-J. Gao and H. Yang, Nat. Commun., 2021, 12, 2608 Search PubMed.
- W. Mtangi, F. Tassinari, K. Vankayala, A. Vargas Jentzsch, B. Adelizzi, A. R. Palmans, C. Fontanesi, E. Meijer and R. Naaman, J. Am. Chem. Soc., 2017, 139, 2794–2798 Search PubMed.
- T. Sun, Z. Tang, W. Zang, Z. Li, J. Li, Z. Li, L. Cao, J. S. Dominic Rodriguez, C. O. M. Mariano and H. Xu, Nat. Nanotechnol., 2023, 18, 763–771 Search PubMed.
- H. Im, S. Ma, H. Lee, J. Park, Y. S. Park, J. Yun, J. Lee, S. Moon and J. Moon, Energy Environ. Sci., 2023, 16, 1187–1199 Search PubMed.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 Search PubMed.
- Z. Shi, X. Zhang, X. Lin, G. Liu, C. Ling, S. Xi, B. Chen, Y. Ge, C. Tan and Z. Lai, Nature, 2023, 621, 300–305 Search PubMed.
- V. I. Parvulescu, F. Epron, H. Garcia and P. Granger, Chem. Rev., 2021, 122, 2981–3121 Search PubMed.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, 1901997 Search PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 Search PubMed.
- Q. Li, X. Li, S. Wageh, A. A. Al-Ghamdi and J. Yu, Adv. Energy Mater., 2015, 5, 1500010 Search PubMed.
- H. Kisch, Angew. Chem., Int. Ed., 2010, 122, 9782–9783 Search PubMed.
- Y. Chen, L. Soler, C. Cazorla, J. Oliveras, N. G. Bastús, V. F. Puntes and J. Llorca, Nat. Commun., 2023, 14, 6165 Search PubMed.
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, M. Bahri, H. Yang, A. Gardner, M. A. Zwijnenburg, N. D. Browning and A. J. Cowan, Angew. Chem., Int. Ed., 2022, 134, e202201299 Search PubMed.
- B. Liu, X. Wang, Y. Zhang, L. Xu, T. Wang, X. Xiao, S. Wang, L. Wang and W. Huang, Angew. Chem., Int. Ed., 2023, 135, e202217346 Search PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 Search PubMed.
- S. He, M. Shen, E. Wu, R. Yin, M. Zhu and L. Zeng, Environ. Sci. Ecotechnol., 2022, 9, 100141 Search PubMed.
- Z. Wang, N. Hu, L. Wang, H. Zhao and G. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202407628 Search PubMed.
- S. He, C. Zhai, M. Fujitsuka, S. Kim, M. Zhu, R. Yin, L. Zeng and T. Majima, Appl. Catal., B, 2021, 281, 119479 Search PubMed.
- S. He, Y. Chen, X. Li, L. Zeng and M. Zhu, ACS ES&T Eng., 2022, 2, 527–546 Search PubMed.
- S. He, L. Ling, Y. Wu, S. Yang, Z. Hua, K. Tang, M. Wang, M. Zhu and J. Fang, Environ. Sci. Technol., 2023, 57, 9055–9063 Search PubMed.
- J. Qu, D. Zhang, Y. Li, P. Wang, Y. Mao, T. Zhang, S. Zhan and Y. Li, Appl. Catal., B, 2024, 340, 123246 Search PubMed.
- S. He, R. Yin, Y. Chen, T. Lai, W. Guo, L. Zeng and M. Zhu, Chem. Eng. J., 2021, 423, 130172 Search PubMed.
- L. Wang, W. Wu, K. Liang and X. Yu, Energy Fuels, 2022, 36, 11278–11291 Search PubMed.
- B.-H. Wang, B. Hu, G.-H. Chen, X. Wang, S. Tian, Y. Li, X.-S. Hu, H. Wang, C.-T. Au and L.-L. Jiang, Chem. Catal., 2024, 4, 101083 Search PubMed.
