
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimulation of time-resolved site-selective X-ray spectroscopy tracing nonadiabatic dynamics in meta-methylbenzophenone
Lorenzo
Restaino†
 ,
Thomas
Schnappinger
and
Markus
Kowalewski
*
,
Thomas
Schnappinger
and
Markus
Kowalewski
*
Department of Physics, Stockholm University, Albanova University Centre, SE-106 91 Stockholm, Sweden. E-mail: markus.kowalewski@fysik.su.se
First published on 9th October 2025
Abstract
Benzophenone and its derivatives are important diaryl ketone building blocks that have applications ranging from UV blockers to organic optoelectronics. This class of substances exhibits efficient intersystem crossing processes that make them a popular choice. However, the ultrafast internal conversion processes that precede the intersystem crossing are less frequently discussed in the literature. This work provides a comprehensive theoretical investigation of these nonadiabatic relaxation events in meta-methylbenzophenone utilizing spectroscopy techniques. Based on a full quantum mechanical description of the nonadiabatic dynamics, we simulate both time-dependent X-ray absorption and off-resonant-stimulated X-ray Raman spectra at the oxygen K-edge. We show that the use of time-resolved X-ray spectroscopy allows for a selective probing of the first singlet excited state by filtering out signals from other excited states with different electronic character. The element sensitivity of oxygen core level spectroscopy restricts the sensitivity to nonadiabatic processes localized at the carbonyl double bond.
I. Introduction
Benzophenone (BP) and its derivatives are aromatic ketones with a rich photochemistry with respect to ultraviolet (UV) excitation. This class of molecules is characterized by an efficient intersystem crossing between the lowest singlet excited state, S1, and several triplet states, which is exploited in many technological applications1–4 and also used in biological contexts.5,6 In our previous work,7 we investigated the nonadiabatic dynamics of the S1 state in BP and its alkyl derivative meta-methyl benzophenone (m-BP), upon UV excitation in nearly degenerate electronic states, S2 and S3. This internal conversion mechanism is an experimentally realistic route to populate S1, as direct transfer of the population from the ground state to S1 is very inefficient.8–11 We used trajectory surface hopping (TSH) dynamics and time-resolved photoelectron spectroscopy to trace nonadiabatic relaxation processes via two conical intersections (CoIns) in the first 500 fs. Mixed quantum-classical methods such as TSH can capture all nuclear degrees of freedom of a molecule, but treat the nuclei as classical particles and do not provide access to vibronic coherences in the molecular system. By analyzing the trajectories, we identified two main coordinates that drive the nonadiabatic dynamics for the initial 120 fs, before additional coordinates become important governing the relaxation processes on the S1 potential energy surfaces (PESs). These coordinates are q1, which corresponds to a puckering motion of the aryl ring, and q2, which corresponds to a dihedral angle motion between aryl rings. We used these two coordinates to construct 2D potential energy surfaces for m-BP. Fig. S1 of the SI provides a visual representation of the two coordinates. The additional methyl group in m-BP breaks the C2 symmetry and lifts the degeneracy between S3 and S2, without fundamentally changing the photochemistry of benzophenone. By including the first four valence states in the full quantum dynamics (QD) description, we were able to qualitatively reproduce the full-dimensional TSH simulations, with smaller quantitative discrepancies arising from the different electronic structure methods employed in the calculations and the reduced subspace used. The QD simulations yield direct access to the vibronic coherences12,13 generated in the system by the nonadiabatic transitions.In this manuscript, the same reduced coordinate space is used to construct the 2D PESs of six core-excited states at the oxygen K-edge. The S1/S2 internal conversion and the vibronic coherences generated at CoIns are probed using state-of-the-art time-resolved X-ray techniques. Our framework employs grid-based quantum dynamics within a reduced nuclear subspace, followed by the calculation of spectra using time-dependent perturbation theory. Another well-established approach to simulating the dynamics of coupled electronic states is the multiconfiguration time-dependent Hartree (MCTDH) method.14 This strategy has been successfully applied in the calculation of nonlinear optical spectra,15 UV/VIS spectra,16 and X-ray spectra,17,18 demonstrating its versatility for studying ultrafast nonadiabatic effects. As an alternative route, transient X-ray spectra can also be simulated from a mixed quantum-classical description of the nonadibatic dynamics.19,20
Extreme ultraviolet (XUV) and soft X-ray pulses have the necessary time and frequency resolution to directly detect the passage of a wave packet through a CoIn.21–30 Moreover, the atomic selectivity of X-rays makes these techniques very advantageous because they are sensitive to the properties of the S1 state, rather than S2 and S3. This occurs because the S1 state of BP, and many of its derivatives, has nπ* character, with transitions localized on the carbonyl double bond. The higher excited states S2 and S3 have ππ* character, with transition densities delocalized over the aryl rings. We employ the two techniques time-resolved X-ray absorption spectroscopy (tr-XAS) and transient redistribution of ultrafast electronic coherences in attosecond Raman signals (TRUECARS)31 which offer a comprehensive picture of the nonadiabatic relaxation processes by providing complementary information.32 With tr-XAS being sensitive to transition dipole moments (TDMs), we can monitor the population dynamics taking place between S2/S3 and the S1 state. Since TRUECARS is probing the polarizability of the molecular system, we have direct access to the vibronic coherence generated by the involved CoIns.
II. Computational details
All calculations, mentioned in the following, were performed in a reproducible computing environment using the Nix package manager together with NixOS-QChem33 (commit ee8700758).A. Ab initio level of theory
The two-dimensional PESs and the corresponding TDMs for 6 core-hole states at the oxygen K-edge of m-BP were calculated utilizing the perturbative approximate variant of the combined density functional theory and multireference configuration interaction method DFT/MRCI(2)34–36 with the GRACI program,37 using the QTP1738 xc-functional, the QE8 Hamiltonian36 and the 6-311G* basis set, within the core–valence separation (CVS) approximation.39–41 The chosen reaction coordinates are: q1, which describes the puckering motion of one of the aryl rings, and q2, which corresponds to a change in the dihedral angle between the two aryl rings. The benchmarking and validation of the level of theory can be found in ref. 7. A visual representation of the two reaction coordinates is available in the SI in Fig. S1. The reference space for the DFT/MRCI calculations is shown in Fig. S2 of the SI. The raw data obtained from the quantum chemistry calculations were interpolated with polyharmonic splines42 onto a fine grid (N1 = N2 = 128 points), for the wave packet dynamics.The X-ray absorption energies of m-BP obtained at the Franck–Condon (FC) were broadened with a Gaussian function (0.4 eV full width at half maximum (FWHM)) to generate a static spectrum. To benchmark the use of CVS-DFT/MRCI(2) for m-BP, we calculated the static X-ray absorption spectrum of benzophenone using XMS-RASPT2 and CVS-DFT/MRCI(2). The comparison of the two spectra and details of the XMS-RASPT2 calculations are reported in Fig. S7 of the SI.
B. Quantum dynamics simulations
To simulate the wave packet dynamics of the photoexcited m-BP, we solved the time-dependent (non-relativistic) Schrödinger equation numerically with the Fourier method,43–45 using the software package QDng.46 The quantum dynamics was performed with quasi-diabatic states.7,47 The corresponding Hamiltonian in the internal coordinates q1, q2 reads: | (1) |
![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) is the kinetic energy operator,
is the kinetic energy operator, ![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) S0, S1, S2 and S3 are the diabatic potentials for the states S0, S1, S2 and S3, respectively. The diabatic couplings are given by S1S2 and S2S3. The interaction with the pump pulse is explicitly included in the Hamiltonian and is described by the TDMs
S0, S1, S2 and S3 are the diabatic potentials for the states S0, S1, S2 and S3, respectively. The diabatic couplings are given by S1S2 and S2S3. The interaction with the pump pulse is explicitly included in the Hamiltonian and is described by the TDMs ![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) S0S2 and S0S3 and the classical electric field εp(t). We employed a Gaussian pulse for the classical electric field
S0S2 and S0S3 and the classical electric field εp(t). We employed a Gaussian pulse for the classical electric field| εp(t) = E0e−t2/2σ2·cos(ωpt), | (2) |
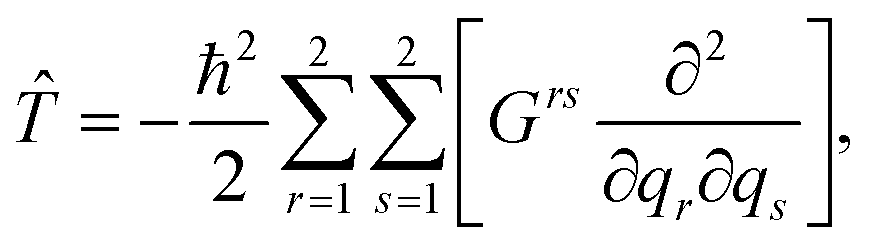 | (3) |
The dynamics in the core-hole states required for tr-XAS is treated by means of time-dependent perturbation theory and the direct propagation of the wave function approach.49 The core-hole lifetime at the oxygen K-edge is approximately 4 fs,50–52 therefore, the wave packet was propagated up to 6 fs on the core-hole state to remain safely beyond the lifetime.
III. Spectroscopic signals
The time- and frequency-resolved tr-XAS signal, described by the diagrams in Fig. 1, is given by ref. 31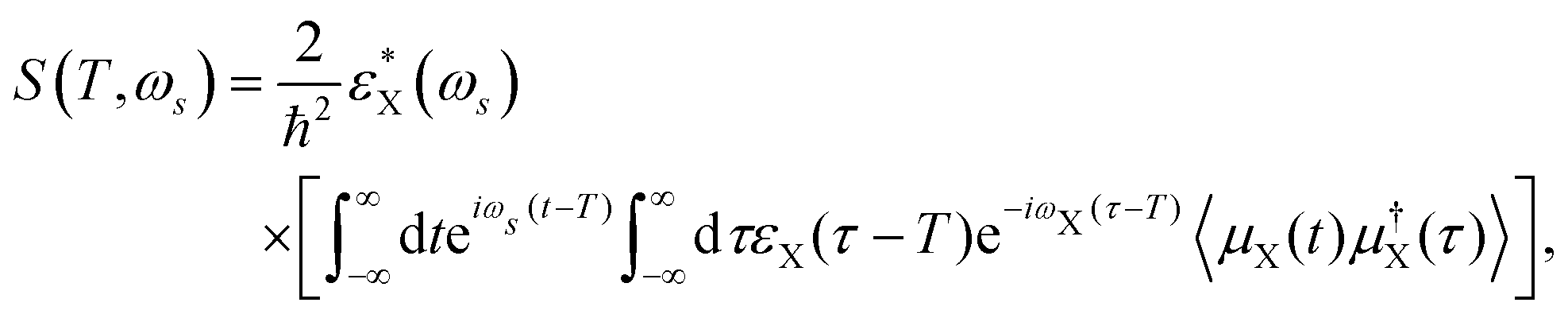 | (4) |
 | ||
| Fig. 1 The loop diagrams (a) and (b) and the corresponding level diagram (c) for the tr-XAS technique. | ||
TRUECARS is an off-resonant X-ray stimulated Raman technique, which is sensitive to coherences rather than populations. This method employs two X-ray pulses, ε1 and ε0, which interact simultaneously with the molecule to drive stimulated Raman transitions between electronic states, as shown in Fig. 2. The time and frequency resolved signal reads as
 | (5) |
![[small alpha, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e101.gif) (t)〉 is the matter correlation function, which consists of the time-dependent expectation value of the X-ray polarizability. The SI provides details on the calculation of the X-ray polarizability tensor alongside a derivation of the correlation function.
(t)〉 is the matter correlation function, which consists of the time-dependent expectation value of the X-ray polarizability. The SI provides details on the calculation of the X-ray polarizability tensor alongside a derivation of the correlation function.
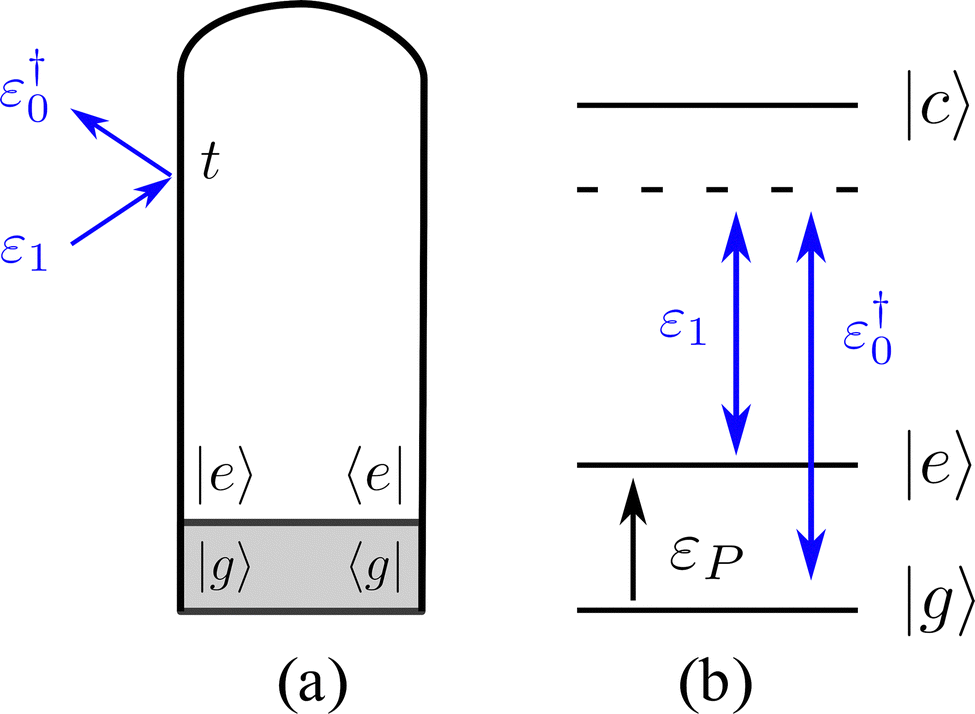 | ||
| Fig. 2 The loop diagram (a) and the corresponding level diagram (b) for the TRUECARS technique. | ||
IV. Results
A. X-ray absorption spectrum
We start by discussing the static X-ray absorption spectrum of m-BP, shown in panel (a) of Fig. 3 with the most important natural transition orbitals (NTO) in panel (b). To the best of our knowledge, no oxygen K-edge spectrum is available for gas-phase BP or m-BP in the literature. Therefore, we used the oxygen K-edge spectrum of acetone as a reference. Ref. 54 reports the transition from oxygen 1s to the carbonyl π* at 531.38 eV. Based on this π* transition, all spectra shown in this paper are shifted by 2.5 eV to account for the missing scalar relativistic effects and parts of the electron correlation. | ||
| Fig. 3 Static X-ray absorption spectrum of m-BP and corresponding natural transition orbitals (NTO); (a) X-ray absorption spectra from four valence states at the FC point. The peaks are color coded according to their initial state. (b) Particle NTO corresponding to a specific peak in the spectrum. The hole NTO is always the oxygen 1s orbital. An isovalue of 0.05 was used to plot the orbitals. | ||
The absorption spectrum of the ground state is characterized by two bright transitions: S0 → C0 at 531.4 eV, labeled (I); and S0 → C4 at 536.1 eV, labeled (II). At the FC point, the peak (I) can be identified as the excitation of an electron from the oxygen 1s to the π* antibonding orbital of the carbonyl group, as shown by the corresponding particle NTO in Fig. 3(b). Peak (II) at approximately 536 eV is much weaker and can be identified as the excitation of an electron into a π* orbital delocalized over the entire molecule.
The absorption spectrum of S1 shows a single peak at 527.6 eV, S1 → C0 labeled (III) in Fig. 3. This signal corresponds to the excitation of a core electron into an oxygen lone pair. In contrast, the spectra of S2 and S3 are characterized by few weaker signals clustered around 531.4 eV. All of these weaker peaks correspond to transitions into delocalized π orbitals, which are single-occupied in S2 and S3.
The diabatic populations of S1, S2 and S3 resulting from full QD simulations are plotted against the pump–probe delay in the upper panel of Fig. 4. The pump-pulse promotes approximately 10% of the ground-state population into a coherent superposition of S2 and S3. After photoexcitation, the population in S3 is transferred to S2via internal conversion. Around 35 fs, the part of the wave packet propagating on the S2 PES reaches the S2/S1 CoIn and branches off, generating a superposition of these two electronic states. It is challenging to detect the change in the diabatic population of S1 from Fig. 4(a). Fig. S9 of the SI shows the temporal evolution of the state S1 alone, confirming that there is a corresponding population transfer due to the CoIn.
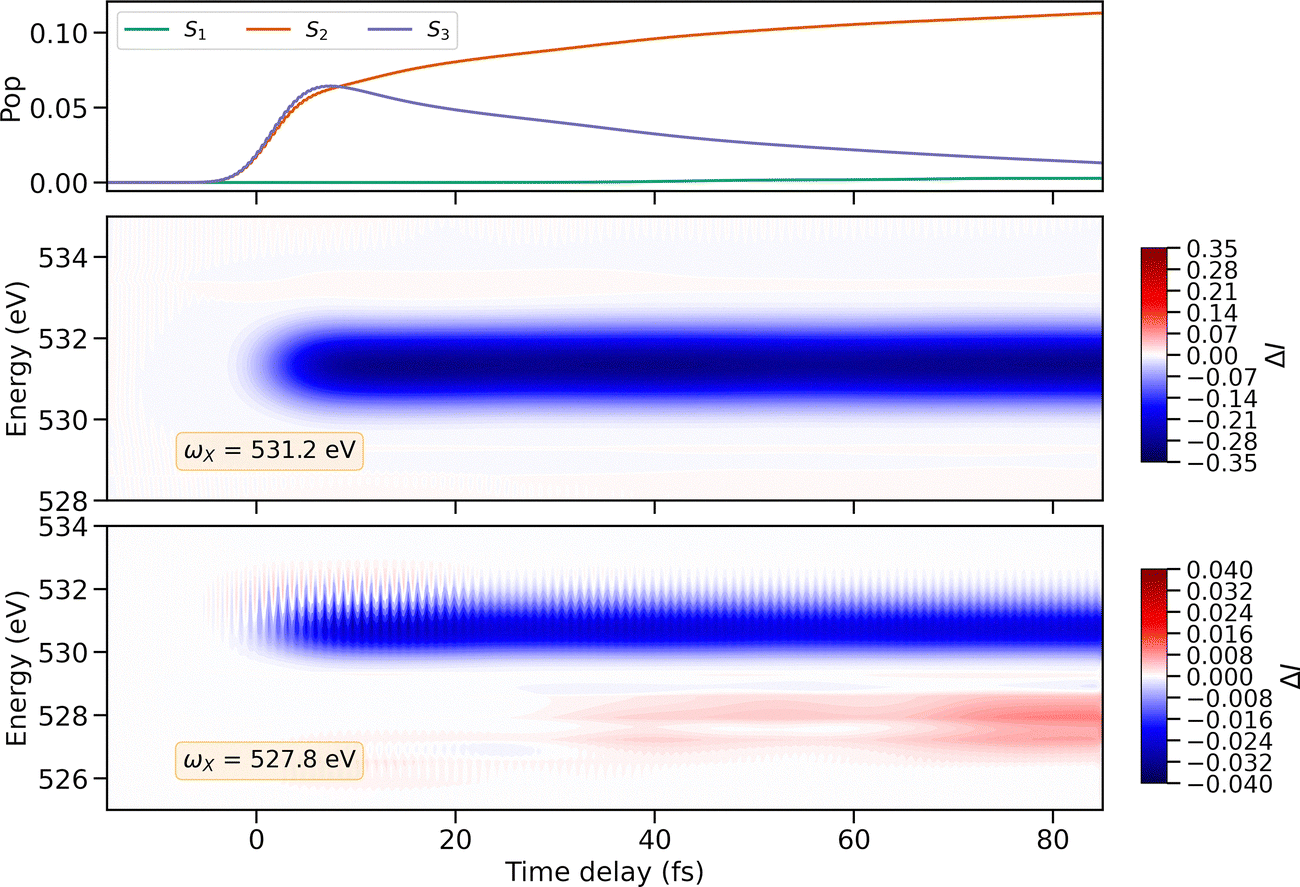 | ||
| Fig. 4 Time-resolved X-ray absorption spectrum of m-BP. Top panel: Diabatic populations of three valence excited states as a function of the pump–probe time delay. Mid panel: Difference spectrum calculated using a Gaussian probe pulse centered at 531.2 eV, with a FWHM of 1.8 eV. Bottom panel: Difference spectrum calculated using a Gaussian probe pulse centered at 527.8 eV, with a FWHM of 1.8 eV. The energy axis was shifted by 2.5 eV. | ||
The middle and lower panels of Fig. 4 present two time-resolved X-ray absorption spectra for m-BP, simulated at two different probe center frequencies ωX. For ωX = 531.2 eV, the probe is resonant with the absorption signal from S0, while for ωX = 527.8 eV, the probe is resonant with the absorption signal from the excited state S1. To emphasize the impact of the pump on the X-ray signal, the spectrum is not displayed directly as calculated from eqn (4). Instead, the spectrum without the pump–pulse (Sref(ωs)) is subtracted from the complete pump–probe spectrum (S(T, ωs)):
