
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Accurate prediction of the energies of the lowest excited states, S1, T1, and T2, of chromophores for improving solar cell applications†
Muhammet Erkan Köseab,
Roshan Khatri a and
Barry D. Dunietz*a
a and
Barry D. Dunietz*a
aKent State University, Department of Chemistry and Biochemistry, Kent, OH 44242, USA. E-mail: bdunietz@kent.edu
bDepartment of Chemistry, Kocaeli University, Izmit, Kocaeli 41001, Turkey
First published on 1st July 2025
Abstract
The lowest singlet excited state and triplet states of acene and polyaromatic hydrocarbon derivatives are calculated using a screened range separated hybrid functional within a polarizable continuum model (SRSH-PCM). Excited state energies are obtained at the time-dependent density functional theory (TDDFT) and the Tamm–Dancoff approximation (TDA) levels. The SRSH-PCM electronic structure protocol successfully incorporates the effect of the electrostatic environment of an active molecule. SRSH-PCM TDDFT excitation energies present a significantly decreased averaged deviation from relevant experimental benchmark energies in comparison to TDA energies. In particular, the energies of the two lowest lying excited triplet states, T1 and T2, are predicted with an average error of 0.06 eV and that of the lowest singlet state, S1, with an average error of 0.11 eV for a molecular test set following a linear fit correction based on a benchmark set. The predictive description of the excited states can be achieved only by properly incorporating effects of the dielectric medium as accomplished by the SRSH-PCM approach. The results highlight the prospect of using SRSH-PCM to uncover molecules bearing optimal properties for singlet fission or triplet–triplet annihilation upconversion applications, where the conditions addressing the energies of these states must be satisfied.
1 Introduction
Photovoltaic cells based on organic semiconducting materials continue to draw research activity to materialize the promise of achieving economical and efficient energy production.1 The technology, however, is fundamentally limited by the Shockley–Queisser relationship,2 with functionality capped by energy loss through competing relaxation processes and material absorption cross-section. To overcome these limiting relationships large scale research efforts pursue the design of materials that invoke complementary photophysical processes such as triplet–triplet annihilation upconversion (TTA-UC)3 and singlet fission (SF).4,5 Furthermore, the design of materials to control and tune triplet excitations can impact such energy generation applications. Light generation applications can also be improved through involving low lying triplet excitations in optoelectronic processes such as thermally activated delayed fluorescence.6–9Through such mechanisms of TTA-UC and SF the material absorption spectral range can be extended.10 Specifically, efficient TTA-UC requires that an intermediate triplet excitation state, T1, energy is higher than or equal to the energy of the absorbing lowest singlet states, S1, where 2 × T1 ≥ S1. Efficient SF, on the other hand, requires that the energy of the S1 state is higher than or equal to the formed triplets, where S1 ≥ 2 × T1.11 Both processes can be limited by triplet–triplet combination to a higher triplet state, T2, and therefore require that their energies satisfy T1 ≤ T2 × 0.5.12 Computational studies can play a decisive role in research efforts to uncover materials that bear excited states that meet these requirements for the excited state energies.
A computational approach of predictive quality is required to impact molecular design efforts.13 For achieving the predictive quality the approach must tackle well challenges in resolving structure–function relationships affecting relevant energy criteria.14 Toward this goal, excited state calculations must go beyond gas phase descriptions, reliably addressing the dielectric environment and molecular packing interactions.12,15,16 In recent years, a DFT framework that addresses the dielectric environment has been developed and benchmarked. In particular, a screened range separated hybrid (SRSH) functional within the polarizable continuum model (PCM), denoted as SRSH-PCM, has been shown to predict energies of frontier orbitals,17 excited states,18–20 and charge transfer (CT) states21 in a condensed phase remarkably well. The SRSH-PCM approach accounts for the effects of the dielectric environment of organic materials in crystal and solution phases, establishing a high quality and efficient approach to predict excited state energies of organic semiconductors in single molecule calculations.8,17,19,22
We point out that the noted energetic relationships determining the SF and TTA-UC efficiencies relate to low lying excited states of molecular dimers. Nevertheless, to demonstrate the potential impact on the design of materials this work focuses on calculating low lying excited states of single molecules using SRSH-PCM. Indeed, only such a framework that addresses excited state energies of molecular systems within a condensed phase at a predictive quality can be used to contribute to molecular design research. Future design efforts will use SRSH-PCM to investigate relevant molecular dimer systems,23 where intermolecular orientation effects are resolved.
In this study, SRSH-PCM is employed to calculate the lowest excited singlet state, S1 and the two lowest triplet states, T1 and T2, in organic semiconducting materials that are involved in SF and TTA processes. Fig. 1 presents a benchmark set of acene derivatives, including tetracene,24 pentacene,25 PDI,26 and DPH,27 that are widely studied in the context of SF activity. These acenes are complemented by several related molecules with reported measured excited state energies. (Molecule acronyms are introduced in the figure.) We address this set of molecules to establish the success of the calculated values to reproduce the experimental benchmark energies through a linear fit with experimental values. We then proceed and use the linear fit to further improve the agreement of calculated S1, T1, and T2 energies with experimental values of molecules outside the benchmark set. We emphasize that the linear fit parameters provide for a correction measure to improve the agreement of calculated energies with experimental values. The correction is solely based on line parameters obtained by correlating calculated values to experimental values of the benchmark set (see ESI,† Table S1 for the listed experimental excitation energies of both sets28–38).
 | ||
| Fig. 1 The training set of molecules. | ||
As shown below, SRSH-PCM calculated excitation energies are found to reproduce well the measured values, which are significantly affected by the polarizable environment. The framework's success stands on imposing meaningful dielectric screening of the electronic interactions at the long range. In particular the approach is found to predict exceptionally well energies of the two low lying triplet states,19 and especially well the important T1–T2 energy gap and the triplet states alignment relative to the lowest singlet excitation, S1. The SRSH-PCM energies are within chemical accuracy of experimental energies without any empirical fitting. The excellent linear fit between the calculated and experimental energies establishes the predictive quality of the approach and at the same time offers a means to further improve the quality of the calculated energies.