- Z. Zhao, D. Wang, R. Gao, G. Wen, M. Feng, G. Song, J. Zhu, D. Luo, H. Tan and X. Ge, Angew. Chem., Int. Ed., 2021, 60, 11910–11918 Search PubMed.
- P. Wang, J. Wang, T. Ming, X. Wang, H. Yu, J. Yu, Y. Wang and M. Lei, ACS Appl. Mater. Interfaces, 2013, 5, 2924–2929 Search PubMed.
- Y. Lei, C. Yang, J. Hou, F. Wang, S. Min, X. Ma, Z. Jin, J. Xu, G. Lu and K.-W. Huang, Appl. Catal., B, 2017, 216, 59–69 Search PubMed.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 Search PubMed.
- W. Yang, L. Zhang, Y. Hu, Y. Zhong, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 11501–11504 Search PubMed.
- H. Yang, Y. Huang, B. Luo, Z. Xie, D. Li, D. Xu, Y. Lei and W. Shi, Chem. Commun., 2024, 60, 1035–1038 Search PubMed.
- R. Hao, G. Wang, H. Tang, L. Sun, C. Xu and D. Han, Appl. Catal., B, 2016, 187, 47–58 Search PubMed.
- W. Wang, J. Fang, S. Shao, M. Lai and C. Lu, Appl. Catal., B, 2017, 217, 57–64 Search PubMed.
- F. Lin, D. Wang, Z. Jiang, Y. Ma, J. Li, R. Li and C. Li, Energy Environ. Sci., 2012, 5, 6400–6406 Search PubMed.
- J. Xiong, J. Di, J. Xia, W. Zhu and H. Li, Adv. Funct. Mater., 2018, 28, 1801983 Search PubMed.
- J. Ran, B. Zhu and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 Search PubMed.
- S. Bai, N. Zhang, C. Gao and Y. Xiong, Nano Energy, 2018, 53, 296–336 Search PubMed.
- J. Hao, X. Xu, H. Fei, L. Li and B. Yan, Adv. Mater., 2018, 30, 1705634 Search PubMed.
- I. Liu, L. A. Lawton and P. K. Robertson, Environ. Sci. Technol., 2003, 37, 3214–3219 Search PubMed.
- W. Pauli, Naturwissenschaften, 1924, 12, 741 Search PubMed.
- A. Pais, Phys. Today, 1989, 42, 34–40 Search PubMed.
- F. Hund, Z. Angew. Phys., 1927, 40, 742–764 Search PubMed.
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1928, 117, 610–624 Search PubMed.
- Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 Search PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 Search PubMed.
- W. H. Lee, M. H. Han, Y.-J. Ko, B. K. Min, K. H. Chae and H.-S. Oh, Nat. Commun., 2022, 13, 605 Search PubMed.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Chem, 2017, 3, 812–821 Search PubMed.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang and I. Zenyuk, Nat. Catal., 2021, 4, 10–19 Search PubMed.
- Y. Liu, X. Liu, Z. Lv, R. Liu, L. Li, J. Wang, W. Yang, X. Jiang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2022, 134, e202117617 Search PubMed.
- M. T. Sougrati, V. Goellner, A. K. Schuppert, L. Stievano and F. Jaouen, Catal. Today, 2016, 262, 110–120 Search PubMed.
- C. Ma, N. Lin, Z. Wang, S. Zhou, H. Yu, J. Lu and H. Huang, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 99, 115401 Search PubMed.
- Q. Lan, X. Zhang, X. Shen, H. Yang, H. Zhang, X. Guan, W. Wang, Y. Yao, Y. Wang and Y. Peng, Phys. Rev. Mater., 2017, 1, 024403 Search PubMed.
- S. A. Bonke, T. Risse, A. Schnegg and A. Brückner, Nat. Rev. Methods Primers, 2021, 1, 33 Search PubMed.
