
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in the photon avalanche luminescence of inorganic lanthanide-doped nanomaterials†
Marcin
Szalkowski
ab,
Agata
Kotulska
a,
Magdalena
Dudek
 a,
Zuzanna
Korczak
a,
Martyna
Majak
a,
Lukasz
Marciniak
a,
Malgorzata
Misiak
a,
Katarzyna
Prorok
a,
Artiom
Skripka
ce,
P. James
Schuck
d,
Emory M.
Chan
*e and
Artur
Bednarkiewicz
*a
a,
Zuzanna
Korczak
a,
Martyna
Majak
a,
Lukasz
Marciniak
a,
Malgorzata
Misiak
a,
Katarzyna
Prorok
a,
Artiom
Skripka
ce,
P. James
Schuck
d,
Emory M.
Chan
*e and
Artur
Bednarkiewicz
*a
aInstitute of Low Temperature and Structure Research, Polish Academy of Sciences, ul. Okolna 2, 50-422 Wroclaw, Poland. E-mail: a.bednarkiewicz@intibs.pl
bNanophotonics Group, Institute of Physics, Faculty of Physics, Astronomy and Informatics, Nicolaus Copernicus University in Toruń, 87-100 Toruń, ul. Grudziądzka 5, Poland
cDepartment of Chemistry, Oregon State University, Corvallis, Oregon 97331, USA
dDepartment of Mechanical Engineering, Columbia University, New York, NY, USA
eThe Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. E-mail: emchan@lbl.gov
First published on 11th December 2024
Abstract
Photon avalanche (PA)—where the absorption of a single photon initiates a ‘chain reaction’ of additional absorption and energy transfer events within a material—is a highly nonlinear optical process that results in upconverted light emission with an exceptionally steep dependence on the illumination intensity. Over 40 years following the first demonstration of photon avalanche emission in lanthanide-doped bulk crystals, PA emission has been achieved in nanometer-scale colloidal particles. The scaling of PA to nanomaterials has resulted in significant and rapid advances, such as luminescence imaging beyond the diffraction limit of light, optical thermometry and force sensing with (sub)micron spatial resolution, and all-optical data storage and processing. In this review, we discuss the fundamental principles underpinning PA and survey the studies leading to the development of nanoscale PA. Finally, we offer a perspective on how this knowledge can be used for the development of next-generation PA nanomaterials optimized for a broad range of applications, including mid-IR imaging, luminescence thermometry, (bio)sensing, optical data processing and nanophotonics.
 Marcin Szalkowski | Marcin Szalkowski received his PhD in Biophysics in 2019 from Nicolaus Copernicus University in Toruń. His doctoral research focused on the application of plasmonically active materials in biosolar cells. In 2020–2022, he worked as a Postdoctoral Fellow in the group of Prof. Artur Bednarkiewicz at the Institute of Low Temperatures and Structure Research, PAS, in Wrocław, where he studied the optical properties of photon avalanching materials. Currently, he continues his research on plasmonics, microscopy techniques and photon avalanche in nanomaterials in the Nanophotonics Group headed by Prof. Sebastian Maćkowski at the Nicolaus Copernicus University in Toruń. |
 Emory M. Chan | Dr Emory Chan is a Career Staff Scientist at the Molecular Foundry, a U. S. Department of Energy nanoscience user facility at Lawrence Berkeley National Laboratory. Dr Chan's research interests include the combinatorial, high-throughput synthesis of nanomaterials with complex compositions, such as semiconductor nanoparticle heterostructures tailored for solid-state lighting and biological imaging. He also investigates nanomaterials that host complex energy transfer networks, such lanthanide-doped upconverting nanoparticles for lasing and super-resolution imaging. Towards this, Dr Chan has developed robotic workflows that increase the reproducibility and throughput of colloidal nanoparticle synthesis, while also enabling machine-learning guided experimentation. |
 Artur Bednarkiewicz | Professor Artur Bednarkiewicz works in the Institute of Low Temperature and Structure Research, Polish Academy of Sciences in Wroclaw, Poland. He is interested in physics, modeling, and spectroscopy of lanthanide-doped materials; developing novel materials, methods and optical instrumentation for imaging and sensing. His current interdisciplinary research focuses on optical nano(bio)spectroscopy, imaging, sensing; novel designs of lanthanide-doped nanoparticles and photon avalanche (bio)labels towards luminescence nano-thermometry and biosensing; upconversion FRET (bio)sensing; reservoir computing. |
1. Introduction
Photon avalanching (PA) is a highly nonlinear upconversion process in which the absorption of a single additional photon by a material produces a cascade of photophysical interactions that ultimately induce the emission of many photons, as shown in Fig. 1a. In PA, weak ground-state absorption (GSA) is followed by a sequence of efficient excited-state absorption (ESA) and energy cross-relaxation (CR) processes that are repeated in a positive feedback loop, which explains the high non-linearity and narrow ‘window’ in which PA is observed as well as the existence of PA threshold and saturation regions (Fig. 1b) in a pump power-dependent luminescence profile. For example, Tm3+-doped NaYF4 PA nanoparticles were recently shown to exhibit nonlinearities with the order S = 30, meaning that above their PA threshold, their luminescence intensity scales with the 30th power of the excitation intensity, similar to a 30-photon process. Thus, doubling the input power results in a staggering 104–106-fold increase in emission intensity. These extreme nonlinearities have been historically investigated and exploited in bulk, lanthanide-doped crystals (e.g., in Pr3+-doped LaCl3 and LaBr3 single crystals1) and fibers to develop new upconversion laser materials and infrared quantum counters. However, the application of PA has been limited by the large size and stringent requirements for observing PA in these materials (e.g., high-quality crystals and cryogenic temperatures). Another characteristic feature of PA emission is its long luminescence rise time, which varies with pump power and excitation history (Fig. 1c). | ||
| Fig. 1 ‘Chain reaction’ mechanism of PA emission (a) and basic features of PA emission – pump power-dependent PA luminescence intensity (b) compared to linear (N = 1) and nonlinear (N = 2, 3, 4, 5) luminescence behaviour, as well as PA luminescence rise times upon switching on photoexcitation (c); curves in (c) correspond to circles in (b) of the respective shade of red colour. | ||
A resurgence of interest in PA has been driven by the recent observation of room-temperature PA in nanoscale materials2,3 and the corresponding advances in the synthesis,2,4–6 heterostructuring,3,5,7–9 modelling,2,3,6,10–13 sensitization,3,7–9,11,14 and biofunctionalization15,16 of lanthanide-doped colloidal nanoparticles that exhibit PA. Scaling PA below 100 nm allows its extreme nonlinearity to be leveraged for applications in sub-diffraction imaging and sensing of temperature, forces, and analytes. In these applications, performance metrics such as spatial resolution and sensitivity are determined by the small size and high nonlinearity of the probe. The colloidal nature of PA nanoparticles also facilitates incorporation of these materials into biological environments and the solution-phase processing of high-density devices for optical storage, computing, and detection. A key advantage of these avalanching nanoparticles (ANPs), which are built upon decades of research into lanthanide-doped upconverting nanoparticles, is that they exhibit excellent photostability, narrowband absorption and emission, high biocompatibility, relatively low critical pump threshold, and extreme susceptibility to signals such as temperature, infrared light, and chemical environment.
This review surveys the recent developments in photon avalanching nanomaterials, with emphasis on their design, synthesis, characterization, and application. This review complements the canonical reviews on PA in bulk materials17–19 to cover the surge in reports on PA nanomaterials within the past 5 years. Initially, we introduce the general principles, mechanism and key features of PA (Section 2) and its brief history in bulk materials (Section 3). Section 4 and Section 5 survey the recent developments in nanomaterials that exhibit PA and discuss their unique optical properties. Section 6 reviews the applications enabled by these PA nanomaterials. Finally, we offer a perspective on the future opportunities and challenges for research in the field of PA nanomaterials in Section 7. By providing a deep understanding of PA and its current state-of-the-art, we hope that this review will stimulate the further development of PA nanomaterials for photonics, biomedicine, sensing, computing, and other applications that would benefit from extreme nonlinearity.
2. Fundamentals of photon avalanche
2.1 Conventional Stokes and anti-Stokes luminescence
PA materials are intriguing because they do not follow the photophysical mechanisms of luminescence found in most of the known emitters, such as organic dyes and quantum dots. Conventional linear emitters exhibit the most basic form of Stokes emission, i.e. fluorescence, in which the GSA of excitation energy quanta (hνEXC) is followed by the emission of a photon (hνEMI) whose energy is lower compared to the incident one (hνEXC > hνEMI). This process is relatively fast, with timescale of 10−10–10−8 s. However, in selected compounds, for example, organic (collagens and lipids) and inorganic crystals, nonlinear anti-Stokes phenomena (hνEXC < hνEMI), such as second-harmonic generation (SHG) and third-harmonic generation (THG) (Fig. 2a), can be observed, where the input frequency (ω) is, respectively, doubled (2ω) or tripled (3ω), resulting from the higher order susceptibility (χ(2) and χ(3)) of the medium. The observation of SHG and THG became possible by the invention of high-intensity, polarized and coherent light sources, lasers. In this case, an electric field with amplitude E(ω) traveling through a medium exhibiting nonlinear susceptibility tensor, χ(2), can be described in terms of polarization at double frequency,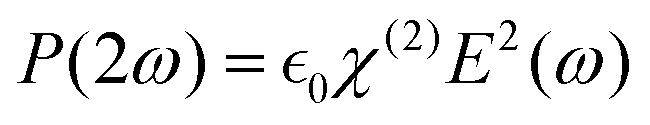 . However, besides a few recently published reports,20 the probability of this process is rather low and can only be observed in non-centrosymmetric crystals under precise phase matching conditions between the laser beam and crystallographic axes of the crystal.21 Another type of anti-Stokes emission can occur following a 2- or 3-photon absorption process (Fig. 2b), in which the excited state of the emitter is reached via the simultaneous absorption of 2 or 3 photons, respectively. While 2- or 3-photon microscopy is feasible and used to enhance the imaging depth and spatial confinement, similar to the previous processes, these mechanisms require excitation with high-power, ultrafast pulsed lasers.
. However, besides a few recently published reports,20 the probability of this process is rather low and can only be observed in non-centrosymmetric crystals under precise phase matching conditions between the laser beam and crystallographic axes of the crystal.21 Another type of anti-Stokes emission can occur following a 2- or 3-photon absorption process (Fig. 2b), in which the excited state of the emitter is reached via the simultaneous absorption of 2 or 3 photons, respectively. While 2- or 3-photon microscopy is feasible and used to enhance the imaging depth and spatial confinement, similar to the previous processes, these mechanisms require excitation with high-power, ultrafast pulsed lasers.
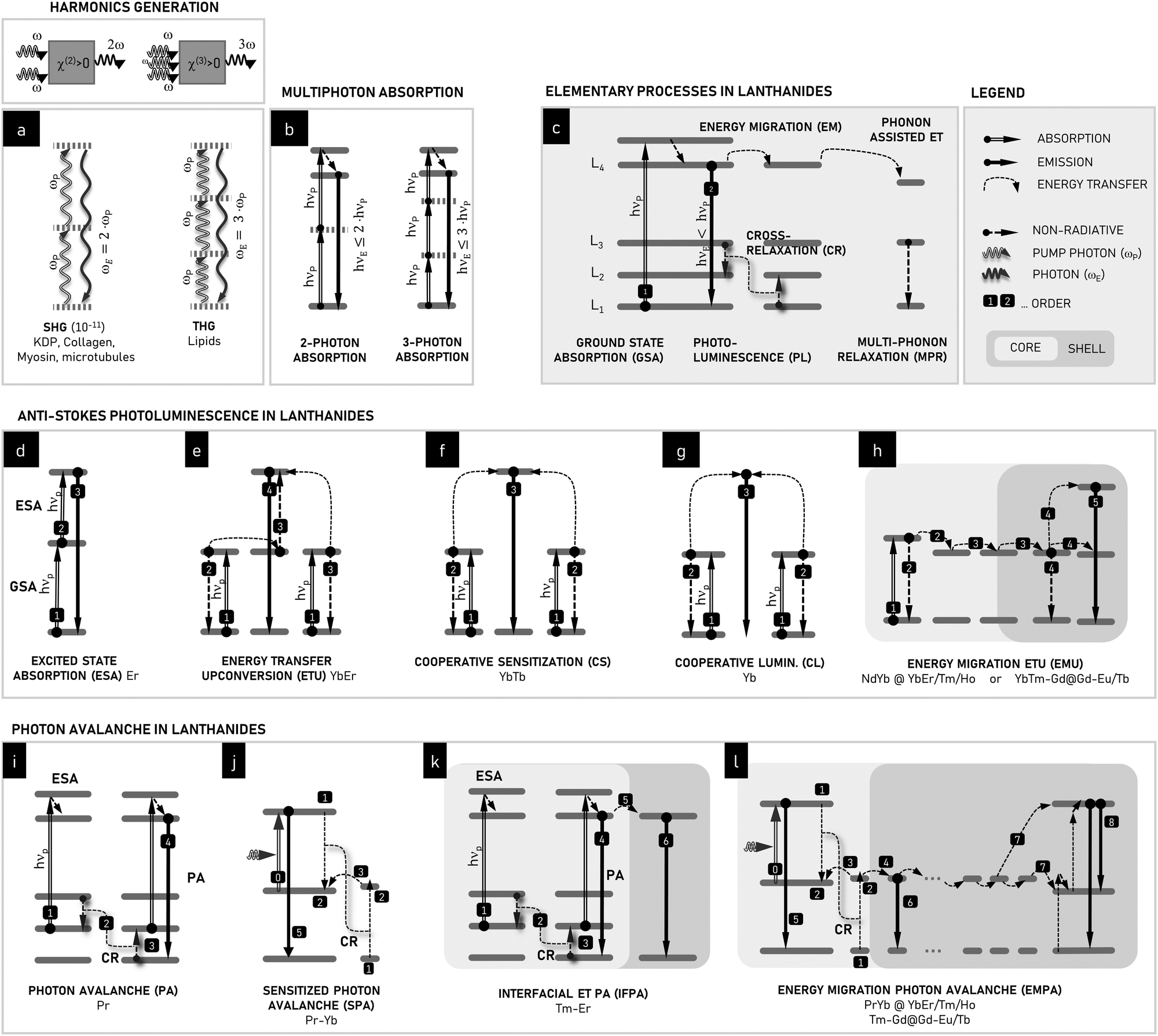 | ||
| Fig. 2 Schematic ET processes leading to anti-Stokes emission in Ln3+ ions compared to nonlinear optical processes such as second and third harmonics generation (a) and 2- and 3-photon absorption (b). In lanthanides, numerous energy transfer (ET) processes may occur (c): ground-state absorption (GSA), Stokes photoluminescence (PL), energy migration (EM), cross-relaxation (CR), phonon-assisted ET, and multi-phonon relaxation (MPR), Numerous anti-Stokes processes can be found in lanthanides: (d) GSA + excited-state absorption (ESA), (e) energy transfer upconversion (ETU), (f) cooperative sensitization (CS), (g) cooperative luminescence (CL), (h) energy migration ETU (EMU), (i) photon avalanche (PA), (j) sensitized photon avalanche (SPA), (k) inter-facial energy transfer photon avalanche (IFPA) and (l) energy migration-assisted PA (EMPA). EMU, IFPA and EMPA are only possible in non-trivially distributed lanthanide ions using core–shell compositional architectures. All lanthanides denote the 3+ ions. | ||
2.2 Upconversion in lanthanide ions
PA is observed in lanthanide ions because they can host anti-Stokes mechanisms that are not accessible to other fluorophores. The forbidden nature of the f–f transitions in trivalent lanthanide ions and ladder-like arrangement of energy levels result in long-lived metastable excited states through which ions can be repeatedly and sequentially excited to high energies. This efficient upconversion in the rich energy level structure of lanthanides is further facilitated in solid state materials by the fact that neighbouring lanthanide ions can interact and transfer energy amongst themselves in processes (Fig. 2c) such as (i) resonant energy migration (EM), (ii) phonon-assisted energy transfer (PAET), and (iii) cross-relaxation (CR). In efficient upconverting materials, these photon excitation and energy transfer (ET) processes outcompete processes that quench the excited states through non-radiatively relaxation, including multiphonon relaxation (MPR), energy quenching on crystal defects, and ET to surface ligands and solvent molecules. CR and MPR are typically considered parasite processes, which limit the brightness of lanthanides. CR results in concentration quenching, preventing the use of higher amounts of emitting lanthanide centers. Alternatively, MPR deactivates excited states non-radiatively to lower intermediate levels or to the ground state, which limits the emission quantum yield significantly. Thus, upconversion in lanthanide-doped materials is the result of a complex, nonlinear network of photophysical interactions,22 involving all the processes summarized in Fig. 2c.Most upconversion applications utilizing lanthanide ions follow the two-photon upconversion processes shown schematically in Fig. 2d and e. The upconversion process in Fig. 2d involves the sequential absorption of two photons by the same ion, i.e., the GSA between the ground energy level and a metastable intermediate level, followed by ESA from the intermediate level to a higher emitting state. A variant of this upconversion scheme, known as ET upconversion (ETU) or APTE (addition de photon par transferts d’energie), utilizes ET to sensitize absorption with a second lanthanide ion such as Yb3+ (Fig. 2e). In the case of both ESA and ETU, it is preferable if the energy of the photon absorption transitions is equal to the energy of the excitation radiation. However, these phenomena may still be observed with a limited amount of energy mismatch with the assistance of phonons, albeit with lower transition probabilities and efficiencies. Thus, as a general rule, resonant photon absorption is desirable for most upconversion applications. The other mechanisms include the much weaker cooperative sensitization (CS) and cooperative luminescence (CL). By exploiting core–shell heterostructured nanomaterials, where the core and the shell are individually optimized and doped with different Ln3+ ions, the combination of EM and ETU has opened new possibilities, such as UC under the more biocompatible 800 nm excitation and multicolour emission.23–26
Stemming from the basic ET processes found in Ln3+ ions (Fig. 2c) and conventional upconversion mechanisms (Fig. 2d–h) in homogenously and heterogeneously doped nanomaterials, various PA mechanisms can be derived, such as the conventional single-dopant PA (Fig. 2i – PA) and sensitized PA (Fig. 2j – SPA), which are the underlying mechanisms for interfacial ET-based (Fig. 2k – IFPA) and energy migration-based (Fig. 2l – EMPA) photon avalanche emission. The major difference between the conventional non-linear (SHG/THG, 2/3PhAbs) or upconversion processes (i.e., ETU, ESA, CS, and CL) and photon avalanche (i.e., PA, SPA, IFPA, and EMPA) is that PA exploits a combination of inefficient GSA with efficient ESA and purposefully augmented CR processes, whose coexistence enables unique PA features to be achieved, but requires a paradigm change in materials design.
2.3 Mechanism of photon avalanching
The mechanism of PA in lanthanide ions (shown in Fig. 2i and 3) is distinct from the other non-linear or upconversion processes described in Fig. 2 because PA specifically occurs when GSA is extremely non-resonant (Fig. 3, process 1′), while the ESA remains resonant and efficient (Fig. 3, process 2). In a conventional upconversion process in which GSA is followed by ESA, slow GSA will limit the overall rate of upconversion given that it will not be able to populate the intermediate level (n2) sufficiently to enable appreciable ESA. However, the unique mechanism of PA circumvents this GSA bottleneck (which occurs due to weak side-band phonon-assisted ground-state absorption) through the use of a type of ET known as cross-relaxation (CR). Here, in the rare circumstance that an Ln3+ ion is excited via ESA, it transfers some of its energy to a neighbouring Ln3+ ion in its ground state, resulting in two Ln3+ ions in their intermediate states. Thus, CR can double the population of the intermediate level, n2, in every iteration of the ESA+CR “energy looping” process (3; 1) → (2; 2). Subsequently, this enhanced n2 population becomes responsible for the enhanced rate of ESA, and the material becomes less transparent to the PA photoexcitation wavelength (λP). Thus, CR bypasses the need for GSA. The enhanced ESA results in increased CR, resulting in a positive feedback loop in which repeated cycles of ESA+CR looping result in the exponential amplification of the population of the n2 and other excited states. This ‘explosion’ of population gives rise to a corresponding ‘avalanche’ of photons emitted from highly excited states, thus giving rise to the PA nomenclature. In short, the mechanism of PA is defined by three main features, as follows: (1) non-resonant GSA, which initially limits luminescence, (2) resonant ESA, and (3) CR, which couples with ESA in a positive feedback loop to nonlinearly populate the intermediate levels from which ESA originates.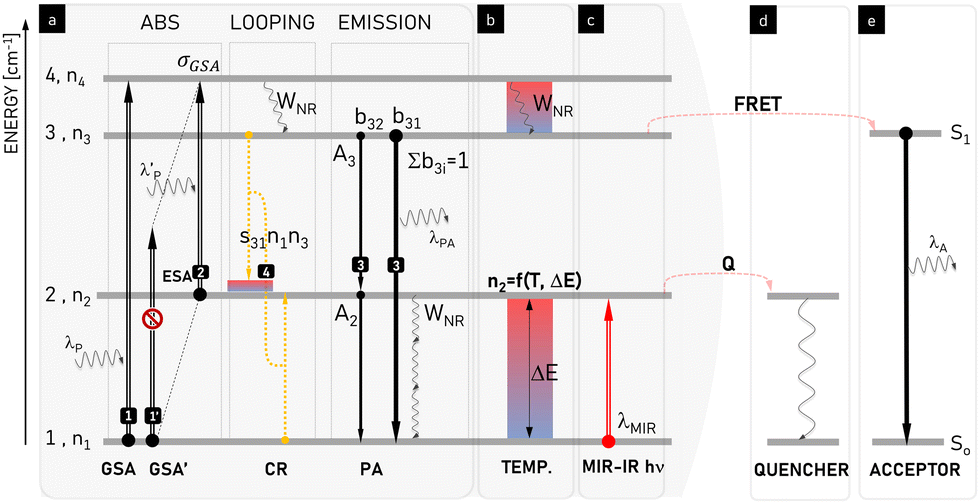 | ||
| Fig. 3 Schematic presentation and explanation of the physical processes involved in photon avalanche emission. Phenomena leading to (a) pure PA, which can be modulated by (b) temperature, (c) illumination with medium-infrared (MIR) photons, and (d) presence of an energy quencher or (e) acceptor. At increased temperature (b) or in the presence of MIR photons (c), the population of the starting level (n2) increases, which modifies PA performance. Temperature may be additionally involved in temperature-sensitive phonon-assisted ET (e.g. cross-relaxation s31(T)) and multi-phonon relaxation (WNR(T)) responsible for energy looping in the system. Because the energy quencher Q (d) or acceptor (e) compete for energy stored in level 2 and 3, the cross-relaxation-based energy looping (4) becomes compromised and the intermediate starting level population (n2) gets smaller. Thus the ESA (2) is hindered, which overall suppresses the PA intensity and increases the PA threshold. λP/λPA/λA denotes the emission wavelength of the pump/photon avalanche/acceptor, s31 defines the temperature-dependent looping rate, WNR denotes the temperature-dependent rate of multiphonon relaxation, b3i is the host-dependent branching ratio of emission between levels 3 and ith, and ΔE is energy gap between levels 1 and 2. | ||
2.4 Prerequisites for observing photon avalanching
Although the mechanistic features listed above are necessary for PA, they are not sufficient to observe the highly nonlinear optical responses characteristic of PA. A common rule-of-thumb for achieving PA is that the ratio of resonant ESA cross section σESA(λP) to non-resonant GSA cross section σGSA(λP) at a given pump wavelength λP follows the condition: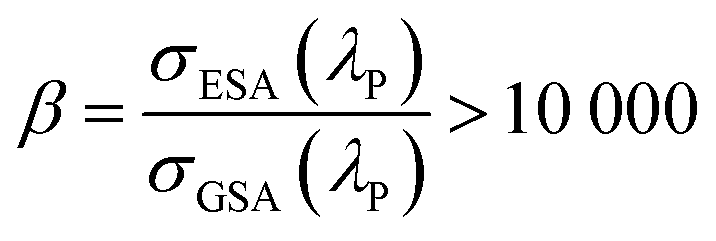 | (2-1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 is typically known as energy looping rather than PA. The primary distinction between energy looping and PA is that PA exhibits a much more nonlinear optical response at the excitation power threshold at which the populations and luminescence suddenly increase. Furthermore, although no clear definitions of avalanche and looping processes exist, based on the present literature, S ∼ 10 seems to be a commonly observed non-linearity that separates the looping (3 < S < 10) regime from the avalanching (S ≥ 10) regime. A β parameter larger than 10000 is commonly assumed to be required, but not a sufficient condition to achieve PA. The second condition is an efficient CR process to multiply the intermediate manifold population, which then enhances the efficiency of ESA. Due to the increased concentration of doping ions, the cross relaxation process between neighbouring ions is competitive with the luminescence in the PA regime, ultimately leading to the avalanche of photons, i.e., the release of energy accumulated in the system during the ESA+CR looping. Above the PA threshold (ITH), rapid luminescence intensity (IL) growth is observed in response to a minute increase in the pump intensity (IP). This pump power-dependent relationship is described by a simple power law,27 as follows:
000 is typically known as energy looping rather than PA. The primary distinction between energy looping and PA is that PA exhibits a much more nonlinear optical response at the excitation power threshold at which the populations and luminescence suddenly increase. Furthermore, although no clear definitions of avalanche and looping processes exist, based on the present literature, S ∼ 10 seems to be a commonly observed non-linearity that separates the looping (3 < S < 10) regime from the avalanching (S ≥ 10) regime. A β parameter larger than 10000 is commonly assumed to be required, but not a sufficient condition to achieve PA. The second condition is an efficient CR process to multiply the intermediate manifold population, which then enhances the efficiency of ESA. Due to the increased concentration of doping ions, the cross relaxation process between neighbouring ions is competitive with the luminescence in the PA regime, ultimately leading to the avalanche of photons, i.e., the release of energy accumulated in the system during the ESA+CR looping. Above the PA threshold (ITH), rapid luminescence intensity (IL) growth is observed in response to a minute increase in the pump intensity (IP). This pump power-dependent relationship is described by a simple power law,27 as follows:| IL ∼ (IP/ITH)S | (2-2) |
2.5 Criteria for confirming photon avalanching
The ability to extract critical parameters from power- and time-dependent luminescence data allows one to objectively determine whether PA is actually occurring. In its most rigorous definition, one can classify a material as undergoing PA when all the following criteria are satisfied simultaneously:(i) Although measuring ESA directly is challenging, the ESA/GSA ratio should exceed β > 104. Given that the ESA and GSA are wavelength dependent, the ability to tune the laser wavelength to maximize the β value is critical and a non-optimal β may be prohibitive to get PA emission or reduce its PA character.
(ii) There are no rigid rules about nonlinearity and PA gain, but the values of PA non-linearity of S ≥ 10 and PA gain of ΔAV ≥ 100 (eqn (2-7)), respectively, are typically considered to indicate PA. Materials that do not meet these criteria but exhibit the same mechanism are considered to be undergoing energy looping. Measuring these values experimentally in a reliable and operator-to-operator variation free manner is often difficult, and thus some hints are presented in Section 2.7. Also, some computer algorithms have been developed to automatically derive S, ITH and ΔAV.6
(iii) A significant slowing down of the rise times at the PA threshold, and their shortening as excitation intensities are increased above the threshold.
2.6 Modelling of PA with rate equations
Numerical modelling of lanthanide photo physics proved critical in the discovery of PA in nanomaterials. In fact, PA in nanomaterials and its utility in sub-diffraction imaging29 were predicted with rate equation modelling several years before experimental realization. The absorption and luminescence processes of the simple system presented in Fig. 3a can be described by the following general rate equations: | (2-3) |
 | (2-4) |
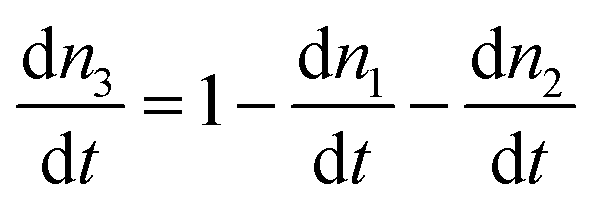 | (2-5) |
 and IGSA, σGSA, denote the pump intensity and the absorption cross section under GSA, while IESA, σESA denote the pump intensity and absorption cross section under ESA; s31,WNR denote the CR rate between level 3 and 1, bai denotes the branching ratio between states
and IGSA, σGSA, denote the pump intensity and the absorption cross section under GSA, while IESA, σESA denote the pump intensity and absorption cross section under ESA; s31,WNR denote the CR rate between level 3 and 1, bai denotes the branching ratio between states  and Aa is the radiative rate from level a (a = 2 and 3), respectively. In the steady state,
and Aa is the radiative rate from level a (a = 2 and 3), respectively. In the steady state,  occurs (for all the levels), which enables the derivation of the n3 population the luminescence intensity (A3n3) as functions of the excitation power density. In the case of PA, analytically solving the set of rate equations may be too complex or even impossible, and thus numerical methods are used.
occurs (for all the levels), which enables the derivation of the n3 population the luminescence intensity (A3n3) as functions of the excitation power density. In the case of PA, analytically solving the set of rate equations may be too complex or even impossible, and thus numerical methods are used.
Due to the inherent nature of PA emission, pump power-dependent luminescence rise times of up to hundreds of milliseconds (τR ∼ 100–1000 ms) have been observed (Fig. 4). Under sufficiently long excitation pulses (t > τR), these kinetic profiles of the PA(t) luminescence intensities enable the extraction of the steady-state luminescence intensity (ISS) and rise time of the luminescence intensity. The inverse of the experimental luminescence decay (τexp) equals the sum of all radiative (kR) and nonradiative (kNR) rates, as follows:
 | (2-6) |
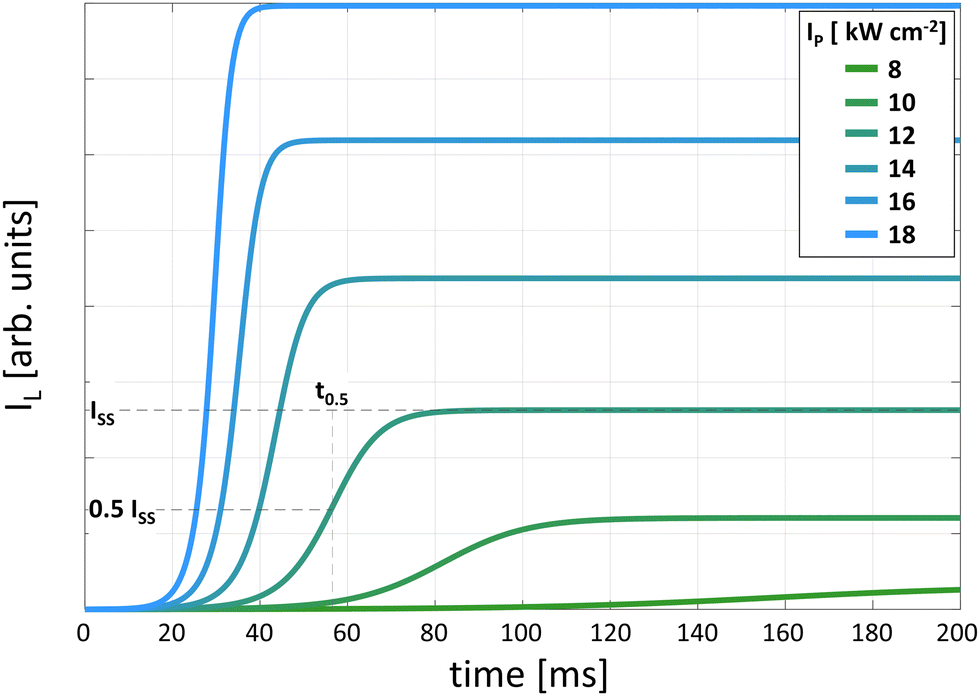 | ||
| Fig. 4 DRE-simulated kinetic profiles of luminescence intensity rise for different excitation intensities. The 50% rise time (t0.5) is shown as the time at which the intensity reaches half of the steady state intensity (ISS). | ||
Differential rate equation (DRE) modelling can be useful to both quantitatively and qualitatively understand the particular PA mechanism (e.g. the role and mechanism of Yb3+ sensitization of Pr3+ avalanching),3,11 and likewise understand some trends in the PA thresholds, slopes or emission quantum yields that occur in response to a variation in the critical PA parameters (e.g. the rates of CR, radiative, non-radiative or absorption cross-section).2,6,11 Moreover, DRE's can be equipped with terms corresponding to additional physical phenomena such as additional ET and relaxations, and has been proven to be an important tool, beside experimental evidence, helping to understand way PA becomes modified in response to a variation in temperature10 or the underlying quenching mechanism affecting the looping and emitting levels.12
2.7 Extraction of key physical parameters of PA from experimental and computational data
Essential PA features can be extracted from the kinetic profiles of PA luminescence intensity (IL(t, IP)) vs. pump power, as shown in Fig. 4. These critical parameters include the steady-state PA emission intensity (ISS = IL(∞)) and rise time measured as t0.5 (defined as IL(tf, IP) = f·ISS(IP) = f·IL(∞, IP), where f ∈ (0; 1〉, with the typical f = 0.5), as shown in Fig. 5, respectively. These standardized methods for the evaluation of the PA spectral, intensity and kinetics should be adopted to enable quantitative comparisons among materials, dopants and labs. | ||
| Fig. 5 Characteristic properties of the PA phenomenon: excitation power-dependent (a) steady-state – IL(IP), (b) nonlinearities (S), (c) risetime – t0.5(IP) and (d) PA gain. The dotted line on (a) represents the experimental values for 8%Tm3+-doped NaYF4 avalanching NPs,2 while the blue lines are generated with the DRE model for an increasing Tm3+ concentration developed in ref. 2 (b)–(d) data have been derived from (a) using algorithms from ref. 6 orange dashed lines show correspondence between the (a)–(d) plots and account for the light blue data line (15%Tm3+). | ||
The rise times (Fig. 4 and 5c) are initially in the micro-second range (very short), then significantly prolong to tens or hundreds of milliseconds (close to the PA threshold), and finally shorten again due to the saturation process with an increase in the pumping intensity.2,29
By plotting the pump intensity (IP)-dependent steady-state PA emission (ISS), one may derive further parameters that characterize the PA emission, such as the pump intensity at which PA occurs (PA threshold, ITH) or saturates (ISAT) and the slope (S) of the power dependence curve (Fig. 5a and b). One may also calculate a pump power-dependent PA gain, which is defined by Lee et al.,2 as follows:
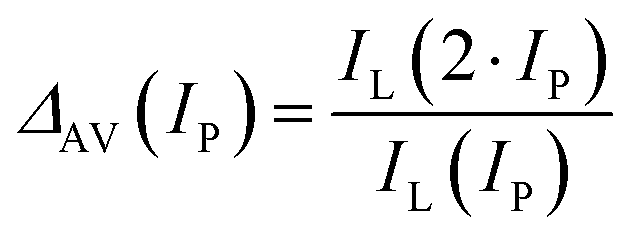 | (2-7) |
One of the most distinctive and unintuitive features of PA is that near the PA pump power threshold, the PA rise time t0.5 gets significantly longer, as long as milliseconds or even seconds. These long rise times are useful both for confirming the presence of PA, and also as critical parameters for evaluating PA materials. These long rise times can be disadvantageous, e.g., typically being viewed as detrimental for achieving fast frame rates during imaging.3 Alternatively, these long rise times allows one to extract ‘pure avalanche photons’ in the time domain,29i.e., the photons that have their origin in the photon avalanche phenomenon and not from the linear absorption and emission (IP ≤ ITH) or from saturated emission (ISAT ≤ IP). These “pure” avalanche photons, IPA, may be quantified by the IPA = I(t2) − I(t1) difference (t1 < t2), where t1 and t2 (0 < t1 < t2) are arbitrarily selected to discriminate long rise times photons, corresponding to PA, from the faster ones, which can be ascribed to the conventional luminescence.
More precisely, t1 denotes the time point when steady-state emission is obtained using an excitation intensity close to the saturation region, significantly exceeding the PA pumping threshold. Alternatively, t2 is the shortest time required to observe steady-state emission from the system excited with the power close to the PA threshold. The “pure” PA photons is a term that may be important for applications where only most nonlinear behaviour is critical, such as in photon avalanche single-beam super-resolution imaging (PASSI).29
2.8 Photon avalanching operating regimes
Depending on the excitation power density, three main operating regimes defined the by relations between the efficiencies of the involved processes can be found, as depicted in Fig. 6a–d. Below the PA threshold (Fig. 6b), photoexcitation results in an increase in the population of the n2 state due to the presence of inefficient GSA.17 At this point, the energy of the pump light is mainly stored by the system in the increasing population of the intermediate level, spreading thorough the network of Ln3+ ions within the host crystal structure with each looping cycle (Fig. 7). However, at this stage, other processes, such as emission, which can be considered a parasitic effect from this perspective, are balancing CRs and one can observe a linear dependence between the excitation and emission intensities (S = 1). With a further increase in the excitation intensity exceeding the PA threshold (Fig. 6c), the emission intensity rapidly increases. This is the power regime where a highly nonlinear relationship can be observed between the pumping power density and the resulting output, and the S parameter reaches the highest value. At the atomic level in the avalanching system, the ESA absorption becomes more efficient due to the increased number of ions in the intermediate energy level, and until the saturation regime, the CRs remain effective, leading to a further increase in this population. | ||
| Fig. 6 Pump power-dependent PA luminescence intensity and population dynamics. (a) Typical S-shape relationship between pump (IP) and luminescence (IL) intensities. (b)–(d) Simulated kinetics of the population of ground (n1), intermediate (n2) and emitting (n3) levels as well as cross-relaxation strength CR·n1·n2 in pre-avalanche (b), avalanche (c) and saturation (d) regions. | ||
 | ||
| Fig. 7 Mechanistic and time-lapse-like explanation of PA emission. The occurrence of several elementary situations is possible in the avalanching system, including the ESA excitation, CR between the ions, local saturation of the system with emitting state ions and, finally, emission (a). In the initial steps of the avalanche mechanism, the single ion is excited by non-GSA-resonant but ESA-resonant photons and undergoes CR (c) with neighbouring ions to double the population of the starting level (b). Upon ESA photoexcitation, steps (b) and (c) are repeated, and the population of the starting level doubles on each loop iteration, as long as the CR rate is much higher than the photon emission or non-radiative depopulation. Red boxes symbolize the pairs of ions (ground state and excited state energy levels) taking a part in the single CR event. Yellow boxes denotes ions in the excited state, which are not capable of cross-relaxing energy with their ground-state neighbours anymore and tend to emit photons. By default, all lanthanide symbols are 3+ ions. | ||
However, certain domains of ions in the emitting level are surrounded by neighbours also excited to the emitting level, making the emission the only possible way to relax the energy. Importantly, after the emission of photons, this ground state ion may be immediately promoted to the intermediate level by the CR with one of the neighbours in the emitting energy level, and then both of them can turn back to the emitting level after capturing the ESA-resonant excitation light. However, a further increase in the illumination power density gradually limits the number of the ground-state ions in the host material, limiting further CRs (Fig. 6d). The dominant process is now the release of accumulated energy in the form of photons (Fig. 6, bottom row). In the power dependence measurements, a linear dependence between the excitation and emission intensities is observed again at this stage (Fig. 6a) because a balance among ESA, CR, kMPR and luminescence processes is reached. Based on the DRE modelling of the PA Tm3+ emission, Fig. 6b–d show the importance of various processes (shown schematically by arrow thickness in energy levels (EL) insets), the average population (AP), as well as the population kinetics (PK) of the ground (n1), intermediate (n2) and emitting (n3) levels, and the contribution of the CR process kinetics (CRK) to the whole PA process. A mechanistic and time-lap like explanation for PA process is schematically presented in Fig. 7.
2.9 Effect of radiative and nonradiative rates on PA
Rate equation models are useful for understanding the design rules for PA materials because they allow the simulation of how the PA parameters will respond to variations in materials properties. For example, in the case of the model depicted in Fig. 3 and described by eqn (2-3)–(2-5), the concentration of dopants, and thus the looping strength may be related to the s31 parameter. Although strong CR is indispensable for efficient PA emission, this process may simultaneously reduce the luminescence intensity. Therefore, by fixing all the other parameters, one may qualitatively study how the concentration of the dopant impacts the PA behavior. Similarly, the non-radiative rate, kNR, is related to the host material. In general, the non-radiative quenching of level 2 is undesirable, but non-radiative transitions are needed to bridge the energy gaps between levels 4 and 3. Under real conditions (not included in the rate equations), these phonons are also required to bridge imperfect energy matches between cross-relaxing energy levels (i.e. when |E3 − E2| ≠ |E2 − E1|).Fig. 8 semi-quantitatively demonstrates the impact of the radiative and non-radiative rates of the 2nd and 3rd levels in the simplified scheme of an exemplary Tm3+ PA material on its power dependence characteristics. It presents the significantly different impact of the dynamics of both levels on the PA performance and its main parameters. In particular, the increasing radiative and non-radiative rates of the 2nd level (the intermediate level during PA) clearly shift the power dependence curve (and the values of the key parameters ITH and ISAT) toward higher powers (Fig. 8a and c).6 A much smaller trend can be observed in the case of the corresponding rates of the 3rd level (the emitting level), as shown in Fig. 8b and d.
 | ||
| Fig. 8 Simulations of pump power-dependent PA emission intensity of NaYF4:8%Tm3+: materials in response to the radiative rates of level 2 (a) and 3 (b) (WR2 and WR3, correspondingly), as well as the impact of non-radiative rates for levels 2 (c) and 3 (d) (WNR2 and WNR3 rates, respectively). Simulations include the pristine rates found for the original NaYF4: 8%Tm3+ material (experimental data are shown as black dots), while varying respective parameters around original values.6 | ||
3. Paradigm shift in designing luminescent materials for PA
3.1 Historical context
To put recent advances in PA nanomaterials into context and demonstrate how the theoretical concepts introduced in the previous section have been applied in real materials, we first discuss the development of PA in bulk materials. The history of PA emission dates back to 1979, when Jay S. Chivian et al. unexpectedly discovered unusual nonlinear luminescence in LaCl3 and LaBr3 crystals doped with Pr3+ (ref. 1) when excited with a green continuous-wave dye laser matching the 3H5 → 3P1 ESA transition in Pr3+. Above a critical pump power threshold (1–12 W cm−2) at temperatures in the range of 20–300 K, the Pr3+ luminescence increased rapidly by two orders of magnitude, which was the first observation of avalanching. Notably, above the PA threshold, the laser extinction increased dramatically although the bulk crystal was transparent to the laser at sub-threshold excitation powers.The discovery of PA stimulated researchers to study different materials from this unconventional point of view, inducing further interest and development of infrared quantum counter detectors1,31 and becoming an impulse for the advancement of upconversion lasers.32–36 In addition to Pr3+-doped LaCl3, PA was obtained with other quantum counters such as LaBr3:Sm3+,37 and CeCl3:Nd3+.38 Other early observations of the PA phenomenon were also reported for bulk materials such as LiYF4:Nd3+,19,32,39 YAG:Tm3+,40 ZrF4–BaF2–LaF3–AlF3–NaF (ZBLAN):Er3+,41 YAG:Ho3+ (ref. 42) and ZBLAN co-doped with Yb3+ and Pr3+.43 PA was also investigated in optical fiber materials, especially in ZBLAN cores doped with Tm3+ (ref. 44) as well as with Er3+ (ref. 45) or co-doped with Yb3+ and Pr3+.46 ZBLAN glasses and YAlO3 doped with Ho3+ were also studied as PA materials.42,47 PA has been reported for YAG waveguides doped with Tm3+ (ref. 17 and 48) and BIGaZYTZr glass doped with Tm3+,49 and for many other materials doped with thulium ions, such as YAlO3,50 Cs2NaGdCl6,51 LaF3,52 Y2O3,53 Y2SiO5,54 KYF4,55 and CdF2.56 Moreover, LiYF4 was identified as a favourable material for hosting PA due to its low phonon energy. Doping this host with Tm3+,57 as well as with Nd3+,32 Er3+ (ref. 58) or Ho3+ (ref. 42 and 59) has been shown to provide suitable conditions to obtain PA. Co-doping materials with a second ion such as Yb3+ was beneficial to obtain the PA phenomenon. In these instances, the Yb3+ ions act as a sensitizer,46 enabling the pumping of the emitting states of the activators via ETU, and take a part in energy cross-relaxation processes, which is known to be crucial for the energy looping and PA mechanism.
An overview of the early work on PA, which was practically limited to bulk crystals, glasses, fibers and ceramics, can be found in a few early review articles.17–19,48 However, despite these successful demonstrations, PA remained a scientific curiosity rather than a mainstream research area, as can be concluded based on several publications from a given year reporting investigations of PA in hosts doped with various Ln3+ ions (Fig. 9). After the first decade since the groundbreaking experiments revealing this new phenomenon, only a few new reports were published. In the next few decades, due to trials on utilizing PA mechanisms to develop new possibilities in the field of laser materials, broadened studies of this subject were performed in several research groups, resulting in over ten new papers per year and leading to a much better understanding of the details of this phenomenon, mainly based on Tm3+-, Er3+- or Nd3+-doped host materials. Nevertheless, the rather specific conditions required to observe PA, often limited to low temperatures, even in bulk materials, have limited the development of this topic, especially compared to the number of publications related to the more general issue of upconversion in lanthanides.
 | ||
| Fig. 9 Historical perspective on the PA emission (WebofScience.com database, update 10.07.2024). | ||
3.2 Materials considerations for PA
Changing the host lattice and doping ion concentration allows the design of nanoparticles with predetermined properties. Many types of materials have been studied as hosts for the upconversion process between lanthanide ions including phosphates,60 vanadates,61,62 sulfides,63 borates,64–66 oxides67 and fluorides. Among the available types of materials, fluorides, ALnFx (A = alkali metal, Ln = lanthanide), such as NaLnF4 and KLnF4, and LnF3, CaF2, and KMnF3 are considered as ideal host candidates. They are characterized by low phonon energies, high chemical stability and good optical transparency over a wide wavelength range; therefore, they are often used as the host materials for upconversion. Additionally, the host lattice based on Na+, Ca2+, and Y3+ cations with an ionic radius close to the lanthanide dopant ions prevents the formation of crystal defects. Among the fluorides, hexagonal phase sodium yttrium fluoride (β-NaYF4) is regarded as the most efficient host materials for upconversion due to the very low characteristic phonon energy for this crystal lattice (350 cm−1).68 The crystal structure of the hexagonal phase NaYF4 has been considered beneficial for upconversion efficiency because two types of relatively low-symmetry cation sites are occupied by Na+ and RE3+ ions.69 Numerous studies have shown that the hexagonal NaYF4 is a much better host lattice for the upconversion emission than its cubic counterpart. Compared to cubic NaYF4:Yb3+,Er3+, the green emission in hexagonal-phase NaYF4:Yb3+,Er3+ is approximately 10 times more efficient. The CaF2 host lattice shows high thermal and chemical stability, wide transmission range and low phonon energy (328 cm−1). However, the introduction of trivalent lanthanide ions in the divalent alkaline earth fluoride MF2 (M = Ca, Sr, Ba), in which the Ln3+ ion substitutes the M2+ ion, potentially leads to the formation of crystal defects and lattice stress due to the difference in charge between the ions.70 Only lanthanum fluoride (LaF3) has intense low-energy phonons (Fig. 11), which are advantageous for reducing the multiphonon quenching of the upconversion emission. These modes are located at 227 and 390 cm−1.71 Other heavy halides (e.g., chlorides, iodides and bromides) exhibit phonon energies less than 300 cm−1, but because of their low chemical stability and hygroscopicity, their potential for application is somewhat limited. The next group of materials that is used as upconversion lattices are oxides (such as Y2O3, Gd2O3, and Lu2O3). Oxide particles are stable in a wide temperature range; moreover, they have good chemical stability and can be doped with a wide variety of lanthanide ions due to the relatively small difference in the ionic radius of the dopant and the same charge as the RE host cations. However, their phonon energy is relatively high (larger than 500 cm−1) due to the stretching vibration of the host lattice. The emission intensity of α-NaYF4 co-doped with Er3+ and Yb3+ was 20 times higher in comparison to Y2O3 co-doped with the same ions.
The choice of low-phonon energy host lattice is crucial for obtaining efficient upconversion or PA. In the high-energy phonon hosts, the luminescence lifetimes, and thus the populations of the intermediate states are reduced by multiphonon relaxation, resulting in low efficiency for upconversion processes. Recently, spectacular PA results have been achieved with ultra phonon energy nanoparticles (KPb2Cl5 and KPb2Br54) and a more conventional tetrafluoride host (NaLuF428), which clearly indicate that low cut-off energy phonons are important, but they are not the only parameters determining the possibility to get highly non-linear PA emission.
The Stark levels belonging to one J manifold of a given lanthanide ion (4fN: 2S+1LJ) in a given host material are usually split by no more than the available maximum host phonon energy. Because the electron–phonon interaction for the 4f optically active electrons (being screened by electrons in the filled 5s and 5p orbitals) in trivalent lanthanides is low, the relaxation rates of the transitions within a single J manifold are usually much faster than the inter-manifold (J ⇝ J', with energy gap ΔEJJ') relaxation rates. A population quasi-equilibrium is established within a single manifold, while inter-manifold transitions are realized by either photon emission (Ar) or sum of all non-radiative transitions (Wnr) accompanied by the generation of matrix phonons.
 | (3-1) |
Based on the Miyakawa–Dexter theory,74 the MPR rates WJJ′(ΔEJJ′) were phenomenologically found to exhibit exponential dependence on the energy gap ΔEJJ′ between the J and J′ manifolds:
| WJJ′(ΔEJJ′) = WJJ′(0)·exp(−α·ΔEJJ′) | (3-2) |
A similar relationship describes PAET processes (e.g. CR, ET between two lanthanide ions, etc.), as follows:
| WPAT(ΔEJJ′) = WPAT(0)·exp(−β·ΔEJJ′) | (3-3) |
Based on theory,10 the multi-phonon-assisted ET (either relaxation or inter ionic ET) can be expressed as follows:
 | (3-4) |
3.3 Challenges limiting the development of PA in bulk materials
The lower degree of investigation of PA in bulk materials may originate from its mechanistic complexity and the narrow optimal conditions over which PA occurs. The PA phenomenon, being a positive feedback system, is highly susceptible to tiny perturbations, especially compared to Stokes emitters. The giant nonlinearity of PA means that these small disturbances in ET pathways lead to disproportionately large changes in luminescence. This sensitivity is exacerbated by the complexity of these ET networks in PA materials, where luminescence quenching may occur via any process that directs energy away from the looping and emitting levels, whose populations are critically important to make the PA material absorb light.12 Thus, the PA phenomenon is very demanding in terms of materials (i.e. dopant type and concentration, type of host, and surface quenching) and experimental methods (i.e. relatively high pump power density, long acquisition times, smooth pump power tuning, etc.).Research in PA utilization is also limited by several practical considerations, such as the need to (i) precisely control the excitation wavelength to ensure resonance or lack of resonance with narrow excitation bands, (ii) pump strongly and continuously or (iii) utilize cryogenic temperatures to diminish MPR. Finally, experimentally confirming the presence of PA is challenging; although there are rigorous criteria for avalanching (Section 2.5), these criteria are debated by some in the community, and ambiguous to others. Consequently, numerous scientific reports on PA luminescence do not definitively confirm all the PA features, and many could be ascribed to pre-PA cases (i.e. energy looping)77 or explained as a combination of ESA and ETU.17 All these facts have contributed to PA remaining a scientific curiosity with niche applications for over 40 years since its discovery.
Compared to conventional Ln3+-doped materials, the PA phenomenon requires a radical change in the paradigms in materials design. For example, in Ln3+-doped bulk materials photoexcited through GSA (Fig. 10a), the effective concentration of doping ions has been typically limited to below 1% due to the belief that higher concentrations result in “concentration quenching” of the luminescence due to CR or energy migration to defects.78 Alternatively, in PA luminophores, the absorption occurs from the first excited level, which originally is almost empty, and thus the sample is initially transparent at the pump wavelength. The population of this level and the resulting absorption coefficient may be increased, but the CR process must be engaged to multiply (double) the number of ions in this state (Fig. 10b). This is achieved by increasing the interaction between neighbouring ions by increasing their concentration. Because the luminescence intensity is proportional to the concentration of the activator (in some range), these two facts make the concentration effects a concentration enhancement rather than concentration quenching. However, it should be noted that by further increasing the dopant concentration, the quenching effects start to outweigh the concentration enhancement. Simultaneously, great care must be devoted to optimizing the selection of the hosts and dopants to reduce the non-radiative depopulation of the starting level and stream the pumping energy to enhance the population of the first excited level or the luminescence. The former is achieved through the selection of the appropriate (typically low) phonon host materials, optimization of the temperature, elimination of the sensitizer ions used typically for upconversion, and selection of the lanthanide activators with a supportive energy level scheme that promotes efficient ESA and CR and reduces non-radiative de-excitation.
 | ||
| Fig. 10 Comparison of the paradigms for conventional (a) and photon avalanche (b) based luminophores. The conventional GSA-based emission is susceptible to CR-caused concentration quenching (a), while in the PA mode, a high dopant concentration and CR are responsible for positive feedback, enhanced ESA absorption and luminescence (b). | ||
4. Considerations for scaling PA to nanoscale materials
4.1 Challenges limiting the development of PA in nanomaterials
Similar to the many challenges limiting research into bulk PA materials, even more obstacles previously hindered the observation of PA in nanoscale materials. The sensitivity of PA is especially troublesome in nanoscale materials, where their surface to volume (SA:V) ratio is high. The large SA:V ratio of nanoscale materials means that a large portion of these materials is exposed to surface quenching from the environment and defects. The high dopant concentrations required for PA can foster rapid energy migration to the surface of nanomaterials, which is more prone to defects and has a higher concentration of quenching species such as ligands and solvent molecules. Colloidal nanoparticles must be stabilized by ligands to maintain the advantages of colloidal dispersion (discussed in Section 4.4). To date, only a few theoretical6,79 and experimental12,80 studies have discussed the impact of surface quenching on PA. Thus, the lack of understanding how to mitigate quenching in nanoscale PA materials has limited their discovery.