| ΔI(T, ωs) = S(T, ωs) − Sref(ωs). | (6) |
In the bottom panel of Fig. 4, the intensity of the ground-state bleach signal shows distinct oscillations with a constant period, but with an amplitude damping over time. These intensity fluctuations are visible because the probe is not resonant with the S0 → C0 transition. However, when the X-ray probe is resonant with the ground-state bleach signal, as in the middle panel of Fig. 4, the fluctuations are concealed. Fig. S10(b) of the SI displays the Fourier transform of the time trace of the ground-state signal, taken at ħωs = 531.5 eV. The Fourier spectrum shows a cluster of peaks around 4.5 eV, corresponding to a period of 0.9 fs. The oscillation pattern is consistent with the temporal behavior of the electronic coherence, ρ, induced by the pump pulse between S0 and the excited states S2 and S3. For comparison see Fig. S11 of the SI, where the real parts of ρS0S2 and ρS0S3 are shown together with their Fourier transforms. Although ρS0S2 decays exponentially, the electronic coherence between S0 and S3 survives much longer, since both the ground state and the S3 wave packets remain confined within the FC region. This can be seen in Fig. S12 of the SI, which shows the time-dependent expectation value of the position operator for all electronic states. Note that the limited dimensionality of our nuclear subspace leads to an overestimation of the electronic coherences in the system, which could decay more rapidly if all nuclear degrees of freedom (DOF) were accessible.55
B. TRUECARS spectrum
The tr-XAS signal at the oxygen K-edge enables a clear, site-selective mapping of the S2/S1 CoIn crossing event by observing the appearance of the S1 band in the spectrum shown in Fig. 4. This mapping is based on the population in the S1 state. In contrast, the TRUECARS signal provides information about the electronic superposition present in the system due to interaction with the pump pulse and the subsequent nonadiabatic dynamics. The 8 fs UV pump pulse naturally induces vibrational coherences in the electronic valence states by launching a nuclear wave packet consisting of a linear combination of vibrational eigenfunctions. The pump pulse also creates a coherent superposition of S2 and S3 that is visible in the TRUECARS spectrum. Furthermore, as discussed in Fig. 4, after photoexcitation there is long-lived electronic coherence between S0 and S3. In addition to the vibronic coherences directly induced by the pump, there are vibronic coherences generated by the branching of the wave packet at the CoIns. The top panel of Fig. 5 shows the temporal evolution of the magnitude of the electronic coherences between S1 and S2. The electronic coherence originating from the S2/S3 CoIn is not visible, since it is completely concealed by the one induced by the UV pump pulse. Moreover, the nuclear wave packet overlap between the two states decays rapidly, as the wave packet on S2 quickly follows the gradient toward S1/S2 CoIn, while the wave packet on S3 remains at the FC region. The S1/S2 electronic coherence is generated almost solely by the bifurcation of the wave packet at the respective CoIn, which begins to build up around 30 fs.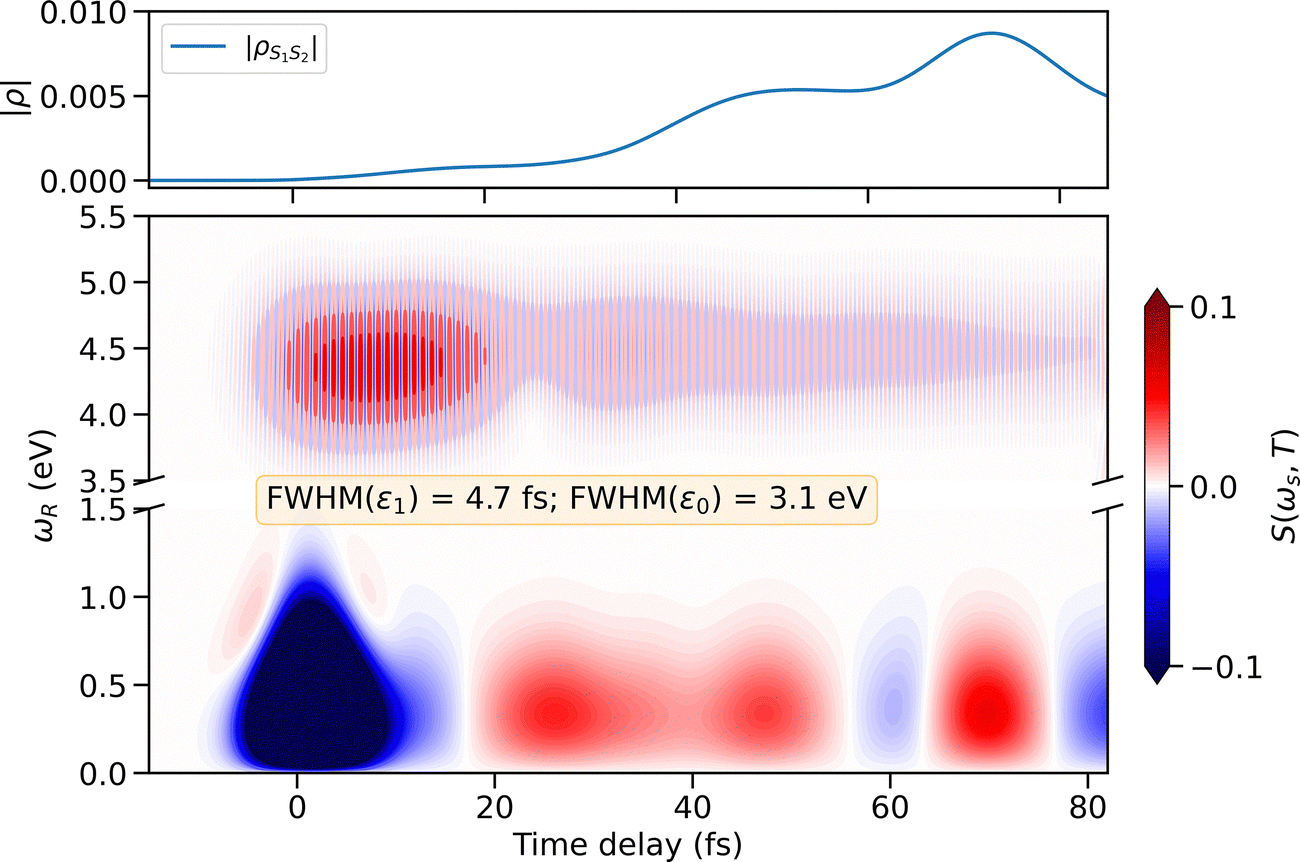 | ||
| Fig. 5 TRUECARS spectrum of m-BP. Top panel: Temporal evolution of the magnitude of the electronic coherences between S1 and S2. Bottom panel: TRUECARS spectrum calculated using a Gaussian probe pulse with a temporal FWHM of 4.7 fs for ε1, and an energy bandwidth (FWHM) of 3.1 eV for ε0. The X-ray polarizability was calculated at ω = 520.0 eV. A broken Raman shift axis is used to highlight two distinct energy regions (0–1.5 eV and 3.5–5.5 eV), excluding an interval where no signal is observed. The total spectrum is normalized. | ||
Fig. 5 shows the TRUECARS spectrum of m-BP calculated using the diagonal components of the X-ray polarizability tensor. Detailed calculations of the polarizability tensor and the resulting signal are discussed in the SI. The αyy is the dominant component. The Raman shift axis is divided into two energy windows: 0–1.5 eV, which contains information about vibronic coherences in the ground and excited states. The 3.5–5.5 eV region contains only information about the electronic coherences involving the electronic ground state S0 and the S2/S3 excited states. These coherences are generated solely by the pump pulse. In the low-energy region at time T = 0 fs, the largest contribution to the signal comes from the vibrational coherence generated by the short pump pulse. Note that this contribution is enhanced during the pump laser field is on, by temporarily dressing the vibrational states. Thus, the first 5 fs of the signal should be excluded from interpretation. These vibrational coherences decrease shortly after the pump pulse is turned off. Then, as the wave packet approaches the S2/S1 CoIn, the TRUECARS signal increases in intensity, with temporal Stokes and anti-Stokes oscillations now visible in the spectrum.
The TRUECARS signal is sensitive to vibronic coherences associated with S0 or S1 states, but is less sensitive to contributions from the S2 and S3 states. This can be understood as follows. The frequency dependent X-ray polarizability—the key physical observable—is derived by slightly detuning below the valence to oxygen core-hole transition. Thus, the TDMs of the respective core-hole transitions dominate the polarizability. This effectively gives less weight to the contributions from S2 and S3 due to their delocalized ππ* character. Fig. S8 in the SI showcases the state sensitivity of the TRUECARS signal by comparing the contributions of each state to the time-dependent expectation value of the X-ray polarizability.
The TRUECARS signal contains information about the energy separations in the Raman shift as well as in the time delay. To extract more detailed information from the oscillation period visible in the spectrum, a temporal trace, S(T), was chosen at ωR = 0.3 eV and its Wigner spectrogram56–58 was calculated (details on the calculation of the Wigner spectrogram and a spectrogram for ωR = 4.4 eV can be found in the SI Section S10). Fig. 6 shows the calculated Wigner spectrogram (bottom panel) and the time trace used to create the spectrogram (top panel). In addition, the time-dependent energy separation between the electronic states is shown. Note, that these are averages and the vibronic wave packet spans a much broader energy range. At ≈20 fs, as the molecule approaches the S2/S1 CoIn, a 0.3 eV broad band begins to build up. This band peaks in intensity at ≈40 fs, when the energy splitting is between S2 and S1 minimal, and then decays rapidly. At around 78 fs the wave packet returns towards the CoIn and another, albeit weak, recurrence can be observed.
 | ||
| Fig. 6 Wigner spectrogram of a selected trace of the TRUECARS signal. Top panel: Temporal trace of the signal, taken at ωR = 0.3 eV; bottom panel: absolute value of the normalized Wigner spectrogram of the top panel. Solid black line is an estimate of the time-dependent adiabatic energy gap between S1 and S2. Solid gray line is an estimate of the time-dependent adiabatic energy gap between S2 and S3. | ||
The spatial overlap between nuclear wave packets on S3 and S2 decays rapidly within the first 20 fs (see SI Fig. S12) and thus no contribution from this state pair is observed. Moreover, the pure vibrational coherences from the S2 or S3 is also suppressed in the Wigner spectrum. A comparison with Fig. 3(a) confirms this finding: using a X-ray Raman scheme that has small detuning with respect to the oxygen 1s core transition, makes the measurement more sensitive to the S0 and S1 states. The contributions from the S2 and S3 states to the polarizability are much smaller in comparison.