- C. Wu, X. Wang, Y. Tang, H. Zhong, X. Zhang, A. Zou, J. Zhu, C. Diao, S. Xi and J. Xue, Angew. Chem., Int. Ed., 2023, 135, e202218599 Search PubMed.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 Search PubMed.
- Z.-D. He, R. Tesch, M. J. Eslamibidgoli, M. H. Eikerling and P. M. Kowalski, Nat. Commun., 2023, 14, 3498 Search PubMed.
- X. Zhang, H. Zhong, Q. Zhang, Q. Zhang, C. Wu, J. Yu, Y. Ma, H. An, H. Wang and Y. Zou, Nat. Commun., 2024, 15, 1383 Search PubMed.
- A. Moroz and C. Barnes, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14272 Search PubMed.
- R. Schaffer, E. K.-H. Lee, B.-J. Yang and Y. B. Kim, Rep. Prog. Phys., 2016, 79, 094504 Search PubMed.
- F. Schwabl, Quantum Mechanics, Springer, 2007 Search PubMed.
- S. Jiao, X. Fu and H. Huang, Adv. Funct. Mater., 2022, 32, 2107651 Search PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 Search PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 Search PubMed.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng and J. Cheng, Nat. Commun., 2021, 12, 1734 Search PubMed.
- M.-H. Whangbo, E. E. Gordon, H. Xiang, H.-J. Koo and C. Lee, Acc. Chem. Res., 2015, 48, 3080–3087 Search PubMed.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, 2003297 Search PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 Search PubMed.
- A. L. Buchachenko and V. L. Berdinsky, Chem. Rev., 2002, 102, 603–612 Search PubMed.
- Y. Li, D. Zhang, P. Wang, J. Qu and S. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2407012121 Search PubMed.
- C. Deng, S. Xie, Y. Li, Y. Zhao, P. Zhou, H. Sheng, H. Ji, C. Chen and J. Zhao, J. Phys. Chem. A, 2023, 127, 2787–2794 Search PubMed.
- Y. Liu, Q. Deng, Z. Yao, T. Liang, S. Zhang, T. Zhu, C. Xing, J. Pan, Z. Yu and K. Liang, J. Colloid Interface Sci., 2024, 664, 500–510 Search PubMed.
- W. Li, Y. Zhang, W. Ran, Y. Wang, F. Tian, F. Zhang, M. Xu, D. Zhang, N. Li and T. Yan, Appl. Catal., B, 2024, 351, 123978 Search PubMed.
- Y. Mao, B. Yu, P. Wang, S. Yue and S. Zhan, Nat. Commun., 2024, 15, 6364 Search PubMed.
- C. Cheng, W.-H. Fang, R. Long and O. V. Prezhdo, JACS Au, 2021, 1, 550–559 Search PubMed.
- C. Zhu, L. Lu, Q. Fang, S. Song, B. Chen and Y. Shen, Adv. Funct. Mater., 2023, 33, 2210905 Search PubMed.
- M. Li, S. Wu, D. Liu, Z. Ye, L. Wang, M. Kan, Z. Ye, M. Khan and J. Zhang, J. Am. Chem. Soc., 2024, 146, 15538–15548 Search PubMed.
- R. Ramprasath, V. Manikandan, S. Aldawood, S. Sudha, S. Cholan, N. Kannadasan, S. Sampath and B. Gokul, Environ. Res., 2022, 214, 113866 Search PubMed.
- X. Zhan, J. Liu, Y. Zhao, Y. Sun, R. Gao, H. Wang and H. Shi, Sep. Purif. Technol., 2022, 302, 122146 Search PubMed.
- Y. Wang, Y. Xie, S. Yu, K. Yang, Y. Shao, L. Zou, B. Zhao, Z. Wang, Y. Ling and Y. Chen, Appl. Catal., B, 2023, 327, 122420 Search PubMed.
- J. Wan, L. Liu, Y. Wu, J. Song, J. Liu, R. Song, J. Low, X. Chen, J. Wang and F. Fu, Adv. Funct. Mater., 2022, 32, 2203252 Search PubMed.