2 Computational details
In range-separated hybrid (RSH) functionals and the SRSH framework the electronic Coulomb interactions are range-partitioned using an error function in the following form:22,39
 | (1) |
 | (2) |
We then invoke the PCM and establish the SRSH-PCM level, where β is reset according to the preset α and the scalar dielectric constant associated with the environment (ε),17,22 by following the expression
| α + β = 1/ε. | (3) |
Using the range separation expression opens the opportunity to align the frontier molecular orbital (MO) energies, the highest occupied MO (HOMO) and the lowest unoccupied MO (LUMO), and match them to the ionization potential (IP) and the electron affinity (EA), respectively. In particular, the range separation parameter, ω, is chosen to minimize the error measure, J(ω),
| J(ω) = [εHOMO(ω) + EIP(ω)]2 + [εLUMO(ω) + EEA(ω)]2. | (4) |
Excited-state energies are calculated at the Tamm–Dancoff approximation (TDA) and full time-dependent density functional theory (TDDFT) levels. The effect of the dielectric environment on excited state densities is evaluated at the perturbative state-specific (ptSS) level43 for TDA states, where the electronic response is parameterized by the optical dielectric constant. The ptSS corrections are expected to increase (in absolute value) with the optical dielectric constant and with the CT character of the excited state. In this work, the corrections are found to be minor at about −0.002 eV, reflecting the relatively weak response of the solute excited electron density to the electrostatic environment. A single exception is found for the S1 state of 1-cyano naphthalene bearing significant CT character leading to a −0.07 eV ptSS correction.
In this study we employ the PBEh as the semilocal functional, therefore addressing the tuned LR-corrected (LRC) hybrid functional (ωPBEh)44 as the RSH level. All geometry optimizations of the molecules used in this study are carried out with the ωB97X-D functional45 and the cc-pVTZ basis set. The calculated excited states are obtained using the cc-pVTZ basis set. The geometry optimizations and the excited state calculations are performed using the software package Q-Chem version 6.0.2.46
The scalar dielectric constant representing the environment is set to 3.5 and the optical dielectric constant parameterizing the ptSS correction is set to 1.4. These values reflect reasonably well the non-polar solvents affecting the considered experimental benchmark data and essentially follow the frameworks affected in related benchmark studies.9,47,48 Scalar dielectric constants of organic crystals are typically in the range of 3–4.49–54 See examples of dielectric constants associated with organic materials in a molecular crystal phase listed in the ESI,† Table S2. We also address the sensitivity of the calculated excited state energies to the employed dielectric constant especially in cases of smaller molecules by resolving the excited state energy and dielectric constant relationship, demonstrating the larger dependence of the singlet state over that of triplet states.
3 Results and discussion
3.1 Representative molecule – anthracene
Trends related to the condensed phase effects parameterized by the dielectric constant in the PCM calculations are presented using anthracene as a representative system. Both ωB97X-D and the RSH predict at the gas phase IP/EA values of anthracene quite satisfactorily with matching frontier orbital energies. The RSH functional is associated with a smaller mean absolute error (MAE) between the IP/EA and frontier orbital energies, reflecting the gas phase optimally tuned ω. See the orbital energy gaps presented in the ESI,† Fig. S1. We also point out that the RSH orbital energies align well with the corresponding experimental gas phase IP55/EA56 energies.We now consider the condensed phase effects by using PCM-based energies. To highlight the effect of invoking dielectric screening we compare SRSH-PCM energies to those obtained using the corresponding unscreened RSH-PCM energies. We also consider energies based on the ωB97X-D functional used in the geometry optimizations which includes an elaborate fitting process to establish the RS parameter.57
Calculated anthracene HOMO/LUMO energies and the corresponding oxidation potentials (OPs) and reduction potentials (RPs) obtained using DFT-PCM are presented in Fig. 2 and compared to the experimentally measured values in the solid state.58 As expected, the frontier orbital energies do not match the –OP/–RP energies obtained using the unscreened functionals, ωB97X-D-PCM and RSH-PCM. Also as expected,17 by introducing screening effects in SRSH-PCM and even without affecting any further tuning the HOMO/LUMO energies nicely match the OP/RP values and appear to also match well the experimental data. Such excellent alignment of the frontier orbital energies with the OP/RP also leads to an improved match of TDDFT calculated excited state energies with measured spectral trends as demonstrated for similar systems.18,19,21
 | ||
| Fig. 2 Anthracene PCM-calculated HOMO, LUMO (H,L) energies, and –OP,–RP energies, compared to experimental solid-state data.58 Only the SRSH-PCM calculated gaps match and reproduce well the experimental solid-state data. | ||
The calculated S1, T1, and T2 energies of anthracene within the PCM and in the gas phase are presented in Fig. 3. (Excited state properties are listed in the ESI,† Table S3.) The lowest excited states in anthracene are predominantly due to HOMO → LUMO transitions (it is important to note that all lowest excitations involve HOMO–LUMO orbital replacements in this study). Consequently, the incorrectly overestimated HOMO–LUMO gaps shown in Fig. 2 using ωB97X-D and RSH in the PCM, where the gaps remain similar to the gas phase values, foreshadow the overestimated excited state energies at these levels, especially for the S1 state. Indeed, these PCM energies, without the dielectric screening in the functional, are quite similar to the gas phase energies. On the other hand, we also point out that TDA excitation energies based on such traditional functionals are known to compare better with experimental energies than those based on full TDDFT.59–62 However, this TDA trend in conventional functionals appears to benefit from fortuitous cancellation of errors associated with gas-phase underestimated orbital gaps and therefore negates the goal of predictive calculations.63 On the other hand, TDDFT in the PCM based on SRSH-PCM reproduces well measured excitation state energies, offering a predictive level of calculations. Here we find a MAE of only 0.05 eV for anthracene S1, T1, and T2 state energies, whereas all other levels are associated with larger than 0.15 eV MAEs.
 | ||
| Fig. 3 Anthracene calculated low lying excited state energies, comparing TDA and TDDFT energies to the solid-state measured energies. Lower panel: MAEs from the experimental value. The SRSH-PCM calculated excited state energies are of the smallest MAE of 0.05 eV. | ||
Before we proceed to consider the set of molecules for comprehensive benchmarking, we present the relationship of the excited state energies to the dielectric constant. The dielectric constant represents the molecular environment affecting the excited state energies. To demonstrate these trends, we list the calculated SRSH-PCM excited states within a range of dielectric constants in Table 1. The S1 energies are found to vary significantly within the range of ε by 0.2 eV, whereas the T1 and T2 energies change by less than 0.1 eV within the same range. Notably RSH-PCM energies without the dielectric screening show essentially the same energies across the considered range of dielectric constants. Such vanishing dielectric constant dependence of the RSH-PCM energies is erroneously established, especially, for the S1 state. See the RSH-PCM calculated energies across the same range of dielectric constants listed in the ESI,† Table S4.
| Solvent | S1 | T1 | T2 | |||||
|---|---|---|---|---|---|---|---|---|
| Name | ε | nD | TDA | TDDFT | TDA | TDDFT | TDA | TDDFT |
| Cyclohexane | 2.02 | 1.426 | 3.650 | 3.419 | 2.176 | 1.801 | 3.565 | 3.306 |
| Benzene | 2.27 | 1.501 | 3.624 | 3.396 | 2.171 | 1.813 | 3.565 | 3.313 |
| Solid state | 3.50 | 1.400 | 3.549 | 3.330 | 2.155 | 1.836 | 3.559 | 3.323 |
| Anisole | 4.33 | 1.517 | 3.521 | 3.305 | 2.149 | 1.843 | 3.557 | 3.327 |
| Chloroform | 4.81 | 1.446 | 3.509 | 3.295 | 2.148 | 1.850 | 3.558 | 3.332 |
| Chlorobenzene | 5.62 | 1.525 | 3.494 | 3.281 | 2.143 | 1.850 | 3.554 | 3.330 |
| THF | 7.58 | 1.407 | 3.470 | 3.260 | 2.138 | 1.855 | 3.553 | 3.333 |
| DCM | 8.93 | 1.424 | 3.459 | 3.250 | 2.136 | 1.858 | 3.552 | 3.335 |
3.2 Benchmark training set
Ground state energy gaps due to frontier orbital energies and the IP and EA are listed in Table 2 for the benchmark set of molecules shown in Fig. 1. We consider first the energy gap trends, where optimal tuning of ω leads to excellent (within 0.02 eV) correspondence of HOMO and LUMO energies to IP and EA, respectively, for all molecules, with, however, a single exception. The quinoxaline molecule presents a weaker match, presumably due to the delocalized nature of the frontier orbitals. We are pursuing means to improve RSH functional performance also for such challenging cases and plan to report such progress in future publications.| Molecule | ω | RSH | RSH-PCM | SRSH-PCM | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HOMO | –OP | LUMO | –EA | HOMO | –OP | LUMO | –RP | HOMO | –OP | LUMO | –RP | ||
| Naphthalene | 0.216 | −8.10 | −8.11 | 0.53 | 0.51 | −8.15 | −6.71 | 0.49 | −0.94 | −6.39 | −6.58 | −1.11 | −0.96 |
| Anthracene | 0.189 | −7.28 | −7.29 | −0.33 | −0.33 | −7.33 | −6.04 | −0.36 | −1.64 | −5.79 | −5.93 | −1.79 | −1.67 |
| Tetracene | 0.170 | −6.74 | −6.75 | −0.91 | −0.90 | −6.78 | −5.61 | −0.94 | −2.11 | −5.24 | −5.35 | −2.37 | −2.27 |
| Pentacene | 0.156 | −6.35 | −6.36 | −1.33 | −1.32 | −6.39 | −5.31 | −1.36 | −2.43 | −5.12 | −5.20 | −2.56 | −2.48 |
| Perylene | 0.177 | −6.87 | −6.86 | −0.70 | −0.72 | −6.92 | −5.75 | −0.74 | −1.93 | −5.51 | −5.62 | −2.06 | −1.97 |
| PDI | 0.156 | −7.82 | −7.81 | −2.46 | −2.45 | −7.62 | −6.54 | −2.27 | −3.29 | −6.35 | −6.44 | −3.43 | −3.33 |
| Quinoxaline | 0.242 | −9.17 | −9.36 | −0.26 | −0.35 | −9.19 | −7.94 | −0.29 | −1.87 | −7.25 | −7.57 | −2.01 | −1.83 |
| DPB | 0.164 | −7.21 | −7.23 | −0.38 | −0.37 | −7.24 | −6.08 | −0.40 | −1.56 | −5.83 | −5.95 | −1.72 | −1.63 |
| DPH | 0.156 | −6.93 | −6.94 | −0.65 | −0.65 | −6.95 | −5.85 | −0.66 | −1.76 | −5.62 | −5.72 | −1.91 | −1.83 |
| (E)-Stilbene | 0.178 | −7.57 | −7.58 | −0.07 | −0.08 | −7.61 | −6.36 | −0.10 | −1.35 | −6.08 | −6.22 | −1.52 | −1.41 |
| Chrysene | 0.176 | −7.52 | −7.52 | 0.01 | −0.01 | −7.57 | −6.37 | −0.03 | −1.24 | −6.09 | −6.21 | −1.41 | −1.31 |
| 1-Cyano naphthalene | 0.206 | −8.57 | −8.59 | −0.42 | −0.43 | −8.49 | −7.11 | −0.35 | −1.74 | −6.80 | −6.98 | −1.90 | −1.75 |
| Triphenylene | 0.176 | −7.89 | −7.88 | 0.33 | 0.31 | −7.94 | −6.73 | 0.28 | −0.94 | −6.44 | −6.56 | −1.11 | −1.01 |
| 1-Methoxy naphthalene | 0.202 | −7.58 | −7.58 | 0.71 | 0.70 | −7.67 | −6.28 | 0.61 | −0.77 | −5.98 | −6.16 | −0.94 | −0.80 |
We address next energies associated with environment effects using PCM calculations. Table 2 lists the calculated energies at the RSH-PCM and SRSH-PCM levels. At the RSH-PCM level the HOMO/LUMO energies remain similar to the gas-phase values.17 The calculated OP and RP, on the other hand, are strongly affected, again as expected, by the polarizable embedding of the solute. Consequently, the energy differences between the HOMO and –OP and between the LUMO and –RP are significant.
At the SRSH-PCM level, on the other hand, the frontier orbital energies realign within 0.15 eV of the –OP/–RP values and notably even without retuning in the PCM. Quinoxaline presents again an exception reflecting the weaker mismatch at the gas phase, where the SRSH-PCM HOMO energy deviates by 0.3 eV from the –OP. Here, we note that retuning ω in the PCM does not improve error measure for orbital energy matching to IP/EA for quinoxaline. Indeed, this trend of the electronic density response to the environment depends and may vary with the size of the molecule and the degree of delocalization of the frontier orbitals.
The SRSH-PCM calculated excited state energies are presented in Fig. 4 in the form of correlation plots to the experimental energies at both the TDDFT and TDA levels. The energies are listed in the ESI,† Table S5. (For completeness, RSH-PCM excitation energies and their correlation plots are provided in the ESI,† Table S6 and Fig. S2, respectively.) More specifically, the calculated energies of each state are fitted to the experimental values, establishing a linear relationship, Eexp = mEcalc + n. The line equations for S1, T1, and T2 states at the TDDFT-SRSH-PCM level are as follows:
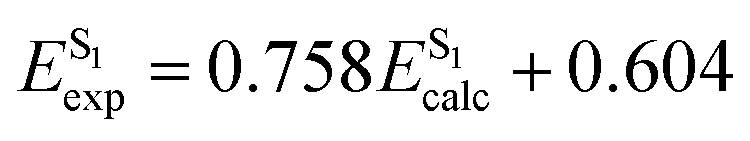 | (5) |
 | (6) |
 | (7) |
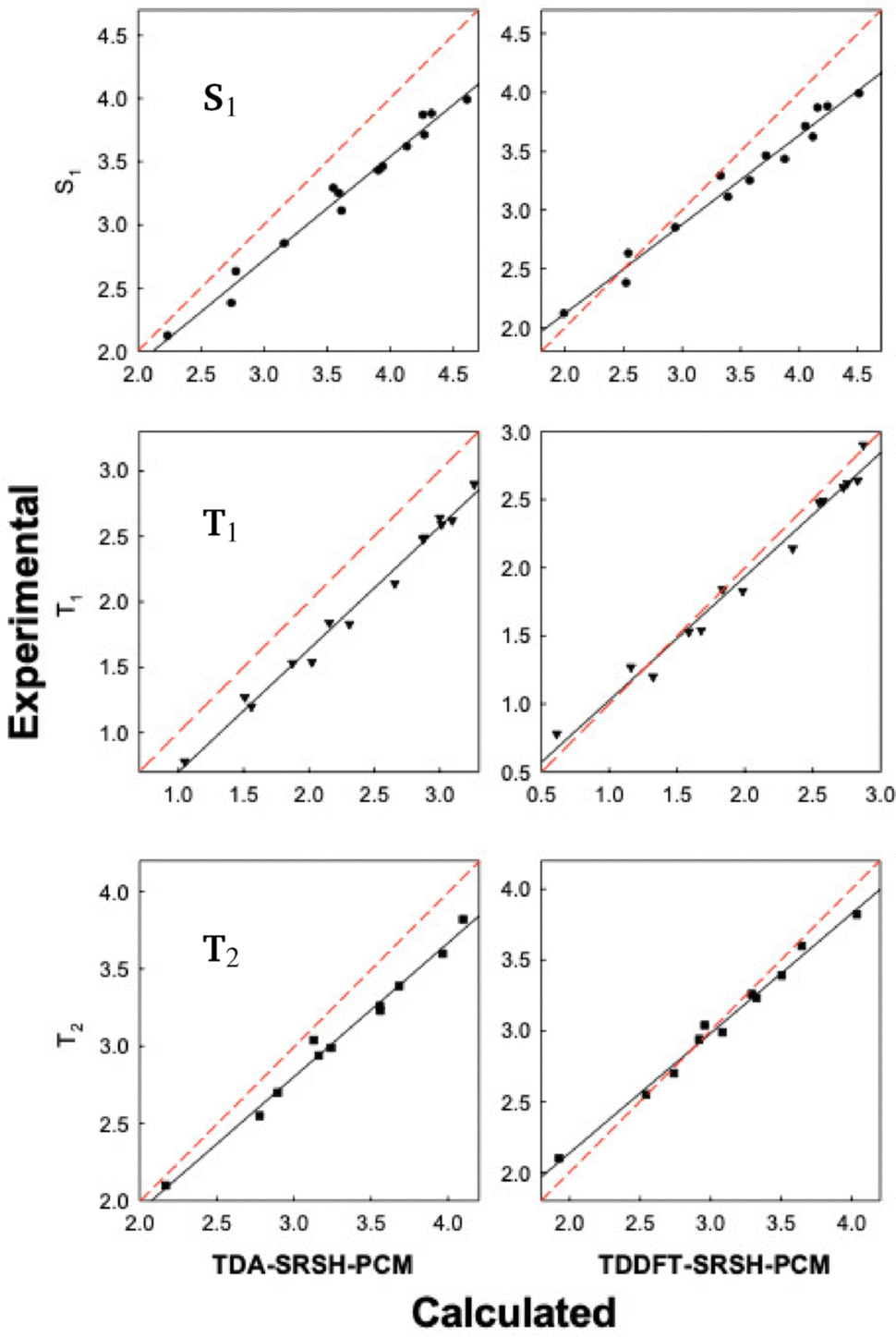 | ||
| Fig. 4 Correlation plots of the calculated excitation energies (eV) vs. the experimental data for the training set molecules (Fig. 1). Red dashed lines of y = x lines represent the perfect fit for comparison. | ||
The linear fit parameters of TDA and TDDFT excited state energies along with the MAEs are listed in Table 3, where we include those based on the RSH-PCM energies as well. Overall, the TDDFT energies present a closer match with experimental values than the TDA energies with significantly smaller MAEs. TDA energies present correlation plots with larger slopes and smaller intercept values than TDDFT. Namely, the TDA tendency to overpredict the excitation energy increases with the energy. TDDFT is found to underpredict the experimental values only at sufficiently low excitation energies. To better gauge the quality of the linear fit we also list the coefficient of determination, R2, which is 1.0 in the case of a perfect fit. Notably, all plots are associated with good fits finding R2 values larger than 0.96. Regardless of whether TDA or TDDFT is used, the SRSH-PCM approach performs much better than RSH-PCM, reflecting the excellent match of frontier orbital energy levels with the corresponding –OP and –RP values.
| Method | S1 | T1 | T2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m | n | R2 | MAE | m | n | R2 | MAE | m | n | R2 | MAE | |
| TDA-RSH-PCM | 0.832 | 0.001 | 0.996 | 0.66 | 0.935 | −0.308 | 0.987 | 0.47 | 0.853 | 0.193 | 0.994 | 0.30 |
| TDA-SRSH-PCM | 0.819 | 0.266 | 0.981 | 0.40 | 0.937 | −0.236 | 0.989 | 0.39 | 0.867 | 0.200 | 0.987 | 0.24 |
| TDDFT-RSH-PCM | 0.760 | 0.405 | 0.982 | 0.50 | 0.864 | 0.331 | 0.961 | 0.11 | 0.804 | 0.579 | 0.974 | 0.10 |
| TDDFT-SRSH-PCM | 0.758 | 0.604 | 0.975 | 0.29 | 0.908 | 0.118 | 0.981 | 0.11 | 0.845 | 0.448 | 0.988 | 0.08 |
More specifically, the TDDFT-SRSH-PCM MAEs for the experimental energies for T1 and T2 energies are 0.11 and 0.08 eV, respectively, smaller than that of 0.29 eV for S1. The relative, while systematic, larger deviation found for the S1 energies reflects the tendency to further overestimate with the increase of the excitation energy. We emphasize first that the S1 energies with the linear fit correction are also within chemical accuracy of 0.1 eV deviation. We relate the overestimated S1 energies to the imposed fixed dielectric constant, where the S1 states present stronger dependence on the dielectric constant than the triplet excitations as demonstrated above in Table 1. The S1 energies are found to vary significantly within the range of ε, for example by 0.2 eV for anthracene, whereas the T1 and T2 energies change by less than 0.1 eV within the same range. In addition, we emphasize that significantly overestimated S1 is indicated only for the smaller molecules with the larger excitation energies. Indeed, the smaller molecules typically require tuning within the PCM or even present a challenge for optimal tuning as noted above for quinoxaline. Indeed, as shown in Table 2, the naphthalene case presents a HOMO energy that indicates a 0.2 eV overestimated IP and one of the more overestimated S1 energies, whereas the larger acenes (anthracene through pentacene) are associated with smaller deviations in both the HOMO from IP and in the calculated S1 energy from the experimental value. Investigating more precisely the smaller molecules establishes a refinement or a challenge that can be addressed in future studies.
For the triplet states, we found a different trend than for the singlet energies. Here the calculated energies show a similar deviation across the considered set of molecules. This reflects success in addressing the challenge of triplet states in the condensed phase and where the triplet states bear a weaker dependence on the polarizing environment than indicated for the singlet states.9,19,64,65
The absence of a transition dipole moment in spin-forbidden vertical transitions for triplet states leads to weak interaction with the polarized solute environment and therefore weaker solvent dependence.
3.3 Molecular test set
We next test the applicability of the TDDFT-SRSH-PCM method, where we also validate the linear fits using the set of molecules shown in Fig. 5. The calculated excited state energies are compared to the experimental data,28–37 presenting a MAE of 0.28 eV for S1, 0.09 eV for T1, and 0.12 eV for T2 states; see the energies listed in Table 4. Such MAEs are quite similar to the values found for the training set listed above in Table 3, confirming a statistically meaningful data set. As established above, improvement of the calculated energies, Ecalc, in reproducing the experimental energies within 0.1 eV accuracy is achieved using the linear fit parameters provided above in eqn (5)–(7). The linear-fit corrected SRSH-PCM excited state S1, T1, and T2 energies present significantly smaller MAEs of 0.06 eV for T1 and T2 and 0.11 eV for S1. The SRSH-PCM energies and linear fit corrected energies for the test set molecules are presented in the form of correlation plots in the ESI,† Fig. S3. | ||
| Fig. 5 The test set of molecules, where we refer to MCT as 5-chlorotetracene, DCT as 5,11-dichlorotetracene, and TCT as 5,6,11,12-tetrachlorotetracene. | ||
| Molecule | RS | SRSH-PCM | Corrected | ||||
|---|---|---|---|---|---|---|---|
| (ω) | S1 | T1 | T2 | S1 | T1 | T2 | |
| Pyrene | 0.190 | 3.81 | 2.18 | 3.51 | 3.49 | 2.10 | 3.41 |
| Phenanthrene | 0.189 | 4.10 | 2.77 | 3.53 | 3.71 | 2.63 | 3.43 |
| Porphyrin | 0.164 | 2.34 | 1.48 | 1.82 | 2.38 | 1.46 | 1.99 |
| Dibenzofuran | 0.198 | 4.58 | 3.21 | 3.71 | 4.08 | 3.03 | 3.58 |
| Phenazine | 0.198 | 3.12 | 1.93 | 2.62 | 2.97 | 1.87 | 2.66 |
| TCT | 0.154 | 2.21 | 0.87 | 2.35 | 2.28 | 0.91 | 2.43 |
| DCT | 0.160 | 2.41 | 1.07 | 2.45 | 2.43 | 1.09 | 2.52 |
| MCT | 0.164 | 2.48 | 1.12 | 2.50 | 2.48 | 1.14 | 2.56 |
| MAE | — | 0.28 | 0.09 | 0.12 | 0.11 | 0.06 | 0.06 |
The energies listed in Table 4 indicate that TCT, DCT, and MCT molecules have excited states energy alignment that satisfies the requirement for effective SF. In particular, TCT with the lowest band gap among the considered chlorinated tetracene derivatives stands out as a promising candidate for the SF application and therefore for potentially enhancing device efficiencies in photovoltaics.
3.4 Discussion
Design and synthesis of novel molecules, characterization, and the subsequent testing in applications such as SF or TTA-UC tend to be quite expensive and labor-intensive.10,11,66 Uncovering optimal molecules for these applications is hindered by the difficulty in resolving the T2 states. There are only a few molecules for which T2 state energies are reported. To address these challenges, computational studies of the excitation energies can significantly impact molecular design efforts. Such studies, however, to be truly predictive and impactful must account for the condensed phase environment even in cases of non-polar molecules and environments.In this regard, SRSH-PCM has evolved as a powerful approach achieving accurate condensed phase-affected excited state energies in single molecule calculations.17,19,20,67,68 In this study we use this polarization consistent framework to demonstrate the predictive quality of the SRSH-PCM framework. Such predictive quality is necessary for reliably highlighting molecules that satisfy either SF or TTA-UC requirements affecting the excited state energies.
This work shows that SRSH-PCM can be used to predict the energies of T1 and T2 states reliably with MAE < 0.1 eV and S1 with MAE < 0.3 eV, noting the larger deviations found for the relatively smaller molecules. Importantly, the calculated SRSH-PCM TDDFT energies can be improved to offer chemical accuracy of less than 0.1 eV deviation also for S1 energies by using linear fit correction (the line fit parameters obtained using the benchamrk set are provided for general utility). This trend is especially impressive, given the moderate cost of SRSH-PCM bearing single molecule calculations and corrections based on a simple linear fit. As noted, the S1 energies present a significant overestimated energy only for the higher energies associated with the smaller molecules. These molecules, while tending to be less relevant as potential candidates for optoelectronic applications, may benefit from pursuing optimal tuning in the PCM.
We also point out that intermolecular interactions in a crystalline environment can affect excited state energies while neglected in a scheme that represents the environment as a dielectric continuum. To fully address such molecular-specific environment effects requires sophisticated embedding protocols as recently reported for the SRSH framework.67 Nevertheless, SRSH-PCM has emerged as a highly effective framework to achieve predictive calculations. SRSH-PCM, therefore, bears potential to impact experimental efforts to design molecules with optimal properties for SF and TTA-UC applications. Future studies can readily explore dimers made of candidate molecules as highlighted in this study (TCT, DCT, and MCT) at varied orientations to consider their applicability through the highly efficient SRSH-PCM calculations. We point out that we do not expect that calculated excitation energies based on a single molecule will significantly be affected by the required ω retuning. Indeed in foreseeing calculation of these excited states in dimer systems, a decrease in the dimer tuned ω is projected. We confirm that recalculating the excited states using such a smaller ω parameter by an assumed 10% decrease results in only a negligible change in the excitation energies; see examples in the ESI,† Table S7.
4 Conclusion
In summary, S1, T1, and T2 state energies of polyaromatic hydrocarbon derivatives are successfully predicted using TDDFT with a SRSH functional within the PCM. The SRSH-PCM framework accounts exceptionally well for effects of the polarizable environment on the electronic density of the embedded molecule. The excited state energies calculated using TDA yield a slightly better correlation against the experiment than TDDFT. Nonetheless, comparison of calculated excited state energies with experiment reveals that TDDFT-SRSH-PCM stands out by accurately predicting S1, T1, and T2 energies with MAEs smaller than 0.1 eV. The linear fitting formulas obtained at the TDDFT-SRSH-PCM level in this work can be applied to polyaromatic hydrocarbon derivatives to further improve calculated predictions of excited state energies. Therefore, SRSH-PCM has emerged as an effective framework with potential to impact efforts to design materials suitable for SF or TTA-UC applications, and therefore promise to reduce the time, cost, and efforts in related experimental studies. We, in particular, highlight TCT as a potential candidate for efficient SF applications.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI† that provides the following: experimental S1, T1, and T2 energies for molecules shown in Scheme S1 (ESI†); excited state energies, orbital transitions, and oscillator strengths of the transitions with various methods for anthracene; calculated RSH-PCM excited state energies within different solvents for tetracene; excited state energies obtained using RSH-PCM and SRSH-PCM for S1, T1, and T2 at TDA and full TDDFT levels; and comparison of calculated frontier orbital energies with IP/EA energies for ωB97X-D and RSH-GAS methods.Acknowledgements
This work was supported by the Basic Energy Sciences, Office of Science, U.S. Department of Energy, award DE-SC0025404 and Kent State University.Notes and references
- C. Battaglia, A. Cuevas and S. De Wolf, Energy Environ. Sci., 2016, 9, 1552–1576 RSC.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- L. Naimovicius, P. Bharmoria and K. Moth-Poulsen, Mater. Chem. Front., 2023, 7, 2297–2315 RSC.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- D. Barman, K. Narang, R. Gogoi, D. Barman and P. K. Iyer, J. Mater. Chem. C, 2022, 10, 8536–8583 RSC.
- S. E. Seo, H.-S. Choe, H. Cho, H.-I. Kim, J.-H. Kim and O. S. Kwon, J. Mater. Chem. C, 2022, 10, 4483–4496 RSC.
- B. Mandal and B. D. Dunietz, J. Phys. Chem. A, 2023, 127, 216–223 CrossRef CAS PubMed.
- R. Khatri and B. D. Dunietz, J. Phys. Chem. C, 2025, 129, 436–446 CrossRef CAS.
- S. Ito, T. Nagami and M. Nakano, Phys. Rep., 2018, 34, 85–120 CAS.
- A. J. Carrod, V. Gray and K. Boerjesson, Energy Environ. Sci., 2022, 15, 4982–5016 RSC.
- D. Padula, O. Omar, T. Nematiaram and A. Troisi, Energy Environ. Sci., 2019, 12, 2412–2416 RSC.
- X. P. Wang, S. Y. Gao, Y. Q. Luo, X. Y. Liu, R. Tom, K. J. Zhao, V. Chang and N. Marom, J. Chem. Phys., 2024, 128, 7841–7864 CAS.
- R. Grotjahn, T. M. Maier, J. Michl and M. Kaupp, J. Chem. Theory Comput., 2017, 13, 4984–4996 CrossRef CAS PubMed.
- C. F. Perkinson, D. P. Tabor, M. Einzinger, D. Sheberla, H. Utzat, T. A. Lin, D. N. Congreve, M. G. Bawendi, A. Aspuru-Guzik and M. A. Baldo, J. Chem. Phys., 2019, 151, 121102 CrossRef PubMed.
- D. López-Carballeira and T. Polcar, J. Comput. Chem., 2021, 42, 2241–2249 CrossRef PubMed.
- S. Bhandari, M. Cheung, E. Geva, L. Kronik and B. D. Dunietz, J. Chem. Theory Comput., 2018, 14, 6287–6294 CrossRef CAS PubMed.
