
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving structure search with hyperspatial optimization and TETRIS seeding
Daviti
Gochitashvili
 a,
Maxwell
Meyers
a,
Cindy
Wang
b and
Aleksey N.
Kolmogorov
*a
a,
Maxwell
Meyers
a,
Cindy
Wang
b and
Aleksey N.
Kolmogorov
*a
aDepartment of Physics, Applied Physics, and Astronomy, Binghamton University-SUNY, Binghamton, New York 13902, USA. E-mail: kolmogorov@binghamton.edu
bBarrington High School, Barrington, Illinois 60010, USA
First published on 24th October 2025
Abstract
Advanced structure prediction methods developed over the past decades include an unorthodox strategy of allowing atoms to displace into extra dimensions. A recently implemented global optimization of structures from hyperspace (GOSH) has shown promise in accelerating the identification of global minima on potential energy surfaces defined by simple interatomic models. In this study, we extend the GOSH formalism to more accurate Behler–Parrinello neural network (NN) potentials, make it compatible with efficient local minimization algorithms, and test its performance on nanoparticles and crystalline solids. For clusters modeled with NN potentials, four-dimensional optimization offers fairly modest improvement in navigating geometric relaxation pathways and incurs increased computational cost largely offsetting the benefit, but it provides a significant advantage in facilitating atom swaps in nanoalloys. In comparison, the introduction of a moderate, controlled bias for generating more physically sensible starting configurations, achieved via TETRIS-inspired packing of atomic blocks, has a more direct impact on the efficiency of global structure searches. The benchmarked systems are Lennard-Jones clusters, Au or Cu–Pd–Ag nanoparticles and binary Sn alloys described by NN potentials, and compounds with covalent B or BC frameworks modeled with density functional theory.
I. Introduction
Accurate and efficient prediction of atomic structures has evolved from an intractable challenge in the 1990s to an integral component of the materials discovery process.1–4 Advanced structure search methods are now routinely used to identify crystalline and nanoscale configurations synthesizable under specific experimental conditions.5–7 Prediction of thermodynamically stable compounds with unique morphologies beyond known prototypes8–11 has been particularly important in the exploration of matter subjected to extreme pressures. Notable predictions that guided materials synthesis or helped interpret experimental results include the ground states of elemental Na12 and B13 exhibiting unusual properties under compression; LiB,14,15 MB4 (M = Cr, Mn, or Fe),16–18 and CaB619,20 metal borides featuring unfamiliar frameworks; Na2IrO3,21 Na3Ir3O8,22 and Cu2IrO323 iridates showcasing rich strongly correlated physics; and SiH4,24,25 H3S,26 and other compressed hydrides boasting record superconducting critical temperatures.The complex optimization problem of locating global minima on multidimensional potential energy surfaces in given chemical spaces has been addressed with a variety of strategies implemented in USPEX,4 XtalOpt,27 CALYPSO,28 AIRSS,29 MAISE,30 and other packages. The methodological diversity reflects the implications of the no-free-lunch theorem, which posits that no single optimization algorithm can outperform all others across every possible system.31 While the developed algorithms are often fine-tuned for specific materials classes, such as metal alloys,32,33 molecular crystals,34,35 or nanoparticles,36,37 their overall efficiency is largely determined by common factors: how structures are generated, evolved, and selected.
Creation of diverse yet physically meaningful starting configurations is critical for a large-scale efficient screening and ranges from purely random to highly constrained. It has been demonstrated that the introduction of atom exclusion zones,38,39 incorporation of lattice parameters from experiment,19 seeding searches with supercells of randomized known ground states,19,22 or generation of crystal structures with randomly chosen symmetries40,41 can speed up the determination of ground states by orders of magnitude.
Equally important is to control bias in structure optimization. Local relaxation of starting configurations with deterministic gradient-based algorithms introduces minimal bias and has been used extensively in ab initio random structure searches (AIRSS).29 Variable degrees of implicit steering toward global minima can be achieved with the minima hopping method,42 particle swarm optimization,43 evolutionary algorithm,4,44,45 and other strategies that allow structure populations to escape from local minima. Modified variable-composition46–48 and variable-size multitribe49 evolutionary algorithms further facilitate the sharing of favorable motifs identified in large configuration spaces to iteratively construct improved configurations. The introduction of fingerprint-based energy penalty for low-symmetry structures has been shown to enhance convergence in various systems.50
Screening candidate structures with classical interatomic potentials can be a promising means of accelerating ab initio structure searches. Inexpensive traditional interaction-specific models have been used extensively in studies of nanoclusters37 but their generally limited transferability can lead to finding suboptimal solutions.49,51 Recently developed machine learning interatomic potentials offer significantly higher accuracy52,53 and have been successfully used to identify more favorable nanocluster configurations49,51 and thermodynamically stable compounds54,55 overlooked in previous ab initio searches.
An out-of-the box idea explored decades ago is based on performing global searches in higher dimensions. It was proposed that introducing extra spatial dimensions could help structures escape local minima by enabling exploration of directions unavailable in 3D space.56,57 In these studies, structures were first minimized in 3D, then displaced into a higher-dimensional configuration space where they performed constant-energy walks, before being returned to 3D for further relaxation. The added degrees of freedom provided access to relaxation pathways that more effectively traverse barriers on the potential energy surface, increasing the likelihood of reaching low-energy configurations. This strategy proved particularly useful for systems with complex landscapes, such as the double-funnel topology of the Lennard-Jones (LJ) 38-atom cluster. The unorthodox approach was investigated further in a recent study.58 The global optimization of structures from hyperspace (GOSH) generates atomic configurations stochastically in higher special dimensions and gradually brings them back to the real 3D space during a local optimization. The algorithm was found to outperform the traditional 3D local relaxation for several non-periodic systems, from unary and binary clusters to linear chains and covalently bonded frameworks, modeled with simple pairwise potentials. The probability enhancement of finding the ground state reached two orders of magnitude in some cases and was especially evident for the connected systems.58 One of the open questions was how effective the hyperdimensional optimization would be in the exploration of energy surfaces defined by more sophisticated potentials.
The present study focuses on implementing and examining two structure generation and optimization factors. First, we investigate the performance of GOSH by extending the formalism to neural network potentials (NNPs). We have modified our MAISE package30 to perform GOSH for clusters and crystals described with more complex interatomic NNPs of the Behler–Parrinello type59 that provide near- ab initio accuracy. Our implementation reproduces and enhances the previously observed efficiency gains for LJ monoatomic clusters but shows little to no advantage over the deterministic 3D optimization in the case of Au clusters or Sn crystalline alloys, especially when the increased computational cost is taken into account. On the other hand, a notable improvement is observed for multicomponent clusters.
Second, we generalize our previously proposed TETRIS generation of atomic configurations49 to periodic materials and benchmark its performance against more stochastic population initialization. The scheme was originally designed for creating compact starting nanoparticle configurations with physically meaningful interatomic distances by shooting atoms toward the cluster's center iteratively and keeping the closest positions. Our present tests reveal that this essentially parameter-free approach offers a better efficiency improvement than GOSH does for monoatomic LJ and multicomponent Cu–Pd–Ag clusters. The implementation of the TETRIS scheme for crystalline compounds enables a controlled injection of bias in the form of atomic blocks and targeted number of bonds. We demonstrate that the method has little effect on the probability of finding ground states in M–Sn alloys (M = Li, Pd, or Ag) with predominantly metallic bonding54,55 but offers significant improvement in locating more stable configurations in a layered metal borocarbide (ambient-pressure LiB2C2)60 and a metal boride (CaB6 at 50 GPa)19 featuring complex covalent networks. The customizable TETRIS seeding protocol, made available in the open-source MAISE package,30 holds promise for accelerating the convergence of global optimization searches via a judicious introduction of beneficial motifs in structure populations.
II. Methods
A. GOSH original formulation
The global optimization method designed by Pickard58 relies on generating atomic arrangements in higher dimensions, following a downhill trajectory on a hypersurface, and eventually forcing the system to materialize in the normal space. The creation of non-periodic structures involves random placement of hard hyperspheres of radius ã0 within a confining hypersphere of volume Ṽ/f, where ã0 is half the expected equilibrium bond length, Ṽ is the total volume of atom centered hyperspheres, and f is the packing fraction. The potential energy surface in the hyperspace of dimension d = d0 + d+ is a combination of the pairwise potential energy Vij(![[r with combining tilde]](https://www.rsc.org/images/entities/i_char_0072_0303.gif) ij) and a harmonic penalty term
ij) and a harmonic penalty termwhere the Euclidian distances are defined as

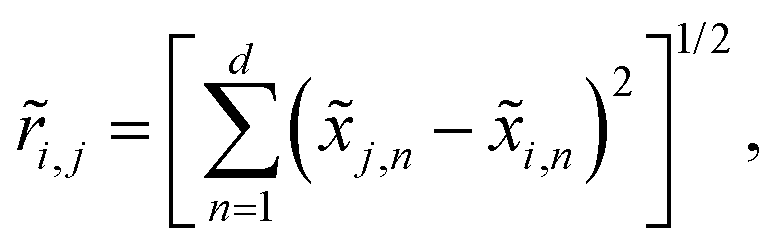

The chosen two-step steepest-decent algorithm61 that does not require line minimization allows for incrementing the spring constant μk+1 = μkβ after each atomic position optimization cycle k. The μ0 = 10 and β = 0.001 values provided an efficient convergence of the hyperspatial energy gradients and ensured the fall of extraspatial coordinates below an acceptable threshold for most systems, while a more conservative μ0 = 1 starting value was used for covalent materials.
B. GOSH implementation in MAISE
As discussed by Pickard,58 GOSH can be naturally generalized for non-pairwise potentials and coupled with other minimization algorithms. In order to make the method compatible with the existing structure generation and optimization capabilities available in MAISE, we introduced a few modifications in the original scheme.First, we enabled access to a wider range of local optimization algorithms with line minimization available in the GSL62 by switching from gradual stepwise to sharp plateau-wise change in the spring constant μ. We break down the maximum number of minimization steps M into N equal groups and set μn = μ0βn. In the last set, we zero the extraspatial coordinates, which are almost always negligible at this stage, to ensure that the structure is fully optimized in the normal space. At each stage n, the relaxation halts if the energy change between successive steps falls below 10−8 eV (the same tight tolerance value is used in pure 3D minimizations). We observed little dependence of the optimization efficiency on these settings and used M = 600, N = 6, μ0 = 2, and β = 2 (for ease of comparison, we keep specifying unitless μ values but scale the parameter as eV Å−2 and set the pairwise LJ energy parameter to 1 eV in our simulations). Choosing a moderate starting spring constant value of μ0 = 2 proved important for converging structures with large initial extraspatial coordinates. Based on the benchmarking tests for Broyden–Fletcher–Goldfarb–Shanno (BFGS2), Fletcher–Reeves conjugate gradient (CG-FR), Polak–Ribiere conjugate gradient (CG-PR), and steepest descent (SD) algorithms63–66 in Fig. 1(a), we chose BFGS2 as the default minimizer.
 | ||
| Fig. 1 Performance of different protocols in hyperdimensional optimization of LJ or Au clusters and Li–Sn crystals. ΔEave represents the average energy of the best half of 2000 structures relative to the corresponding ground state. (a) The local minimization algorithms were tested for MAP0 and Δ+ = 1.0 Å. (b) The mapping and displacement variants were tested with BFGS2. The MAP2 scheme is independent of Δ+. | ||
Second, we implemented alternative procedures for extending structures beyond the normal space. For clusters, we aimed to retain the possibility of initializing configurations with the efficient TETRIS algorithm or generating offspring with various mutation or crossover operations in evolutionary searches.30,49,51 Therefore, instead of creating clusters with equally randomized atomic coordinates in all dimensions, we enabled random shifts into the extra dimensions for clusters with already defined 3D atomic positions. Protocol MAP0 illustrated in Fig. 2(a) involves uncorrelated random displacements of atoms into each extra dimension up to Δ+ (for d0 = 2 and d+ = 1, this operation transforms a 2D disk into a fuzzy flat cylinder). Optionally, the normal coordinates are uniformly contracted to compensate for the increase of the interatomic distances in the hyperspace (MAP1 in Fig. 2(b)). Fig. 1(b) illustrates a good performance of the modified GOSH for Δ+ ∼ 1 Å. The last tested protocol, MAP2 in Fig. 2(c), treats the normal and extra dimensions on a more equal footing and maintains a roughly uniform atomic density via a correlated adjustment of the normal and hyper coordinates (for d0 = 2 and d+ = 1, it converts a 2D disk into a near-spherical 3D shape). If the maximum distance to the origin in a cluster with N atoms is R0 in the normal space and becomes ![[R with combining tilde]](https://www.rsc.org/images/entities/i_char_0052_0303.gif) 0 upon expansion into one extra dimension, the normal space radial distance
0 upon expansion into one extra dimension, the normal space radial distance 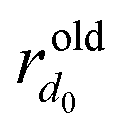 for each atom can be mapped approximately as
for each atom can be mapped approximately as

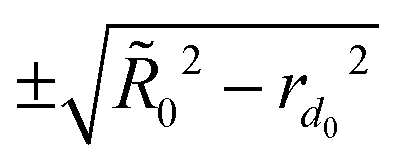 range. The 0/R0 ratios, assessed by rearranging N atoms on uniform grids in d0 and d0 + 1 dimensions, are 1.110N−1/6 for d0 = 2 and 1.082N−1/12 for d0 = 3. Since Fig. 1(b) illustrates a generally good performance of the MAP0 protocol with Δ+ = 1 Å, we have used it in all present simulations unless specified otherwise.
range. The 0/R0 ratios, assessed by rearranging N atoms on uniform grids in d0 and d0 + 1 dimensions, are 1.110N−1/6 for d0 = 2 and 1.082N−1/12 for d0 = 3. Since Fig. 1(b) illustrates a generally good performance of the MAP0 protocol with Δ+ = 1 Å, we have used it in all present simulations unless specified otherwise.