- C. Zhu, Q. Fang, R. Liu, W. Dong, S. Song and Y. Shen, Environ. Sci. Technol., 2022, 56, 6699–6709 Search PubMed.
- J. Luo, C. Feng, C. Fan, L. Tang, Y. Liu, Z. Gong, T. Wu, X. Zhen, H. Feng and M. Yan, J. Catal., 2022, 413, 1132–1145 Search PubMed.
- X. Zhang and G. Lu, Carbon, 2016, 108, 215–224 Search PubMed.
- Y. Wang, W. Xu, Y. Zhang, Y. Wu, Z. Wang, L. Fu, F. Bai, B. Zhou, T. Wang and L. Cheng, Nano Energy, 2021, 83, 105783 Search PubMed.
- Q. Wang, X. Deng, H. Pen, F. Liu, M. Song, P. Chen and S.-F. Yin, Nano Res., 2023, 16, 4225–4232 Search PubMed.
- Y. Lin, Z. Jiang, C. Zhu, X. Hu, X. Zhang, H. Zhu, J. Fan and S. H. Lin, J. Mater. Chem. A, 2013, 1, 4516–4524 Search PubMed.
- C. Zhu, L. Lu, J. Xu, S. Song, Q. Fang, R. Liu, Y. Shen, J. Zhao, W. Dong and Y. Shen, Chem. Eng. J., 2023, 451, 138537 Search PubMed.
- X. Yang, F. Li, W. Liu, L. Chen, J. Qi, W. Sun, F. Pan, T. Duan and F. Sun, Appl. Catal., B, 2023, 324, 122202 Search PubMed.
- J. He, L. Hu, C. Shao, S. Jiang, C. Sun and S. Song, ACS Nano, 2021, 15, 18006–18013 Search PubMed.
- W. Wu and L. Yang, J. Catal., 2024, 429, 115283 Search PubMed.
- Z. Jin, J. Zhang, J. Qiu, Y. Hu, T. Di and T. Wang, J. Colloid Interface Sci., 2023, 652, 122–131 Search PubMed.
- Z. Qi, J. Chen, Q. Li, N. Wang, S. A. Carabineiro and K. Lv, Small, 2023, 19, 2303318 Search PubMed.
- J. Xu, L. Lu, C. Zhu, Q. Fang, R. Liu, D. Wang, Z. He, S. Song and Y. Shen, J. Colloid Interface Sci., 2023, 630, 430–442 Search PubMed.
- J. Li, Q. Pei, R. Wang, Y. Zhou, Z. Zhang, Q. Cao, D. Wang, W. Mi and Y. Du, ACS Nano, 2018, 12, 3351–3359 Search PubMed.
- K. Sun, Y. Huang, Q. Wang, W. Zhao, X. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2024, 146, 3241–3249 Search PubMed.
- G.-Z. Huang, Y.-S. Xia, F. Yang, W.-J. Long, J.-J. Liu, J.-P. Liao, M. Zhang, J. Liu and Y.-Q. Lan, J. Am. Chem. Soc., 2023, 145, 26863–26870 Search PubMed.
- T. H. Kim, K. Cho, S. H. Lee, J. H. Kang, H. B. Park, J. Park and Y.-H. Kim, Chem. Eng. J., 2024, 492, 152095 Search PubMed.
- Y. Sun, J. Suriyaprakash, L. Shan, H. Xu, J. Zhang, G. Chen, Y. Zhang, H. Wu, X. Li and L. Dong, Appl. Catal., B, 2024, 355, 124209 Search PubMed.
- J. Li, D. Hu and Q. Chen, Ceram. Int., 2024, 50, 5293–5310 Search PubMed.
- G. Li, X. Sun, P. Chen, M. Song, T. Zhao, F. Liu and S.-F. Yin, Nano Res., 2023, 16, 8845–8852 Search PubMed.
- W. Gao, X. Zhao, T. Zhang, X. Yu, Y. Ma, E. C. dos Santos, J. White, H. Liu and Y. Sang, Nano Energy, 2023, 110, 108381 Search PubMed.