Another challenge in achieving PA in nanoscale materials has been the need to develop reliable methods for synthesizing colloidal nanoparticles with robust and reproducible properties. Fortunately, the field of lanthanide-doped nanoparticles has been maturing since 2004, when Haase et al.68 and soon after Gue et al.,81 Yan et al.82 and Capobianco et al.83 managed to develop low-temperature methods for the synthesis of monodisperse NaYF4 nanoparticles. In the 20 years since these seminal reports, researchers have developed robust methods for synthesizing high-quality lanthanide upconverting nanoparticles, e.g., using thermal decomposition.83 These methods allow control of the crystal size, shape, and crystal phases, e.g., low-phonon-energy matrices such as β-NaYF4,84,85 which is well-known to host higher upconversion efficiencies than its α-NaYF4 analogue. Additional upconversion enhancement is realized through the synthesis24,25,85–87 of core–shell nanoparticle heterostructures.88–91 Undoped shells passivate the doped cores by preventing deleterious ET to surfaces, while the doped “active” shells can be used to sensitize absorption,25 segregate incompatible dopants, and mediate ET between domains,23 thereby allowing fine control of the complex ET networks among lanthanide ions. The ability to grow controlled shells has enabled new doping compositions and paradigms, particularly highly doped and alloyed nanoparticles.2,77,92–95 In addition to methods for minimizing surface quenching,93,96–98 biofunctionalization protocols have been developed. All these advancements contributed significantly to the current state of research into lanthanide-doped nanoparticles. In principle, ANPs offer similar advantages together with higher upconversion efficiencies and potential use in applications that exploit the extremely nonlinear response, although several challenges remain. Below, we discuss important considerations when adapting PA for nanoscale materials.
4.2 Size effects and scaling laws
Decreasing the size of crystals down to single nanometres decreases the number of luminescent centres and simultaneously increases their surface to volume ratio, SA:V. Moreover, the distribution of luminescent ions is non-homogenous within the volume of NPs, which may impact the upconversion rates.99 A significant amount of active ions in nanosized materials is located close to the surface,100 and thus their excited states are susceptible to quenching by crystal structure defects, the local chemical environment of the active ions, ligands or the solvent molecules.101 For example, for spherical particles with a diameter of 3 nm, nearly 50% of their atoms are present on the surface. In the case of 10 nm particles, this decreases to 20%, while for a particle with a diameter of 1 μm, only ∼0.15% of its atoms are found on its surface. The increasing amount of superficial lanthanide ions makes them susceptible to non-radiative relaxation of both emitting and intermediate levels, occurring in the course of interaction with solvent and ligands molecules. The higher SA:V also increases the relative contribution of surface defects, which cause the additional luminescence quenching. As a consequence, the quantum yield for NaYF4:2%Er3+, 20%Yb3+ nanoparticles with a size in the range of 10 to 100 nm is in the range of 0.005–0.3%, while for bulk materials, this parameter can be considerably higher (3%).100 Direct evidence of the impact of SA:V and its possibly thermal effects on PA was shown by Deng, et al.102 In their work, isolated nanoparticles did not exhibit PA, but clear avalanching phenomena emerged when microscale aggregates of their nanocrystals were formed, which may be a sign of thermal issues. Approaches to circumvent surface quenching have been extensively studied for conventional UCNPs, and thus some fundamental knowledge exists. There are several methods to reduce the detrimental role of MPR and augment the emission quantum yields of up-converting nanoparticles. These methods include optimization of the dopant and co-dopant concentrations,103,104 intentional altering of the local chemical and structural environment by adding passive co-dopants or changing the ‘structural’ cations, and managing the distribution of active ions in the host materials by the core–shell approach.
4.3 Heterostructures
One of the most effective strategies to improve the luminescent properties of nanoparticles is the utilization of core–shell structures, where the shell layers are grown around the luminescent core.105,106 The protective undoped shell both reduces the surface quenching and changes the balance in multiple nonradiative pathways. By coating the shell, the total intensity improves from several times to several hundred times in various upconversion nanoparticle structures.107 Additionally, nanoparticles doped with lanthanide ions have been used in multicolour labelling and multiplexed bio-detection. The strategy for tuning the emission colour was achieved by manipulating dopant-host combinations and the doping lanthanide ion concentration. For example, the emission spectra of Er3+-doped NaYF4 and KMnF3 nanoparticles differ, owing to the distinct ET pathways stemming from the different dopant-host interactions.108,109 The concentration, and thus the distance-dependent interaction between the dopants and between the dopants and sensitizers play a key role and actually define the upconversion efficiency. The combinations of different upconverting lanthanide ions and changes in the concentrations thereof cause the tuning of the fluorescence emission wavelengths from the visible to near-infrared (NIR) regions. However, co-doping nanoparticles with several different emissive ions may facilitate the ET process, which leads to deleterious CRs between the adjacent dopant ions, often resulting in the quenching of the excitation energy, and thus in a reduction in the luminescence intensity from the doped ions.78,110 This limitation can be overcome by providing the spatial isolation of the emissive ions in the core–shell structure by selective doping of the core and shell. The advantages of spatial positioning of the lanthanide ions in the core and shell were first demonstrated by Qian et al.84 The core (NaYF4:Yb3+,Er3+) nanoparticles coated with active shells (NaYF4:Yb3+,Tm3+) allowed a reduction in the quenching of the fluorescence from the active ions through an increase in the distance between them.4.4 Ligands and coatings
Currently, nanoparticles are coated not only with other inorganic materials but also with a variety of materials such as silica,111–113 polymers85,114,115 and metal nanoparticles116–118 (Fig. 11). Coating the surface of nanoparticles not only allows an improvement in their luminescent properties but also their functionalization for biomedical applications and the creation of multifunctional structures. Alternatively, the capping ligands influence the chemical, physical and optical properties of upconverting nanoparticles. However, their presence on the surface of nanoparticles is necessary to stabilize the surface and prevent the agglomeration of the particles during their synthesis.119 This is necessary, especially when nanoparticles are to be used in biology and medicine and characterized by a narrow size distribution, suitable shape, high aqueous dispersability, and biocompatibility, and thus require a special method for their synthesis. The most effective method for the synthesis of nanoparticles allowing shape and size control is the thermal decomposition reaction of organic metal salts in high-boiling point solvents, typically with polar capping groups and long hydrocarbon chains (such as oleic acid and oleylamine).83,120 These molecules can selectively bind to the surface cations of nanocrystals through their carboxylic groups and form a layer of surface ligands. Carboxylic acids are considered good complexing agents of rare earth ions; however, these molecules may also act as surface oscillators, which significantly quench the upconversion emission of lanthanide ions due to the high energy stretching vibration of their long carbon chain (CH2)n (about 2800–2950 cm−1) and polar COOH groups (about 3000–3500 cm−1).121 Due to the presence of a hydrophobic ligand-stabilizing surface, nanoparticles are generally hydrophobic, only dispersable in nonpolar solvents and requiring post-synthetic functionalization to transfer them to aqueous colloids to finally achieve biocompatibility. To enable the dispersibility and stability of nanoparticles in aqueous media, it is indispensable to make them hydrophilic and conjugatable with various biomolecules and functional groups (e.g., carboxyl, amine, thiol, and phosphate).122 Different surface engineering strategies to transfer hydrophobic nanoparticles from the organic to aqueous phase have been reported including ligand exchange and ligand attraction reactions using a number of different ligands such as citric acid, poly(acrylic acid) (PAA), poly(ethylene glycol) (PEG), poly(ethyleneimine) (PEI), polyvinylpyrrolidone (PVP), and encapsulation in a silica shell. Hydrophilic groups on the surface of nanoparticles provide water-solubility and enable their further bioconjugation. The ‘dark’ side of the surface functionalization of NPs is that the vibrational states of chemical bonds such as O–H, C–H and N–H match the phonon states of the host lattice and may severely quench the emitting and intermediate levels of the luminescent, long living lanthanide levels.123 The quenching of the lanthanide excited states occurs by most chemical ligands; however, those comprised of light elements (such as –OH and –CH groups) quench the nanoparticle luminescence more effectively.101 Simultaneously, O–H vibrations have been reported to be highly effective compared to the C–H vibrational quenching of the excited states. Typically, the upconversion from Yb3+ sensitizers to Er3+/Ho3+ co-doped nanoparticles results in an intense green emission together with weak red emission. The citrate ligands and OH− groups present on the surface of NPs after their hydrothermal synthesis change their green to red emission ratio. Additionally, the high energy vibrational modes of the OH− groups (3200–3600 cm−1) typical for water significantly reduce the emission efficiency compared to oleate-stabilized nanoparticles dispersed in organic solvents.98 | ||
| Fig. 11 Phonon properties of the crystalline host and ligands used to stabilize colloidal nanoparticles. Band absorption of vibration modes from solvents and surface ligands. The maximum vibrational phonon energies of commonly used inorganic lattice, based on digitized data for CaF2,124 NaYF4,125 Y2O3,126 YVO4,127 LaPO4,128 YAG,129 LaF3,129 LaCl3,130 KPb2Br5 and KPb2Cl5.4 | ||
5. Photon avalanching in lanthanide-doped nanomaterials
Due to the challenges of investigating PA materials in nanoscale size regimes, the development of nanomaterials whose luminescence meet the strict criteria for avalanching has been realized only in recent years. These advances have been driven by the development of high-throughput modelling and synthesis of lanthanide-doped nanoparticles. PA emission has been observed in nanoparticles with compositions including NaYF4:Tm3+,2 LiYF4:Tm3+ (ref. 6) and KPb2Cl5:Nd3+ (ref. 4) (Fig. 14), as well as in Yb3+ co-doped with Pr3+.3,11,14 Table S10 (ESI†) summarizes the studies in the literature on lanthanide-doped nanostructures that claim nanoscale PA emission. Although PA is claimed to occur in other classes of materials (e.g., gold and silver nanowires and gold nanorod-nanoparticle hybrids131–133), we focus our survey on lanthanide-doped nanoparticles. The summary in Table S10 (ESI†) lists the essential parameters of the experiments, such as the sizes and compositions of the materials, the used excitation and emission wavelengths, and the key parameters characterizing the PA phenomenon, including the slopes of the emission intensity power dependence or emission dynamics parameters. The table is supplemented by additional comments about the feasibility of achieving PA under the presented conditions and possible alternative explanations based on the available (not always full) characterization presented in the given articles. These comments were based on clearly defined conditions (efficient ESA with negligible GSA and efficient energy looping) and spectroscopic criteria (i.e. clear threshold and steep power dependence with S > 10, followed by saturation of emission intensity vs. pump intensity, pump power dependent rise times, and PA gain of >100), which should be expected from PA emission. To satisfy the biomedical application of PA, some potential NIR excitation lines are predicted beyond that proposed and studied in bulk materials. However, precise ESA wavelengths are often difficult to define and seem critical within the spectral resolution ϕ ± 5 nm.This ESI† focuses on papers that either hypothesize or directly show photon avalanche behaviour in nanoparticles. There are many reports ascribing luminescence behaviour to PA (given that slopes are higher than 4 and simple ESA/ETU is not sufficient to explain the UC process), but most claims are unsupported and are not formally considered PA because they do not meet the strict criteria outlined in Section 2.5, i.e. (i) β > 104 and observation of (ii) clear threshold behavior, and (iii) elongation of rise times near the threshold. The term, “photon avalanche-like behaviour,” can be also found in the literature, which typically indicates a situation in which only part of the expected PA features is observed and the authors suspect other mechanisms than pure PA (e.g. thermal effects, multiple CR and ESA) play a role. Owing to the set of specific requirements necessary to observe PA, the efficiency of PA varies between different dopant types, host types and compositions, as well as the presence of external quenchers or temperature. These considerations, supplemented with a collection of the most up-to-date literature on PA emission in nanomaterials are presented in Table S10 (ESI†).
5.1 Establishing the foundation for the discovery of PA in nanomaterials
5.2 PA in singly doped nanoparticles
The first confirmed reports of lanthanide-based PA in single nanomaterials (i.e., not aggregates) used singly doped colloidal nanoparticles including NaYF4:Tm3+ NPs,2 LiYF4:Tm3+,6 KPb2Cl5:Nd3+ (ref. 4) and NaLuF4:Tm3+ (ref. 28) (Fig. 12). In 2021, Lee et al. demonstrated the first rigorously validated PA in a single nanomaterial using core–shell NaYF4 nanoparticles highly doped with Tm3+ (8–100 mol%) excited at 1064 nm. These avalanching nanoparticles (ANPs) exhibited nonlinearity orders as high as S = 22–31 at threshold powers of ca. 20 kW cm−2 (Fig. 14) and were shown to meet all three stringent criteria for classifying PA (Section 2.5), including the extension of the rise times to 608 ms. The authors attributed their ability to access PA to the enhancement in cross-relaxation resulting from these high dopant concentrations, together with the thick, 3–9 nm NaY0.8Gd0.2F4 shells, which prevented quenching of PA on the surface of the ANPs. Subsequently, avalanching was observed upon doping Tm3+ (3–8%) in LiYF4 NPs, with slopes as high as 126 and extremely high values of >500 for NaLuF4:Tm3+.28 One may note the huge variability in (with thresholds of above 90 kW cm−2) the achieved non-linearities in the Tm3+-doped hosts, from 12 to over 30 and up to 500, which occur in the tetrafluoride fluoride materials LiYF4, NaYF4, and NaLuF4, respectively. These observations confirm the capricious character of photon avalanche emission, given that changes in the local crystallographic sites and crystal field effects, and possibly tiny variation in the excitation laser line may significantly modify the basic PA features. This variance may also indicate that the methodology of PA measurements and characterization are not sufficiently standardized, and cross-lab verifications in the near future must be performed to understand the origin of these discrepancies. | ||
| Fig. 12 Overview graph of PA processes that have been observed or can be predicted in singly (a) and co-doped (b) lanthanide ions. The detailed studies on PA in Pr3+,135 Nd3+,29 Sm3+,136 Ho3+,137 Er3+,58 and Tm3+2 ions and ion-combinations for Yb3+–Pr3+,135,138 Yb3+–Ho3+,139 Yb3+–Er3+ and Yb3+–Tm3+140 are provided in more detail in the summary tables (available in ESI†). The (1), (2), (3), etc. symbols indicate the sequence of ET steps in the Yb co-doped photon avalanching pairs. The line named ‘GSA’ indicates the energy mismatch between the photoexcitation PA photons in relation to the energy levels. | ||
5.3 Sensitized PA via co-doping in nanoparticles
Besides the classical PA scheme (Fig. 2 and 3), in which energy-looping occurs between the same type of dopant (Fig. 12a), PA has also been proposed and reported in co-doped systems, e.g., utilizing different sensitizing and emitting dopants in the same crystalline domain (Fig. 12b). In the sensitized PA scheme reported by Liang et al.,3 and later Dudek et al.,11 Yb3+ ions were used to sensitize Pr3+ emitters. Core/shell nanoparticles of NaYF4:0.5%Pr3+,15%Yb3+@ NaYF4 exhibited PA emission bands at 607 nm and 482 nm, with a slope of 26 and thresholds as low as 60 kW cm−2.3In these systems, the excitation wavelength of the sensitizer remains off-resonance with the wavelengths used to excite the emitter ions through resonant ESA. Owing to the high absorption cross-section and limited concentration quenching of Yb3+ ions, these ions accept energy from the looping emitter ions and (i) store it in their long-lived 2F5/2 levels or (ii) spread this energy efficiently in the host. Then, these excited Yb3+ ions are capable, as in conventional upconversion, of sequentially donating this energy back to neighbouring emitting ions, and thus build the population of intermediate excited states. This is in contrast to the conventional upconversion, in which Yb3+ ions are excited directly with 940–980 nm light. Thus, these sensitized PA schemes may be envisioned to occur (similar to conventional UC scheme) between Yb3+ sensitizers and 3H4 → 1G4 transitions in Pr3+; 5I8 → 5I5, 5I7 → 5F5 and 5I6 → 5S2 transitions in Ho3+; 4I15/2 → 4I11/2 and 4I11/2 → 4F7/2 transitions in Er3+ and 3F4 → 3F3, 3H4 → 1G4 and 1G4 → 1D2 transitions in Tm3+ ions. The preliminary results135,141 for the sensitized approach support the feasibility of achieving PA emission at a much wider selection of emission lines. For example, under 854 nm, Yb3+,Pr3+ PA behaviour was observed, showing high pump power dependence slopes and pump power-dependent rise times. The 854 nm wavelength is resonant with 1G4 → 3P0,1,2,1I6 ESA in Pr, and the two CR processes between the Pr–Pr pair (Pr: 3P0,1,2, 1I6; Pr: 3H4) → (Pr: 1G4, Pr: 1G4) and between Pr and Yb (Pr: 3P0,1,2, 1I6; Yb: 2F7/2) → (Pr3+: 1G4; Yb: 2F5/2) drive energy looping (Fig. 12b). Ultimately, Yb can enhance the Pr: 1G4 population through the Yb: 2F5/2 → Pr: 1G4 transition. Interestingly, the described Yb3+-sensitized Pr3+ PA emission was observed in the LiYF4 single-crystal matrix, but not in the YAlO3 crystal host. This is explained by the fact that the spectral overlap between ESA in Pr3+ and GSA in Yb3+ is non-negligible, and higher ET rates between Yb3+ and Pr3+ and within the Pr3+ ions were present as well as slower decays found for YAlO3 (i.e. 50/11/2000 μs for YLF and 12/4/650 ms for YAlO3 for the Pr3+: 3P0/Pr: 1G4 and Yb3+: 2F5/2). These facts show the sensitivity of PA to nuances related to the absorption cross section for ESA and GSA, the metastable character of the intermediate levels, and the host phonon energies. Similar PA schemes may be derived for other lanthanide pairs, in which Yb sensitizes photon avalanching in Er3+, Ho3+ or Tm3+.
5.4 ANP nano-heterostructures – expanding the spectral range of ANPs with energy migration
Despite achieving moderate nonlinearities (ca. 9) in pure Yb3+/Pr3+ co-doped nanoparticles, most attempts to achieve PA in homogeneously co-doped systems result in sharply quenched emission due to the cross-relaxation between the ions. This CR is particularly detrimental to PA because it disrupts the delicate and critical balance between ESA, looping, and emission, an effect that is strongly amplified by the nonlinearity of PA. To minimize this CR, researchers have borrowed the strategy of sequestering sensitizing and emitting dopants into different domains of hetero-structured core–shell nanoparticles. In this scheme, the nonlinear behaviour (i.e. high slopes, long rise times, and larger dopants content) of the avalanching ions in one crystalline domain is transferred (typically via resonant ET) to the emitting ions in a neighbouring domain, broadening the range of emitters that can exhibit PA emission (Fig. 2l).Two of these schemes have been proposed recently for UCNPs, exploiting resonant energy migration through either Yb3+ or Gd3+ ions in UCNPs. Both schemes have been demonstrated experimentally for avalanching nanoparticles.3,5 In 2022, Liang et al.37 first reported this “migrating” PA (Fig. 13) with slopes as high as 28 in a four-layer nanoparticle heterostructure consisting of an NaYF4:Yb3+/Ho3+ (3/4%) active shell sandwiched between a core and 2nd shell doped with Yb3+/Pr3+ (15/0.5%), with an undoped outer shell of NaLuF4. When Ho3+ was replaced with Tm3+ in these 4-layer heterostructures, high slopes of 46 were observed in the 452 nm emission of Tm3+. In these heterostructures, the 852 nm excitation wavelength is resonant with the ESA of the Pr3+ ions, and is simultaneously off-resonance with Yb3+ and with GSA of Pr3+. The 852 nm pump is not absorbed by the Ho3+ or Tm3+-doped active layers; instead, PA occurs in the Yb3+/Pr3+-co-doped layers. The PA-enhanced population is transferred via resonant energy migration through Yb3+ ions to the Yb3+/Ho3+- or Yb3+/Tm3+-doped active layers. Notably, the emission from all of the doping ions, Pr3+, Tm3+ and Ho3+, exhibited features of avalanche emission at many lines in the visible spectrum (e.g. Tm3+: 452 nm; Ho3+: 541, 646 nm; and Pr3+: 484, 609 nm), providing avenues for multicolour PA emission at visible wavelengths and PASSI microscopy.
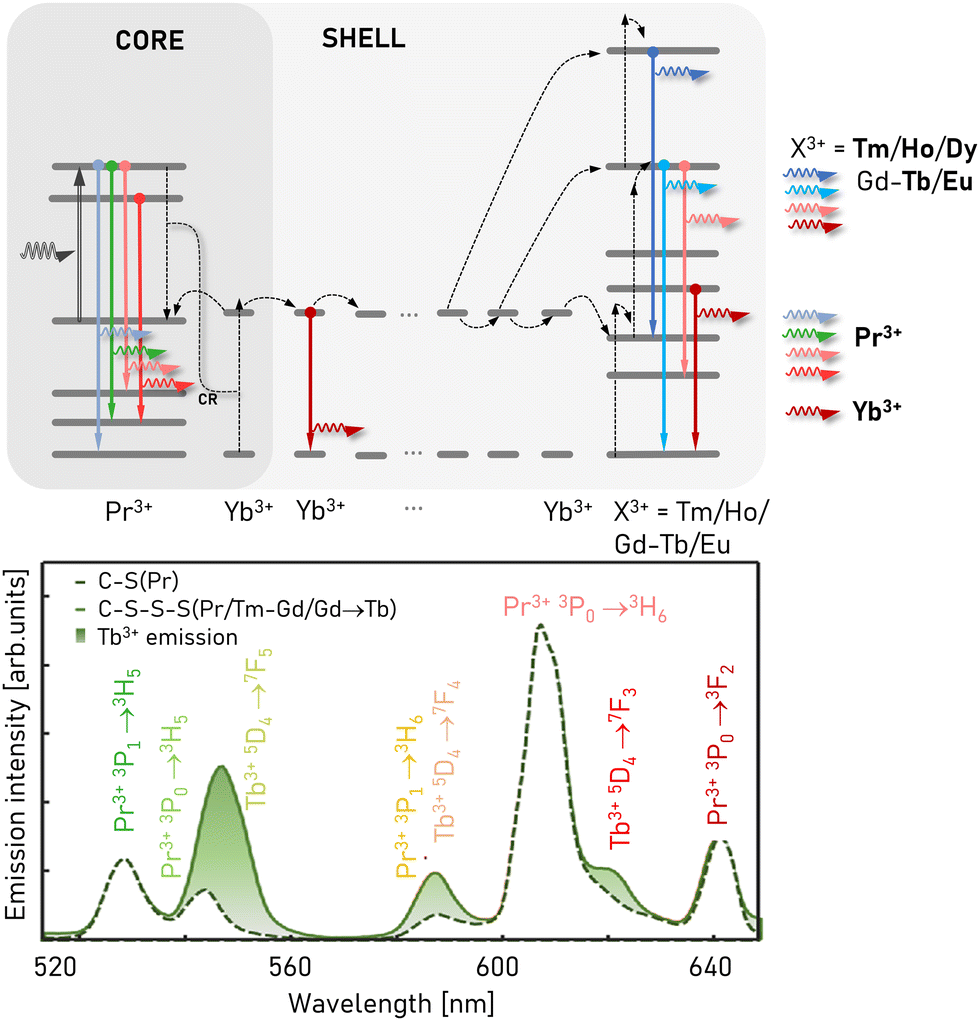 | ||
| Fig. 13 Basic principles of migrating PA emission. The Yb–Pr pair undergoes sensitized PA mechanism, which makes the Yb3+ intermediate migrating ions transfer this behaviour to other lanthanide ions that they sensitize (e.g. Tm3+ and Ho3+).3,142 Some emission of emitting ions, which stems from the Yb–Pr3+ PA workhorse, spectrally overlap with the Pr3+ emission bands. Adapted with permission from ref. 142 Copyright 2024, SPIE and Chinese Laser Press (https://creativecommons.org/licenses/by/4.0/). | ||
One disadvantage of using the Yb3+/Pr3+ avalanching pair is the overlap of multiple Pr3+ emission lines with the multiple visible emission of other intermediate lanthanide dopants (Fig. 13), such as Tm3+, Ho3+, Tb3+ and Eu3+.3,9,142 This may pose some technical challenges in distinguishing these spectral fingerprints from various PA labels in complex biological samples. As an alternative to migration through the Yb3+ sublattice, Skripka et al.5 used migration through an intermediate Gd3+-doped 1st shell to transfer the excited-state populations generated by PA in the NaGdF4:Tm3+-doped core to Eu3+, Tb3+, Er3+, and Ho3+ emitters in a 2nd shell of NaGdF4, which was overcoated with a passivating NaYF4 shell (Fig. 14). Exciting the Tm3+ avalanching dopants at 1064 nm resulted in visible emission with nonlinearities of 14.6, 17.2, 10.7, and 11.5, respectively. Notably, the visible emission lines for these emitting dopants do not overlap with that of the Tm3+ avalanching dopants. Furthermore, the use of Eu3+ and Tb3+ is significant because these dopants are typically thought to be less suitable for anti-Stokes emitters.
 | ||
| Fig. 14 Mechanisms of multicolour PA emission and examples of PA systems capable of generating PA emission at various wavelengths. (1st and 2nd row) The base approach with a single dopant ion, i.e. Tm3+ (ref. 2, 6 and 13) or Nd3+.4 Adapted with permission from ref. 4 Copyright 2023, John Wiley and Sons. (3rd row) Sensitized photon avalanche emission obtained for Pr3+, Yb3+.3,10,14 (4th and 5th row) New emission lines enabled by multi-shell architecture powered by energy migration from avalanching core.3–5 Reprinted with permission from ref. 5 Copyright 2023, the American Chemical Society. | ||
In addition to imprinting PA behaviour onto other lanthanide dopants, Skripka et al.5 also demonstrated the first transfer of PA nonlinearity to non-lanthanide emitters. NaGdF4:20%Tm3+@NaGdF4 core shell ANPs were used to transfer high energy excitation to CdS/CdSe/CdS quantum dot (QD) heterostructures, which exhibited nonlinearities of 10.5 at the 630 nm QD emission wavelength under sub-band gap 1064 nm excitation. The experience gained with these examples is important because the stringent conditions for PA emission to occur are much more difficult to satisfy compared to simple ET upconversion. The ability to exploit the work-horse photon avalanche ‘engine’ to achieve PA characteristics broadens the selection of emission spectra to any fluorophore or luminophore, e.g. for multiplexing applications.
However, another contribution to multicolour PA emission was recently shown by C. Wang et al.,8 who employed a Tm3+ ‘PA engine’ and Yb3+ migrating layer to achieve >60 nonlinearities and Ho3+ (S up to 37), Tb3+ (S = 48), Eu3+ (S = 37), Dy3+ (S = 35) and Nd3+ emission bands in the PA mode in NaYF4:Tm (8%)@NaGdF4:Yb/Tm (10/1%)@NaGdF4:X (X = Tb, Eu, Dy, Nd)@NaYF4 materials. Although multicolour PA emission was shown, the emission spectra were obviously composed of strong Tm3+ emission bands at 450 and 800 nm, which are overlapped with additional weaker bands characteristic for the additional ions.
5.5 PA materials with alternative host matrices: chlorides, bromides, and alloys
The choice of an appropriate host lattice for avalanching lanthanide ions seems to be of critical importance for either PA availability or nonlinearity. For example, the low-phonon energy host lattice minimizes the MPR, increases the occupation of the metastable levels and changes the dynamics of the ET distribution. Oppositely, high-energy phonon hosts are responsible for efficient MPR, which shorten the luminescence lifetimes and reduce the population of intermediate states, ultimately reducing the efficiency of upconversion energy processes. Typically, tetrafluoride Na(Y/Gd)F4 hexagonal nanocrystals have been used, mostly because of their low phonon energies (<400 cm−1), and great advancements have been made in the synthesis of heterostructured nanomaterials for upconversion. These hosts were suitable to show PA phenomenon for the first time in nanomaterials. Recently, colloidal KPb2Br5 and KPb2Cl5 nanocrystals have been developed, with ultralow 250 and 175 cm−1 phonon cut-off energies.4 Although their synthesis is demanding, these nanoparticles reduced the multiphonon relaxation rates and enabled Nd3+ photon avalanche under 1064 nm photoexcitation in the PA regime. This is something that has been postulated previously,29 but not achieved experimentally in other low-phonon NaYF4 or LiYF4 hosts. It must be noted that although ultralow phonon energy Ln3+-doped materials reduce the multiphonon relaxation, they simultaneously reduce the efficiency of other phonon-assisted processes, such as CR and ET, which in some specific PA schemes may be prerequisite to achieving efficient energy looping.Beside engineering of MPR, the local crystal field affects the Stark level splitting and has been shown to have a profound importance in PA. For example, replacing the Y3+ structural ions with smaller Lu3+ ions led to the contraction of the unit cell, and a slight but negligible blue shift in the available phonons and modification of the local crystal field.28 The spectacularly high, monotonic rise in nonlinearity from S = 41 to over S = 150 was noticed for Lu3+ progressively (from 0 to 85%) replacing the Y3+ structural ions (at a constant 15%Tm doping of avalanching ions), and a single particle-to-particle variable of S > 400 (up to 525) at 500–600 kW cm−2 PA thresholds was explained by the modified magnetic–dipole to electric–dipole transition strength. Even if the results are early and their explanations debatable, this enormously high nonlinearity combined with much shorter rise times of around 9 ms enables the further prediction of new applications in imaging or all-optical data processing. This high nonlinearity enabled the study of S in the center and the edge of 172 nm diameter NPs, confirming the substantial role of surface quenching in PA emission, given that S dropped from 450 in the NP center down to 186 close to the edge.
6. Applications of photon avalanching nanoparticles
Since its first demonstration in 1979, the PA phenomenon has been studied, aiming for applications in upconversion laser technology17,36,143,144 or for MIR photon counters.1,17,31,145 However, the potential to observe this phenomenon in nanoscale materials and at room temperature opens a new range of possible applications that are not achievable with homogenously doped, bulk or microcrystal materials (Fig. 15). Here, we briefly present selected research and technological areas in which ANPs have already realized significant advancements.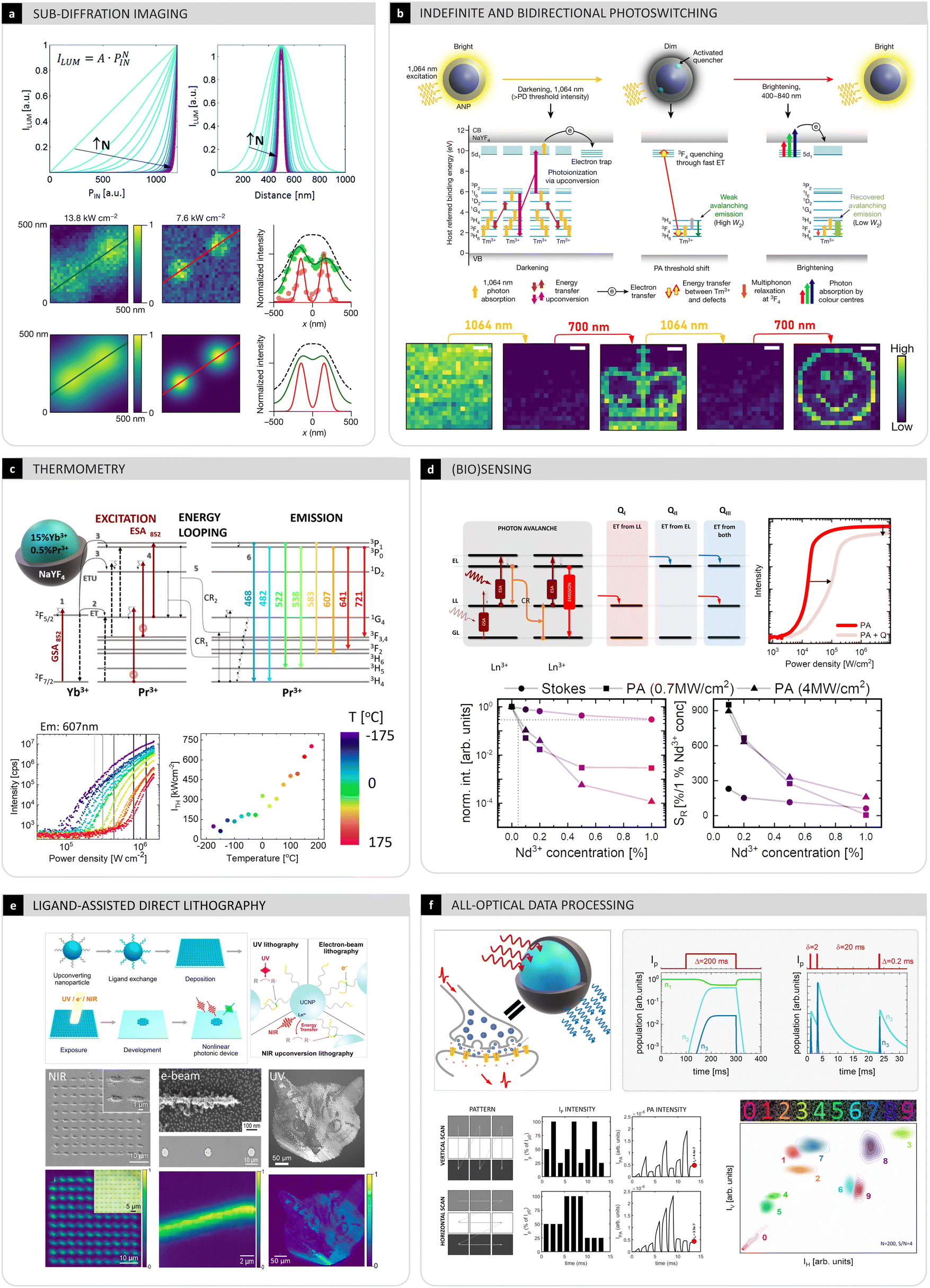 | ||
| Fig. 15 Novel applications of PA (nano)materials. (a) Highly nonlinear pump power dependence enables a reduction in the effective point spread function volume and allows for sub-diffraction imaging of individual ANPs with single excitation raster scanned beam.2,29 Reprinted with permission from ref. 29. Copyright 2019, the Royal Society of Chemistry. Reprinted with permission from ref. 2. Copyright 2021, Springer Nature. (b) Efficient energy looping and ESA may help reach high energy levels close to the conduction band of the host material and enable reversible, indefinite and bidirectional photo switching by changing the PA threshold.146 Reprinted with permission from ref. 146. Copyright 2023, Springer Nature. (c) Positive energy loop is also susceptible to disruption of the energy looping due to changes in temperature and possibly other physical and chemical factors.10,14 Reprinted with permission from ref. 14. Copyright 2023, AIP Publishing. (d) Positive energy looping within avalanching mechanism is extremely susceptible to quenching by either specific or unspecific quenchers, leading to disruption of both emitting and looping level, and consequently quenching of PA luminescence being significantly more efficient than in the conventional Stokes photoluminescence mode.12 Reprinted with permission from ref. 12. Copyright 2024, the Royal Society of Chemistry (https://creativecommons.org/licenses/by/3.0/). (e) By chemical functionalization of the surface of the UCNP, including also ANPs, with photosensitive ligands tending to agglomerate irradiated NCs, UV/NIR or e-beam lithography is possible.147 Reprinted with permission from ref. 147. Copyright 2024, the American Chemical Society. (f) All optical data processing that mimics functioning of biological synapses; the slow kinetics of the ground (n1), looping (n2) and emitting (n3) levels combined with ESA photoexcitation, lead to paired pulse facilitation and photoexcitation history dependence, which enables optical data analysis such as raster scanned image (e.g. of digits) feature extraction.13 Reprinted with permission from ref. 13. Copyright 2023, John Wiley and Sons. | ||
Compared to conventional non-linear (i.e., SHG/THG, 2/3PhAbs) or upconversion (i.e., ETU, ESA, CS, and CL) processes, PA shows some features that are critically important and key to enable numerous applications, as follows:
• PA brightness is gained from significantly increased concentration of luminescence centers, which enhance both the absorption and emission intensity.
• PA non-linearity is significantly higher and easier to access (in terms of pump power densities) than other anti-Stokes processes.
• PA nanomaterials stem from UCNP technology, which has made advances in many areas including biofunctionalization, surface passivation, and the development of models that yield a deep understanding of internal ET processes that occur between lanthanide dopants.
Simultaneously, the high non-linearity of the PA luminescence intensity makes it extremely vulnerable to the pump source stability and infinitesimal quenching, which put higher demands on the pump sources and purity of PA materials compared to UC materials, respectively. Although the PA pump power thresholds are relatively moderate (10–100 of kW cm−2) considering sensitive biomedical applications, they are still higher than in conventional upconverting materials (single W cm−2). Moreover, PA materials are much more sensitive to the excitation wavelength selection compared to UC, and suitable compact and cheap laser sources are not always available on the market. Therefore, it is important to explore new applications concurrently with the continuing design of new and improved PA materials.
6.1 PA-based sub-diffraction imaging
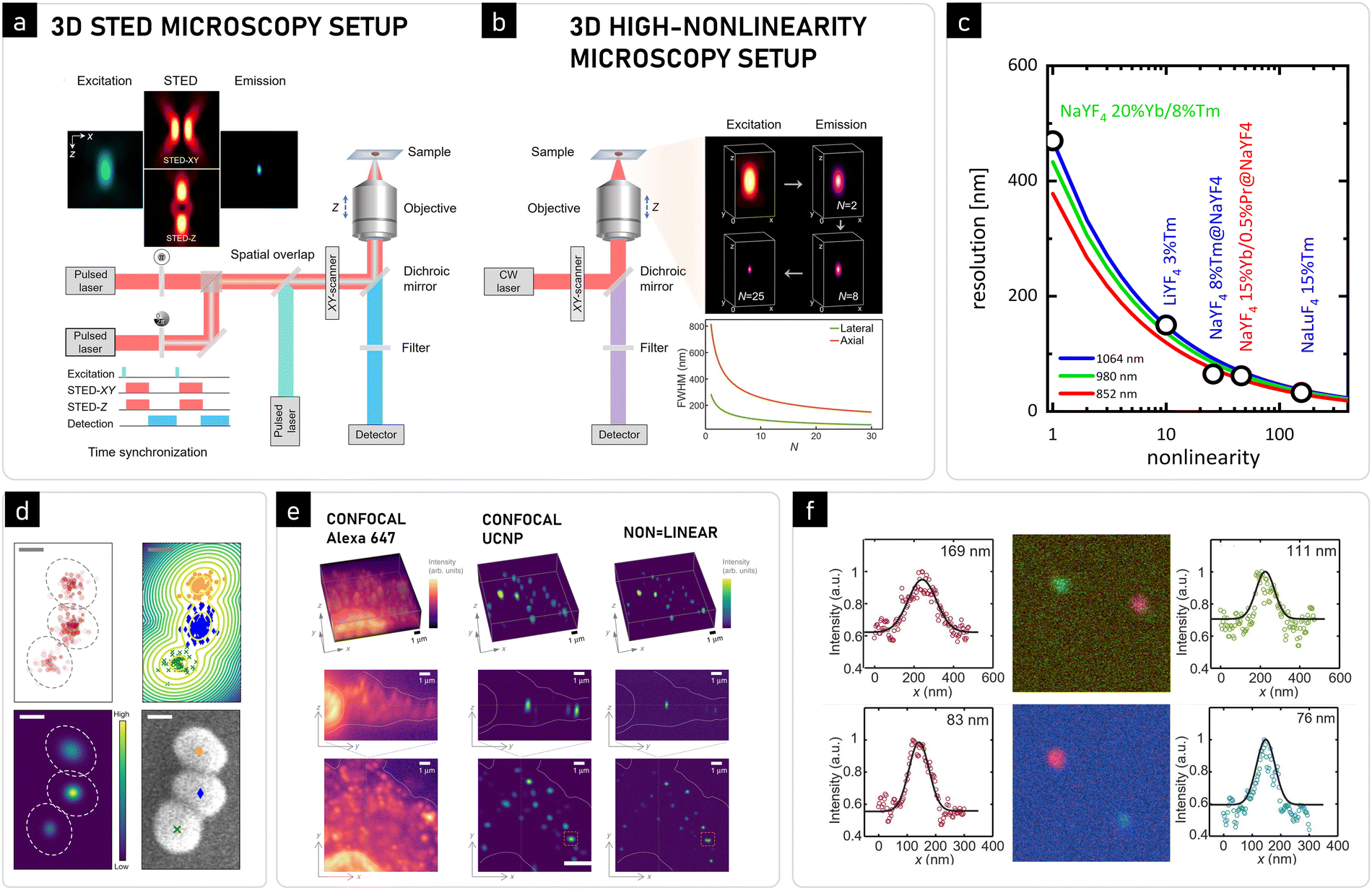 | ||
| Fig. 16 Sub-diffraction imaging with avalanching nanoparticles. (a) Comparison of instrumentation required for conventional raster scanning STED (left) and (b) sub-diffraction imaging using highly nonlinear avalanching nanomaterials (right).9 Reprinted with permission from ref. 9. Copyright 2024, Elsevier. (c) Resolution enhancement originating from highly non-linear emission ref. 28. (d) Photodarkening-based imaging of ANPs demonstrated 2 nm localization precision146 (scale bar: 20 nm). Reprinted with permission from ref. 146. Copyright 2023, Springer Nature. (e) First demonstration of in vitro 3D sub-diffraction imaging with super-linear LnNP labels compared to confocal Stokes and UCNP-based imaging.160 Adapted with permission from ref. 160. Copyright 2019, Springer Nature (https://creativecommons.org/licenses/by/4.0/). (f) Two colour photon avalanche single beam sub-diffraction imaging.9 Reprinted with permission from ref. 9. Copyright 2024, Elsevier. | ||
The need to finely control the optical properties of super-resolution fluorophores has led to the use of nanoparticles for super-resolution imaging.150–152 In particular, lanthanide-doped nanoparticles have been successfully used as luminescent probes in several super-resolution microscopy techniques, including STED,95 nonlinear structured illumination microscopy,161 and near-infrared emission saturation nanoscopy.162 These excellent demonstrations exploit the unique features of lanthanides, such as long luminescence lifetimes (facilitating depletion), perfect photostability and narrowband excitation end visible/NIR emission bands, with relatively low excitation/depletion power density thresholds and cheap and easily available CW light sources.
One of the first STED-inspired approaches for the application of lanthanide-doped nano labels was realized by Kolesov et al.,163 where Pr3+-doped YAG NPs were imaged with a system composed of two beams, a 609 nm pumping pulse and 532 nm depleting one, giving a spatial resolution of 50 nm for the image collected for UV emission. A different operating scheme for application of lanthanide-doped NPs for STED imaging was proposed by Liu et al.95 In this realization, NaYF4 nanocrystals co-doped with Yb3+ and Tm3+ were imaged with pumping with a 980 nm laser beam to observe an emission from the 1D2 state, while an 808 nm donut-shaped beam was utilized for inducing stimulated emission (3H4 → 3H6 transition), and therefore depleting the emission from higher energetic levels. As a result, fluorescence imaging with a spatial resolution of 28 nm was achieved. A similar mechanism in Tm3+-doped hosts was shown to give imaging resolutions of 50 nm for laser scanning through tens of micrometers thick tissue.162
Although STED-based techniques result in an impressive quality of direct super-resolution imaging, these methods still require rather complex dedicated optical systems operating with two concentric beams, one with a Gaussian profile and the other (depleting beam) characterized by a cylindrical donut shape. These two beams must also be properly synchronized in time to most effectively confine the point spread function of the STED microscope, which is not trivial when femtosecond pulses are used (Fig. 16a, left). In contrast, stochastic methods such as PALM and STORM are not necessarily demanding in terms of complex optical setup, but the sequence of excitation, acquisition, and analysis of fluorescence images at single-photon sensitivity is demanding for the photodetectors and requires significant computational power. From a user perspective, the need to computationally reconstruct images means that these images cannot be viewed and interpreted in real time during acquisition.
Extending the concept of using nonlinear materials for improving the optical resolution, Caillat et al.167 proposed the use of lanthanide-doped upconversion, and over a 2-fold resolution improvement was found for Yb3+,Tm3+ upconverting nanocrystals characterized with a nonlinearity order equal to S = 4. However, this value is the ultimate limit, given that the higher the upconversion order, the dimmer and more blue-shifted (into UV region) the lanthanide luminescence. Extending this concept for enhanced optical resolution was possible with photon avalanche, where high nonlinearity orders are enabled (Fig. 16a, right). Moreover, it is achieved without any blueshift in the emission band, in contrast to other multiphoton processes.
In recent years, several imaging methods have validated the theory of PA single-beam super-resolution imaging (PASSI).2,3,6,9,142 Table S11 (ESI†) compares the previously demonstrated super-resolution imaging using lanthanide-doped nanoparticles with currently presented examples of PASSI imaging and its modifications. The first experimental realization of PASSI was the 2021 report by Lee et al.,2 who used Tm3+-doped ANPs with nonlinearities as high as 31 to achieve resolutions finer than 70 nm at 800 nm emission. Notably, these resolutions, which were 7.5-fold finer than the diffraction limit of the 1064 nm excitation, precisely match that calculated ones using the revised Abbe diffraction equation.29 Later, in 2022, multicolour core–shell-shell migrating PA heterostructures with a reported 46th-order nonlinearity were used to label HeLa cells and image them with resolutions reported as high as 62 nm (Fig. 16d–f) at 550 nm (NaGdF4:Yb/Pr (25/0.5%)@NaGdF4:Yb/Tm (8/2%)@NaGdF4:Tb (20%)@NaYF4 ANPs) and 452 nm (NaYF4:Yb/Pr (25/0.5%)@Yb, Tm(10/4%)@Yb/Pr (25/0.5%)@NaYF4).9 Recently, due to the extreme non-linearity of S > 500, FWHM = 33 nm and 36 nm localization precision were achieved for a single, 27-nm diameter ANP.28
These experimental realizations of PASSI and subsequent reports are directly analogous to multi-photon microscopy methods, with the major difference being the ability of ANPs to realize the resolution enhancement of >30–500 photon processes with minimal anti-Stokes shift. Most importantly, these exquisite <30–100 nm resolutions were achieved with inexpensive, commonly available continuous wave lasers and standard confocal microscopes.152,168 These advantages suggest that PASSI has the potential to democratize sub-diffraction-resolution imaging, although some challenges remain to be solved, such as the size of the ANP label compared to biological species, bio-functionalization, and corona formation around inorganic particles entering living tissues.
The ability to switch the bright and dark states of ANPs on demand is reminiscent of photoswitchable fluorophores used for stochastic localization microscopy techniques such as PALM and STORM. Lee et al.146 identified power densities that stochastically photobrightened a sub-population of ANPs such that the centroid positions of individual ANPs could be localized without obfuscation from neighbouring ANPs (which were most likely dark). Due to the robustness of the photobrightening/photodarkening cycle of ANPs, the ANPs could be cycled indefinitely without permanent photobleaching. This indefinite cyclability of ANPs is a critical feature given that the accuracy of photon localization methods such as PALM scales inversely with the square root of the number of collected photons, which is the current limiting factor for the resolution of PALM and STORM. Lee et al. used repeated cycling and localization of ANPs to demonstrate indefinite photon avalanche localization microscopy (INPALM), exhibiting <1 Å localization accuracy even with ANPs that were touching (Fig. 16b).146
The ability to use the same Tm3+-doped ANPs to achieve two different modes of sub-diffraction imaging (<70 nm resolution with PASSI, and <1 Å accuracy with INPALM) demonstrates the unique and powerful applications of PA nanomaterials.
6.2 Photon avalanche-based sensing and biosensing
One of the most promising applications of the PA phenomenon is imaging, diagnosis and sensing biological activity. Luminescent nanomaterials have found countless of applications in biosensing of proteins, DNA/RNA fragments, antigens, cell components, and eukaryotic/prokaryotic cells.170–172 There are several requirements that should be satisfied by the given materials that are suitable for this type of utilization. Firstly, they should feature (intrinsic or biofunctionalized) biorecognition and biospecificity, and they should allow for remote optical probing using signals easy to distinguish from the background or tissue autofluorescence. These labels should additionally be non-toxic and small in size, given that their sensitivity is most often proportional to the surface-to-volume ratio. All of these requirements are, in first approximation, fulfilled by lanthanide-doped NPs, including ANPs. From a functional perspective, the biosensing capabilities are considered either biospecific passive labelling of the target objects (e.g. cells and cell structures, and proteins) or active sensing of the presence or concentration of the target moieties (e.g. proteins and DNA/RNA fragments). In the former case, the luminescent reporter is just a lantern, which is biofunctionalized to specifically anchor to these molecules and report about their presence and location. In the latter case, some specific optical response is expected from the luminescent reporters, such as a change in luminescence colour, intensity or luminescence kinetics.Lanthanide emission is perfectly photostable, which is considered one of the most important advantages of these labels and accounts for either Stokes, upconverting or avalanching nanoparticles. This is critical for biosensing capabilities, given that it not only enables the signal to be reliably acquired over longer dwell times to improve the photon budget at a significantly lower background, but most of all it enables long-term time lapse imaging to be performed over hours of continuous illumination. Although this possibility has not answered any serious biological questions to date (spreading of NPs in the blood stream, small organism development, etc.), these types of studies will definitely gain interest in the near future. Combining cellular component targeting with multiplexed labels and bio-sensing or super-resolution imaging, these novel luminescence labels may offer unprecedented tools for developmental biology or in situ continuous sensing.
In conventional Ln3+ luminescence, short-wavelength excitation beams are required, which simultaneously photoexcite organic fluorescent molecules, and thus increase the background.180 The anti-Stokes emission in UCNPs diminish these problems, but to sensitize their upconversion, Yb3+ ions are used with an excitation wavelength at 980 nm, which matches the absorption spectrum of water, and thus may cause undesired sample heating. With the development of core–shell inorganic UCNP nanomaterials, the possibility to sensitize their upconversion with Nd3+ ions at ca. 800 nm excitation was demonstrated with efficiency and brightness comparable to excitation with Yb3+, and less overheating.181 Thus far, PA has been observed under a few excitation wavelengths in the visible and NIR (e.g., 852 and 1064 nm) regions, which is promising for biomedical sensing. For example, PA was observed in Tm3+-doped ANPs under biocompatible excitation at 1064 nm using power densities typical or smaller than in conventional confocal microscopes.2 These high pump power densities are too high for wide-field imaging, but are acceptable for raster scanning and should not harm biological samples.
In contrast, PA emission should be susceptible to the presence of acceptor molecules in a spectacular manner79 because a small perturbation to the energy looping should be evidenced by a significant perturbation to the photon avalanche emission. In this context, the PA does not change the physics of the dipole–dipole interaction between the donor and the acceptor but enables the quantification of minute FRET signals occurring at distances beyond 100 nm, where the FRET effectiveness drops down below 1% in conventional approaches. Moreover, the volume of the ANPs that interacts with acceptors would increase or the surface passivation (to protect ANPs from unspecific quenching from ligands) would favour the bio-specific response of the ANPs to the presence of appropriate acceptors. This is in contrast to conventional UCNPs used for FRET sensing, where only superficial lanthanide dopants have a chance to interact with the acceptors (Fig. 17) and undergo surface passivation, although to improve the brightness, the acceptors need to be moved further away from the surface of the UCNPs and the donor lanthanides ions.
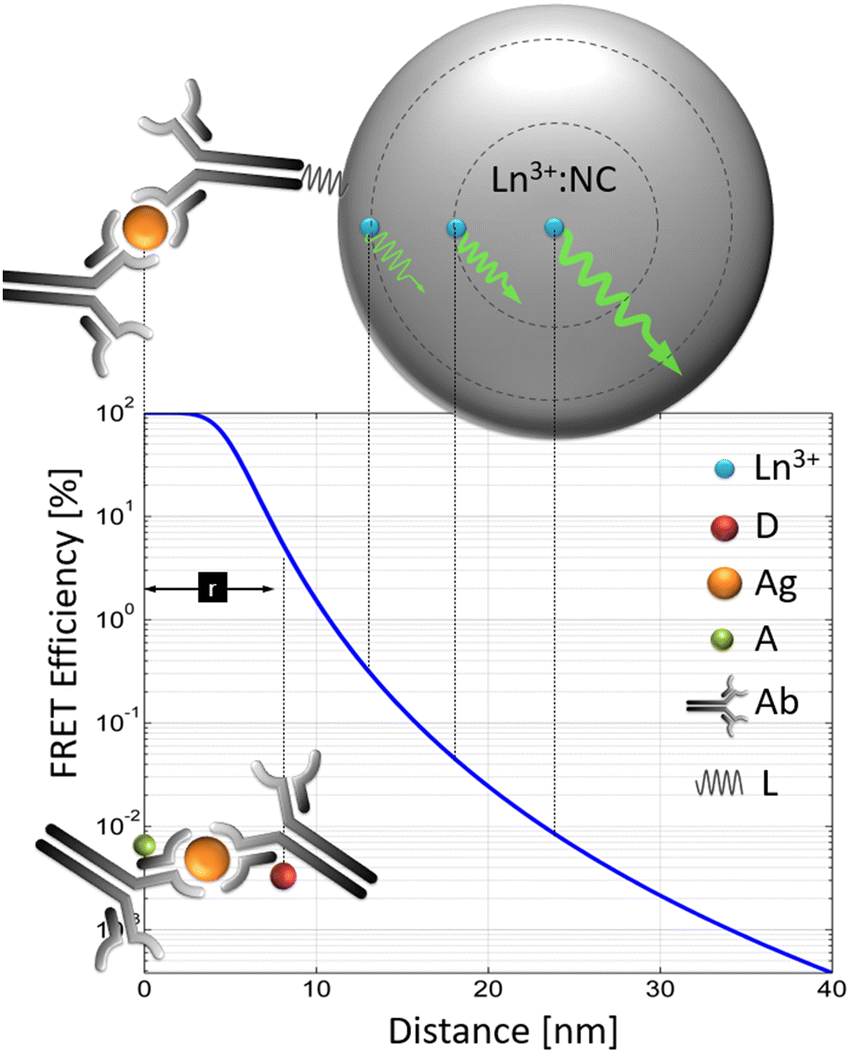 | ||
| Fig. 17 Issues related to UC-FRET.79 In molecular FRET, the resonant ET between the donor (D) and acceptor (A) occurs owing to the presence of an antigen. The FRET becomes enabled in carefully selected D and A moieties by a D–A distance (rDA) of a few nanometers and spectral overlap of the D emission with A absorption. In upconverting nanoparticles, not only a single UCNP contains 100–1000 of donor lanthanide ions that participate in RET to multiple A molecules, but these individual D ions remain in statistically variable distance from the UCNP surface, where the A molecules can attach to. Obviously, this complicates the analysis. PA emission may become an alternative type of D nanoparticles, which will augment the FRET efficiency, enable surface passivation and biofunctionalization without compromising the FRET sensitivity. Adapted with permission from ref. 79. Copyright 2020, the Royal Society of Chemistry (https://creativecommons.org/licenses/by/3.0/). | ||
Although no PA-based biosensors have been experimentally demonstrated beyond simulations,79 some progress has been made recently in understanding the susceptibility of the PA emission to the presence and concentration of PA quenchers (see Section 7.1.2).12,80 These studies pave a way to develop PA transducers that can detect specific D–A interactions at increased length scales.
PA performance is expected to be strongly affected by the temperature, resulting in a shift in the PA threshold and in variations of the nonlinearity of the emission (similarly to Fig. 15c).10 These macroscale effects are a result of the complex interplay between opposite microscopic effects and especially in PA materials may modify the luminescence in a non-trivial way. Interestingly, in the case of PA materials, high temperature relative sensitivity, SR, was predicted between 4% K−1 and over 35% K−1, with a useful temperature range (where SR > 1% K−1) of ca. 150 K. Moreover, the range with the highest sensitivity may be tuned by the selection of the pump power used for probing. Optimizing the system for higher sensitivity ranges results in limited temperature useful regions. As a result, two opposite effects can be observed. On the one hand, the capability to select a particular calibration curve (T = IL−1 (T)) tailored for the expected sensitivity and broadest temperature sensitivity range provides the unique ability to dynamically adjust the optical thermometry performance for the given utilization and for its specific requests. Nevertheless, this effect should most probably limit the practical application of PA luminescence thermometry of these materials due to the necessary use of a matrix of pump power and temperature-dependent calibration curves. The other limitation in the utilizing of these types of materials in thermometry is their thermal stability and thermal decomposition at >580 K.199 Other effects, such as nanoscale effects, presence of ligands and solvents (discussed in Section 4.4), as well the presence of quenchers (discussed in Section 6.2.2), will further make the PA susceptible to the external chemical environment. Thus, the understanding and proper management of these factors are critical for the further development of new PA nanomaterials and their applications.
Therefore, alternative luminescent thermometers and reliable methods for temperature-dependent spectral properties quantification are sought, and the PA concept may offer new possibilities (Fig. 3b). In this case, the so-called single-band ratiometric (SBR) approach,200–203 the luminescence intensity of a single emission band can be monitored under two, ground and excited-state absorption photoexcitation lines. This approach is advantageous given that it is technically less challenging and cheaper to all-electronically switch between 2 laser/LED light sources and record two (GSA and ESA based) images within a single spectral channel than the opposite of recording two spectral images under a single photoexcitation line. Additionally, by using the same spectral range of emission, the risk of disturbing the temperature readout due to the influence of the medium will be eliminated. The unique configuration of the 4f energy levels of Ln3+ opens a wide variety of different ions for which the ESA process can be observed. The former, temperature-dependent ESA-based thermometry has been recently proposed and studied in Nd3+, Eu3+, Tb3+- and Pr3+-doped nanoparticles204–207 In the case of materials with thermal coupling between the ground and excited states, they enable an enhancement in the ESA-excited luminescence by an increase in the population of the upper energetic state according to the Boltzmann distribution. However, versatile studies enabled the understanding that the interionic interactions, usually via the CR process, lead to boosting of the thermal response of the luminescence signal. Therefore, the optimization of the dopant concentration has led to the development of luminescent thermometers with sensitivity to temperature changes as high as >10% K−1. Although the probability of the CR process is temperature independent, the splitting energy of the particular 4f multiplets involved in this process may shift the CR process out of resonance. Therefore, the assistance of phonons will lead to the strong thermal dependence of the CR process (see Section 3.2.2). Given that the splitting energy of the 4f levels is dependent on the crystal field strength of the host material, by deliberate selection of the host material stoichiometry, the probability of this process can be modified. Considering the essential dependence of the PA process probability on the CR performance, the sensitivity of luminescent thermometers using photon avalanching nanoparticles will be unprecedently high and should definitely exceed the values reported thus far for thermometers based on the ESA/GSA SBR approach.208,209 The relative sensitivity of PA-based luminescent thermometers reaching tens of % K−1 in a broad and tuneable (by tuning the excitation power) temperature range was predicted recently with the numerical simulations employing the phenomenological model of an avalanching system.10
Beside technical issues, metrology factors are also important and the two emission wavelengths (in traditional ratiometric imaging) or the two excitation wavelengths (in ESA/GSA thermometry concept) may be affected by the sample in different ways, which is strongly related to the sample spectral properties (e.g. dispersive tissue absorption and scattering coefficients).209 These aspects are difficult to predict, given that the sample-to-sample and patient-to-patient variability may hinder simple external temperature calibration.203,209 A solution can be offered by the temperature-dependent kinetics of the luminescence rise or decay times. In the case of luminescence decay, various luminescent compounds have been proposed, whose lifetimes are typically shortened with an increase in temperature due to thermal quenching. However, in the case of thermal imaging, the acquisition time needs to be shorter than the decay itself. Therefore, the shortening of the decay observed at elevated temperatures may affect the number of photons absorbed, and thus the reliability of the temperature estimation. On the other hand, in case of the PA process, for the threshold conditions (excitation density and temperature), the rise time is extended. Therefore, kinetic-based thermometry may beneficially influence the signal-to-noise ratio by facilitating a reliable temperature measurement. Although this approach has not been confirmed experimentally to date, its applicative potential for remote temperature determination from the theoretical perspective is indubitably appealing.
As can be noted in Fig. 3b, temperature may contribute to the PA emission by enhancing the population of the starting level. An educated guess leads to the conclusion that an increase in temperature should reduce the PA threshold, but due to the rich energy level scheme of most lanthanides, temperature may also negatively impact the PA emission, leading to thermal quenching by enhancing multiphonon relaxation rates.10 Moreover, the rise times of luminescence under PA photoexcitation and looping conditions is not only affected by temperature, but also dependent on the excitation intensity, IEXC, and due to the sample properties, IEXC is actually difficult to be determined. Therefore, the latter features may hinder the practical implementation of PA-based thermometry at the nanoscale in ‘difficult’ environments, such as absorbing and scattering samples (e.g. tissue and cells).
The fact that a slight change in temperature can affect both the spectral broadening of the absorption bands and the activation of energy bridging the CR processes means that even a small change in temperature can cause rapid (up to a dozen or even orders of magnitude) changes in the intensity of luminescence. In this case, the expected rapid changes in thermometric parameters should take place in a narrow temperature range. However, a wide range of possibilities of selecting the appropriate energy levels and activating various physical processes through the appropriate selection of the luminescent host material and Ln3+ admixture ions offered by the unique configuration of the 4f energy levels can enable the design of a wide range of super-sensitive luminescence thermometers/manometers based on the PA process, whose operating range can cover a wide range of temperatures/pressures. A necessary but not sufficient condition for the occurrence of the PA phenomenon is an efficient ESA process. The first attempts to create luminescence thermometers using the ESA process in nanocrystals, glasses and microcrystalline materials doped with lanthanide ions such as Nd3+, Pr3+, Tb3+, and Eu3+ confirmed the high potential of this type of materials for remote temperature reading, which was confirmed by the sensitivity of nearly 15% K−1. Moreover, the favourable role of CR processes on the variability in temperature parameters was confirmed, which indicates that for the conditions required for the occurrence of PA, high relative sensitivities will be obtained. A limitation of this type of thermometer may be the fact that it will be necessary to provide a reference signal enabling a ratiometric temperature reading. This can be done using the SBR approach, which due to the need to change the wavelength, may cause some experimental difficulties. Another possibility is the use of the emission band of the co-dopant ion, for which the excitation wavelength used to initiate the PA process, allowing luminescence to be obtained by GSA. This concept has been proposed for ESA/GSA processes but is unpopulated for the PA process thus far. This is because interionic interactions can disturb the equilibrium conditions for PA occurrence. Nevertheless, the appropriate selection of dopant ions and the optimization of their concentration may enable the creation of this type of thermometer in the future. The undoubtful profits coming from the utilization of PA-based luminescence thermometry resulting in the achievements of the nano-spatial and 10−3 K thermal resolution of thermal imaging are sufficient motivation, initiating research studies devoted to this in the near future.
Thus far, beside homogenous temperature-dependent PA studies (Fig. 15c)14,203,209,210 and theoretical modelling of temperature dependence of PA emission,10 no experimental evidence of temperature quantification at the single ANP level has been demonstrated.
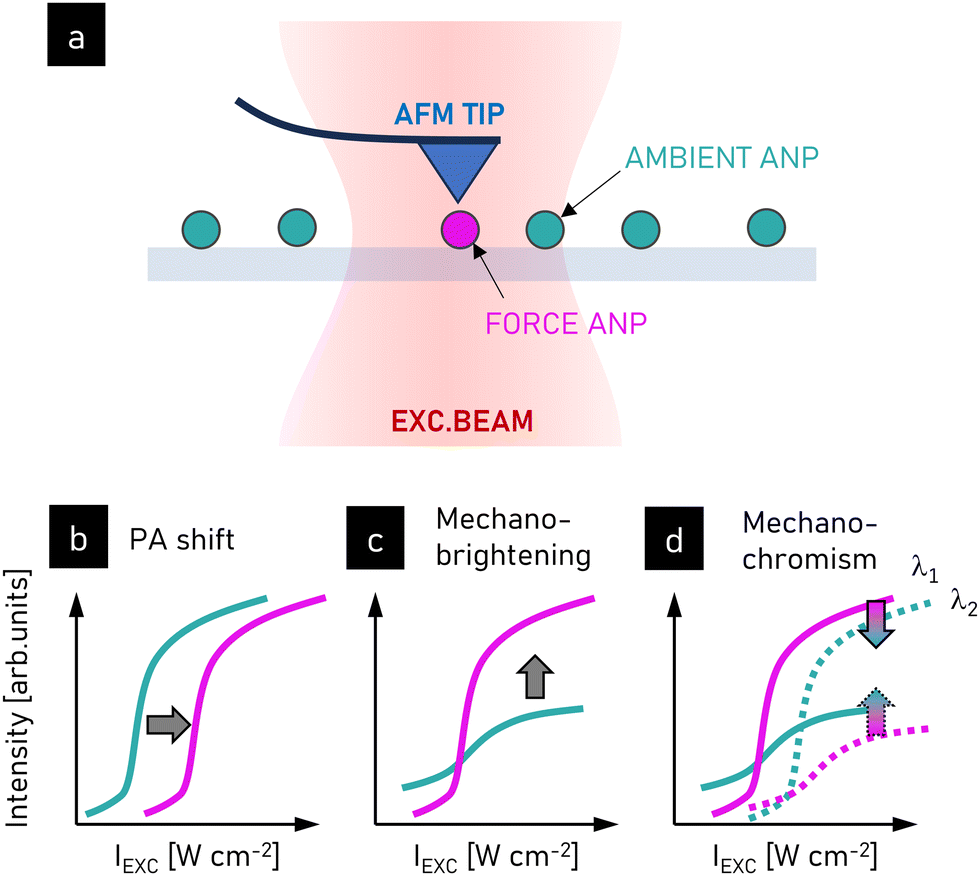 | ||
| Fig. 18 Photon avalanche-based force sensing. (a) Scheme of the experiment, measurement of the impact of the mechanic force applied by an atomic force microscope tip on avalanche emission of single ANPs; (b) force-induced shifting of the power dependence observed for ANPs (doped with 4.5–8% of Tm3+); (c) force-induced avalanching of pre-ANPs (doped with 4% of Tm3+); and (d) mechano-chromic effect observed for piezo-chromic ANPs (doped with 15% of Tm3+).211 Adapted with permission from ref. 211 (https://creativecommons.org/licenses/by/4.0/). | ||
The application of PA to force sensing was demonstrated in experiments where Tm3+-doped NCs were studied spectroscopically with forces applied in situ using an atomic force microscope tip. Importantly, the impact of the force on the emission varied with the doping concertation of the NPs under study. In the case of ANPs doped with 4.5–8% of Tm3+ ions, for which the full-performance PA is observed in a single ANP (ϕ ∼ 25 nm), applying force (from the hundreds of pN to few μN range) resulted in a shift in the power dependence curve towards higher values (Fig. 18b). This enables force sensing based on the luminescence intensity from a single ANP. Alternatively, reducing the doping concentration (to 4%) results in ‘pre-avalanching’ nanoparticles that exhibit energy looping but not the steep emission intensity rise characteristic for PA. In such pre-ANPs (Fig. 18c), applied mechanical force and reduction in the average distances between the photoactive ions in the NCs pushes these nanoparticles into the avalanching regime, resulting in much brighter luminescence. In a third force-sensing modality, much heavier doping (15%) enables remote force sensing based on the modulation of emission peak ratios with force, known as mechano- or piezochromism. This approach has been demonstrated with the emission from two Tm3+ energy levels (3F2,3 and 3H4), whose relative intensities respond differently to applied mechanical force (Fig. 18d). It is believed that a force-induced increase in the nonradiative rates results in the relaxation of energy from the higher level (3F2,3, emission at 700 nm) to the lower one (3H4, emission at 800 nm). This enables sensing of forces with single digit nN resolution.
6.3 All-optical computing and data storage
The switching properties of ANPs146,212 also provide a path towards optical computing and data storage, as a valuable alternative to microelectronics, which are approaching their physical limits in terms of the size of transistors, heat dissipation, and dielectric breakdown. In optical computing devices, input data are delivered remotely (with no physical wires) and coded as the modulation of light (e.g., its intensity or phase). The output data are also read remotely, which so far has hindered the concatenation of data to the inputs of subsequent processing units. The main advantage of optical computing is its fast and possibly massively parallel operation with high energy efficiency and critically reduced heating. However, it still is problematic to integrate optical computing devices with other functional blocks, such as electronic input–output blocks and memory units. One of the leading approaches is based on waveguide systems composed of Mach-Zehnder interferometer units, in which the phase-sensitive interference of the beams enables the desired operation.213 However, the recent advances in ANPs open a new field of applications for lanthanide-doped materials in all-optical data processing13 and memory effects.146,2146.3.1.1 Non-volatile memory. ANPs have demonstrated two different modes of switching, which provide avenues for the long- and short-term optical storage of information. The defect-based switching reported by Lee et al.146 allows long-term information coding through photoswitching into persistent photodarkening or photobrightened states. Although appropriate optical stimulation of PA materials is needed to induce reversible switching between the two physically distinct bright and dark states, optical pumping is not needed to maintain these states. Thus, Tm3+-doped ANPs have been utilized for persistent non-volatile storage, akin to phase change materials in Blu-ray discs. Lee et al.146 illustrated the ability to write, erase, and rewrite two- and three-dimensional patterns of bits in films of Tm3+-doped ANPs. These write/rewrite cycles have been repeated over 1000 cycles for a single bit, demonstrating the robustness of ANPs for optical storage. The sub-diffraction spot sizes afforded by the giant nonlinearity of PA also suggest that bits smaller than 70 nm can be written in films of ANPs. In fact, photo-switchable ANP memory devices have been patterned by switching the solubility of ANPs through the use of photoactive ligands that can cross-link or undergo redox reactions when stimulated with UV, NIR, or electron-beam radiation. Pan et al.147 demonstrated the patterning of features as small as 100 nm and photoswitching of 5 μm patterned arrays of Tm3+-doped NaYF4 ANP memory devices. This reversible optical storage of information can be used in optoelectronics for optical control of the system properties. It also has the capability for long-term volumetric addressable data storage option in all-optical signal processing systems.
6.3.1.2 Volatile memory. In contrast to the persistent storage of photodarkened/photobrightened Tm3+-doped ANPs, Skripka et al.212 demonstrated an orthogonal optical storage method based on the intrinsic optical bistability (IOB) of Nd3+-doped KPb2Cl5 ANPs at low temperature. In this IOB phenomenon, the ANPs having two discrete stable luminescence states (dark and bright) that could be treated as logical 0 and 1 bits, respectively. Unlike Tm3+-doped ANPs, whose bright and dark states are physically distinct, bright and dark IOB ANPs have an identical physical structure and operate under identical excitation conditions. The only difference between the two bistable states is their excited state populations, which are distinct due to their differing excitation histories. Skripka et al. observed hysteresis in the power-dependent luminescence of Nd3+-doped KPb2Cl5 ANPs, similar to that exhibited by magnetic domains. The dark state was observed before the PA threshold, while the bright state appeared when the intensity of the 1064 nm laser exceeded a power threshold associated with the initiation of PA, which exhibited nonlinearities of S > 200 at 77 K. Importantly, when the power was subsequently reduced to intermediate levels, the bright state was still maintained; thus, luminescence, or logical 1, may be read out at powers lower than that required to write it. Thus, the history dependence, or hysteresis, of ANPs allows them to store the optically encoded information. Notably, the bright state and all the stored information disappear below a power threshold lower than the PA threshold observed during power ramp up. Given that the minimum excitation power must be maintained, this IOB-storage is analogous to volatile memory.
Besides non-volatile memory, the photoswitching properties of Nd3+-doped KPb2Cl5 ANPs have been leveraged as prototype optical transistors. Skripka et al. demonstrated the ability of these bistable ANPs to move between their dark and bright states without changing the intensity of the 1064 nm pump laser.212 A low-power (<100 nW) 808 nm laser pulse was used to cross the instability region that separates the bright and dark states of these ANPs at intermediate powers (thereby creating the hysteresis). This instability crossing switched the ANP emission from a dark to bright state, with the latter maintained by the continuously applied 1064 nm bias. In this configuration, many ANPs may be excited with a diffusely spread 1064 nm laser and flipped from a dark to a bright state by raster scanning with an 808 nm laser in any arbitrary pattern. This low-power, fast, and flexible photoswitching is particularly attractive in developing all-optical processors, memory, and interconnects, although advances in these IOB nanomaterials are needed to increase their operating temperatures to ambient conditions.
This application leverages the photoexcitation history dependence, which can be especially pronounced in PA given that it is severely dependent on the initial population of the looping level. In a simplified perspective, to observe PA emission, a certain seed population of the looping level must be established through processes of GSA and ESA. A larger level population is facilitated by significantly longer lifetimes, typically in the order of tens of milliseconds,6 in contrast to other levels with decay times measured in the tens to hundreds of microsecond range. Although the emitting level exhibits short electron lifetimes and can be depopulated easily through both radiative and non-radiative relaxation, as well as other non-radiative processes such as ET, the looping level may retain a residual population even after a few tens of ms. This long-lived population of the looping level can be likened to a form of system short-term memory. By exciting the PA material with a pulsed light source, with a time gap between the pulses smaller than the lifetime of the looping level, one can observe that the emission is dependent on the train of photo-excitation subsequent pulses, and the first pulse facilitates the emission achieved in the course of the second and subsequent pulses (Fig. 19). The facilitation effect stems from the fact that with the first few pulses, the looping level electron seed population is established, and the subsequent pulses start with non-zero population and contribute to further population growth, which enhances the emission intensity with each subsequent pulse (Fig. 19). The intensity of this emission, which is dependent on the photoexcitation history, can be controlled in situ by adjusting the pulse frequency, pulse and gap duration, and pulse power.13
 | ||
| Fig. 19 Complex nature of luminescence originating from the multiple level and multiple dopant interactions in PA mode, where ESA absorption depends on the residual population of the looping level (n2), and thus, two subsequent excitation pulses (a) induce a pulse-gap dependent emission and the so-called (b) paired-pulse signal facilitation (PPF).13 Adapted with permission from.13 Copyright 2023, John Wiley and Sons (https://creativecommons.org/licenses/by/4.0/). | ||
This memory-like behaviour of PA resembles the operational principles of biological synapses, given that it demonstrates in situ plasticity, inherent short-term memory, and signal facilitation across multiple subsequent pulses. This similarity of the observed behaviour of PA upon pulsed excitation to neural circuits holds particular interest from the perspective of all-optical, training-free, reservoir computing, an alternative computational paradigm aiming to mimic brain function, to reduce the computation time and energy consumption in analysing temporal signals. Additionally, owing to their short memory and high nonlinearity, PA materials can function as optical, analogue AND logic gates with multiple optical inputs.13
There are also other characteristic features of the synaptic operation that have been evidenced, i.e., the threshold intensity of the stimulus below which it is neglected, as well as the paired pulse facilitation effect. Both of these features match the characteristics of avalanching materials. One of the required features thereof is a metastable long-living intermediate energy level, given that the 3H4 level in Tm3+ is characterized with a lifetime of around 16 ms,6,13 which is much longer compared to the lifetimes of the levels from where avalanche emission occurs. One of the imminent features of the avalanching is slowing down of the intensity rise dynamics for excitation powers close to the PA threshold. However, the presence of the oddment population in the metastable level at the beginning of illumination leads to a significant reduction in the rise time required to achieve a steady-state emission intensity. Thus, during pulsed excitation with time gaps between the pulses comparable in duration to the lifetime of the metastable level, at the beginning of each of the consecutive pulses, the starting population in the metastable level is a bit higher compared to the previous one, facilitating faster build-up of the emission intensity and providing an increased population of metastable level for the next pulse. This effect can be understood as the short-term (on the timescale of the lifetime of the metastable level, up to tens of milliseconds, typically) memory required for the realization of the paired pulse facilitation effect, in which the response to the stimulus appearing shortly after former stimulus will be enhanced.
This effect, with operation at some points similar to that found in natural synaptic systems, allows all-optical data processing.13 This includes pulse counting enabled by the short-memory effect caused by maintaining the oddment population in the intermediate state of the avalanching system between the pulses. Thus, for each next pulse, the response from the system (emission intensity) depends on the number of these pulses. However, this effect may be further developed by adding additional parameters modulating the rise dynamics of the avalanche emission. In general, this dynamic not only depends on the frequency of the pulses and their duration and width, but also on the sequence of these parameter changes. This sensitivity to the excitation history opens other fields of application, including pattern recognition, features amplification or extraction, and phase-sensitive detection.13
7. Outlook and perspectives
PA emission is indeed a spectacular luminescence phenomenon. The unique features of lanthanide ions, including multiple, long-living real electronic levels with multiple up- and down energy transitions and exchange, fulfils sophisticated conditions to let PA occur in bulk, micro, and down to nanoscale materials. The highly nonlinear response of the luminescence intensity to the pumping intensities and factors such as the history of photoexcitation, the presence and strength of numerous physical and chemical factors open new possibilities for further scientific discoveries and promise an implementation in all-optical metrology, sensing, imaging and data processing. However, to further advance the applicability of these PA materials, additional studies are needed in several areas.Although the discovery of the PA phenomenon was the result of serendipity, actual progress in the intentional development of PA materials was triggered by understanding the fundamental differences between UC and PA and the shift in paradigms such as an increase in the concentration of the dopants (against conventional thinking about concentration quenching in lanthanide-doped phosphors) and the positive role of energy CR. Therefore, despite their many similarities, these new PA (nano)materials require de novo optimization of their composition. Obviously, the developments in PA materials stem from the last two decades of intensive progress and optimization of upconverted materials and technology. The most prominent contributions of UCNP to ANP technology are as follows: (i) the existence of reliable protocols for the synthesis of colloidal nanoparticles, (ii) ligand stabilization of individual nanoparticles, (iii) understanding the surface quenching mechanisms and (iv) clustering of dopants, resulting in great interest in single nanoparticle studies and further enabling the development of core-multiple-shell designs. Concerning core–shell nanoparticles, surface passivation with undoped homo- or heterogeneous shells was the key technology enabling the practical implementation of UCNPs in biosensing and bioimaging. Another great step forward was the studies dedicated to a compositional architecture, i.e. intentionally doped core and individual shell layers with various compositions of co-dopants, which enabled the engineering of the luminescent properties of these nanoparticles on demand. These fundamental achievements will further stimulate work and discoveries related to interfacial energy migration and lead to improved control of the luminescence colour and lifetimes of PA nanomaterials.
7.1 Current challenges in PA research on the nanoscale
Depending on the driving force of a particular application, skillful optimization of PA materials must be considered. Whether this is a lower PA threshold, higher PA gain range, either smooth or step-like transition from PA to saturation regime, photostability or susceptibility to energy trapping (photodarkening), all these scenarios and requirements may require various and maybe sometimes contradictory solutions. In this view, PA definitely needs many fundamental, experimental and theoretical studies aiming to understand the role of individual (chemical and physical) factors on the PA performance.4,6,80 Because the PA phenomenon ‘encloses’ multiparameter dependent non-ground state absorption and energy circulation within a continuous positive loop, it will be enormously sensitive to numerous factors and processes that disrupt this machinery.12 The examples include the impact of the concentration of dopants on the balance between (positive) energy looping and energy migration, the impact of MPR (stemming from temperature, crystal field, and phonons) on the energy redistribution (i.e. emptying or populating-required energy levels) or external factors such as temperature, pressure and electric/magnetic field as a few properties or features. These properties will not only affect the possibility to achieve PA emission characteristic features, but will determine the brightness, nonlinearity, PA gain range and lifetime of the looping level, which will define the possible scope of future applications.Because of the slow rise time of PA emission, some technological aspects must be also developed, such as pre-pumping (to speed up the raster scanning) and parallelization of multiple excitation spots (to image and localize ANPs in many areas simultaneously) similar to localization microscopy or Nipkow-disk approaches.217 Optimization, i.e. shortening of the PA emission rise time, is another way to circumvent the slow frame rates of current PASSI-type imaging.3,28
Moreover, PA sensitization with Yb3+ co-doping can potentially be beneficial to improve the brightness and reduce the PA threshold and rise times of ANPs. The use of Yb3+ ions enables connotation with core–shell nanomaterials, where the Yb3+ ions are responsible for the energy migration between the absorbing core and emissive shell, which can potentially increase the number of available PA colours, aiming to perform multiplexed bio-detection or bio-imaging.
 | ||
| Fig. 20 Impact of donor–acceptor ET on luminescence. Simplified schemes of quenching of conventional luminescence (a) and photon avalanche upconversion (b). The color arrows indicate GSA (orange arrow) and ESA (green arrow) excitation, looping (yellowish), emission (red), D–A RET quenching. For PA scheme, either the emitting level (mechanism QI) or looping level (mechanism QII) can be quenched, which both affect the cross-relaxation, energy looping and PA emission. | ||
In the case of conventional luminescence, the presence of Q reduces the emission output of the donor (D) (IDQ) and shortens the luminescence lifetime of the donor (tDQ) compared to bare donor (ID and tD, respectively) proportionally to the D–Q distance, rD–Q, which can be quantified using the well-known Förster relations and theory.182 In contrast to RET occurring in the conventional photoexcitation scheme (Fig. 20a), for the PA process, the presence of Q may affect the emitting (n3), looping (n2), or both levels, while both levels participate in the energy looping and ESA. In mechanism I (QI), the quenching occurs to the emitting level, which cannot participate in the CR with neighbouring ions in the ground state. In mechanism II (QII), the quenching occurs to the looping level, which hinders ESA. These two mechanisms should affect the PA features in different manners.12
The impact of the quencher on the emitting level has been theoretically considered by Bednarkiewicz et al.79 The impact of the acceptor was defined as the relative ratio of WRET to the CR rate, WCR, existing in an unperturbed PA system. Next, the steady-state luminescence ratio ΩSS of D accompanied by the acceptor, which is related to the luminescence intensity of the donor alone, was defined as  enabling the quantification of how the PA emission reacts to an increase in the D–Q effectiveness when assuming that the FRET mechanism (R0 = 5 nm) is responsible for this ET distance. These simulations indicated the significant susceptibility of the PA emission intensity to perturbations introduced by the quenching species, and consequently it was hypothesized that the interaction distance between D and Q can be extended by a few-fold (2–6) beyond the conventional Förster distance. To some extent, these simulations have been recently confirmed experimentally by studies devoted to unspecific quenching80 and by cooping the Nd3+ quencher in the avalanching Tm3+ ions.12
enabling the quantification of how the PA emission reacts to an increase in the D–Q effectiveness when assuming that the FRET mechanism (R0 = 5 nm) is responsible for this ET distance. These simulations indicated the significant susceptibility of the PA emission intensity to perturbations introduced by the quenching species, and consequently it was hypothesized that the interaction distance between D and Q can be extended by a few-fold (2–6) beyond the conventional Förster distance. To some extent, these simulations have been recently confirmed experimentally by studies devoted to unspecific quenching80 and by cooping the Nd3+ quencher in the avalanching Tm3+ ions.12
The unspecific surface quenching has been demonstrated and modelled at the single-nanocrystal level to uncover the design-dependent heterogeneity in the ANP threshold intensity.80 The 8%Tm3+-doped NaYF4 core of the Rc radius was passivated by the undoped NaYF4 shell with a variable thickness (from ca. 3 to ca. 9 nm). As expected, a correlation was found between the mean threshold pump power and the ANP shell thickness (d ± δ, where δ is the standard deviation of the shell thickness) up to ca. 9 nm, beyond which the ANP behaviour did not depend on the ‘unspecific’ ligand field anymore. It was found that when the CR rate is large, the total depopulation rate, A2 + k2, of the first excited 3F4 level in Tm3+ is proportional to the product of the PA pump threshold and ESA cross-section. Interestingly, the non-radiative quenching through the surface (k2) was well described by the surface-quenching model, i.e. k2 was found to be dependent on 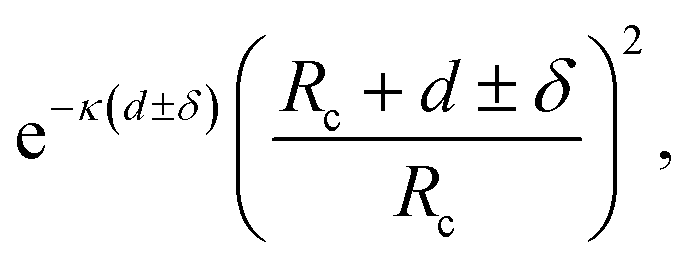 where κ [nm−1] is the exponential passivation improvement with shell thickness (Fig. 21a and b).218 FRET theory was considered in this work; however, it provided less accurate results compared to the surface-quenching model, which most probably originates from the differences between the specific (FRET) and non-specific quenching.
where κ [nm−1] is the exponential passivation improvement with shell thickness (Fig. 21a and b).218 FRET theory was considered in this work; however, it provided less accurate results compared to the surface-quenching model, which most probably originates from the differences between the specific (FRET) and non-specific quenching.
 | ||
| Fig. 21 Impact of quenching on PA emission. (a) Unspecific quenching of Tm3+–Tm3+ avalanching pair by surface effects in Tm3+-doped NaYF4 ANPs, (b) mean ± variance pump power threshold versus the shell thickness for 2.6, 5.6 and 8.6 nm shell thickness. (b) Correlation between shell thickness and the mean ± variance of single-ANP Ith distributions.80 Reprinted with permission from ref. 80. Copyright 2021, the American Chemical Society. (c) Specific quenching of the Tm3+–Tm3+ avalanching pair by volumetric Nd3+ ions. (d) Pump power-dependent luminescence of Tm3+ ions for an increasing concentration of the quencher in LiYF4 microcrystals and intensity profiles (inset) at fixed pump powers.12 Adapted with permission from ref. 12. Copyright 2024, the Royal Society of Chemistry (https://creativecommons.org/licenses/by/3.0/). | ||
The specific RET quenching of the emitting and looping levels of the looping Tm3+ ions has been recently tackled by incorporating Nd3+ acceptor ions within the LiYF4 host material12 (Fig. 21c and d). The microcrystals were proposed to exclude surface effects and unspecific quenching, which in principle enabled control of RET in a more precise way by varying the average donor–acceptor distance, which was controlled by the acceptor concentration. The 2H9/2,4S5/2 and 4I13/2 levels of Nd3+ remained resonant to the 3H4 (emitting) and 3F4 (looping) levels in Tm3+ ions, respectively. The GSA and ESA-based photoexcitation were directly compared, which evidenced many important aspects of PA behaviour. Firstly, a gradual increase in the acceptor concentration from 0.1% to 1% led to gradual quenching of the Tm3+ emission was observed both in the steady-state and kinetic domains, but while the GSA only led to a 4 times decrease in the Stokes emission intensity, in the ESA mode, the PA emission dropped by 4 orders of magnitude. Simultaneously, the luminescence decay dropped from 240 μs to ca. 80 μs (for 1%Nd3+ co-doping), which confirmed the non-radiative RET mechanism. By pump–probe measurement, the luminescence lifetime of the looping level at ca. 6000 cm−1 was dramatically shortened from 13.6 ms for the pristine Tm3+ only-doped crystals to 5.2 ms for as little as 0.1%Nd3+ co-doping, which obviously disrupted the energy looping, resulting in a chance to achieve PA emission. Higher acceptor concentrations prohibited the further kinetic characterization of the looping levels. These experimental studies were supplemented with further theoretical modelling based on the phenomenological model of Tm3+ PA in NaYF4 nanoparticles,2 which remains a very useful method to evaluate the impact of the phenomenological parameters on the PA performance.6,10 Here, this model and the rate equations describing both the looping and emitting levels were extended with the RET mechanism. These simulations enabled the impact of the acceptor individually on the emitting and the looping levels to be distinguished. These theoretical considerations are extremely valuable because it is difficult to find and quantitatively compare the impact of the specific quenching of the looping level because it is typically located in the range of 2000–6000 cm−1. Moreover, the Nd3+ levels are suitable to quench both the looping and emitting levels of Tm3+ ions, and because within single ANP there are hundreds to thousands of avalanching Tm3+ ions, the output luminescence is the sum of luminescence from all the ions exposed to quenchers staying at the average distance. There, the simulations clearly showed distinct effects for the two quenching mechanisms, i.e. through quenching of the emitting (QI) and the looping (QII) levels. The QII mechanism increased the PA threshold but in principle did not affect the nonlinear behaviour and brightness in the saturation regime. The QI mechanism reduced the nonlinearity but additionally reduced the brightness. The combination of both processes should be actually observed in real systems.
These results suggest that PA emission is significantly susceptible to the presence of acceptor/quencher molecules at distances, which are a few-fold larger than that expected from the Förster distance calculated for the same D–A pair. This susceptibility is not related to any change in the properties of the D or A alone or the fundamental mechanisms behind RET, but originates from the extreme susceptibility of the donor excitation to minute perturbations in photoexcitation and energy distribution on the looping and emitting levels. The predicted and demonstrated susceptibility is of critical importance because of the practical design of in situ bio-sensors.
Another reason to minimize the excitation power is to avoid inadvertent photodarkening. Although photodarkening has been leveraged for photo switching and sub-diffraction imaging,169 in other applications, photodarkening can be a major disadvantage of ANPs compared to conventional UCNPs, which are generally stable under most excitation powers. Further research into the mechanism of photodarkening in different PA nanosystems is needed, given that there are studies on the mitigation of photodarkening, e.g., through the development of synthetic methods and structures that suppress the defect states in the host matrix.
It is also critical to mention that the optical setup used to characterize the PA properties should be extremely well evaluated and optimized because any unconscious variation in the pump power in the PA regime (originating from the laser diode intensity/laser optical mode switching/positioning of the diffraction limited beam against the sample, short and long term instabilities, switching optical components in the light path) can be a source of significant errors. In view of the possible variation in the derived parameters depending on the pump power intensities (due to photodarkening,214 sample density and way of preparation, single NPs versus a monolayer of NPs versus aggregates of ANPs), the experimental conditions should be precisely reported. Moreover, for single ANP studies, a change in the ANP position against the laser beam should be verified before and after the experimental evaluation, given that tiny sample shifts in XYZ space occurring at the time of the studies may significantly contribute to the outcome and interpretation of the PA results. Moreover, thermal issues should also be avoided and evaluated in the analysis of the results. Typically, large aggregates of particles and a high-power laser with a large beam size will make the samples generate excessive heat, which will be evidenced with broadened emission lines up to white/yellow broadband emission generation and irreversible sample burning and damage.
In any biosensing system, the bio-specific interaction between the optode and the analyte should be translated to a variation in some spectral properties capable of being quantified. In luminescence, they are typically luminescence spectra (or emission intensities at specific wavelength(s)) or luminescence kinetics. In the photon upconversion mode, the typical short wavelength photoexcitation from the commonly applied light sources (lamps with filters, LEDs or sometimes lasers) must be exchanged with stronger NIR lasers matching the sensitizing Yb3+ (at 980 nm) or Nd3+ (at 800 nm) ion absorption. This imposes further modifications in the spectral filtering of the detection system. Firstly, anti-Stokes emission requires short-pass filters to replace long-pass ones. The large Stokes shift simplifies filtering out the laser excitation, but the small QY translates to a large discrepancy between the excitation and emission line, and thus the dichroic filters must be effective in reflecting the NIR excitation and transmitting the weak visible luminescence. Beneficially NIR photoexcitation undergoes scattering to a lesser extent compared to short wavelength photoexcitation. Alternatively, the tens of microseconds to milliseconds long kinetics are technically much easier to be measured compared to the very demanding time-correlated single-photon counting techniques required for (sub)-nanosecond-long kinetics. However, these slower kinetics are related to dimmer luminescent labels, and ultimately slower fluorescence imaging, e.g. during raster scanned imaging. In summary, these properties are distinct from conventional fluorophores and require customization of the detection instruments (e.g. well plate readers, and microscopes), but it is clear that the benefits of UCNPs justifies this work. Furthermore, the requirements needed to generate and utilize photon avalanche are not very different than that valid for the conventional UCNPs. With current developments in semiconductor lasers, available photodetectors and appropriate filters, all these modifications are easily within reach. PA requires relatively high pump power (in the order of tens to hundreds of kW cm−2), which precludes wide-field imaging. Nevertheless, ANPs offer other unprecedented features such as single-beam photon avalanche super-resolution imaging (Section 6.1.3) at no additional cost. This means that converting any confocal microscope to the anti-Stokes luminescence mode is not only beneficial for an improved signal-to-background ratio, but with no charge, enables imaging below the diffraction limit with resolutions of ca. 60 nm. Combining these features with the expected bio-sensitivity of ANPs should enable functional super-resolution imaging in the near future.
Despite the engineering of materials with different visual colors,221 a limited number of spectral fingerprints can be effectively considered when lanthanide-doped nanoparticles are not overlapping spatially. Otherwise, two labels that differ only in proportion between, e.g. the green and red emission band ratio, will not be distinguished if they co-localize in space (e.g. within the point spread function of the confocal photoexcitation beam). A number of attempts tried to provide spectrally pure emission by precise optimization of the co-doping with various lanthanides223 or by co-doping the upconverting lanthanides with additional ions (e.g. Mn2+ ions) that can quench some of the energy levels, e.g. green Er3+ emission.224 Moreover, although lanthanides produce characteristic (lanthanide and host dependent) emission bands, recognizing these labels is not trivial and requires high-resolution spectrally resolvable photodetectors, which make these detection systems more complex, costly, slower and computationally (due to spectral deconvolution) demanding.
The multiple band emissions from lanthanides have been demonstrated to be suitable for the so-called ratiometric sensing, which exploits the inner filter effect. For example, Er3+-doped nanoparticles emit simultaneously at 520–540 nm (green) and 650 nm (red). When only green emission intensity is modulated by external factors, for example by pH-sensitive dye, whose absorption spectrum varies in this green spectral region, the erbium red emission may serve as a reference for the pH (indirectly)-dependent green emission, and after a simple calibration procedure this green-to-red ratio pH probe can be easily detected or imaged. It should be noted there is no direct interaction between the emitting erbium ions and the pH-sensitive dye, but the erbium green emission from the nanoparticle is spectrally filtered (modulated) by the dye anchored at the UCNP surface. PA emission should show similar features, but the very steep nonlinearity of ANPs to the pump power may complicate the technical side of the measurements. Alternatively, PA offers an unprecedented referencing feature because the PA emission under ESA photoexcitation may be used as a FRET sensor reporter, while the emission at the same wavelength but excited in GSA mode may provide the reference signal. Alternatively, the very slow and pump power-dependent PA rise times have not been explored at all thus far. All these possibilities will surely be studied in the future.
In contrast to multicolour spectral fingerprints, fingerprints that enable various luminescence labels to be distinguished in the time domain have been also proposed and demonstrated. For example, by varying the concentration quenching labels that emit photons of the same energy (colour), but whose luminescence lifetimes are designed such that the luminescence kinetics may be analysed and ascribed. PA emission is quite specific and different from the conventional upconversion. All the problems with creating multilabel (multicolour or time tags) emission are also valid for ANPs, but the specific features of PA emission may enable the generation of additional emission lines or the capability to emit only at a specific photoexcitation wavelength. There is some expectancy in sensitized avalanche emission and core–shell ANP designs, which under a single excitation wavelength may generate numerous new emission lines. However, experimental demonstrations are missing thus far, which will gather interest in the near future.
Multiplexing, i.e. the ability to target multiple analytes in liquid or complex samples such as cells of tissues, requires unique and easily quantifiable spectral codes. Considering the recent emergence of PA nanomaterials, the range of emission wavelengths and excitation wavelengths for ANPs is still limited, presenting an opportunity for the continued development of methods to expand the library of ANPs. A good example of tunability for multiplexing is quantum dots, where the quantum confinement effect produces a spectrally distinct shift in the emission bands. Despite their efficient absorption and emission, improved photostability and small size, QDs demonstrate relatively broad and overlapping emission bands, and ubiquitous short wavelength excitation, which overlap with endogenous chromophores in the absorption, scattering and excitation regions.
To realize the multiplexed imaging of multiple labels, a wider range of spectrally distinct ANP labels must be developed. There are four viable strategies for using lanthanide-doped ANPs as luminescence labels, as follows:
1. The first is displacing these labels in space or sample volume, aiming to address them one-by-one by raster scanning confocal imaging. However, due to light diffraction, the best spatial resolution offered does not go below a few hundreds of nanometers. Therefore, despite the multicolour emission, achieving many unique fingerprints with UCNP LnNP labels remains a challenge.
2. The second approach relies on creating pure, single emission band labels, which enable the detection of various LnNP labels in the spectral domain even when the spatial resolution is limited. However, due to the nature of the Ln3+ emission, this approach can only generate a limited number of spectral codes, often reducing the brightness.223
3. The third approach relies on time domain labels. However, although the lifetimes of LnNPs are long (μs–ms) and they can be engineered to design up to a few time tags,225 the possibility to distinguish them in complex samples containing multiple labels within the diffraction limited volumes is in practice difficult for numerous reasons, e.g., brightness under short pulses becomes weak and results in a poor signal to noise ratio.
4. The last possibility is using various excitation wavelengths suitable to excite a particular LnNP. However, although short wavelength excitation is often suitable to excite multiple Ln3+ nano-labels, it is typically undesirable in bio-applications because it stimulates bio-sample autofluorescence and enhances the background signal. In the case of upconverting labels, NIR excitation (at 920, 976, 800 or 1530 nm) can initiate visible emission, but this remains a technical challenge to switch between light sources, avoid crosstalk between these excitation lines and optically align multispectral images under various excitations.
Therefore, it remains highly desirable to develop ANPs as nanoscale labels, exhibiting multiple pure colour emission spectral fingerprints under a single excitation laser line. However, thus far, only a limited number of singular lanthanide ions in a limited number of host materials under well-adjusted excitation wavelength of sufficient excitation pump power density has been capable of generating PA emission at the nanoscale.
7.2 Novel opportunities for PA studies and applications
Despite the fact that the physical mechanism responsible for PA is relatively well understood and that PA emission has been experimentally demonstrated in single-crystals, further studies and optimization of PA in nano- and micro-scale materials may bring a whole plethora of new features and benefits, and thus may generate active elements for new optical and photonic applications. Here, we briefly discuss some of these application areas, where potentially highly controllable, scalable PA-hosting materials with customizable performance, can stimulate great progress and may promise new possible applications.There is great demand for the detection of MIR photons due to the importance of this radiation in many applications, such as spectroscopy, metrology, (bio)imaging, molecular analysis of gases and astronomy.238–242 Unfortunately, the detection of MIR radiation in a sensitive and cost-effective way is challenging, mostly due to the insufficient sensitivity of the available detectors in this wavelength range.242 Moreover, its detection is hampered by the high noise signal due to the natural emission above absolute zero in the infrared range. Thus, there is a substantial need to develop new MIR photon counters allowing to bypass the limitations resulting from the low energy of the analysed radiation, and hence the high thermal background.243
One of the proposed methods for the detection of the MIR signal is its conversion to visible radiation, where the detectors are much more sensitive, cheap and widely available. An example is the upconversion process, which in the most well-known case shifts the NIR photons to a higher energy in the visible, and thus it can be detected with the generally available detectors.243 In 1959, Bloembergen proposed an IR quantum counter (IRQC), the operation of which is based on a two-step excitation process.244 In the Bloembergen IRQC, the operation scheme includes the first step when infra-red radiation excites the atoms to a metastable level, followed by the next step in which the intense pump excites the atoms to a higher emission level. Therefore, the IR signal is incoherently upconverted to emission in the spectral range where very sensitive and much cheaper detectors are commonly available.
Based on the Bloembergen approach, in 1968, Esterowitz et al. proposed an ideal five-level IRQC system (Fig. 22a), where the IR signal transition (1 → 3) is followed by rapid depopulation via a nonradiative transition to the lower lying metastable level (2).145 Subsequently, the pump transition (2 → 5) occurs, which is followed by a fast nonradiative transition to the fluorescent lower lying level (4). The proposed system allows the problems occurring in the simplest IRQC system with three energy levels to be bypassed, in particular to eliminate the reabsorption of the resulting radiation by the active ions and pump saturation. Moreover, additional levels allow the electron to be recycled, and thus for each absorbed IR photon, more than one visible photon may be emitted. In their work, the authors reported 165 schemes of IRQC found in trivalent rare earth ions in various host materials, the amount of which, as indicated, can be greatly increased by the addition of another co-dopant ion (e.g. Yb3+). However, most of the reported IRQC schemes were established for IR radiation shorter than 3 μm. Recently, Liang et al.238 proposed an approach with an inverted sequence, with continuous visible pumping responsible for promoting the ions from the ground to the excited level, which is followed by fast energy relaxation to the lower energy levels, and then NIR emission. However, presence of the MIR photons results in the ions being excited back from this emitting level to a higher one, from where also higher energy emission occurs. Thus, the intensity of the MIR radiation affects the luminescence intensity of these two emitting bands.
 | ||
| Fig. 22 MIR photon counting (a) five-level quantum counter scheme (hvp – pump, hvs – signal, hvf – fluorescence. Wavy arrows indicate non-radiative transitions).145 (b) IR detection scheme based on Pr3+.245 Dashed arrows are possible cross relaxations (CR) responsible for photon avalanche phenomenon. Transparent lines are additional possible CR and fluorescence transitions not shown in ref. 245. | ||
PA medium infrared quantum counter detectors (MIRQC) were first reported in 1979 by Chivian et al., who while studying materials for IR quantum counters, discovered the PA phenomenon in Pr3+-doped LaCl3 and LaBr3 hosts.1 The underlying concept was simple, i.e., the materials were pre-pumped with wavelength matching ESA (3H5 → 3PJ) instead of GSA (3H4 → 3PJ) excitation. Above some pump power density threshold (ca. 1.2–12.2 W cm−2), a sharp 100-fold increase in the red luminescence intensity at 600–640 nm (3P0 → 3H6, 3P1 → 3F2, 3P0 → 3F2) was observed. When the system was close to the PA threshold, the 4.5 μm photons (matching ground 3H4 to 3H5 level of Pr3+ ions) at 1–10 mW cm−2 intensities were sufficient to trigger PA emission, and consequently a 4-fold increase in the sensitivity of MIR photon detection was demonstrated.1 The interpretation of the obtained results was based on the sequence of ion interactions, in which the absorption of one photon in the infrared region led to the generation of multiple excitations.
In 1983, Krasutsky presented the first crystal quantum counter working at a wavelength longer than 5 μm. This system, based on the LaBr3:Sm3+ crystal, was sensitive in the spectral range of 2 to 10 μm due to the utilization of a low phonon material.37 The authors described the pump-dependent background noise as the main limitation of the studied system detectivity. The PA, observed above pump power densities of 3 kW cm−2, was considered a limitation mainly due to difficulties in performing experiments and ensuring pump stability and intensity. Consequently, the PA phenomenon set an upper limit on the maximum pump power density that could be used for IRQC experiments.
Although benefits arising from the utilization of PA were not fully recognized initially, in 2003, D. B. Gatch et al. showed that due to PA, the signal-to-noise ratio for the IRQC can be improved.245 This was shown by both experimental results and numerical modelling. Moreover, this experiment was carried out at room temperature. The authors used an LaCl3:8%Pr3+ crystal pumped by a focused dye laser beam (560 nm), while the IR signal was provided by a tungsten-halogen lamp. They demonstrated an enhancement in the detection of the IR signal corresponding to the three transitions, i.e., 3H4 → 3H6/3FJ/1G4, in the range of 1.0–2.3 μm for monitored PA emission. Below the critical threshold of pump power (30 mW) and with the addition of an 11 mW IR signal, an increase in the visible emission was observed with a signal-to-noise ratio greater than one. In contrast, for the same IR signal but for a pump power above the threshold, an increase in the visible emission was observed with a signal-to-noise ratio of less than unity. Moreover, the signal-to-noise ratio decreased to 0.5 at the threshold power of 30 mW when the IR signal was reduced to 5 mW.
Fig. 22b presents a schematic illustration of the IRQC based on Pr3+ ions. When the ion concentration is increased, cross-relaxation (CR) processes loop the energy between the excited and ground states of two neighbouring Pr3+ ions, leading to doubling of the population of intermediate excited level in every iteration of the loop and an increase in resonant photon absorption of pump photons, hνP. Consequently, a visible emission was observed from the 3P0 level, and the intensity thereof is dependent on the initial population of the 3H5 level. The population of the latter level depends on the flux of MIR photons, and thus the visible emission is nonlinearly dependent on the MIR light detected by this quantum converter.
Among the methods for detecting single photons in MIR, one can mention the technique based on a crystal detector utilizing the sum frequency generation phenomenon. Photons are upconverted to near infrared radiation, and then detected by avalanche photodiodes.246 Another method for MIR detection is the use of the nonlinear optics process of three-wave mixing, but this technique requires precise phase matching and high pulse powers, which are only achievable with femto- and picosecond lasers.246 InGaAs semiconductor materials provide technical feasibility to detect or image MIR photons, but these detectors are not very sensitive and are also very expensive.247 The mid-infrared light detection by optomechanical frequency upconversion is a novel advanced method and was first theoretically studied by P. Roelli et al.248 and further developed by W. Chen et al.242 and A. Xomalis et al.249 In this method the frequency upconversion was achieved by the utilization of the IR absorption and Raman activity of molecular vibrations. The coherent interaction of the electronic state of a molecule with its vibrational states through IR light, together with specially designed antennas and gold nanospheres confining the MIR energy to a narrow slot allowed the detection of the MIR signal with a power of less than a microwatt. However, despite their enormous potential, the presented devices showed limited stability due to the use of organic molecules. The lifetime of these device can exceed one month only if proper encapsulation that excludes the presence of oxygen is provided.243
The broad application field of MIR detectors is constantly motivating researchers to develop new sensitive, cost-effective detection schemes. IRQC based on the PA phenomenon, as detailed in this review, can bring new possibilities of measurements at room temperature with a reduced signal-to-noise ratio and, through ladder-like energy levels, prevent rapid saturation of the level from which emission occurs. The development of new PA materials based on various lanthanide ions and nanoscale host materials together with advancements in measuring systems may also open new possibilities of MIR detection at the nano-microscale in biology through optical microscopes that traditionally work in the visible spectral range.1,37,145,243,245,246,250
Extending the concept of photon counting with PA nanomaterials, a straightforward extrapolation is IR imaging instead of simple intensity measurements counting from one single point or total intensity emitted from the bulk volume of a sample. The involvement of PA as a workhorse enables the reaping of the essential benefits connected to it, such as great sensitivity and signal amplification accompanied with translation of the detected MIR photons into visible or NIR photons, which are more suitable to detect even with simple and cheap photodetectors. Two valuable architectures of the PA-based optically active element can be proposed. The first, utilizing a bulk monocrystal, acting as a screen or photographic film, inside which the image constructed by the incoming MIR photons is transferred into the visible projection, making it possible to map with optical microscopy techniques. An analogous operating blueprint can be also found for the second approach, where the active element is composed of a film of nanocrystals, and each of them can operate as an independent pixel (or voxel), providing better resolution due to the limited cross-talk (owing to energy migration) between neighbouring regions. Moreover, this design opens the possibility of sensitivity at multiple spectral bands owing to the use of mixtures of several ANP. Nevertheless, in these approaches for narrowband sensitivity, MIR detection is inherently connected with several technical difficulties. Firstly, a wide area of the avalanching material should be pumped homogeneously with a stable power density slightly below the threshold value. This can be challenging because tens or hundreds of kW cm−2 are required for the most well-known materials featuring efficient PA. Furthermore, the MIR spectral sensitivity can require the application of various PA ions or PA schemes, and thus different pumping wavelengths or thresholds may apply. These issues can be at least partially overcome, similarly as some challenges arising from the point of view of sub diffraction imaging, thermometry or FRET sensors, by the development of materials featuring efficient PA and characterized by a low power threshold value.
PA pumping of lasers has been observed since 1990 in Pr3+ (7%)-doped LaCl3 material,252 where 677 nm input photons were converted into a 644 nm laser output, giving an efficiency of 25%. Other early reports presented lasing in bulk materials and fibers46 doped with Nd3+ (ref. 32) and Tm3+ (ref. 253) and co-doped with combinations such as Pr3+/Yb3+.254 A broad range of visible emissions is covered by PA lasers, with most of the laser systems (and all the ones exhibiting efficiencies above 10%) produced based on Pr3+ or Pr3+/Yb3+ co-doped materials. However, the great majority of the known reports on this topic were published in the 90s and early 2000s, which had no chance to gain from the current knowledge and technologies developed and provided by the nanotechnology field in the last decade.
The use of upconverting nanomaterials as gain media for lasers has opened new applications for lasers, such as microscale lasers as ultrasharp emitters for bioimaging. Unlike bulk materials, UCNPs can be readily coated onto or assembled into resonators and cavities (including the whispering gallery modes),255–257 microcavities or plasmonic array nanocavities,258,259 Promising results have been reported using NaYF4 core–shell nanoparticles doped with Tm3+ ions, which are known for hosting energy looping77 and PA. These nonlinear processes promote population inversion through the avalanche-like building of the population of the 3H4 metastable state.95 These nanoparticles, assembled on 5 μm polystyrene microspheres, emit at the room temperature whispery gallery mode-enhanced continuous wave laser light at 800 nm and 450 nm.260 The avalanche-like pumping (with a slope of ∼7 or ∼4 for emissions at 800 nm and 450 nm, respectively, and the average threshold value of 44 ± 23 kW cm−2) was presented with a 1064 nm ESA-resonant excitation beam. Moreover, controlled assembling of the UCNP layer on the microcavities enabled the production of systems with a down-shifted threshold power density to 0.7–0.9 kW cm−2, with demonstrated potential for biosensing.261 Other types of lasing with avalanche-like characteristics (slope of 4.4 and threshold of emission intensity at 70 W cm−2) have been reported for NaYF4 core–shell nanoparticles, with their core co-doped with Er3+ and Yb3+ ions, assembled on an array of plasmonic Ag nanopillars.262 The studies of the highly nonlinear response of materials and possibility of laser emission in micro or nano laser cavities can not only provide insights into laser light and interaction at the molecule level but also for photonics devices or mechanisms on the nanoscale.
Although PA-based lasers preserve all the key advantages of conventional lanthanide ion-based upconverting lasers, they offer new possibilities. Firstly, they significantly extend the selection of available combinations of pumping wavelengths, which are not resonant with GSA transitions (like in most common pumping schemes) and have to be well fitted to the desired ESA transition. The resulting laser lines often provide emission and amplification in the visible range, similar to the case of Pr3+-doped systems. Also important, in conventional UC mechanisms, efficient energy CR is a parasitic effect hindering population inversion and attenuating the laser emission, and therefore higher dopant concentrations are neither recommended nor assure the optimal performance. However, an increase in the concentration of dopants in the case of the PA scheme is highly desirable because energy looping, which is governed by the CR mechanism, is responsible for fuelling the multiplication of ions in the intermediate metastable state. Not only a ca. 10-fold larger absorption cross section (for excitation powers exceeding the threshold value) can be obtained compared to laser materials operating in other schemes, but the brightness is enhanced as well with an increase in the number of emitting ions. The nature of the PA phenomenon exhibits a highly nonlinear relation between pumping power and resulting output efficiency. Consequently, PA lasers provide a high in-out efficiency performance; however, they require pumping power densities exceeding the threshold value for their optimal operation.251 Finally, the fact that the PA has become available at the nanoscale level opens new possibilities in the fields of nano-bio-technology, biology or environment sensing, nano-photonics, telecommunication and optical computing.251
8. Conclusion
This review surveyed the fundamentals of photon avalanche, the development of lanthanide-doped photon avalanching nanoparticles, their application, challenges and opportunities for their further investigation. As a nascent field, the examples of true PA in nanomaterials are sparse, together with their practical applications. However, these few examples have been groundbreaking, with their impacts and applications far beyond the immediate field of lanthanide-doped upconverting nanoparticles. The unprecedented optical feedback within these ANPs makes them the most nonlinear materials reported thus far, a fact that has captured the imagination of scientists across a broad range of disciplines. These diverse applications include sub-diffraction imaging, mechanical sensing, optical computing, and machine learning. We envision that through collaborations within these disciplines, further new applications will arise from the basic concepts developed thus far and reviewed herein. Similar to the mechanism of PA, the initial seeds of progress in the ANP research reviewed herein will explode into outsized scientific contributions to any field that can utilize the unique optical nonlinear responses of these materials.Abbreviations
| AF | Autofluorescence |
| ANPs | Photon avalanche nanoparticles |
| APD | Avalanche photodiodes |
| Bkg | Background signal (e.g. autofluorescence) |
| CET | Cooperative energy transfer |
| CL | Cooperative luminescence |
| CR | Cross relaxation |
| CS | Cooperative sensitization |
| CW | Continuous wave |
| C@S | General naming strategy for core@shell nanoparticles |
| EM | Energy migration |
| EMU | Energy migration-based energy transfer upconversion |
| ESA | Excited state absorption |
| ET | Energy transfer |
| ETU | Energy transfer upconversion or APTE addition de photon par transferts d’energie |
| FED | Fluorescence emission difference |
| FRET | Förster resonant energy transfer |
| GSA | Ground state absorption |
| IRQC | The infrared quantum counter |
| PMT | Photomultiplier |
| LIR | Luminescence intensity ratio |
| LN | Nonlinear luminescence |
| LnNP | Lanthanide doped nanoparticle |
| LT | Luminescence lifetime |
| MIR | Medium infrared |
| MPA | Migrating photon avalanche |
| MPR | Multiphoton relaxation |
| MRI | Magnetic resonance imaging |
| NGRER | Non-GSA-resonant ESA-resonant |
| NIR | Near infrared |
| NIRES | Near-infrared emission saturation |
| NR | Nonradiative relaxation |
| NSI | Nonlinear structured illumination microscopy |
| PA | Photon avalanche |
| PAA | Poly(acrylic acid) |
| PAET | Photon assisted energy transfer |
| PALM | Photoactivated localization microscopy |
| PANP or ANP | Photon avalanching nanoparticles, |
| PA-RET | Photon avalanche resonant energy transfer |
| PASSI | Photon avalanche single beam super resolution imaging |
| PAT | Photon avalanche thermometry |
| PEG | Poly(ethylene glycol) |
| PEI | Poly(ethyleneimine) |
| PVP | Polyvinylpyrrolidone |
| qCW | Quasi continuous wave |
| QD | Quantum dot |
| RS | Raster scanning |
| RE ions | Rare earth ions |
| RET | Resonant energy transfer |
| SAT | Saturation nanoscopy |
| SBR | Single band ratiometric |
| SFG | Sum frequency generation |
| SHG | Second harmonic generation, |
| SIM | Structured illumination microscopy |
| SPA | Sensitized photon avalanche |
| STED | Super-resolution stimulated emission depletion microscopy |
| STORM | Stochastic optical reconstruction microscopy |
| THG | Third harmonic generation |
| UC | Up-conversion |
| UCNP | Upconverting nanoparticle |
| UC RET | Upconversion RET |
| uSEE | Super-linear excitation–emission |
| WNR | Non-radiative rates |
| VIS | Visible radiation |
| ZBLAN | ZrF4–BaF2–LaF3–AlF3–NaF |
Author contributions
Conceptualization, resources and funding acquisition (AB, EC, JS, AS, MSz), writing original draft (AB, MSz, EC), writing – review & editing and visualisation (all authors), supervision (AB).Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from NCN, Poland, grant numbers NCN 2018/31/B/ST5/01827 (AB, MSz, ZK, MD), 2021/43/B/ST5/01244 (AB, MMa, MMi, KP). Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences (BES), of the U.S. Department of Energy (DOE) under Contract No. DE-AC02-05CH11231. A. S. acknowledges partial support from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement No. 895809 (MONOCLE). A. S. and E. M. C. were partially supported by the DOE Office of Science, BES, through the Chemical Sciences, Geosciences, and Biosciences Division, Separations Program under Contract no. DE-AC02-05CH11231. P. J. S. acknowledges support from the National Science Foundation grant CHE-2203510.References
- J. S. Chivian, W. E. Case and D. D. Eden, Appl. Phys. Lett., 1979, 35, 124–125 CrossRef CAS.
- C. Lee, E. Z. Xu, Y. Liu, A. Teitelboim, K. Yao, A. Fernandez-Bravo, A. M. Kotulska, S. H. Nam, Y. D. Suh, A. Bednarkiewicz, B. E. Cohen, E. M. Chan and P. J. Schuck, Nature, 2021, 589, 230–235 CrossRef CAS.
- Y. Liang, Z. Zhu, S. Qiao, X. Guo, R. Pu, H. Tang, H. Liu, H. Dong, T. Peng, L.-D. Sun, J. Widengren and Q. Zhan, Nat. Nanotechnol., 2022, 2022, 1–7 Search PubMed.
- Z. Zhang, A. Skripka, J. C. Dahl, C. Dun, J. J. Urban, D. Jaque, P. J. Schuck, B. E. Cohen and E. M. Chan, Angew. Chem., Int. Ed., 2023, 62, e202212549 CrossRef CAS.
- A. Skripka, M. Lee, X. Qi, J. A. Pan, H. Yang, C. Lee, P. J. Schuck, B. E. Cohen, D. Jaque and E. M. Chan, Nano Lett., 2023, 23, 7100–7106 CrossRef CAS PubMed.
- M. Dudek, M. Szalkowski, M. Misiak, M. Ćwierzona, A. Skripka, Z. Korczak, D. Piątkowski, P. Woźniak, R. Lisiecki, P. Goldner, S. Maćkowski, E. M. Chan, P. J. Schuck and A. Bednarkiewicz, Adv. Opt. Mater., 2022, 2201052 CrossRef CAS.
- C. Wang, Z. Wen, R. Pu, B. Pan, B. Wang, K. Zheng, Y. Du and Q. Zhan, Adv. Mater., 2023, 2, e2307848 Search PubMed.
- C. Wang, Z. Wen, R. Pu and Q. Zhan, Laser Photonics Rev., 2024, 2400290 CrossRef CAS.
- Z. Zhu, Y. Liang, Q. Zhao, H. Wu, B. Pan, S. Qiao, B. Wang and Q. Zhan, Sci. Bull., 2023, 69, 458–465 CrossRef.
- M. Szalkowski, M. Dudek, Z. Korczak, C. Lee, Ł. Marciniak, E. M. Chan, P. J. Schuck and A. Bednarkiewicz, Opt. Mater.: X, 2021, 12, 100102 CAS.
- M. Dudek, Z. Korczak, K. Prorok, O. Bezkrovnyi, L. Sun, M. Szalkowski and A. Bednarkiewicz, Nanoscale, 2023, 15, 18613–18623 RSC.
- M. Majak, M. Misiak and A. Bednarkiewicz, Mater. Horiz., 2024, 4791–4801 RSC.
- A. Bednarkiewicz, M. Szalkowski, M. Majak, Z. Korczak, M. Misiak and S. Maćkowski, Adv. Mater., 2023, 35, 2304390 CrossRef CAS.
- Z. Korczak, M. Dudek, M. Majak, M. Misiak, Ł. Marciniak, M. Szalkowski and A. Bednarkiewicz, Low Temp. Phys., 2023, 49, 322 CrossRef CAS.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526–1560 RSC.
- A. Hlaváček, Z. Farka, M. J. Mickert, U. Kostiv, J. C. Brandmeier, D. Horák, P. Skládal, F. Foret and H. H. Gorris, Nat. Protoc., 2022, 2022, 1–45 Search PubMed.
- M.-F. F. Joubert, Opt. Mater., 1999, 11, 181–203 CrossRef CAS.
- S. Guy, M. F. Joubert and B. Jacquier, J. Alloys Compd., 1998, 275–277, 186–190 CrossRef CAS.
- M. F. Joubert, S. Guy, B. Jacquier and C. Linarés, Opt. Mater., 1994, 4, 43–49 CrossRef CAS.
- M. Runowski, P. Woźny, I. R. Martín, K. Soler-Carracedo, T. Zheng, H. Hemmerich, F. Rivera-López, J. Moszczyński, P. Kulpiński and S. Feldmann, Adv. Funct. Mater., 2024, 34, 2307791 CrossRef CAS.
- R. M. Corn and D. A. Higgins, Chem. Rev., 1994, 94, 107–125 CrossRef CAS.
- A. Teitelboim, B. Tian, D. J. Garfield, A. Fernandez-Bravo, A. C. Gotlin, P. J. Schuck, B. E. Cohen and E. M. Chan, J. Phys. Chem. C, 2019, 123, 2678–2689 CrossRef CAS.
- H. Wen, H. Zhu, X. Chen, T. F. Hung, B. Wang, G. Zhu, S. F. Yu and F. Wang, Angew. Chem., Int. Ed., 2013, 52, 13419–13423 CrossRef CAS PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS.
- S. Liu, L. Yan, J. Huang, Q. Zhang and B. Zhou, Chem. Soc. Rev., 2022, 51, 1729–1765 RSC.
- M. Pollnau, D. R. Gamelin, S. R. Luthi, H. U. Gudel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337–3346 CrossRef CAS.
- X. Liu, J. Chen, C. Liu, S. Xi, S. Tan Tan, Q. He and L. Liang, Extreme Optical Nonlinearity (>500) at Room Temperature through Sublattice Reconstruction, 2024 Search PubMed.
- A. Bednarkiewicz, E. M.-Y. E. M. E. M. Chan, A. M. Kotulska, L. Marciniak and K. Prorok, Nanoscale Horiz., 2019, 4, 881–889 RSC.
- A. M. Kotulska, A. Pilch-Wróbel, S. Lahtinen, T. Soukka and A. Bednarkiewicz, Light: Sci. Appl., 2022, 11, 1–14 CrossRef.
- N. J. Krasutsky, W. E. Case and J. S. Chivian, J. Appl. Phys., 1979, 50, 3142–3145 CrossRef CAS.
- W. Lenth and R. M. M. Macfarlane, J. Lumin., 1990, 45, 346–350 CrossRef CAS.
- H. Zellmer, P. Riedel and A. Tünnermann, Appl. Phys. B, 1999, 69, 417–421 CrossRef CAS.
- R. Scheps, Prog. Quantum Electron., 1996, 20, 271–358 CrossRef CAS.
- T. Sandrock, H. Scheife, E. Heumann and G. Huber, Opt. Lett., 1997, 22, 808–810 CrossRef CAS.
- P. Goldner and F. Pelle, Opt. Mater., 1996, 5, 239–249 CrossRef CAS.
- N. J. Krasutsky, J. Appl. Phys., 1983, 54, 1261–1267 CrossRef CAS.
- N. Pelletier-Allard and R. Pelletier, Opt. Commun., 1991, 81, 247–250 CrossRef CAS.
- M. F. Joubert, S. Guy and B. Jacquier, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10031–10037 CrossRef CAS PubMed.
- B. P. Scott, F. Zhao, R. S. F. Chang and N. Djeu, Opt. Lett., 1993, 18, 113 CrossRef CAS PubMed.
- L. Yang, B. Zhang, K. Yin, T. Wu, Y. Zhao and J. Hou, Photonics Res., 2018, 6, 417 CrossRef CAS.
- M. Malinowski, A. Wnuk, Z. Frukacz, G. Chadeyron, R. Mahiou, S. Guy and M. F. Joubert, Journal of Alloys and Compounds, Elsevier, 2001, vol. 323–324, pp. 731–735 Search PubMed.
- T. R. Gosnell, Electron. Lett., 1997, 33, 411 CrossRef CAS.
- S. Guy, D. P. Shepherd, M. F. Joubert, B. Jacquier and H. Poignant, J. Opt. Soc. Am. B, 1997, 14, 926 CrossRef CAS.
- F. Auzel and Y. H. Chen, J. Non-Cryst. Solids, 1995, 184, 57–60 CrossRef CAS.
- P. Xie and T. R. Gosnell, Opt. Lett., 1995, 20, 1014 CrossRef CAS.
- O. K. Liu, Y. H. Chen and J. V. Beitz, J. Lumin., 1999, 81, 7–12 CrossRef.
- S. Guy, M. F. Joubert and B. Jacquier, J. Lumin., 1997, 72–74, 65–67 CrossRef CAS.
- M. F. Joubert, S. Guy, C. Linarès, B. Jacquier and J. L. Adam, J. Non-Cryst. Solids, 1995, 184, 98–102 CrossRef CAS.
- H. Ni and S. C. Rand, Opt. Lett., 1991, 16, 1424 CrossRef CAS PubMed.
- M. F. Joubert, S. Guy, S. Cuerq and P. A. Tanner, J. Lumin., 1997, 75(4), 287–293 CrossRef CAS.
- B. C. Collings and A. J. Silversmith, J. Lumin., 1994, 62, 271–279 CrossRef CAS.
- I. R. Martín, C. Goutaudier, S. Guy, Y. Guyot, G. Boulon, M.-T. Cohen-Adad and M.-F. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 7252–7257 CrossRef.
- E. Bielejec, E. Kisel and A. Silversmith, J. Lumin., 1997, 72–74, 62–64 CrossRef CAS.
- N. Kodama and T. Park, J. Phys.: Condens. Matter, 1999, 11, 4775–4782 CrossRef.
- M. Bouffard, J. P. Jouart and G. Mary, Phys. Status Solidi B, 1996, 193, 239–245 CrossRef CAS.
- A. W. Kueny, W. E. Case and M. E. Koch, J. Opt. Soc. Am. B, 1993, 104(10), 1834–1839 CrossRef.
- F. Auzel and Y. Chen, J. Lumin., 1995, 65, 45–56 CrossRef CAS.
- S. Kück and I. Sokólska, Chem. Phys. Lett., 2000, 325, 257–263 CrossRef.
- A. Tymiński and T. Grzyb, J. Lumin., 2017, 181, 411–420 CrossRef.
- A. Tymiński, T. Grzyb and S. Lis, J. Am. Ceram. Soc., 2016, 99, 3300–3308 CrossRef.
- T. V. Gavrilović, D. J. Jovanović, V. Lojpur and M. D. Dramićanin, Sci. Rep., 2014, 4, 4209 CrossRef PubMed.
- K. Kadono, H. Higuchi, M. Takahashi, Y. Kawamoto and H. Tanaka, J. Non-Cryst. Solids, 1995, 184, 309–313 CrossRef CAS.
- I.-I. Oprea, H. Hesse and K. Betzler, Phys. Status Solidi B, 2005, 242, R109–R111 CrossRef CAS.
- S. Sinha, M. K. Mahata and K. Kumar, New J. Chem., 2019, 43, 5960–5971 RSC.
- Y. Liu, Y. Chen, Y. Lin, Q. Tan, Z. Luo and Y. Huang, J. Opt. Soc. Am. B, 2007, 24, 1046–1052 CrossRef CAS.
- M. Back, E. Trave, N. Mazzucco, P. Riello and A. Benedetti, Nanoscale, 2017, 9, 6353–6361 RSC.
- S. Heer, K. Kömpe, H. U. Güdel and M. Haase, Adv. Mater., 2004, 16, 2102–2105 CrossRef CAS.
- B. Szefczyk, R. Roszak and S. Roszak, RSC Adv., 2014, 4, 22526–22535 RSC.
- C. dos S. Bezerra and M. E. G. Valerio, Phys. B, 2016, 501, 106–112 CrossRef.
- S. Janssens, G. V. M. Williams and D. Clarke, J. Appl. Phys., 2011, 109, 511 CrossRef.
- M. J. Weber, Phys. Rev. B: Condens. Matter Mater. Phys., 1973, 8, 54–64 CrossRef CAS.
- A. A. Kaminskiĭ, Crystalline lasers: physical processes and operating schemes, 1996 Search PubMed.
- T. Miyakawa and D. L. Dexter, Phys. Rev. B: Condens. Matter Mater. Phys., 1970, 1, 2961–2969 CrossRef.
- N. Yamada, S. Shionoya and T. Kushida, J. Phys. Soc. Jpn., 1972, 32, 1577–1586 CrossRef CAS.
- J. M. Collins and B. Di Bartolo, J. Lumin., 1996, 69, 335–341 CrossRef CAS.
- E. S. Levy, C. A. Tajon, T. S. Bischof, J. Iafrati, A. Fernandez-Bravo, D. J. Garfield, M. Chamanzar, M. M. Maharbiz, V. S. Sohal, P. J. Schuck, B. E. Cohen and E. M. Chan, ACS Nano, 2016, 10, 8423–8433 CrossRef CAS.
- N. J. J. J. Johnson, S. He, S. Diao, E. M. Chan, H. Dai and A. Almutairi, J. Am. Chem. Soc., 2017, 139, 3275–3282 CrossRef CAS PubMed.
- A. Bednarkiewicz, E. M. Chan and K. Prorok, Nanoscale Adv., 2020, 2, 4863–4872 RSC.
- K. W. C. Kwock, C. Lee, A. Teitelboim, Y. Liu, K. Yao, S. B. Alam, B. E. Cohen, E. M. Chan and P. J. Schuck, J. Phys. Chem. C, 2021, 125, 23976–23982 CrossRef CAS.
- G. Yi, H. Lu, S. Zhao, Y. Ge, W. Yang, D. Chen and L. H. Guo, Nano Lett., 2004, 4, 2191–2196 CrossRef CAS.
- H.-X. Mai, Y.-W. Zhang, R. Si, Z.-G. Yan, L. Sun, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2006, 128, 6426–6436 CrossRef CAS.
- J. C. Boyer, F. Vetrone, L. A. Cuccia and J. A. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444–7445 CrossRef CAS PubMed.
- H. S. Qian and Y. Zhang, Langmuir, 2008, 24, 12123–12125 CrossRef CAS PubMed.
- G. Yi and G. Chow, Chem. Mater., 2007, 19, 341–343 CrossRef CAS.
- X. Chen, D. Peng, Q. Ju and F. Wang, Chem. Soc. Rev., 2015, 44, 1318–1330 RSC.
- C. Song, Z. Ye, G. Wang, J. Yuan and Y. Guan, ACS Nano, 2010, 4, 5389–5397 CrossRef CAS.
- X. Liu, C. H. Yan and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1299–1301 RSC.
- J. W. Stouwdam and F. C. J. M. Van Veggel, Langmuir, 2004, 20, 11763–11771 CrossRef CAS PubMed.
- M. M. Lezhnina, T. Jüstel, H. Kätker, D. U. Wiechert and U. H. Kynast, Adv. Funct. Mater., 2006, 16, 935–942 CrossRef CAS.
- X. Li, F. Zhang and D. Zhao, Chem. Soc. Rev., 2015, 44, 1346–1378 RSC.
- C. C. S. Pedroso, V. R. Mann, K. Zuberbühler, M. F. Bohn, J. Yu, V. Altoe, C. S. Craik and B. E. Cohen, ACS Nano, 2021, 15, 18374–18384 CrossRef CAS PubMed.
- D. J. Gargas, E. M. Chan, A. D. Ostrowski, S. Aloni, M. V. P. Altoe, E. S. Barnard, B. Sanii, J. J. Urban, D. J. Milliron, B. E. Cohen and P. J. Schuck, Nat. Nanotechnol., 2014, 9, 300–305 CrossRef CAS.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef.
- Y. Liu, Y. Lu, X. Yang, X. Zheng, S. Wen, F. Wang, X. Vidal, J. Zhao, D. Liu, Z. Zhou, C. Ma, J. Zhou, J. A. Piper, P. Xi and D. Jin, Nature, 2017, 543, 229–233 CrossRef CAS.
- K. Prorok, A. Bednarkiewicz, B. Cichy, A. Gnach, M. Misiak, M. Sobczyk and W. Strek, Nanoscale, 2014, 6, 1855–1864 RSC.
- Q. Su, S. Han, X. Xie, H. Zhu, H. Chen, C. K. Chen, R. S. Liu, X. Chen, F. Wang and X. Liu, J. Am. Chem. Soc., 2012, 134, 20849–20857 CrossRef CAS.
- R. Arppe, I. Hyppänen, N. Perälä, R. Peltomaa, M. Kaiser, C. Würth, S. Christ, U. Resch-Genger, M. Schäferling and T. Soukka, Nanoscale, 2015, 7, 11746–11757 RSC.
- C. Dong, J. Pichaandi, T. Regier and F. C. J. M. van Veggel, J. Phys. Chem. C, 2011, 115, 15950–15958 CrossRef CAS.
- J.-C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 1417–1419 RSC.
- F. T. Rabouw, P. T. Prins, P. Villanueva-Delgado, M. Castelijns, R. G. Geitenbeek and A. Meijerink, ACS Nano, 2018, 12, 4812–4823 CrossRef CAS.
- H. Deng, S. Yang, S. Xiao, H. M. Gong and Q. Q. Wang, J. Am. Chem. Soc., 2008, 6, 2032–2040 CrossRef.
- M. Misiak, K. Prorok, B. Cichy, A. Bednarkiewicz and W. Stręk, Opt. Mater., 2013, 35, 1124–1128 CrossRef CAS.
- K. Prorok, A. Gnach, A. Bednarkiewicz and W. Stręk, J. Lumin., 2013, 140, 103–109 CrossRef CAS.
- Y. Wang, Nanoscale, 2019, 11, 10852–10858 RSC.
- B. Zhou, L. Tao, Y. H. Tsang and W. Jin, J. Mater. Chem. C, 2013, 1, 4313–4318 RSC.
- G. Chen, J. Shen, T. Y. Ohulchanskyy, N. J. Patel, A. Kutikov, Z. Li, J. Song, R. K. Pandey, H. Ågren, P. N. Prasad and G. Han, ACS Nano, 2012, 6, 8280–8287 CrossRef CAS.
- Y. Shang, S. Hao, W. Shao, T. Chen, Y. Zhu and C. Yang, J. Mater. Chem. C, 2020, 8, 2847–2851 RSC.
- J. Wang, F. Wang, C. Wang, Z. Liu and X. Liu, Angew. Chem., Int. Ed., 2011, 50, 10369–10372 CrossRef CAS PubMed.
- Z. Wang and A. Meijerink, J. Phys. Chem. C, 2018, 122, 26298–26306 CrossRef CAS PubMed.
- Z. Li, L. Wang, Z. Wang, X. Liu and Y. Xiong, J. Phys. Chem. C, 2011, 115, 3291–3296 CrossRef CAS.
- X. Kang, Z. Cheng, C. Li, D. Yang, M. Shang, P. Ma, G. Li, N. Liu and J. Lin, J. Phys. Chem. C, 2011, 115, 15801–15811 CrossRef CAS.
- L. Liang, Y. Liu, C. Bu, K. Guo, W. Sun, N. Huang, T. Peng, B. Sebo, M. Pan, W. Liu, S. Guo and X.-Z. Zhao, Adv. Mater., 2013, 25, 2174–2180 CrossRef CAS PubMed.
- B. Zhang, J. Meng, X. Mi, C. Zhang, Z. Zhang and H. Zheng, RSC Adv., 2018, 8, 37618–37622 RSC.
- J. Zhao, T. Jia, X. Wang and X. Kong, J. Nanomater., 2014, 2014, 821706 CrossRef.
- P. Yuan, Y. H. Lee, M. K. Gnanasammandhan, Z. Guan, Y. Zhang and Q.-H. Xu, Nanoscale, 2012, 4, 5132–5137 RSC.
- B. Dong, S. Xu, J. Sun, S. Bi, D. Li, X. Bai, Y. Wang, L. Wang and H. Song, J. Mater. Chem., 2011, 21, 6193–6200 RSC.
- Z. Wang, C. Wang, Q. Han, G. Wang, M. Zhang, J. Zhang, W. Gao and H. Zheng, Mater. Res. Bull., 2017, 88, 182–187 CrossRef CAS.
- Y. Chen, C. D’Amario, A. Gee, H. T. T. Duong, O. Shimoni and S. M. Valenzuela, Acta Biomater., 2020, 102, 384–393 CrossRef CAS.
- X. Liu, X. Zhang, G. Tian, W. Yin, L. Yan, L. Ruan, Z. Yang, D. Xiao and Z. Gu, CrystEngComm, 2014, 16, 5650–5661 RSC.
- S. Niu, Y. Zhou, H. Yu, C. Lu and K. Han, Energy Convers. Manage., 2017, 149, 495–504 CrossRef CAS.
- L. Guerrini, R. A. Alvarez-Puebla and N. Pazos-Perez, Materials, 2018, 11, 1154 CrossRef.
- S. F. Himmelstoß and T. Hirsch, Part. Part. Syst. Charact., 2019, 36, 1900235 CrossRef.
- P. Dolcet, A. Mambrini, M. Pedroni, A. Speghini, S. Gialanella, M. Casarin and S. Gross, RSC Adv., 2015, 5, 16302–16310 RSC.
- C. Dubey, A. Yadav, D. Baloni, S. Kachhap, S. K. Singh and A. K. Singh, RSC Adv., 2023, 13, 20975–20983 RSC.
- A. Kruk, Materials, 2020, 13, 4928 CrossRef CAS PubMed.
- J. Mitrić, U. Ralević, M. Mitrić, J. Ćirković, G. Križan, M. Romčević, M. Gilić and N. Romčević, J. Raman Spectrosc., 2019, 50, 802–808 CrossRef.
- D. Errandonea, O. Gomis, P. Rodríguez-Hernández, A. Muñoz, J. Ruiz-Fuertes, M. Gupta, S. N. Achary, A. Hirsch, F. J. Manjon, L. Peters, G. Roth, A. K. Tyagi and M. Bettinelli, J. Phys.: Condens. Matter, 2018, 30, 065401 CrossRef CAS.
- S. Kostić, Z. Lazarević, V. Radojević, A. Milutinović, M. Romčević, N. Romčević and A. Valčić, Mater. Res. Bull., 2015, 63, 80–87 CrossRef.
- A. K. Singh, K. Kumar, A. C. Pandey, O. Parkash, S. B. Rai and D. Kumar, Appl. Phys. B, 2011, 104, 1035–1041 CrossRef CAS.
- H. M. Gong, Z. K. Zhou, S. Xiao, X. R. Su and Q. Q. Wang, Plasmonics, 2008, 3, 59–64 CrossRef CAS.
- Z. Ma, Y. Yu, S. Shen, H. Dai, L. Yao, Y. Han, X. Wang, J.-B. Han and L. Li, Sci. Rep., 2016, 6, 18857 CrossRef CAS.
- Q. Q. Wang, J. B. Han, D. L. Guo, S. Xiao, Y. B. Han, H. M. Gong and X. W. Zou, Nano Lett., 2007, 7, 723–728 CrossRef CAS PubMed.
- A. Fernandez-Bravo, K. Yao, E. S. Barnard, N. J. Borys, E. S. Levy, B. Tian, C. A. Tajon, L. Moretti, M. V. Altoe, S. Aloni, K. Beketayev, F. Scotognella, B. E. Cohen, E. M. Chan and P. J. Schuck, Nat. Nanotechnol., 2018, 13, 1 CrossRef.
- S. Kück, A. Diening, E. Heumann, E. Mix, T. Sandrock, K. Sebald and G. Huber, J. Alloys Compd., 2000, 300, 65–70 CrossRef.
- N. J. Krasutsky, J. Appl. Phys., 1983, 54, 1261–1267 CrossRef CAS.
- M. Rathaiah, I. R. Martín, P. Babu, K. Linganna, C. K. Jayasankar, V. Lavín and V. Venkatramu, Opt. Mater., 2015, 39, 16–20 CrossRef CAS.
- E. Osiac, S. Kück, E. Heumann, G. Huber, E. Sani, A. Toncelli and M. Tonelli, Opt. Mater., 2003, 24, 537–545 CrossRef CAS.
- P. Babu, I. R. Martín, K. Venkata Krishnaiah, H. J. Seo, V. Venkatramu, C. K. Jayasankar and V. Lavín, Chem. Phys. Lett., 2014, 600, 34–37 CrossRef CAS.
- M. Plöschner, D. Denkova, S. de Camillis, M. Das, L. M. Parker, X. Zheng, Y. Lu, S. Ojosnegros and J. A. Piper, Opt. Express, 2020, 28, 24308 CrossRef PubMed.
- T. Sandrock, E. Heumann, G. Huber, V. Lupei, B. Bejan and E. Osiac, Optical Society of America. Australian Council on Quantum Electronics. International Quantum Electronics Conference (20th: 1996: Sydney, N.S.W.), Optical Society of America, Sydney, 1996, p. ThQ7 Search PubMed.
- H. Wu, B. Pan, Q. Zhao, C. Wang, R. Pu, C. Liu, Z. Chen, Z. Luo, J. Huang, W. Wei, T. Chen and Q. Zhan, Adv. Photonics, 2024, 6(5), 056010 Search PubMed.
- M. E. Koch, A. W. Kueny and W. E. Case, Appl. Phys. Lett., 1990, 56, 1083–1085 CrossRef CAS.
- E. Osiac, E. Heumann, G. Huber, S. Kuck, E. Sani, A. Toncelli and M. Tonelli, Conference on Lasers and Electro-Optics Europe – Technical Digest, 2003 Search PubMed.
- A. Schnitzler, J. Bahler, J. Noonan and L. Esterowitz, Appl. Opt., 1968, 7, 2053–2070 CrossRef.
- C. Lee, E. Z. Xu, K. W. C. Kwock, A. Teitelboim, Y. Liu, N. Fardian-Melamed, C. C. S. Pedroso, H. S. Park, J. Kim, S. D. Pritzl, S. H. Nam, T. Lohmueller, P. Ercius, Y. D. Suh, B. E. Cohen, E. M. Chan and P. J. Schuck, Nature, 2023, 618, 951–958 CrossRef CAS PubMed.
- J.-A. Pan, A. Skripka, C. Lee, X. Qi, A. L. Pham, J. J. Woods, R. J. Abergel, P. J. Schuck, B. E. Cohen and E. M. Chan, J. Am. Chem. Soc., 2024, 146, 7487–7497 CrossRef CAS.
- L. Schermelleh, R. Heintzmann and H. Leonhardt, J. Cell Biol., 2010, 190, 165–175 CrossRef CAS.
- G. Vicidomini, P. Bianchini and A. Diaspro, Nat. Methods, 2018, 15, 173–182 CrossRef CAS.
- D. Jin, P. Xi, B. Wang, L. Zhang, J. Enderlein and A. M. van Oijen, Nat. Methods, 2018, 15, 1–9 CrossRef.
- L. Francés-Soriano, J. Ferrera-González, M. González-Béjar and J. Pérez-Prieto, Anal. Bioanal. Chem., 2022, 414, 4291–4310 CrossRef.
- W. Li, G. S. Kaminski Schierle, B. Lei, Y. Liu and C. F. Kaminski, Chem. Rev., 2022, 122, 12495–12543 CrossRef CAS PubMed.
- M. G. L. Gustafsson, J. Microsci., 2000, 198, 82–87 CrossRef CAS PubMed.
- M. G. L. Gustafsson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13081–13086 CrossRef CAS.
- J. M. Gerton, L. A. Wade, G. A. Lessard, Z. Ma and S. R. Quake, Phys. Rev. Lett., 2004, 93, 180801 CrossRef.
- N. Mauser and A. Hartschuh, Chem. Soc. Rev., 2014, 43, 1248–1262 RSC.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780 CrossRef CAS PubMed.
- D. Denkova, M. Ploschner, M. Das, L. M. Parker, X. Zheng, Y. Lu, A. Orth, N. H. Packer and J. A. Piper, Nat. Commun., 2019, 10, 1–12 CrossRef CAS.
- B. Liu, C. Chen, X. Di, J. Liao, S. Wen, Q. P. Su, X. Shan, Z.-Q. Xu, L. A. Ju, C. Mi, F. Wang and D. Jin, Nano Lett., 2020, 20, 4775–4781 CrossRef CAS.
- C. Chen, F. Wang, S. Wen, Q. P. Su, M. C. L. L. Wu, Y. Liu, B. Wang, X. Shan, M. Kianinia, I. Aharonovich, M. Toth, S. P. Jackson, P. Xi and D. Jin, Nat. Commun., 2018, 9, 4–9 CrossRef.
- R. Kolesov, R. Reuter, K. Xia, R. Stöhr, A. Zappe and J. Wrachtrup, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 1–4 CrossRef.
- F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932–940 CrossRef CAS PubMed.
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73–76 CrossRef CAS.
- M. Oheim, E. Beaurepaire, E. Chaigneau, J. Mertz and S. Charpak, J. Neurosci. Methods, 2001, 111, 29–37 CrossRef CAS PubMed.
- L. Caillat, B. Hajj, V. Shynkar, L. Michely, D. Chauvat, J. Zyss and F. Pellé, Appl. Phys. Lett., 2013, 102, 143114 CrossRef.
- S. Lamon, H. Yu, Q. Zhang and M. Gu, Light: Sci. Appl., 2024, 13, 252 CrossRef CAS PubMed.
- S. Li, F. Li, N. Kong, J. Liu and X. Zhu, Adv. Healthcare Mater., 2023, 12, e2302276 CrossRef.
- J. Yao, M. Yang and Y. Duan, Chem. Rev., 2014, 114, 6130–6178 CrossRef CAS.
- T.-M. T. M. Liu, J. Conde, T. Lipiński, A. Bednarkiewicz and C.-C. C. C. Huang, Prog. Mater. Sci., 2017, 88, 89–135 CrossRef CAS.
- T.-M. T. M. Liu, J. Conde, T. Lipiński, A. Bednarkiewicz and C.-C. C. C. Huang, NPG Asia Mater., 2016, 8, 1–25 CrossRef.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS.
- W. Park, D. Lu and S. Ahn, Chem. Soc. Rev., 2015, 44, 2940–2962 RSC.
- S. Hao, J. Liu, M. Tan and G. Chen, Upconversion Enhancement Using Epitaxial Core–Shell Nanostructures, CRC Press, 1st edn, 2016 Search PubMed.
- S. Han, R. Deng, Q. Gu, L. Ni, U. Huynh, J. Zhang, Z. Yi, B. Zhao, H. Tamura, A. Pershin, H. Xu, Z. Huang, S. Ahmad, M. Abdi-Jalebi, A. Sadhanala, M. L. Tang, A. Bakulin, D. Beljonne, X. Liu and A. Rao, Nature, 2020, 587, 594–599 CrossRef CAS.
- D. J. Garfield, N. J. Borys, S. M. Hamed, N. A. Torquato, C. A. Tajon, B. Tian, B. Shevitski, E. S. Barnard, Y. D. Suh, S. Aloni, J. B. Neaton, E. M. Chan, B. E. Cohen and P. J. Schuck, Nat. Photonics, 2018, 12, 402–407 CrossRef CAS.
- A. D. Ostrowski, E. M. Chan, D. J. Gargas, E. M. Katz, G. Han, P. J. Schuck, D. J. Milliron and B. E. Cohen, ACS Nano, 2012, 6, 2686–2692 CrossRef CAS PubMed.
- M. Kaiser, C. Würth, M. Kraft, I. Hyppänen, T. Soukka and U. Resch-Genger, Nanoscale, 2017, 9, 10051–10058 RSC.
- B. E. Cohen, Nature, 2010, 467, 407–408 CrossRef CAS PubMed.
- Y. Zhong, G. Tian, Z. Gu, Y. Yang, L. Gu, Y. Zhao, Y. Ma and J. Yao, Adv. Mater., 2014, 26, 2831–2837 CrossRef CAS.
- I. Medintz and N. Hildebrandt, FRET – Förster Resonance Energy Transfer: From Theory to Applications, Wiley-VCH Verlag, 2013 Search PubMed.
- C. Wang, H. Lin, Z. Xu, Y. Huang, M. G. Humphrey and C. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 6621–6628 CrossRef CAS PubMed.
- E. Carrasco, B. del Rosal, F. Sanz-Rodríguez, Á. J. de la Fuente, P. H. Gonzalez, U. Rocha, K. U. Kumar, C. Jacinto, J. G. Solé and D. Jaque, Adv. Funct. Mater., 2015, 25, 615–626 CrossRef CAS.
- D. Ruiz, B. del Rosal, M. Acebrón, C. Palencia, C. Sun, J. Cabanillas-González, M. López-Haro, A. B. Hungría, D. Jaque and B. H. Juarez, Adv. Funct. Mater., 2016, 27, 1604629 CrossRef.
- E. Carrasco, B. del Rosal, F. Sanz-Rodríguez, Á. J. de la Fuente, P. H. Gonzalez, U. Rocha, K. U. Kumar, C. Jacinto, J. G. Solé and D. Jaque, Adv. Funct. Mater., 2015, 25, 615–626 CrossRef CAS.
- J. P. Feist, A. L. Heyes and S. Seefelt, Proc. Inst. Mech. Eng., Part A, 2003, 217, 193–200 CrossRef CAS.
- C. Abram, B. Fond and F. Beyrau, Prog. Energy Combust. Sci., 2018, 64, 93–156 CrossRef.
- R. G. Geitenbeek, A.-E. Nieuwelink, T. S. Jacobs, B. B. V. Salzmann, J. Goetze, A. Meijerink and B. M. Weckhuysen, ACS Catal., 2018, 8, 2397–2401 CrossRef CAS PubMed.
- D. Jaque and F. Vetrone, Nanoscale, 2012, 4, 4301–4326 RSC.
- C. D. S. Brites, A. Millán and L. D. Carlos, Handbook on the Physics and Chemistry of Rare Earths, Elsevier B.V., 2016, vol. 49, pp. 339–427 Search PubMed.
- M. Dramićanin, Luminescence Thermometry, 2018 Search PubMed.
- A. Rabhiou, J. Feist, A. Kempf, S. Skinner and A. Heyes, Sens. Actuators, A, 2011, 169, 18–26 CrossRef CAS.
- X. D. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- C. D. S. Brites, P. P. Lima, N. J. O. Silva, A. Millán, V. S. Amaral, F. Palacio and L. D. Carlos, Nanoscale, 2012, 4, 4799–4829 RSC.
- B. del Rosal, E. Ximendes, U. Rocha and D. Jaque, Adv. Opt. Mater., 2017, 5, 1600508 CrossRef.
- C. D. S. Brites, A. Millán and L. D. D. Carlos, Handb. Phys. Chem. Rare Earths, 2016, 49, 339–427 CAS.
- A. Bednarkiewicz, L. Marciniak, L. D. D. Carlos and D. Jaque, Nanoscale, 2020, 12, 14405–14421 RSC.
- A. Bednarkiewicz, D. Wawrzynczyk, A. Gagor, L. Kepinski, M. Kurnatowska, L. Krajczyk, M. Nyk, M. Samoc and W. Strek, Nanotechnology, 2012, 23, 145705 CrossRef CAS.
- J. Drabik, R. Kowalski and L. Marciniak, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- K. Trejgis, F. Tian, J. Li, A. Bednarkiewicz and L. Marciniak, Mater. Res. Bull., 2021, 139, 111288 CrossRef CAS.
- K. Trejgis, K. Ledwa, K. Maciejewska, L. Li and L. Marciniak, Sci. Rep., 2022, 12, 1–14 CrossRef.
- L. Marciniak, A. Bednarkiewicz and K. Elzbieciak, J. Mater. Chem. C, 2018, 6, 7568–7575 RSC.
- K. Trejgis, K. Maciejewska, A. Bednarkiewicz and L. Marciniak, ACS Appl. Nano Mater., 2020, 3, 4818–4825 CrossRef CAS.
- K. Trejgis, R. Lisiecki, A. Bednarkiewicz and L. Marciniak, J. Lumin., 2020, 224, 117295 CrossRef CAS.
- J. Drabik, K. Ledwa and Ł. Marciniak, Nanomaterials, 2020, 10, 1333 CrossRef CAS.
- K. Trejgis, R. Lisiecki, A. Bednarkiewicz and L. Marciniak, J. Lumin., 2020, 224, 117295 CrossRef CAS.
- J. Drabik and L. Marciniak, ACS Appl. Nano Mater., 2020, 3, 3798–3806 CrossRef CAS.
- K. Trejgis, A. Bednarkiewicz and Ł. Marciniak, Nanoscale, 2020, 12, 4667–4675 RSC.
- A. Bednarkiewicz, K. Trejgis, J. Drabik, A. Kowalczyk and L. Marciniak, ACS Appl. Mater. Interfaces, 2017, 9(49), 43081–43089 CrossRef CAS.
- N. Fardian-Melamed, A. Skripka, C. Lee, B. Ursprung, T. Darlington, A. Teitelboim, X. Qi, M. Wang, J. Gerton, B. E. Cohen, E. M. Chan and P. J. Schuck, Nature, DOI:10.1038/s41586-41024-08221-41582, preprint available at https://arxiv.org/abs/42404.02026.
- A. Skripka, Z. Zhang, X. Qi, B. Ursprung, P. Ercius, B. E. Cohen, P. J. Schuck, D. Jaque and E. M. ChanNat. Photonics DOI:10.1038/s41566-024-01577-x, preprint available at https://arxiv.org/abs/2403.04098.
- I. V. Dyakonov, I. A. Pogorelov, I. B. Bobrov, A. A. Kalinkin, S. S. Straupe, S. P. Kulik, P. V. Dyakonov and S. A. Evlashin, Phys. Rev. Appl., 2018, 10, 044048 CrossRef CAS.
- C. Lee and P. J. Schuck, Annu. Rev. Phys. Chem., 2023, 74, 415–438 CrossRef CAS PubMed.
- S. Zanella, M. A. Hernández-Rodríguez, L. Fu, L. D. Carlos, R. A. S. Ferreira, C. D. S. Brites, S. Zanella, M. A. Hernández-Rodríguez, L. Fu, L. D. Carlos, R. A. S. Ferreira and C. D. S. Brites, Adv. Opt. Mater., 2022, 2200138 CrossRef CAS.
- H. Bian, X. Qin, Y. Wu, Z. Yi, S. Liu, Y. Wang, C. D. S. Brites, L. D. Carlos and X. Liu, Adv. Mater., 2022, 34, 2101895 CrossRef CAS.
- Y. Wang, B. Liu, L. Ding, C. Chen, X. Shan, D. Wang, M. Tian, J. Song, Z. Zheng, X. Xu, X. Zhong and F. Wang, Laser Photonics Rev., 2024, 2400746 CrossRef CAS.
- S. Fischer, N. D. Bronstein, J. K. Swabeck, E. M. Chan and A. P. Alivisatos, Nano Lett., 2016, 16, 7241–7247 CrossRef CAS.
- A. Gnach and A. Bednarkiewicz, Nano Today, 2012, 7, 532–563 CrossRef CAS.
- U. Kostiv, Z. Farka, M. J. Mickert, H. H. Gorris, N. Velychkivska, O. Pop-Georgievski, M. Pastucha, E. Odstrčilíková, P. Skládal and D. Horák, Biomacromolecules, 2020, 21, 4502–4513 CrossRef CAS.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5 Search PubMed.
- H. H. Gorris and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 3584–3600 CrossRef CAS PubMed.
- E. M. Chan, G. Han, J. D. Goldberg, D. J. Gargas, A. D. Ostrowski, P. J. Schuck, B. E. Cohen and D. J. Milliron, Nano Lett., 2012, 12, 3839–3845 CrossRef CAS.
- G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, S. Li, Y. Zhao, G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, Y. Zhao and S. Li, Adv. Mater., 2012, 24, 1226–1231 CrossRef CAS PubMed.
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2014, 8, 32–36 CrossRef CAS.
- H. Chen, Z. Jiang, H. Hu, B. Kang, B. Zhang, X. Mi, L. Guo, C. Zhang, J. Li, J. Lu, L. Yan, Z. Fu, Z. Zhang, H. Zheng and H. Xu, Nat. Photonics, 2022, 16, 651–657 CrossRef CAS.
- S. Schietinger, T. Aichele, H. Q. Wang, T. Nann and O. Benson, Nano Lett., 2010, 10, 134–138 CrossRef CAS.
- S. N. Sanders, T. H. Schloemer, M. K. Gangishetty, D. Anderson, M. Seitz, A. O. Gallegos, R. C. Stokes and D. N. Congreve, Nature, 2022, 604, 474–478 CrossRef CAS PubMed.
- E. Z. Xu, C. Lee, S. D. Pritzl, A. S. Chen, T. Lohmueller, B. E. Cohen, E. M. Chan and P. J. Schuck, Opt. Mater.: X, 2021, 12, 100099 CAS.
- S. H. Nam, Y. M. Bae, Y. Il Park, J. H. Kim, H. M. Kim, J. S. Choi, K. T. Lee, T. Hyeon and Y. D. Suh, Angew. Chem., Int. Ed., 2011, 50, 6093–6097 CrossRef CAS.
- B. Tian, A. Fernandez-Bravo, H. Najafiaghdam, N. A. Torquato, M. V. P. Altoe, A. Teitelboim, C. A. Tajon, Y. Tian, N. J. Borys, E. S. Barnard, M. Anwar, E. M. Chan, P. J. Schuck and B. E. Cohen, Nat. Commun., 2018, 9, 4–11 CrossRef.
- F. Ş. Boydağ, Sh. V. Mamedov, V. A. Alekperov and Y. Lenger Özcanli, Opt. Spectrosc., 2003, 95, 225–229 CrossRef.
- S. Bhuckory, E. Hemmer, Y.-T. Wu, A. Yahia-Ammar, F. Vetrone and N. Hildebrandt, Eur. J. Inorg. Chem., 2017, 5186–5195 CrossRef CAS.
- Y. Wang, K. Liu, X. Liu, K. Dohnalová, T. Gregorkiewicz, X. Kong, M. C. G. Aalders, W. J. Buma and H. Zhang, J. Phys. Chem. Lett., 2011, 2, 2083–2088 CrossRef CAS.
- L. Mattsson, K. D. Wegner, N. Hildebrandt and T. Soukka, RSC Adv., 2015, 5, 13270–13277 RSC.
- K. Kuningas, T. Rantanen, T. Ukonaho, T. Lövgren and T. Soukka, Anal. Chem., 2005, 77, 7348–7355 CrossRef CAS.
- N. Sirkka, A. Lyytikäinen, T. Savukoski and T. Soukka, Anal. Chim. Acta, 2016, 925, 82–87 CrossRef CAS PubMed.
- L. Liang, C. Wang, J. Chen, Q. J. Wang and X. Liu, Nat. Photonics, 2022, 16, 712–717 CrossRef CAS.
- J. S. Dam, P. Tidemand-Lichtenberg and C. Pedersen, Nat. Photonics, 2012, 6, 788–793 CrossRef CAS.
- A. Khalid, C. Xie, D. R. S. Cumming, I. G. Thayne, M. Sorel, M. Steer, M. Aziz and V. Pusino, Optica, 2017, 4, 1498–1502 CrossRef.
- I. M. Pavlovetc, K. Aleshire, G. V. Hartland and M. Kuno, Phys. Chem. Chem. Phys., 2020, 22, 4313–4325 RSC.
- W. Chen, P. Roelli, H. Hu, S. Verlekar, S. P. Amirtharaj, A. I. Barreda, T. J. Kippenberg, M. Kovylina, E. Verhagen, A. Martínez and C. Galland, Science, 2021, 374, 1264–1267 CrossRef CAS.
- R. Gordon, Science, 2021, 374, 1201–1202 CrossRef CAS PubMed.
- N. Bloembergen, Phys. Rev. Lett., 1959, 2, 84 CrossRef CAS.
- D. B. Gatch, W. M. Dennis and W. M. Yen, Appl. Opt., 2003, 42, 615–620 CrossRef CAS.
- G. Temporão, S. Tanzilli, H. Zbinden, N. Gisin, T. Aellen, M. Giovannini and J. Faist, Opt. Lett., 2006, 31, 1094–1096 CrossRef.
- H. C. Liu, C. Y. Song, E. Dupont, P. Poole, P. H. Wilson, B. J. Robinson and D. A. Thompson, Electron. Lett., 1999, 35, 2055–2056 CrossRef CAS.
- P. Roelli, D. Martin-Cano, T. J. Kippenberg and C. Galland, Phys. Rev. X, 2020, 10, 031057 CAS.
- A. Xomalis, X. Zheng, R. Chikkaraddy, Z. Koczor-Benda, E. Miele, E. Rosta, G. A. E. Vandenbosch, A. Martínez and J. J. Baumberg, Science, 2021, 374, 1268–1271 CrossRef CAS PubMed.
- K. Ueno, E. G. Camargo, T. Katsumata, H. Goto, N. Kuze, Y. Kangawa and K. Kakimoto, Jpn J. Appl. Phys., 2013, 52, 092202 CrossRef.
- P. Goldner and F. Pellé, Opt. Mater., 1996, 5, 239–249 CrossRef CAS.
- M. E. Koch, A. W. Kueny and W. E. Case, Appl. Phys. Lett., 1990, 56, 1083–1085 CrossRef CAS.
- T. Hebert, R. Wannemacher, R. M. MacFarlane and W. Lenth, Appl. Phys. Lett., 1992, 60, 2592–2594 CrossRef CAS.
- T. Sandrock, E. Heumann, G. Huber and B. H. T. Chai, Advanced Solid State Lasers (1996), paper PM1, The Optical Society, 2017, p. PM1 Search PubMed.
- H. Zhu, X. Chen, L. M. Jin, Q. J. Wang, F. Wang and S. F. Yu, ACS Nano, 2013, 7, 11420–11426 CrossRef CAS PubMed.
- T. Wang, H. Yu, C. K. Siu, J. Qiu, X. Xu and S. F. Yu, ACS Photonics, 2017, 4, 1539–1543 CrossRef CAS.
- T. Wang, C. K. Siu, H. Yu, Y. Wang, S. Li, W. Lu, J. Hao, H. Liu, J. H. Teng, D. Y. Lei, X. Xu and S. F. Yu, Inorg. Chem., 2018, 57, 8200–8204 CrossRef CAS PubMed.
- P. Molina, E. Yraola, M. O. Ramírez, C. Tserkezis, J. L. Plaza, J. Aizpurua, J. Bravo-Abad and L. E. Bausá, Nano Lett., 2016, 16, 895–899 CrossRef CAS PubMed.
- Z. Chen, G. Dong, G. Barillaro, J. Qiu and Z. Yang, Prog. Mater. Sci., 2021, 121, 100814 CrossRef CAS.
- A. Fernandez-Bravo, K. Yao, E. S. Barnard, N. J. Borys, E. S. Levy, B. Tian, C. A. Tajon, L. Moretti, M. V. Altoe, S. Aloni, K. Beketayev, F. Scotognella, B. E. Cohen, E. M. Chan, P. J. Schuck, B. Tian, C. A. Tajon, L. Moretti, M. V. Altoe, S. Aloni, K. Beketayev, F. Scotognella, B. E. Cohen, E. M. Chan and P. J. Schuck, Nat. Nanotechnol., 2018, 13, 1 CrossRef.
- Y. Liu, A. Teitelboim, A. Fernandez-Bravo, K. Yao, M. V. P. Altoe, S. Aloni, C. Zhang, B. E. Cohen, P. J. Schuck and E. M. Chan, ACS Nano, 2020, 14, 1508–1519 CrossRef CAS PubMed.
- A. Fernandez-Bravo, D. Wang, E. S. Barnard, A. Teitelboim, C. Tajon, J. Guan, G. C. Schatz, B. E. Cohen, E. M. Chan, P. J. Schuck and T. W. Odom, Nat. Mater., 2019, 18, 1172–1176 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Overview of PA in various lanthanide ions, comparison table of PA In nano- and micromaterials, comparison table of sub-diffraction imaging with PA nanoparticles. See DOI: https://doi.org/10.1039/d4cs00177j |
| This journal is © The Royal Society of Chemistry 2025 |