- H. Aksu, A. Schubert, E. Geva and B. D. Dunietz, J. Phys. Chem. B, 2019, 123, 8970–8975 CrossRef CAS PubMed.
- K. Begam, S. Bhandari, B. Maiti and B. D. Dunietz, J. Chem. Theory Comput., 2020, 16, 3287 CrossRef CAS PubMed.
- C. Chakravarty, H. Aksu, J. A. Martinez, P. Ramos, M. Pavanello and B. D. Dunietz, J. Phys. Chem. Lett., 2022, 13, 4849 CrossRef CAS PubMed.
- S. Bhandari and B. D. Dunietz, J. Chem. Theory Comput., 2019, 15, 4305 CrossRef CAS PubMed.
- R. Khatri and B. D. Dunietz, J. Chem. Phys., 2023, 159, 071103 CrossRef CAS PubMed.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, K. Johnson, S. Gélinas, R. di Pietro, J. Clark and R. H. Friend, J. Am. Chem. Soc., 2013, 135, 16680–16688 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, B. Ehrler and R. H. Friend, Acc. Chem. Res., 2013, 46, 1330–1338 CrossRef CAS PubMed.
- C. Schierl, A. Niazov-Elkan, L. J. W. Shimon, Y. Feldman, B. Rybtchinski and D. M. Guldi, Nanoscale, 2018, 10, 20147–20154 RSC.
- O. Millington, S. Montanaro, A. Leventis, A. Sharma, S. A. Dowland, N. Sawhney, K. J. Fallon, W. X. Zeng, D. G. Congrave, A. J. Musser, A. K. Rao and H. Bronstein, J. Am. Chem. Soc., 2023, 125, 2499–2510 CrossRef PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handb. Photochem., Taylor & Francis Group, LLC, Boca Raton, 3rd edn, 2006 Search PubMed.
- C. C. Ladwig and R. S. H. Liu, Chem. Phys. Lett., 1975, 35, 563–565 CrossRef CAS.
- R. E. Kellogg, J. Chem. Phys., 1966, 44, 411–412 CrossRef CAS.
- C. Burgdorff, T. Kircher and H. G. Lohmannsroben, Spectrochim. Acta, Part A, 1988, 44, 1137–1141 CrossRef.
- B. J. Baer and E. L. Chronister, J. Chem. Phys., 1994, 100, 23–27 CrossRef CAS.
- F. Lewitzka, H. G. Lohmannsroben, M. Strauch and W. Luttke, J. Photochem. Photobiol., A, 1991, 61, 191–200 CrossRef CAS.
- R. Ahmed and A. K. Manna, J. Phys. Chem., 2022, 126, 6594–6603 CrossRef CAS PubMed.
- H. Agren, B. F. Minaev and S. Knuts, J. Phys. Chem., 1994, 98, 3943–3949 CrossRef.
- F. Vogeler, S. Siegert, C. M. Marian and R. Weinkauf, ChemPhysChem, 2011, 12, 1948–1956 CrossRef CAS PubMed.
- N. Nishi, K. Matsui, M. Kinoshita and J. Higuchi, Mol. Phys., 1979, 38, 1–24 CrossRef CAS.
- C. Burgdorff, S. Ehrhardt and H. G. Lohmannsroben, J. Phys. Chem., 1991, 95, 4246–4249 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- H. T. Sun, S. Ryno, C. Zhong, M. K. Ravva, Z. R. Sun, T. Körzdörfer and J. L. Brédas, J. Chem. Theory Comput., 2016, 12, 2906–2916 CrossRef CAS PubMed.
- S. Refaely-Abramson, S. Sharifzadeh, N. Govind, J. Autschbach, J. B. Neaton, R. Baer and L. Kronik, Phys. Rev. Lett., 2012, 109, 226405 CrossRef PubMed.
- D. A. Egger, S. Weissman, S. Refaely-Abramson, S. Sharifzadeh, M. Dauth, R. Baer, S. Kümmel, J. B. Neaton, E. Zojer and L. Kronik, J. Chem. Theory Comput., 2014, 10, 1934–1952 CrossRef CAS PubMed.
- Z. Q. You, J. M. Mewes, A. Dreuw and J. M. Herbert, J. Chem. Phys., 2015, 143, 204104 CrossRef PubMed.
- M. A. Rohrdanz, K. M. Martins and J. M. Herbert, J. Chem. Phys., 2009, 130, 054112 CrossRef PubMed.
- T. Körzdörfer and J. L. Brédas, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- E. Epifanovsky, A. T. B. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff, Z. Gan, D. Hait, P. R. Horn, L. D. Jacobson, I. Kaliman, J. Kussmann, A. W. Lange, K. U. Lao, D. S. Levine, J. Liu, S. C. McKenzie, A. F. Morrison, K. D. Nanda, F. Plasser, D. R. Rehn, M. L. Vidal, Z.-Q. You, Y. Zhu, B. Alam, B. J. Albrecht, A. Aldossary, E. Alguire, J. H. Andersen, V. Athavale, D. Barton, K. Begam, A. Behn, N. Bellonzi, Y. A. Bernard, E. J. Berquist, H. G. A. Burton, A. Carreras, K. Carter-Fenk, R. Chakraborty, A. D. Chien, K. D. Closser, V. Cofer-Shabica, S. Dasgupta, M. de Wergifosse, J. Deng, M. Diedenhofen, H. Do, S. Ehlert, P.-T. Fang, S. Fatehi, Q. Feng, T. Friedhoff, J. Gayvert, Q. Ge, G. Gidofalvi, M. Goldey, J. Gomes, C. E. Gonzalez-Espinoza, S. Gulania, A. O. Gunina, M. W. D. Hanson-Heine, P. H. P. Harbach, A. Hauser, M. F. Herbst, M. Hernandez Vera, M. Hodecker, Z. C. Holden, S. Houck, X. Huang, K. Hui, B. C. Huynh, M. Ivanov, A. Jasz, H. Ji, H. Jiang, B. Kaduk, S. Kähler, K. Khistyaev, J. Kim, G. Kis, P. Klunzinger, Z. Koczor-Benda, J. H. Koh, D. Kosenkov, L. Koulias, T. Kowalczyk, C. M. Krauter, K. Kue, A. Kunitsa, T. Kus, I. Ladjanszki, A. Landau, K. V. Lawler, D. Lefrancois, S. Lehtola, R. R. Li, Y.-P. Li, J. Liang, M. Liebenthal, H.-H. Lin, Y.-S. Lin, F. Liu, K.-Y. Liu, M. Loipersberger, A. Luenser, A. Manjanath, P. Manohar, E. Mansoor, S. F. Manzer, S.-P. Mao, A. V. Marenich, T. Markovich, S. Mason, S. A. Maurer, P. F. McLaughlin, M. F. S. J. Menger, J.-M. Mewes, S. A. Mewes, P. Morgante, J. W. Mullinax, K. J. Oosterbaan, G. Paran, A. C. Paul, S. K. Paul, F. Pavosevic, Z. Pei, S. Prager, E. I. Proynov, A. Rak, E. Ramos-Cordoba, B. Rana, A. E. Rask, A. Rettig, R. M. Richard, F. Rob, E. Rossomme, T. Scheele, M. Scheurer, M. Schneider, N. Sergueev, S. M. Sharada, W. Skomorowski, D. W. Small, C. J. Stein, Y.-C. Su, E. J. Sundstrom, Z. Tao, J. Thirman, G. J. Tornai, T. Tsuchimochi, N. M. Tubman, S. P. Veccham, O. Vydrov, J. Wenzel, J. Witte, A. Yamada, K. Yao, S. Yeganeh, S. R. Yost, A. Zech, I. Y. Zhang, X. Zhang, Y. Zhang, D. Zuev, A. Aspuru-Guzik, A. T. Bell, N. A. Besley, K. B. Bravaya, B. R. Brooks, D. Casanova, J.-D. Chai, S. Coriani, C. J. Cramer, G. Cserey, A. E. DePrince, R. A. DiStasio, A. Dreuw, B. D. Dunietz, T. R. Furlani, W. A. Goddard, S. Hammes-Schiffer, T. Head-Gordon, W. J. Hehre, C.-P. Hsu, T.-C. Jagau, Y. Jung, A. Klamt, J. Kong, D. S. Lambrecht, W. Liang, N. J. Mayhall, C. W. McCurdy, J. B. Neaton, C. Ochsenfeld, J. A. Parkhill, R. Peverati, V. A. Rassolov, Y. Shao, L. V. Slipchenko, T. Stauch, R. P. Steele, J. E. Subotnik, A. J. W. Thom, A. Tkatchenko, D. G. Truhlar, T. Van Voorhis, T. A. Wesolowski, K. B. Whaley, H. L. Woodcock, P. M. Zimmerman, S. Faraji, P. M. W. Gill, M. Head-Gordon, J. M. Herbert and A. I. Krylov, J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- L. Kunze, A. Hansen, S. Grimme and J. Mewes, J. Phys. Chem. Lett., 2021, 12, 8470–8480 CrossRef CAS PubMed.
- T. Froitzheim, S. Grimme and J. Mewes, J. Chem. Theory Comput., 2022, 18, 7702–7713 CrossRef CAS PubMed.
- L. Packman, N. Mallo, A. Raynor, M. L. Gao, M. Babazadeh, H. Jin, D. M. Huang, P. L. Burn, I. R. Gentle and P. E. Shaw, Phys. Chem. Chem. Phys., 2023, 25, 23867–23878 RSC.
- D. Datta and S. Kumar, Sol. Energy Mater. Sol. Cells, 2010, 94, 420–424 CrossRef CAS.
- J. Brebels, J. V. Manca, L. Lutsen, D. Vanderzande and W. Maes, J. Mater. Chem., 2017, 5, 24037–24050 RSC.
- K. Ishii, M. Kinoshita and H. Kuroda, Bull. Chem. Soc. Jpn., 2006, 46, 3385–3391 CrossRef.
- C. H. Kim, O. Yaghmazadeh, D. Tondelier, Y. B. Jeong, Y. Bonnassieux and G. Horowitz, J. Appl. Phys., 2011, 109, 083710 CrossRef.
- U. Kaatze, R. Pottel and P. Schmidt, J. Phys. Chem., 1988, 92, 3669–3674 CrossRef CAS.
- J. W. Hager and S. C. Wallace, Anal. Chem., 1988, 60, 5–10 CrossRef CAS.
- N. Ando, M. Mitsui and A. Nakajima, J. Chem. Phys., 2007, 127, 234305 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- P. I. Djurovich, E. I. Mayo, S. R. Forrest and M. E. Thompson, Org. Electron., 2009, 10, 515–520 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 302, 375–382 CrossRef CAS.
- M. J. Peach, M. J. Williamson and D. J. Tozer, J. Chem. Theory Comput., 2011, 7, 3578–3585 CrossRef CAS PubMed.
- M. J. Peach and D. J. Tozer, J. Phys. Chem. A, 2012, 116, 9783–9789 CrossRef CAS PubMed.
- Y. Shao, Y. Mei, D. Sundholm and V. R. I. Kaila, J. Chem. Theory Comput., 2020, 16, 587–600 CrossRef PubMed.
- M. Zheng, Y. Li, Y. Wei, L. Chen, X. Zhou and S. Liu, J. Phys. Chem. Lett., 2022, 13, 2507–2515 CrossRef CAS PubMed.
- S. Sarkar, H. P. Hendrickson, D. Lee, F. Devine, J. Jung, E. Geva, J. Kim and B. D. Dunietz, J. Phys. Chem. C, 2017, 121, 3771–3777 CrossRef CAS.
- K. Begam, H. Aksu and B. D. Dunietz, J. Phys. Chem. B, 2024, 128, 4315–4324 CrossRef CAS PubMed.
- T. Ullrich, P. Pinter, J. Messelberger, P. Haines, R. Kaur, M. M. Hansmann, D. Munz and D. M. Guldi, Angew. Chem., Int. Ed., 2020, 59, 7906–7914 CrossRef CAS PubMed.
- C. Chakravarty, H. Aksu, B. Maiti and B. D. Dunietz, J. Phys. Chem. A, 2021, 125, 7625–7632 CrossRef CAS PubMed.
- H. Aksu, A. Schubert, S. Bhandari, A. Yamada, E. Geva and B. D. Dunietz, J. Phys. Chem. B, 2020, 124, 1987–1994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp02127h |
| This journal is © the Owner Societies 2025 |