- M. Li, S. Wu, D. Liu, Z. Ye, C. He, J. Wang, X. Gu, Z. Zhang, H. Li and J. Zhang, ACS Catal., 2024, 14, 14098–14109 Search PubMed.
- C. Xing, Z. Yu, N. A. Al-Dhabi, Z. Yao, T. Zhu, Y. Liu, J. Pan, W. Tang and Y. Hou, Sep. Purif. Technol., 2025, 354, 129030 Search PubMed.
- C.-H. Chiang, C.-C. Lin, Y.-C. Lin, C.-Y. Huang, C.-H. Lin, Y.-J. Chen, T.-R. Ko, H.-L. Wu, W.-Y. Tzeng and S.-Z. Ho, J. Am. Chem. Soc., 2024, 146, 23278–23288 Search PubMed.
- F. Wu, X. Zhang, L. Wang, G. Li, J. Huang, A. Song, A. Meng and Z. Li, Small, 2024, 20, 2309439 Search PubMed.
- S. Wei, J. Zhang, L. Zhang, Y. Wang, H. Sun, X. Hua, Z. Guo and D. Dong, Appl. Catal., B, 2024, 358, 124406 Search PubMed.
- G. Yao, S. Yang, S. Jiang, C. Sun and S. Song, Appl. Catal., B, 2022, 315, 121569 Search PubMed.
- Y. Wang, W. Xu, Y. Zhang, C. Zeng, W. Zhang, L. Fu, M. Sun, Y. Wu, J. Hao and W. Zhong, Energy Environ. Mater., 2023, 6, e12390 Search PubMed.
- F. Li, T. Huang, F. Sun, L. Chen, P. Li, F. Shao, X. Yang and W. Liu, Appl. Catal., B, 2022, 317, 121725 Search PubMed.
- Z. He, K. Lin, N. H. Wong, J. Sunarso, Y. Xia, X. Fu, B. Tang, Z. Huang, Y. Wang and H. Yang, Nano Energy, 2024, 124, 109483 Search PubMed.
- I. Zanella, S. Guerini, S. Fagan, J. Mendes Filho and A. Souza Filho, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 073404 Search PubMed.
- A. Zhang, Y. Liang, H. Zhang, Z. Geng and J. Zeng, Chem. Soc. Rev., 2021, 50, 9817–9844 Search PubMed.
- V.-H. Do and J.-M. Lee, ACS Nano, 2022, 16, 17847–17890 Search PubMed.
- A. Srivastava, M. Sidler, A. V. Allain, D. S. Lembke, A. Kis and A. Imamoğlu, Nat. Phys., 2015, 11, 141–147 Search PubMed.
- L. Jiao, J. Wang and H.-L. Jiang, Acc. Chem. Res., 2021, 2, 327–339 Search PubMed.
- K. Chae, N. A. R. C. Mohamad, J. Kim, D.-I. Won, Z. Lin, J. Kim and D. H. Kim, Chem. Soc. Rev., 2024, 53, 9029–9058 Search PubMed.
- B. P. Bloom, Y. Paltiel, R. Naaman and D. H. Waldeck, Chem. Rev., 2024, 124, 1950–1991 Search PubMed.
- S. Ma, H. Lee and J. Moon, Adv. Mater., 2024, 36, 2405685 Search PubMed.
- W. Fu, L. Tan and P.-P. Wang, ACS Nano, 2023, 17, 16326–16347 Search PubMed.
- M. V. Mukhina, V. G. Maslov, A. V. Baranov, A. V. Fedorov, A. O. Orlova, F. Purcell-Milton, J. Govan and Y. K. Gun’ko, Nano Lett., 2015, 15, 2844–2851 Search PubMed.
- C.-C. Chen, C. Zhu, E. R. White, C.-Y. Chiu, M. Scott, B. Regan, L. D. Marks, Y. Huang and J. Miao, Nature, 2013, 496, 74–77 Search PubMed.