 | ||
Fig. 2 Atom distributions in 20 50-atom 2D clusters (in gray) expanded into 3D (in black) using different mapping protocols. The hyperdimensional x3 coordinate is plotted as a function of the 2D radial distance normalized to the largest radius R0. (a) x3 is generated randomly in [−Δ+; Δ+]. (b) After the random x3 generation, the 2D coordinates are contracted. (c) The 2D coordinates are remapped and x3 is generated randomly in the  range as detailed in the main text. range as detailed in the main text. | ||
For crystals, the normal and extra dimensions are inherently different because the periodicity over hyperspatial coordinates, once introduced, cannot be naturally phased out. Therefore, we chose to extend crystalline structures with 3D periodicity by introducing random hyperspatial positions up to ±Δ+ = ±1 Å and uniformly shrinking the 3D volume by 0.8. It should be noted that the normal space contraction compensating for the average bond elongation due to the random shifts in the extra dimensions may bring some atoms too close to each other. To avoid that, we perform a series of hyperspatial coordinate adjustments with a simple repulsive potential for both clusters and crystals until all interatomic distances are larger than 0.7 of the expected equilibrium length, just as done for making random 3D structures.30,49
C. TETRIS algorithm
MAISE features several options for generating initial structures.30 Our default approach for constructing clusters, denoted BLOB in the code,30 relies on randomly placing atoms around the origin and tweaking their positions to ensure realistic interatomic distances with a combination of a repulsive pairwise potential and attractive springs that keep atoms within a targeted cluster radius. An alternative TETRIS-inspired algorithm involves sending atoms from random directions toward the origin one by one and keeping the closest position out of N3/2 tries to create spherical or ellipsoidal shapes.49 Our previous studies illustrated benefits of initializing evolutionary searches with TETRIS-generated configurations. Fig. S367 confirms that TETRIS clusters are more compact than BLOB ones, with the volume definition adopted from ref. 68, even after the latter procedure is tightened with a compression factor of 1.3.We now extend the method to crystals and introduce an option to seed structures with predefined motifs. While packing algorithms are popular for determining molecular crystal ground states,34,35 the use of atomic building blocks is uncommon in explorations of inorganic solid-state materials. Our TETRIS-based generation proceeds as follows: (i) create random lattice vectors; (ii) extend the c lattice vector to temporarily disable periodicity along this direction; (iii) randomly select atomic blocks, natural for the material's bonding type and producing the desired composition, from a user-defined set; (iv) optionally perform a random 3D rotation of each block using quaternions; (v) send blocks one by one from a random lateral position along the c axis, trying all possible orientations in the x–y plane at each downward step, until they either reach the bottom or come into close contact with another block; (vi) repeat the previous step for several starting lateral positions and keep the position with the lowest z; and (vii) once all the blocks are dropped, turn the periodicity along c back on and shrink the axis until atom separations match the shortest allowed distance.
Due to the observed insignificant influence of implementation details on the method's overall performance, the degree of introduced bias can be controlled by customizing the set of atomic blocks. Additionally, one can specify the targeted number of nearest neighbors within a cutoff sphere for each species to discard configurations with unphysical local environments. Our tests illustrate that the designed algorithm works particularly well for generating layered covalent frameworks.
D. DFT and NN calculations
For metal borides and borocarbides, DFT calculations were performed with VASP69 using projector augmented wave potentials70 and the optB88-vdW functional.71 A 500 eV plane-wave cutoff and dense Monkhorst–Pack k-meshes72 with Δk ∼ 2π × 0.025 Å−1 ensured numerical convergence to typically within 1 meV per atom.Detailed information about our previously developed NN interatomic potentials for Au, Cu–Pd–Sn, and Sn alloys, available on GitHub, can be found in ref. 30, 49, 51, 54 and 55. The local atomic environments were represented with 51 Behler–Parrinello symmetry functions per element, with the explicit expressions provided in Note SII67 and the parameters detailed in ref. 49. The details of NN models of different systems studied in this paper are summarized in Table 1. NNs were fitted to PBE-level energies and forces and extensively tested in global structure searches. The iterative generation of reference structure sets and NN training were done with the automated MAISE-NET wrapper.30
| Chemical system | # of weights | # of E data | # of F data | Testing error E | Testing error F | Reference study |
|---|---|---|---|---|---|---|
| Au | 641 | 2912 | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 621 621 |
6.5 | 37 | 51 |
| Cu–Pd | 3162 | 3725 | 32223 |
6.6 | 63 | 49 |
| Cu–Ag | 3162 | 3724 | 32034 |
3.5 | 38 | 49 |
| Pd–Ag | 3162 | 3705 | 32166 |
4.8 | 58 | 49 |
| Cu–Pd–Ag | 8853 | 2191 | 29163 |
5.2 | 53 | 49 |
| Li–Sn | 3162 | 6046 | 46410 |
10.2 | 49 | 54 |
| Pd–Sn | 3162 | 5507 | 46272 |
9.6 | 52 | 55 |
| Ag–Sn | 3162 | 5410 | 37548 |
8.4 | 33 | 55 |
III. Results and discussion
A. Lennard-Jones clusters
Over a century, the LJ potential has served as a canonical model for systems with isotropic interactions and negligible many-body effects. Despite the simplicity of the functional form, the ground states of LJ clusters, established at least up to at N = 150 atoms,42,74,75 exhibit surprising structural complexity and have become standard benchmarks for developing and tuning global optimization algorithms.42,44,50,58,74–77We applied our implementations of the TETRIS and BLOB structure generation methods along with hyperdimensional optimization to five LJ clusters that feature ground states with different point group symmetries: 37 (C1), 38 (Oh), 39 (C5v), 47 (C1), and 55 (Ih). These sizes were selected due to their nontrivial geometries, such as LJ38 known for its double-funnel energy landscape.78Fig. 3 shows the success rates of global minimum searches using various structure generation protocols and optimization strategies. To ensure statistical significance and direct comparison with the original GOSH study, we generated sufficient structure sets, up to 2.5 × 105 per size, to reach 100 ground state hits and picked the highest reported success rates obtained with AIRSS for packing fractions ranging between 0.1 and 0.3, as denser packing was determined to be not universally beneficial for locating ground states.58 In fact, the consistently high compressions achieved with our TETRIS generation scheme (Fig. S367) were apparently detrimental for all considered sizes, as starting configurations constructed with the BLOB protocol converged to global minima ∼2.3 times more frequently.
 | ||
| Fig. 3 Success rates of finding global minima for LJ clusters using different structure generation protocols (BLOB versus TETRIS) and optimization methods (3D versus 4D). The AIRSS results were taken from ref. 58. Each run contained between 104 and 2.5 × 105 structures that ensured the location of the ground state at least 100 times. Lower −log10pe values mean higher success rates. | ||
In contrast to the reported GOSH results, the addition of the extra coordinate through MAP0 improved the convergence across all considered sizes and seed generation strategies, effectively doubling the average probabilities of finding the LJ global minima. Unsurprisingly, the highest success rates, up to 1 in 100 tries, were observed for LJ39 and LJ55 with the most symmetric global minimum configurations. Histograms of local minima energies collected in Fig. S467 establish that the extra dimension helps LJ clusters transform into more favorable configurations not just near the bottom of the potential energy surface but across the full landscape. The presented findings for different sizes and structure initializations demonstrate that systematic gains in hyperspatial optimization of LJ clusters can be attained through relatively modest ±1 Å maximum displacements of preconstructed 3D configurations into the extra dimension.
The net performance should be assessed by taking into account the increased computational cost of the introduced structure generation and optimization algorithms. According to our estimates, the BLOB and TETRIS operations take one to two orders of magnitude less time than typical local optimizations with the LJ potential. However, hyperspatial optimization can lead to significant slowdowns due to the overhead introduced by the extra spatial dimension and the increased number of energy and force evaluations required for convergence. For the LJ potential, the dimensional overhead added only about 8%, while the average number of relaxation steps increased by a factor of 413/212 ≈ 1.9, offsetting but not fully negating the benefit of 4D optimization.
B. Au clusters modeled with NNPs
As the first testbed for benchmarking hyperdimensional optimization on a chemically realistic many-body system, we chose small Au clusters described by a NNP. Nanosized Au has a complex potential energy surface due to relativistic effects and strong d-orbital hybridization, which lead to low-symmetry ground states including planar, hollow, and amorphous morphologies.79–82 Accurate structure prediction is particularly relevant for nanoparticles of this noble metal, as their rich catalytic properties are known to be highly sensitive to geometry.83,84 Extensive tests in our previous study established that the conventional Gupta potential or embedded atom model are inadequate for guiding ab initio structure searches, with Gupta-potential minima dispersing by nearly 30 meV per atom upon DFT relaxations.51 In contrast, our NNP trained to 6.5 meV per atom accuracy enabled refinement of ground-state candidates across the 30 ≤ N ≤ 80 range and helped identify a more stable amorphous configuration for Au55.Since NNPs are roughly two orders of magnitude more expensive than the LJ potential and their ground states are subject to particular parametrization, we adopted a different benchmark strategy for Au clusters. Rather than measuring success rates based on the successful identification of global minima, we generated 5000 structures for each cluster size and method and calculated the NN average energy of the lowest 40% (2000) minima relative to the best NN-relaxed structure obtained in this dataset. This metric reflects the sampling efficiency of each method while remaining robust to uncertainty in the true ground-state configurations. For example, the combined 2 × 104 tries for each size summarized in Fig. 4, reproduced the putative NN ground state for Au47, converged to suboptimal minima for Au32 and Au55, and identified a more favorable configuration, by ∼1.7 meV per atom, for Au38 compared to the prior multi-tribe evolutionary searches with 2.5 × 104 local optimizations.51
 | ||
| Fig. 4 Average energies (between 6 and 14 meV per atom) and standard deviations (between 1.8 and 3.6 meV per atom) of the best 2000 structures relative to the ground states for Au clusters modeled with NNPs. Each set generated with different protocols (random versus TETRIS) and optimized with different methods (3D versus 4D) consisted of 5000 structures. The full ΔE histograms for Au38 and Au55 are shown in Fig. S5.67 The asterisk denotes CPU-time-adjusted average as explained in the text. | ||
Across all tested cluster sizes and initialization strategies, 4D optimization yielded average energy reductions of approximately 2.5 meV per atom compared to 3D relaxation. However, this gain is counterbalanced by an average 1.63-fold increase in the number of iterations leading to an average 1.78-fold increase in total wall time per relaxation. To account for this difference in computational cost, we reevaluated the effective sampling performance by proportionally reducing the size of the low-energy pool used for averaging. Namely, we adjusted the 3D data by computing the average over the lowest 2000/1.78 structures instead of 2000. When this correction is applied, the apparent advantage of 4D optimization largely vanishes, indicating that the observed improvements stem from extended local relaxation rather than more efficient exploration.
C. Cu–Pd–Ag clusters modeled with NNPs
Nanoalloys display a broader spectrum of tunable catalytic, optical, and magnetic properties due to their diverse composition-dependent core–shell, Janus, and mixed-species arrangements. To evaluate the performance of the hyperdimensional and TETRIS methods in identifying the most favorable configurations of multicomponent nanoparticles, we focused on 50-atom binary and ternary Cu–Pd–Ag clusters. Our previous investigation of the system with the evolutionary algorithm coupled with NNPs revealed a highly frustrated energy landscape across the composition range, making it a challenging benchmark for global optimization.49 The reported most favorable Cu–Pd–Ag configurations have core–shell morphologies, with the larger metals preferentially residing on the surface (see Fig. 5). | ||
| Fig. 5 Average NN energies of the best 2000 out of 5000 structures relative to the ground states for 50-atom clusters found in ref. 49 and displayed with blue (Cu), yellow (Pd), and silver (Ag) spheres. The solid, crossed, and open symbols correspond to clusters generated with TETRIS using different species sequences as explained in the text. Visual representations of atomic structures were created with VESTA.85 | ||
Fig. 5 illustrates how the choice of local optimization strategy and initial population seeding influences search efficiency. All starting structures, 5000 for each data point, were generated using the TETRIS approach, but species assignment during cluster assembly followed three different schemes. In the unbiased case, the order in which atoms of different species were added was completely random (e.g., BACABC for A2B2C2). In the random core variant, atoms were grouped by species, but the sequence of species was randomized (e.g., CCAABB). In the select core protocol, species were ordered by atomic size, from smallest to largest (i.e., AABBCC if rA < rB < rC). Since the introduction of extraspatial coordinates introduces additional randomness to the atomic distributions, the resulting nine sets comprised 5000 different initial structures for each composition. We chose to average the energies over the best 2000 minima, i.e., 40% of each set, to clarify the performance of the random core generation for binary or ternary nanoalloys which have 1/(2!) = 50% or 1/(3!) ≈ 17% chances of having the optimal core–shell sequences.
In unbiased searches, 4D optimizations consistently produced lower-energy structures than standard 3D relaxations across all tested nanoalloys. The average energies above the global minima decreased by ∼10% with MAP0 and ∼30% for MAP2, indicating that the added dimension, with larger average displacements in the latter case, allows the system to escape inefficient local geometries during relaxation. As in the Au case, the additional computational overhead, now measured at 2.24 for MAP0 and 3.25 for MAP2, diminishes these gains but the full factors behind this improvement become clearer when considered alongside the results with the other two structure initialization protocols. Growing clusters by grouping incoming atoms by species, either randomly or selectively, dramatically improved the search outcomes for both 3D and 4D optimizations. The moderate randomization of the extra dimensional coordinate up to ±1 Å in MAP0 continued to provide a ∼10% improvement, while the cluster ‘spherization’ in all dimensions in MAP2 likely promoted a certain degree of species mixing and led to less favorable minima than in the standard 3D case. The random and select core schemes yielded fairly similar results for binary clusters, which is consistent with the 50% probability of obtaining the correct species sequence when two species are randomly ordered. In contrast, the difference was more pronounced for ternary clusters, where the chance of realizing the correct sequence under random grouping drops to about 17%. Compared to our previous evolutionary searches with 5 × 104 local optimizations per nanoalloy starting with unbiased TETRIS configurations,49 the present local relaxations totaling 4.5 × 104 tries yielded comparable best minima for binary clusters, 0.1 meV per atom higher for Cu30Pd20, the same for Cu15Ag35, and 0.3 meV per atom lower for Pd15Ag35, but a significantly less optimal minimum, by 7.5 meV per atom, for Cu15Pd20Ag15.
These trends suggest that the key limitation of standard 3D relaxation lies not in geometric constraints, but in its inability to reassign species to more favorable environments. Just as in the optimization of binary chains,58 GOSH circumvents this by effectively enabling atom reordering during relaxation, but the same benefit in the more practical case of multicomponent nanoparticles can be attained more directly through incorporation of chemical knowledge in the initial population. These observations align with alternative efforts in the field to address compositional frustration in multicomponent clusters. Several methods have been developed to enable or approximate species swaps during optimization, including identity-exchange Monte Carlo steps in basin-hopping,77 symmetry-constrained genetic algorithms,86 and multi-agent search protocols with specialized roles such as the Flying–Landing–Hiking framework.87
D. Li–Sn, Pd–Sn, and Ag–Sn modeled with NNPs
To test the performance of global optimization in periodic systems, we selected M–Sn intermetallics known to display a wide range of morphologies. As a group XIV element capable of covalent bonding, Sn readily forms directional extended frameworks and cage-like motifs, especially when alloyed with electropositive metals.54,55,88–90 For example, Sn-rich alloys can be represented with particular sequences of Kepler nets, while M-rich intermetallics often appear as simple decorations of common metallic lattices with isolated or only weakly connected Sn atoms. Our recent NNP-guided exploration of M–Sn binaries (M = Li, Na, Ca, Cu, Pd, and Ag) led to the identification of over 30 new phases stable under different (T,P) conditions.54,55 They include tI36-PdSn2 with an A°B°A−A°B°A+ stacking of Pd atoms between Sn layers with rotated squares and hR75-Li19Sn6 with a34,54 stacking of layered units defining the bcc decoration.We focused on Li7Sn9, Li9Sn6, Pd4Sn12, Pd14Sn2, and Ag14Sn4 compositions determined in our prior evolutionary searches to have complex ground states with sizes between 15 and 18 atoms per unit cell. To gauge the capability of the TETRIS method, we tried only broadly applicable structural fragments, such as M–M, M–Sn, and Sn–Sn dimers or bcc blocks, avoiding overly material-dependent structural motifs that could bias the searches toward previously known solutions. Our tests (not shown) demonstrated little advantage of block seeding over random structure generation, and we chose the latter in our investigation of the hyperspatial optimization.
Fig. 6 compares the average energies of the 2000 lowest structures out of 5000 relative to the ground state, evaluated by neural network potentials. Unlike in nanoalloys, 4D optimization did not yield systematic improvements in periodic crystals. Both 3D and 4D relaxations sampled similar energy distributions across the lowest 40% of minima and delivered comparable success in locating the lowest-energy states identified in earlier evolutionary runs. For Li7Sn9 and Pd14Sn2, both methods repeatedly recovered the ground state. For Pd4Sn12, only 4D found the ground state, with 3D reaching a structure 2 meV per atom higher. For Ag14Sn4, the ground state was found only by 3D, with the closest 4D result 0.5 meV per atom less stable. The lack of significant advantage observed for 4D relaxation in these periodic systems may result from the indirect constraints imposed by lattice vectors, which evolve primarily in response to effective modulation of three-dimensional interatomic distances by the additional hyperspatial coordinates.
 | ||
| Fig. 6 Average energies of the best 2000 structures relative to the ground states for Li–Sn, Pd–Sn, and Ag–Sn crystals modeled with NNPs. Each set optimized with different methods (3D and 4D) consisted of 5000 structures. | ||
E. LiB2C2 and CaB6 modeled with DFT
The usefulness of the TETRIS adaptation to periodic systems can be illustrated with ab initio searches for ground states in complex covalent materials, such as metal borides and borocarbides known to display a variety of 2D and 3D frameworks.91,92 To collect sufficient data for benchmarking different protocols at the DFT level, we generated 1000 or 2000 medium-sized configurations and assessed each variant's performance by calculating the average energy of the best 2% of locally optimized candidates. The dataset sizes were chosen to approximate fractions of random motifs injected into population during typical evolutionary runs totaling 4000–6000 local optimizations.19,30 Our recent screening of layered metal borocarbides for conventional high-Tc superconductors synthesizable under ambient pressure revealed that high levels of hole doping destabilize the honeycomb layer morphology in MxBC (M = Li, Na, or Mg) essential for strong quasi-2D electron–phonon coupling.60,93 At the Li1/2BC composition, our evolutionary searches identified an mS20 (Cm) structure with a 3D framework ∼70 meV per atom below the best oI10 (Imm2) layered counterpart.We performed our present tests for Li3B6C6 and referenced the resulting 8 × 1000 structures to the mS15 (Pm) polymorph with 3D morphology determined previously to have the lowest energy among 15-atom unit cells. Fig. 7 shows the effectiveness of generating low-energy motifs as a function of the introduced bias. For each protocol, we checked whether the additional condition on the number of nearest neighbors, 3–4 within 1.9 Å around B/C atoms expected to be in the sp2 or sp3 environments, leads to better starting configurations. The constraint indeed consistently lowered the average energy, with the largest improvement of ∼40 meV per atom noted for the random scheme. The three TETRIS sets with different numbers of atomic blocks may help find a desired balance for bias in structure generation. A simple switch from a random placement of atoms within a unit cell to the TETRIS packing of individual atoms resulted in a noticeable drop of the average energy by ∼50 meV per atom. The introduction of B and C atoms in pairs to promote a natural alternating order in heteronuclear frameworks further lowered the average energy by ∼75 meV per atom. The larger four-atom preassembled units link into honeycomb layers far more efficiently while not prohibiting interlayer bridging, as we observed low-energy configurations with sp3-bonded C atoms.
 | ||
| Fig. 7 Performance of random and TETRIS structure generation methods in the search for most favorable Li3B6C6 configurations at the DFT level. The number and types of user-defined TETRIS blocks per set are indicated at the top. The sets denoted with diamonds were generated with an additional constraint for B and C atoms to have 2–4 neighbors within 1.9 Å. The energy is referenced to the most favorable mP15-Li3B6C6 phase with 3 formula units found in our previous study,60 with the dashed line corresponding the lowest-energy layered configuration. | ||
Investigation of CaB6 under pressure provides information on the methods’ performance for non-layered configurations. Our previous studies revealed exceptional complexity of pressure-induced structural transformations in this compound, which starts as a simple cP7 at ambient pressure, becomes dynamically unstable above 23 GPa, and displays a variety of more stable exotic 3D frameworks above 13 GPa.19,20 The concurrent synthesis work indicated the formation of a new phase above 31 GPa but its structure could not be established from the collected XRD data and was solved with evolutionary searches without any structural input.19 Our benchmark tests demonstrated that the construction of the 28-atom tI56 ground state could not be accomplished with unbiased random searches in 10000 tries and required either randomization of the cP7 supercells or use of the evolutionary algorithm.
Here, we considered smaller 14-atom Ca2B12 unit cells at 50 GPa, determined to have oS28 (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) as the lowest-enthalpy configuration with up to two formula units,19 and allowed an additional 3D random rotation of the TETRIS blocks with quaternions in their starting position. Fig. 8 displays the average enthalpy of the best 40 structures and shows that 2000 tries were sufficient to locate oS28 with each protocol. In fact, the TETRIS algorithm offered no improvement over the random method when we used blocks with one-three B atoms, a preassembled unit with a uniform distribution of six B atoms around Ca, or a five-atom fragment of the B octahedron (only the last two sets are shown). An appreciable enthalpy drop of nearly 80 meV per atom was observed only when full octahedron units were used.
m) as the lowest-enthalpy configuration with up to two formula units,19 and allowed an additional 3D random rotation of the TETRIS blocks with quaternions in their starting position. Fig. 8 displays the average enthalpy of the best 40 structures and shows that 2000 tries were sufficient to locate oS28 with each protocol. In fact, the TETRIS algorithm offered no improvement over the random method when we used blocks with one-three B atoms, a preassembled unit with a uniform distribution of six B atoms around Ca, or a five-atom fragment of the B octahedron (only the last two sets are shown). An appreciable enthalpy drop of nearly 80 meV per atom was observed only when full octahedron units were used.
 | ||
| Fig. 8 Performance of random and TETRIS structure generation methods in the search for most favorable Ca2B12 configurations at 50 GPa at the DFT level. The number and types of TETRIS blocks are indicated at the top. The enthalpy is referenced to the most favorable oS28-Ca2B12 phase found in our previous study,19 with the dashed line corresponding to the cP7 ground state at ambient pressure. | ||
We would like to stress that we view the TETRIS generation not as a stand-alone optimization method but rather as a protocol for seeding global structure searches with plausible configurations. The presented results of ab initio searches demonstrate that steering global exploration toward layered configurations can be accomplished naturally with minimal bias, such as heteronuclear dimers. Construction of complex 3D frameworks may require the use of more specific building blocks favorable for this material's class but the TETRIS packing is not constrained to supercells of known prototypes to generate periodic configurations.
IV. Summary
We present a systematic extension and examination of two complementary strategies for improving global minima searches in nanoscale and crystalline systems: structure generation with physically biased seeding and hyperspatial optimization. To enable treatment of chemically realistic systems with a variety of structure initialization and minimization schemes, we expanded Behler–Parrinello-type NNPs to extra dimensions and reformulated several elements of the GOSH algorithm. Specifically, we introduced two coordinate mapping schemes, MAP0 and MAP2, that displace physically meaningful configurations constructed in the normal 3D space into extra dimensions using distinct extension strategies. MAP2 mimics the original GOSH approach by generating randomized displacements that effectively treat all spatial directions equally, producing near-hyperspherical distributions. In contrast, MAP0 introduces moderate, uncorrelated shifts of up to ±1 Å into the extra dimensions. Additionally, we adjusted the protocol for changing the spring constant associated with extraspatial coordinates to plateau-wise ramp-up during local optimizations, enabling the use of advanced minimization algorithms, such as BFGS2.Our benchmark tests showed that both the modified mapping scheme and enhanced relaxation protocol were essential for attaining consistent gains in complex systems. In multicomponent Cu–Pd–Ag clusters initialized with unbiased species ordering, 4D optimization with the conservative MAP0 approach reduced the average energy above the global minimum by ∼10%, while the more aggressive MAP2 scheme yielded up to ∼30% reductions, highlighting the benefit of using extra dimensions to facilitate atom swaps. However, a chemically informed TETRIS initialization, which assembles clusters with species grouped by atomic size to favor core–shell formation, led to significantly better convergence even with standard 3D optimization, and performed even more effectively when paired with MAP0. This balance between geometric flexibility and controlled species placement was especially important for ternary systems, where the probability of randomly assembling optimal core–shell configurations is inherently low. For bulk alloys, these factors had little impact, as neither hyperspatial optimization nor species ordering strategies produced measurable gains. In contrast, TETRIS-based seeding, by assembling unit cells from chemically sensible atomic blocks, substantially improved convergence in covalently bonded systems modeled with DFT.
This study provides the first evidence that hyperspatial optimization, previously applied to simple potential energy surfaces,56–58 can yield benefits in systems modeled with sophisticated many-body potentials. By independently varying the coordinate mapping and initialization strategies, we were able to disentangle the contributions of geometric relaxation flexibility and species ordering, a distinction particularly important for nanoalloys. These insights offer a path toward improving hyperspatial optimization across a broader range of materials systems.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study are available within the article and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp02412a.Additional data are available from the corresponding author upon request.
Acknowledgements
The authors acknowledge support from the National Science Foundation (NSF) (Award No. DMR-2320073). This work used the Expanse system at the San Diego Supercomputer Center via allocation TG-DMR180071. Expanse is supported by the Extreme Science and Engineering Discovery Environment (XSEDE) program94 through NSF Award No. ACI-1548562.References
- S. M. Woodley and R. Catlow, Crystal structure prediction from first principles, Nat. Mater., 2008, 7, 937–946, DOI:10.1038/nmat2321.
- D. J. Wales and H. A. Scheraga, Global optimization of clusters, crystals, and biomolecules, Science, 1999, 285, 1368–1372, DOI:10.1126/science.285.5432.1368.
- E. Zurek, Discovering new materials via A Priori crystal structure prediction, Rev. Comput. Chem., 2016, 29, 274–326, DOI:10.1002/9781119148739.ch5.
- A. R. Oganov and C. W. Glass, Crystal structure prediction using ab initio evolutionary techniques: Principles and applications, J. Chem. Phys., 2006, 124, 244704, DOI:10.1063/1.2210932.
- R. J. Needs and C. J. Pickard, Perspective: Role of structure prediction in materials discovery and design, APL Mater., 2016, 4, 053210, DOI:10.1063/1.4949361.
- A. R. Oganov, C. J. Pickard, Q. Zhu and R. J. Needs, Structure prediction drives materials discovery, Nat. Rev. Mater., 2019, 4, 331–348, DOI:10.1038/s41578-019-0101-8.
- S. M. Woodley, G. M. Day and R. Catlow, Structure prediction of crystals, surfaces and nanoparticles, Philos. Trans. R. Soc., A, 2020, 378, 20190600, DOI:10.1098/rsta.2019.0600.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards and S. Dacek, et al., Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002, DOI:10.1063/1.4812323.
- S. Curtarolo, G. L. W. Hart, M. B. Nardelli, N. Mingo, S. Sanvito and O. Levy, The high-throughput highway to computational materials design, Nat. Mater., 2013, 12, 191–201, DOI:10.1038/nmat3568.
- J. E. Saal, S. Kirklin, M. Aykol, B. Meredig and C. Wolverton, Materials design and discovery with High-Throughput density functional theory: The open quantum materials database (OQMD), JOM, 2013, 65, 1501–1509, DOI:10.1007/s11837-013-0755-4.
- G. Bergerhoff and I. Brown, Crystallographic databases, ed. F. H. Allen, et al., International Union of Crystallography, Chester, 1987 DOI:10.1063/1.4812323.
- X. Dong, A. R. Oganov, A. F. Goncharov, E. Stavrou, S. Lobanov and G. Saleh, et al., A stable compound of helium and sodium at high pressure, Nat. Chem., 2017, 9, 440–445, DOI:10.1038/nchem.2716.
- A. R. Oganov, J. Chen, C. Gatti, Y. Ma, Y. Ma and C. W. Glass, et al., Ionic high-pressure form of elemental boron, Nature, 2009, 457, 863–867, DOI:10.1038/nature07736.
- A. N. Kolmogorov and S. Curtarolo, Prediction of different crystal structure phases in metal borides: A lithium monoboride analog to MgB2, Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 73, 180501, DOI:10.1103/PhysRevB.73.180501.
- A. N. Kolmogorov, S. Hajinazar, C. Angyal, V. L. Kuznetsov and A. P. Jephcoat, Synthesis of a predicted layered lib via cold compression, Phys. Rev. B:Condens. Matter Mater. Phys., 2015, 92, 144110, DOI:10.1103/PhysRevB.92.144110.
- A. N. Kolmogorov, S. Shah, E. R. Margine, A. F. Bialon, T. Hammerschmidt and R. Drautz, New superconducting and semiconducting fe-b compounds predicted with an ab initio evolutionary search, Phys. Rev. Lett., 2010, 105, 217003, DOI:10.1103/PhysRevLett.105.217003.
- A. F. Bialon, T. Hammerschmidt, R. Drautz, S. Shah, E. R. Margine and A. N. Kolmogorov, Possible routes for synthesis of new boron-rich Fe–B and Fe1−xCrxB4 compounds, Appl. Phys. Lett., 2011, 98, 081901, DOI:10.1063/1.3556564.
- A. Van Der Geest and A. Kolmogorov, Stability of 41 metal–boron systems at 0 GPa and 30 GPa from first principles, Calphad, 2014, 46, 184–204, DOI:10.1016/j.calphad.2014.03.005.
- A. N. Kolmogorov, S. Shah, E. R. Margine, A. K. Kleppe and A. P. Jephcoat, Pressure-Driven Evolution of the Covalent Network in CaB6, Phys. Rev. Lett., 2012, 109, 075501, DOI:10.1103/physrevlett.109.075501.
- S. Shah and A. N. Kolmogorov, Stability and superconductivity of Ca-B phases at ambient and high pressure, Phys. Rev. B:Condens. Matter Mater. Phys., 2013, 88, 014107, DOI:10.1103/physrevb.88.014107.
- S. K. Choi, R. Coldea, A. N. Kolmogorov, T. Lancaster, I. I. Mazin and S. J. Blundell, et al., Spin Waves and Revised Crystal Structure of Honeycomb Iridate Na2IrO3, Phys. Rev. Lett., 2012, 108, 127204, DOI:10.1103/PhysRevLett.108.127204.
- F. Sun, H. Zheng, Y. Liu, E. D. Sandoval, C. Xu and J. Xu, et al., Electronic and structural response to pressure in the hyperkagome-lattice Na3Ir3O8, Phys. Rev. B, 2018, 98, 085131, DOI:10.1103/PhysRevB.98.085131.
- G. Fabbris, A. Thorn, W. Bi, M. Abramchuk, F. Bahrami and J. H. Kim, et al., Complex pressure-temperature structural phase diagram of the honeycomb iridate Cu2IrO3, Phys. Rev. B, 2021, 104, 014102, DOI:10.1103/PhysRevB.104.014102.
- C. J. Pickard and R. J. Needs, Structures at high pressure from random searching, Phys. Status Solidi B, 2009, 246, 536–540, DOI:10.1002/pssb.200880546.
- C. J. Pickard, Ephemeral data derived potentials for random structure search, Phys. Rev. B, 2022, 106, 014102, DOI:10.1103/PhysRevB.106.014102.
- Y. Li, L. Wang, H. Liu, Y. Zhang, J. Hao and C. J. Pickard, et al., Dissociation products and structures of solid h2S at strong compression, Phys. Rev. B, 2016, 93, 020103, DOI:10.1103/PhysRevB.93.020103.
- D. C. Lonie and E. Zurek, Xtalopt: An open-source evolutionary algorithm for crystal structure prediction, Comput. Phys. Commun., 2011, 182, 372–387, DOI:10.1016/j.cpc.2010.07.048.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Calypso: A method for crystal structure prediction, Comput. Phys. Commun., 2012, 183, 2063–2070, DOI:10.1016/j.cpc.2012.05.008.
- C. J. Pickard and R. J. Needs, Ab initio random structure searching, J. Phys.:Condens. Matter, 2011, 23, 053201, DOI:10.1088/0953-8984/23/5/053201.
- S. Hajinazar, A. Thorn, E. D. Sandoval, S. Kharabadze and A. N. Kolmogorov, MAISE: Construction of neural network interatomic models and evolutionary structure optimization, Comput. Phys. Commun., 2021, 259, 107679, DOI:10.1016/j.cpc.2020.107679.
- D. Wolpert and W. Macready, No free lunch theorems for optimization, IEEE Trans. Evol. Comput., 1997, 1, 67–82, DOI:10.1109/4235.585893.
- C. Wolverton and A. Zunger, Prediction of li intercalation and battery voltages in layered vs. cubic lix coo2, J. Electrochem. Soc., 1998, 145, 2424, DOI:10.1149/1.1838653.
- T. Mueller and G. Ceder, Bayesian approach to cluster expansions, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 80, 024103, DOI:10.1103/PhysRevB.80.024103.
- G. R. Desiraju, Crystal Engineering: From Molecule to Crystal, J. Am. Chem. Soc., 2013, 135, 9952–9967, DOI:10.1021/ja403264c.
- L. M. Hunnisett, et al., The seventh blind test of crystal structure prediction: structure generation methods, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2024, 80, 517–547, DOI:10.1107/S2052520624007492.
- R. L. Johnston, Evolving better nanoparticles: Genetic algorithms for optimising cluster geometries, Dalton Trans., 2003, 4193–4207, 10.1039/B305686D.
- R. Ferrando, J. Jellinek and R. L. Johnston, Nanoalloys: from theory to applications of alloy clusters and nanoparticles, Chem. Rev., 2008, 108, 845–910, DOI:10.1021/cr040090g.
- S. M. Woodley, P. D. Battle, J. D. Gale and C. R. A. Catlow, Prediction of inorganic crystal framework structures part 1: Using a genetic algorithm and an indirect approach to exclusion zones, Phys. Chem. Chem. Phys., 2004, 6, 1815–1822, 10.1039/B315066F.
- S. M. Woodley, Prediction of inorganic crystal framework structures part 2: Using a genetic algorithm and a direct approach to exclusion zones, Phys. Chem. Chem. Phys., 2004, 6, 1823–1829, 10.1039/B315216M.
- P. Avery and E. Zurek, Randspg: An open-source program for generating atomistic crystal structures with specific spacegroups, Comput. Phys. Commun., 2017, 213, 208–216, DOI:10.17632/v2nzgmpd37.1.
- S. Fredericks, K. Parrish, D. Sayre and Q. Zhu, Pyxtal: A python library for crystal structure generation and symmetry analysis, Comput. Phys. Commun., 2021, 261, 107810, DOI:10.1016/j.cpc.2020.107810.
- D. J. Wales and J. P. K. Doye, Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms, J. Phys. Chem. A, 1997, 101, 5111–5116, DOI:10.1021/jp970984n.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal structure prediction via particle-swarm optimization, Phys. Rev. B:Condens. Matter Mater. Phys., 2010, 82, 094116, DOI:10.1103/PhysRevB.82.094116.
- D. Daven, N. Tit, J. Morris and K. Ho, Structural optimization of Lennard-Jones clusters by a genetic algorithm, Chem. Phys. Lett., 1996, 256, 195–200, DOI:10.1016/0009-2614(96)00406-X.
- S. M. Woodley, A. A. Sokol and C. R. A. Catlow, Structure prediction of inorganic nanoparticles with predefined architecture using a genetic algorithm, Z. Anorg. Allg. Chem., 2004, 630, 2343–2353, DOI:10.1002/zaac.200400338.
- G. Trimarchi, A. J. Freeman and A. Zunger, Predicting stable stoichiometries of compounds via evolutionary global space-group optimization, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 80, 092101, DOI:10.1103/PhysRevB.80.092101.
- Q. Zhu, V. Sharma, A. R. Oganov and R. Ramprasad, Predicting polymeric crystal structures by evolutionary algorithms, J. Chem. Phys., 2014, 141, 154102, DOI:10.1063/1.4897337.
- S. V. Lepeshkin, V. S. Baturin, Y. A. Uspenskii and A. R. Oganov, Method for Simultaneous Prediction of Atomic Structure and Stability of Nanoclusters in a Wide Area of Compositions, J. Phys. Chem. Lett., 2019, 10, 102–106, DOI:10.1021/acs.jpclett.8b03510.
- S. Hajinazar, E. D. Sandoval, A. J. Cullo and A. N. Kolmogorov, Multitribe evolutionary search for stable Cu–Pd–Ag nanoparticles using neural network models, Phys. Chem. Chem. Phys., 2019, 21, 8729–8742, 10.1039/c9cp00837c.
- S. Tao, X. Shao and L. Zhu, Accelerating Structural Optimization through Fingerprinting Space Integration on the Potential Energy Surface, J. Phys. Chem. Lett., 2024, 15, 3185–3190, DOI:10.1021/acs.jpclett.4c00275.
- A. Thorn, J. Rojas-Nunez, S. Hajinazar, S. E. Baltazar and A. N. Kolmogorov, Toward ab Initio Ground States of Gold Clusters via Neural Network Modeling, J. Phys. Chem. C, 2019, 123, 30088–30098, DOI:10.1021/acs.jpcc.9b08517.
- J. Behler, Perspective: Machine learning potentials for atomistic simulations, J. Chem. Phys., 2016, 145, 170901, DOI:10.1063/1.4966192.
- A. P. Bartók, S. De, C. Poelking, N. Bernstein, J. R. Kermode and G. Csányi, et al., Machine learning unifies the modeling of materials and molecules, Sci. Adv., 2017, 3, e1701816, DOI:10.1126/sciadv.1701816.
- S. Kharabadze, A. Thorn, E. A. Koulakova and A. N. Kolmogorov, Prediction of stable Li-Sn compounds: boosting ab initio searches with neural network potentials, npj Comput. Mater., 2022, 8, 136, DOI:10.1038/s41524-022-00825-4.
- A. Thorn, D. Gochitashvili, S. Kharabadze and A. N. Kolmogorov, Machine learning search for stable binary Sn alloys with Na, Ca, Cu, Pd, and Ag, Phys. Chem. Chem. Phys., 2023, 25, 22415–22436, 10.1039/D3CP02817H.
- D. B. Faken, A. F. Voter, D. L. Freeman and J. D. Doll, Dimensional strategies and the minimization problem: Barrier-avoiding algorithms, J. Phys. Chem. A, 1999, 103, 9521–9526, DOI:10.1021/jp9920949.
- A. S. Bayden and K. D. Jordan, Use of extended dimensions in global optimization, Chem. Phys. Lett., 2004, 385, 101–104, DOI:10.1016/j.cplett.2003.12.059.
- C. J. Pickard, Hyperspatial optimization of structures, Phys. Rev. B, 2019, 99, 054102, DOI:10.1103/PhysRevB.99.054102.
- J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy surfaces, Phys. Rev. Lett., 2007, 98, 146401, DOI:10.1103/PhysRevLett.98.146401.
- C. R. Tomassetti, G. P. Kafle, E. T. Marcial, E. R. Margine and A. N. Kolmogorov, Prospect of high-temperature superconductivity in layered metal borocarbides, J. Mater. Chem. C, 2024, 12, 4870–4884, 10.1039/D4TC00210E.
- J. Barzilai and J. M. Borwein, Two-point step size gradient methods, IMA J. Nume. Anal., 1988, 8, 141–148, DOI:10.1093/imanum/8.1.141.
- M. Galassi, et al., Gnu scientific library reference manual, 2018, https://www.gnu.org/software/gsl/ Search PubMed.
- R. Fletcher and C. M. Reeves, Function minimization by conjugate gradients, Comput. J., 1964, 7, 149–154, DOI:10.1093/comjnl/7.2.149.
- R. Fletcher, A new approach to variable metric algorithms, Comput. J., 1970, 13, 317–322, DOI:10.1093/comjnl/13.3.317.
- R. Klessig and E. Polak, Efficient implementations of the polak–ribière conjugate gradient algorithm, SIAM J. Control, 1972, 10, 524–549, DOI:10.1137/0310040.
- J. Fliege and B. F. Svaiter, Steepest descent methods for multicriteria optimization, Math. Methods Oper. Res., 2000, 51, 479–494, DOI:10.1007/s001860000043.
- See SI for Notes I and II, Fig. S1–S5. A description for each figure is given in the body of the paper, wherever ref. 67 is cited.
- S. Chiriki, S. Jindal and S. S. Bulusu, Neural network potentials for dynamics and thermodynamics of gold nanoparticles, J. Chem. Phys., 2017, 146, 084314, DOI:10.1063/1.4977050.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169, DOI:10.1103/PhysRevB.54.11169.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953, DOI:10.1103/PhysRevB.50.17953.
- J. Klimeš, D. R. Bowler and A. Michaelides, Chemical accuracy for the van der Waals density functional, J. Phys.:Condens. Matter, 2009, 22, 022201, DOI:10.1088/0953-8984/22/2/022201.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188, DOI:10.1103/PhysRevB.13.5188.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865, DOI:10.1103/PhysRevLett.77.3865.
- R. H. Leary and J. P. K. Doye, Tetrahedral global minimum for the 98-atom Lennard-Jones cluster, Phys. Rev. E:Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 60, R6320–R6322, DOI:10.1103/PhysRevE.60.R6320.
- R. H. Leary, Global Optimization on Funneling Landscapes, J. Global Optim., 2000, 18, 367–383, DOI:10.1023/A:1026500301312.
- R. Brüning, D. A. St-Onge, S. Patterson and W. Kob, Glass transitions in one-, two-, three-, and four-dimensional binary Lennard-Jones systems, J. Phys.:Condens. Matter, 2008, 21, 035117, DOI:10.1088/0953-8984/21/3/035117.
- G. G. Rondina and J. L. F. Da Silva, Revised Basin-Hopping Monte Carlo Algorithm for Structure Optimization of Clusters and Nanoparticles, J. Chem. Inf. Model., 2013, 53, 2282–2298, DOI:10.1021/ci400224z.
- J. P. K. Doye, M. A. Miller and D. J. Wales, Evolution of the potential energy surface with size for Lennard-Jones clusters, J. Chem. Phys., 1999, 111, 8417–8428, DOI:10.1063/1.480217.
- I. L. Garzón and A. Posada-Amarillas, Structural and vibrational analysis of amorphous Au55 clusters, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11796–11802, DOI:10.1103/PhysRevB.54.11796.
- M. P. Johansson, D. Sundholm and J. Vaara, Au32: A 24-carat golden fullerene, Angew. Chem., Int. Ed., 2004, 43, 2678–2681, DOI:10.1002/anie.200453986.
- J. Wang, J. Jellinek, J. Zhao, Z. Chen, R. B. King and P. von Ragué Schleyer, Hollow Cages versus Space-Filling Structures for Medium-Sized Gold Clusters: The Spherical Aromaticity of the Au50 Cage, J. Phys. Chem. A, 2005, 109, 9265–9269, DOI:10.1021/jp052414q.
- N. Tarrat, M. Rapacioli, J. Cuny, J. Morillo, J.-L. Heully and F. Spiegelman, Global optimization of neutral and charged 20- and 55-atom silver and gold clusters at the dftb level, Comput. Theor. Chem., 2017, 1107, 102–114, DOI:10.1016/j.comptc.2017.01.022 . Structure prediction of nanoclusters from global optimization techniques: computational strategies.
- N. Lopez, J. Nørskov, T. Janssens, A. Carlsson, A. Puig-Molina and B. S. Clausen, et al., The adhesion and shape of nanosized Au particles in a Au/TiO2 catalyst, J. Catal., 2004, 225, 86–94, DOI:10.1016/j.jcat.2004.03.036.
- P. Suchomel, L. Kvitek, R. Prucek, A. Panacek, A. Halder and S. Vajda, et al., Simple size-controlled synthesis of Au nanoparticles and their size-dependent catalytic activity, Sci. Rep., 2018, 8, 4589, DOI:10.1038/s41598-018-22976-5.
- K. Momma and F. Izumi, VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276, DOI:10.1107/S0021889811038970.
- S. Han, G. Barcaro, A. Fortunelli, S. Lysgaard, T. Vegge and H. A. Hansen, Unfolding the structural stability of nanoalloys via symmetry-constrained genetic algorithm and neural network potential, npj Comput. Mater., 2022, 8, 121, DOI:10.1038/s41524-022-00807-6.
- D. Rapetti, C. Roncaglia and R. Ferrando, Optimizing the Shape and Chemical Ordering of Nanoalloys with Specialized Walkers, Adv. Theory Simul., 2023, 6, 2300268, DOI:10.1002/adts.202300268.
- J. Nylén, F. J. García García, B. D. Mosel, R. Pöttgen and U. Häussermann, Structural relationships, phase stability and bonding of compounds PdSnn (n = 2, 3, 4), Solid State Sci., 2004, 6, 147–155, DOI:10.1016/j.solidstatesciences.2003.09.011.
- J. Engelkemier, V. M. Berns and D. C. Fredrickson, First-principles elucidation of atomic size effects using dft-chemical pressure analysis: Origins of Ca36Sn23's long-period superstructure, J. Chem. Theory Comput., 2013, 9, 3170–3180, DOI:10.1021/ct400274f.
- J. Shao, C. Beaufils and A. N. Kolmogorov, Ab initio engineering of materials with stacked hexagonal tin frameworks, Sci. Rep., 2016, 6, 28369 CrossRef CAS PubMed.
- J. Kouvetakis, R. B. Kaner, M. L. Sattler and N. Bartlett, A novel graphite-like material of composition BC3, and nitrogen–carbon graphites, J. Chem. Soc., Chem. Commun., 1986, 1758–1759, 10.1039/C39860001758.
- P. F. Rogl, J. Vreštál, T. Tanaka and S. Takenouchi, The B-rich side of the B–C phase diagram, Calphad, 2014, 44, 3–9, DOI:10.1016/j.calphad.2013.07.016 . Special Issue: TOFA 2012 – Discussion Meeting on Thermodynamics of Alloys.
- C. R. Tomassetti, D. Gochitashvili, C. Renskers, E. R. Margine and A. N. Kolmogorov, First-principles design of ambient-pressure MgxB2C2 and NaxBC superconductors, Phys. Rev. Mater., 2024, 8, 114801, DOI:10.1103/PhysRevMaterials.8.114801.
- J. Towns, T. Cockerill, M. Dahan, I. Foster, K. Gaither and A. Grimshaw, et al., XSEDE: Accelerating scientific discovery, Comput. Sci. Eng., 2014, 16, 62–74, DOI:10.1109/MCSE.2014.80.
| This journal is © the Owner Societies 2025 |