Even tough the electronic energy separation can not be read off directly from the Wigner spectrum, there is a clear indication of the appearance of the S1/S2 CoIn. Note, that at the CoIn the vibrational and electronic states are heavily mixed, creating a broad band of vibronic states. A comparison with only off-diagonal polarizabilties, i.e. only the electronic contributions to the signal, shows a pattern similar to Fig. 6 (see SI Section S11).
V. Conclusions
In summary, we presented simulated time-resolved spectra of the ultrafast nonadiabatic relaxation events that take place in the singlet manifold of meta-methyl benzophenone, triggered by an 8-fs UV pump pulse. Time-resolved X-ray absorption spectroscopy at the oxygen K-edge can unequivocally trace the crossing of the conical intersection between S2 and S1, by detecting the S1 band in the signal. The localized nπ* character of the carbonyl bond makes the signal sensitive to the appearance of S1 state population. Due to the delocalized ππ* character of the S2 and S3 states, the contribution of these states to the tr-XAS signal is strongly suppressed. State-of-the-art time-resolved X-ray experiments have access to sub-20-fs pump pulses both in the IR and UV, and in general even sub-5-fs pump pulses are experimentally realized to trigger wave packet dynamics in the excited state.59–65 Therefore, the transient oxygen K-edge absorption spectrum of m-BP in the gas phase could be measured with state-of-the-art light sources.The analysis of the TRUECARS signal shows that the atomistic selectivity may also be harnessed in an X-ray Raman scheme. While the experimental realization of TRUECARS has not yet been achieved, there has been notable progress in the past years in measuring conical intersection-induced vibronic coherences, both theoretically13,58 and experimentally.66,67 Here, we have chosen a photon energy for the probe lasers that is a few electron volts below the oxygen K-edge transition. The resulting spectrum shows an increased sensitivity to coherences, involving the S0 and S1 states, over the S2 and S3 states. The Wigner spectrogram of the TRUECARS shows clear indications of the S1/S2 CoIn, by highlighting the broad vibronic bands, that occur in its vicinity.
For both techniques, tr-XAS and TRUECARS, the spectra show a selectivity towards states that involve excitations that are localized on the carbonyl bond. X-ray techniques are powerful tools for unraveling complex relaxation and reaction pathways, due to their element sensitivity and site selectivity. In recent years, these techniques have been used to study molecular relaxation in various systems, including photorelaxation in uracil at the oxygen edge,68,69 proton motion in hydrogen bonds at the nitrogen edge,70 and CH activation through transition metals by probing the metal center.71 Our results for metamethyl benzophenone demonstrate that this technique can track nonadiabatic dynamics in aromatic ketones and may motivate future experiments applicable to a large class of similar aromatic systems.
Author contributions
Lorenzo Restaino: data curation (lead); investigation (lead); visualization (lead); writing – original draft (lead); writing – review & editing (equal). Thomas Schnappinger: conceptualization (equal); investigation (supporting); supervision (equal); writing – original draft (supporting); writing – review & editing (equal). Markus Kowalewski: conceptualization (equal); funding acquisition (lead); project administration (lead); supervision (equal); writing – original draft (supporting); writing – review & editing (equal).Conflicts of interest
There are no conflicts to declare.Data availability
Data supporting the findings of this study are available from the corresponding author on a reasonable request.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5cp03072b.
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 852286). Support from the Swedish Research Council (Grant No. VR 2022-05005 and VR 2024-04299) is acknowledged.References
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed
.
- S. Jhulki, S. Seth, A. Ghosh, T. J. Chow and J. N. Moorthy, ACS Appl. Mater. Interfaces, 2016, 8, 1527–1535 CrossRef CAS PubMed
- C.-X. Li, W.-W. Guo, B.-B. Xie and G. Cui, J. Chem. Phys., 2016, 145, 074308 CrossRef PubMed
- N. G. K. Wong, C. D. Rankine and C. E. H. Dessent, J. Phys. Chem. Lett., 2021, 12, 2831–2836 CrossRef CAS PubMed
- M. C. Cuquerella, V. Lhiaubet-Vallet, J. Cadet and M. A. Miranda, Acc. Chem. Res., 2012, 45, 1558–1570 CrossRef CAS PubMed
- E. Dumont, M. Wibowo, D. Roca-Sanjuán, M. Garavelli, X. Assfeld and A. Monari, J. Phys. Chem. Lett., 2015, 6, 576–580 CrossRef CAS PubMed
- L. Restaino, T. Schnappinger and M. Kowalewski, J. Chem. Phys., 2025, 162, 084301 CrossRef CAS PubMed
- B. K. Shah, M. A. J. Rodgers and D. C. Neckers, J. Phys. Chem. A, 2004, 108, 6087–6089 CrossRef CAS
- G. Spighi, M.-A. Gaveau, J.-M. Mestdagh, L. Poisson and B. Soep, Phys. Chem. Chem. Phys., 2014, 16, 9610–9618 RSC
- M. Marazzi, S. Mai, D. Roca-Sanjuán, M. G. Delcey, R. Lindh, L. González and A. Monari, J. Phys. Chem. Lett., 2016, 7, 622–626 CrossRef CAS PubMed
- L. Favero, G. Granucci and M. Persico, Phys. Chem. Chem. Phys., 2016, 18, 10499–10506 RSC
- D. Jadoun and M. Kowalewski, J. Phys. Chem. Lett., 2021, 12, 8103–8108 CrossRef CAS PubMed
- L. Restaino, D. Jadoun and M. Kowalewski, Struct Dyn, 2022, 9, 034101 CrossRef CAS PubMed
- M. Beck, Phys. Rep., 2000, 324, 1–105 CrossRef CAS
- F. Segatta, D. A. Ruiz, F. Aleotti, M. Yaghoubi, S. Mukamel, M. Garavelli, F. Santoro and A. Nenov, J. Chem. Theory Comput., 2023, 19, 2075–2091 CrossRef CAS PubMed
- Y. Liu, L. Martínez-Fernández, J. Cerezo, G. Prampolini, R. Improta and F. Santoro, Chem. Phys., 2018, 515, 452–463 CrossRef CAS
- M. Pápai, T. Rozgonyi, T. J. Penfold, M. M. Nielsen and K. B. Møller, J. Chem. Phys., 2019, 151, 104307 CrossRef PubMed
- F. Segatta, D. Aranda, F. Aleotti, F. Montorsi, S. Mukamel, M. Garavelli, F. Santoro and A. Nenov, J. Chem. Theory Comput., 2024, 20, 307–322 CrossRef CAS PubMed
- F. Rott, M. Reduzzi, T. Schnappinger, Y. Kobayashi, K. F. Chang, H. Timmers, D. M. Neumark, R. de Vivie-Riedle and S. R. Leone, Struct. Dyn, 2021, 8, 034104 CrossRef CAS PubMed
- D. Furlanetto and J. O. Richardson, J. Phys. Chem. Lett., 2025, 16, 6794–6800 CrossRef CAS PubMed
- M. C. E. Galbraith, S. Scheit, N. V. Golubev, G. Reitsma, N. Zhavoronkov, V. Despré, F. Lépine, A. I. Kuleff, M. J. J. Vrakking, O. Kornilov, H. Köppel and J. Mikosch, Nat. Commun., 2017, 8, 1018 CrossRef CAS PubMed
- T. J. A. Wolf, R. H. Myhre, J. P. Cryan, S. Coriani, R. J. Squibb, A. Battistoni, N. Berrah, C. Bostedt, P. Bucksbaum, G. Coslovich, R. Feifel, K. J. Gaffney, J. Grilj, T. J. Martinez, S. Miyabe, S. P. Moeller, M. Mucke, A. Natan, R. Obaid, T. Osipov, O. Plekan, S. Wang, H. Koch and M. Gühr, Nat. Commun., 2017, 8, 29 CrossRef CAS PubMed
- J. Yang, X. Zhu, T. Wolf, Z. Li, J. P. Nunes, R. Coffee, J. Cryan, M. Gühr, K. Hegazy, T. Heinz, K. Jobe, R. Li, X. Shen, T. Veccione, S. Weathersby, K. Wilkin, C. Yoneda, Q. Zheng, T. Martínez, M. Centurion and X. Wang, Science, 2018, 361, 64–67 CrossRef CAS PubMed
- S. P. Neville, M. Chergui, A. Stolow and M. S. Schuurman, Phys. Rev. Lett., 2018, 120, 243001 CrossRef PubMed
- N. Rohringer, Philos. Trans. R. Soc., A, 2019, 377, 20170471 CrossRef CAS PubMed
- K. Sun, W. Xie, L. Chen, W. Domcke and M. F. Gelin, J. Chem. Phys., 2020, 153, 174111 CrossRef CAS PubMed
- K. F. Chang, M. Reduzzi, H. Wang, S. M. Poullain, Y. Kobayashi, L. Barreau, D. Prendergast, D. M. Neumark and S. R. Leone, Nat. Commun., 2020, 11, 4042 CrossRef CAS PubMed
- K. S. Zinchenko, F. Ardana-Lamas, I. Seidu, S. P. Neville, J. van der Veen, V. U. Lanfaloni, M. S. Schuurman and H. J. Wörner, Science, 2021, 371, 489–494 CrossRef CAS PubMed
- Y. Nam, D. Keefer, A. Nenov, I. Conti, F. Aleotti, F. Segatta, J. Y. Lee, M. Garavelli and S. Mukamel, J. Phys. Chem. Lett., 2021, 12, 12300–12309 CrossRef CAS PubMed
- D. T. Matselyukh, F. Rott, T. Schnappinger, P. Zhang, Z. Li, J. O. Richardson, R. de Vivie-Riedle and H. J. Wörner, Nat. Commun., 2025, 16, 7211 CrossRef CAS PubMed
- M. Kowalewski, B. P. Fingerhut, K. E. Dorfman, K. Bennett and S. Mukamel, Chem. Rev., 2017, 117, 12165–12226 CrossRef CAS PubMed
- T. Schnappinger, D. Jadoun, M. Gudem and M. Kowalewski, Chem. Commun., 2022, 58, 12763–12781 RSC
- M. Kowalewski and P. Seeber, Int. J. Quantum Chem., 2022, 122, e26872 CrossRef CAS
- S. Grimme and M. Waletzke, J. Chem. Phys., 1999, 111, 5645–5655 CrossRef CAS
- S. P. Neville and M. S. Schuurman, J. Chem. Phys., 2022, 157, 164103 CrossRef CAS PubMed
- T. S. Costain, V. Ogden, S. P. Neville and M. S. Schuurman, J. Chem. Phys., 2024, 160, 224106 CrossRef CAS PubMed
-
S. Neville and M. Schuurman, GRaCI: General Reference Configuration Interaction, 2021, https://github.com/schuurman-group/graci Search PubMed
- Y. Jin and R. J. Bartlett, J. Chem. Phys., 2018, 149, 064111 CrossRef PubMed
- I. Seidu, S. P. Neville, M. Kleinschmidt, A. Heil, C. M. Marian and M. S. Schuurman, J. Chem. Phys., 2019, 151, 144104 CrossRef PubMed
- I. Seidu, S. P. Neville, M. Kleinschmidt, A. Heil, C. M. Marian and M. S. Schuurman, J. Chem. Phys., 2019, 151, 144104 CrossRef PubMed
- T. S. Costain, J. B. Rolston, S. P. Neville and M. S. Schuurman, J. Chem. Phys., 2024, 161, 114117 CrossRef CAS PubMed
- M. Kowalewski, E. Larsson and A. Heryudono, J. Chem. Phys., 2016, 145, 084104 CrossRef PubMed
- M. D. Feit, J. A. Fleck and A. Steiger, J. Comput. Phys., 1982, 47, 412–433 CrossRef CAS
- D. Kosloff and R. Kosloff, J. Comput. Phys., 1983, 52, 35–53 CrossRef CAS
-
D. J. Tannor, Introduction to quantum mechanics: a time-dependent perspective, 2007 Search PubMed
-
M. Kowalewski and R. de Vivie-Riedle, QDng: A Grid Based Molecular Quantum Dynamics Package, 2024, https://zenodo.org/records/10944497 Search PubMed
- S. P. Neville and M. S. Schuurman, J. Chem. Phys., 2024, 160, 234109 CrossRef CAS PubMed
- J. Stare and G. G. Balint-Kurti, J. Phys. Chem. A, 2003, 107, 7204–7214 CrossRef CAS
- M. Kowalewski and S. Mukamel, J. Chem. Phys., 2015, 143, 044117 CrossRef PubMed
- T. X. Carroll and T. D. Thomas, J. Chem. Phys., 1992, 97, 894–899 CrossRef CAS
- T. X. Carroll and T. D. Thomas, J. Chem. Phys., 1990, 92, 7171–7177 CrossRef CAS
- M. Neeb, J.-E. Rubensson, M. Biermann, W. Eberhardt, K. J. Randall, J. Feldhaus, A. L. D. Kilcoyne, A. M. Bradshaw, Z. Xu, P. D. Johnson and Y. Ma, Chem. Phys. Lett., 1993, 212, 205–210 CrossRef CAS
- M. Kowalewski, K. Bennett, K. E. Dorfman and S. Mukamel, Phys. Rev. Lett., 2015, 115, 193003 CrossRef PubMed
- K. C. Prince, R. Richter, M. de Simone, M. Alagia and M. Coreno, J. Phys. Chem. A, 2003, 107, 1955–1963 CrossRef CAS
- C. Arnold, O. Vendrell and R. Santra, Phys. Rev. A: At., Mol., Opt. Phys., 2017, 95, 033425 CrossRef
- D. Keefer, T. Schnappinger, R. de Vivie-Riedle and S. Mukamel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 24069–24075 CrossRef CAS PubMed
- S. M. Cavaletto, D. Keefer, J. R. Rouxel, F. Aleotti, F. Segatta, M. Garavelli and S. Mukamel, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2105046118 CrossRef CAS PubMed
- S. M. Cavaletto, D. Keefer and S. Mukamel, Phys. Rev. X, 2021, 11, 011029 CAS
- M. Kowalewski, S. Kahra, G. Leschhorn, T. Schätz and R. de Vivie-Riedle, EPJ Web Conf., 2013, 41, 02028 CrossRef CAS
- L. Barreau, A. D. Ross, V. Kimberg, P. Krasnov, S. Blinov, D. M. Neumark and S. R. Leone, Phys. Rev. A, 2023, 108, 012805 CrossRef CAS
- A. D. Ross, D. Hait, V. Scutelnic, D. M. Neumark, M. Head-Gordon and S. R. Leone, Commun. Phys., 2024, 7, 304 CrossRef CAS PubMed
- D. T. Matselyukh, F. Rott, T. Schnappinger, P. Zhang, Z. Li, J. O. Richardson, R. de Vivie-Riedle and H. J. Wörner, Nat. Commun., 2025, 16, 7211 CrossRef CAS PubMed
- K. J. Gannan, L. B. Drescher, R. Quintero-Bermudez, N. Rana, C. Huang, K. Schafer, M. B. Gaarde and S. R. Leone, Phys. Rev. B, 2025, 111, 134306 CrossRef CAS
- X. Ou, S.-Y. Yang, X. Chen, L. Hu and B. Z. Tang, Nat. Rev. Chem., 2025, 9, 206–207 CrossRef PubMed
- S. Severino, F. Aleotti, L. Mai, A. Crego, F. Medeghini, F. Frassetto, L. Poletto, M. Lucchini, F. Segatta, M. Reduzzi, M. Nisoli, A. Nenov and R. Borrego-Varillas, J. Am. Chem. Soc., 2025, 147, 30785–30793 CrossRef CAS PubMed
- C. Guo, A. Harth, S. Carlström, Y.-C. Cheng, S. Mikaelsson, E. Märsell, C. Heyl, M. Miranda, M. Gisselbrecht and M. B. Gaarde,
et al.
, J. Physiol. Biochem., 2018, 51, 034006 Search PubMed
- P. Rupprecht, F. Montorsi, L. Xu, N. G. Puskar, M. Garavelli, S. Mukamel, N. Govind, D. M. Neumark, D. Keefer and S. R. Leone, arXiv, 2025, preprint, arXiv:2504.10674 DOI:10.48550/arXiv.2504.10674.
- L. Bäuml, F. Rott, T. Schnappinger and R. de Vivie-Riedle, J. Phys. Chem. A, 2023, 127, 9787–9796 CrossRef PubMed
- D. Faccialà, M. Bonanomi, B. N. C. Tenorio, L. Avaldi, P. Bolognesi, C. Callegari, M. Coreno, S. Coriani, P. Decleva, M. Devetta, N. Došlić, A. De Fanis, M. Di Fraia, F. Lever, T. Mazza, M. Meyer, T. Mullins, Y. Ovcharenko, N. Pal, M. N. Piancastelli, R. Richter, D. E. Rivas, M. Sapunar, B. Senfftleben, S. Usenko, C. Vozzi, M. Gühr, K. C. Prince and O. Plekan, J. Am. Chem. Soc., 2025, 147, 30694–30707 CrossRef PubMed
- S. K. Das, M.-O. Winghart, P. Han, D. Rana, Z.-Y. Zhang, S. Eckert, M. Fondell, T. Schnappinger, E. T. J. Nibbering and M. Odelius, J. Phys. Chem. Lett., 2024, 15, 1264–1272 CrossRef CAS PubMed
- R. M. Jay, A. Banerjee, T. Leitner, R.-P. Wang, J. Harich, R. Stefanuik, H. Wikmark, M. R. Coates, E. V. Beale, V. Kabanova, A. Kahraman, A. Wach, D. Ozerov, C. Arrell, P. J. M. Johnson, C. N. Borca, C. Cirelli, C. Bacellar, C. Milne, N. Huse, G. Smolentsev, T. Huthwelker, M. Odelius and P. Wernet, Science, 2023, 380, 955–960 CrossRef CAS PubMed
Footnote |
| † Science Institute and Faculty of Physical Sciences, University of Iceland, Reykjavík, Iceland. |
| This journal is © the Owner Societies 2025 |