- Z. Dong and Y. Ma, Nat. Commun., 2020, 11, 1588 Search PubMed.
- J. K. Choi, B. E. Haynie, U. Tohgha, L. Pap, K. W. Elliott, B. M. Leonard, S. V. Dzyuba, K. Varga, J. Kubelka and M. Balaz, ACS Nano, 2016, 10, 3809–3815 Search PubMed.
- J. Cheng, J. Hao, H. Liu, J. Li, J. Li, X. Zhu, X. Lin, K. Wang and T. He, ACS Nano, 2018, 12, 5341–5350 Search PubMed.
- Y. Li, J. Cheng, J. Li, X. Zhu, T. He, R. Chen and Z. Tang, Angew. Chem., Int. Ed., 2018, 130, 10393–10397 Search PubMed.
- V. A. Kuznetsova, E. Mates-Torres, N. Prochukhan, M. Marcastel, F. Purcell-Milton, J. O’Brien, A. K. Visheratina, M. Martinez-Carmona, Y. Gromova and M. Garcia-Melchor, ACS Nano, 2019, 13, 13560–13572 Search PubMed.
- H.-E. Lee, H.-Y. Ahn, J. Mun, Y. Y. Lee, M. Kim, N. H. Cho, K. Chang, W. S. Kim, J. Rho and K. T. Nam, Nature, 2018, 556, 360–365 Search PubMed.
- L. Ohnoutek, J.-Y. Kim, J. Lu, B. J. Olohan, D. M. Răsădean, G. Dan Pantos, N. A. Kotov and V. K. Valev, Nat. Photonics, 2022, 16, 126–133 Search PubMed.
- B. Ni, M. Mychinko, S. Gómez-Graña, J. Morales-Vidal, M. Obelleiro-Liz, W. Heyvaert, D. Vila-Liarte, X. Zhuo, W. Albrecht and G. Zheng, Adv. Mater., 2023, 35, 2208299 Search PubMed.
- W. Yan, L. Xu, C. Xu, W. Ma, H. Kuang, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 15114–15121 Search PubMed.
- X. Lan, X. Lu, C. Shen, Y. Ke, W. Ni and Q. Wang, J. Am. Chem. Soc., 2015, 137, 457–462 Search PubMed.
- A. Kuzyk, R. Schreiber, H. Zhang, A. O. Govorov, T. Liedl and N. Liu, Nat. Mater., 2014, 13, 862–866 Search PubMed.
- L. Ma, Y. Liu, C. Han, A. Movsesyan, P. Li, H. Li, P. Tang, Y. Yuan, S. Jiang and W. Ni, Nano Lett., 2022, 22, 4784–4791 Search PubMed.
- M. Dc, R. Grassi, J.-Y. Chen, M. Jamali, D. Reifsnyder Hickey, D. Zhang, Z. Zhao, H. Li, P. Quarterman and Y. Lv, Nat. Mater., 2018, 17, 800–807 Search PubMed.
- G. Long, R. Sabatini, M. I. Saidaminov, G. Lakhwani, A. Rasmita, X. Liu, E. H. Sargent and W. Gao, Nat. Rev. Mater., 2020, 5, 423–439 Search PubMed.
- W. Weng and J. Guo, J. Am. Chem. Soc., 2024, 146, 13201–13209 Search PubMed.
- X. Han, C. Yuan, B. Hou, L. Liu, H. Li, Y. Liu and Y. Cui, Chem. Soc. Rev., 2020, 49, 6248–6272 Search PubMed.
- S. Liu, L. Han, Y. Duan, S. Asahina, O. Terasaki, Y. Cao, B. Liu, L. Ma, J. Zhang and S. Che, Nat. Commun., 2012, 3, 1215 Search PubMed.
- Y. Chang, C. Dong, D. Zhou, A. Li, W. Dong, X.-Z. Cao and G. Wang, ACS Nano, 2021, 15, 14174–14184 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |