
Harnessing electrochemical CO2 reduction and assisted water electrolysis via constrained thermodynamic modeling†
Jinuk
Choi‡
 a,
Hyojung
Lim‡
a,
Subramani
Surendran
a,
Seonghyeon
Park
a,
Junho
Shim
a,
Gyoung Hwa
Jeong
ab and
Uk
Sim
*abc
a,
Hyojung
Lim‡
a,
Subramani
Surendran
a,
Seonghyeon
Park
a,
Junho
Shim
a,
Gyoung Hwa
Jeong
ab and
Uk
Sim
*abc
aHydrogen Energy Technology Laboratory, Korea Institute of Energy Technology (KENTECH), 58330 Naju, Republic of Korea. E-mail: usim@kentech.ac.kr
bResearch Institute, NEEL Sciences, INC, Naju, Republic of Korea
cCollege of Chemistry and Chemical Engineering, Henan Key Laboratory of Function-Oriented Porous Materials, Luoyang Normal University, Luoyang 471934, China
First published on 18th June 2025
Abstract
Electrochemical CO2 reduction reaction (CO2RR) and assisted water electrolysis (AWE) using organic compounds offer promising pathways for sustainable energy conversion. However, the thermodynamic feasibility and efficiency of these processes are strongly influenced by CO2 phase transitions (both gaseous and aqueous) and operating conditions, such as temperature and pH. This study systematically examines the thermodynamic behavior of CO2RR and AWE by calculating Gibbs free energy (ΔG), enthalpy (ΔH), and theoretical potentials (ETN and ERE) over a broad temperature range (0–1000 °C) and varying pH conditions. Pourbaix diagrams for key CO2-derived products, including CO, hydrocarbons, organic acids, and alcohols, are constructed to assess their stability across different electrochemical environments. The analysis reveals that in aqueous-phase CO2 systems, equilibrium potentials shift due to the effects of CO2 speciation. In alkaline conditions, dissolved CO2 undergoes sequential conversion into HCO3− and CO32−, resulting in increased overpotentials in CO2RR. Conversely, gaseous CO2 maintains a stable equilibrium potential, mitigating pH-induced fluctuations that could hinder reaction selectivity and efficiency. In AWE, the phase transition during reaction conditions lowers oxidation potentials, resulting in enhanced energy efficiency. The calculated VTN and VRE values demonstrate that organic oxidation reactions in AWE require substantially lower energy inputs than conventional oxygen evolution reactions, providing a thermodynamic advantage for energy-efficient hydrogen production. This study establishes a comprehensive thermodynamic framework for CO2 electrochemical conversion, integrating Pourbaix diagrams and temperature-dependent electrochemical modeling to optimize reaction conditions and energy efficiency. These insights contribute to the rational design of electrocatalytic systems and the development of scalable CO2 conversion technologies for industrial applications.
Introduction
The transition toward sustainable energy systems has become a critical challenge, driven by the urgent need to mitigate greenhouse gas emissions and establish carbon-neutral production pathways. Among various approaches, electrochemical CO2 conversion has gained significant attention due to its ability to integrate renewable energy sources and enable decentralized chemical synthesis of value-added carbon-based products.1 However, despite its potential, electrochemical CO2 reduction reaction (CO2RR) and assisted water electrolysis (AWE) still face significant energy efficiency and selectivity challenges.2,3Electrochemical CO2RR enables transformation of CO2 into fuels and valuable chemicals, such as carbon monoxide (CO), hydrocarbons, organic acids, and alcohols.4,5 However, its practical implementation remains hindered by high energy requirements, slow reaction kinetics, and issues with product selectivity. The efficiency and selectivity of CO2RR are strongly influenced by key thermodynamic factors, including CO2 phase transitions (gaseous vs. aqueous), temperature variations, and the composition of the electrolyte.6–8 While previous studies have explored CO2RR under different electrolyte conditions, the systematic thermodynamic impact of CO2 phase transitions and temperature changes has not been thoroughly analyzed.
AWE has been investigated as an alternative to the conventional oxygen evolution reaction (OER) to enhance the overall energy efficiency of the water electrolysis systems.9,10 In AWE, organic oxidation reactions replace OER, reducing the overall voltage requirement for hydrogen production. The feasibility of AWE depends on the oxidation potential of organic compounds, which varies with pH and temperature. While CO2RR and AWE are fundamentally distinct processes, they share common thermodynamic principles that allow a systematic comparison within the same framework.
A critical challenge in energy conversion research, including CO2RR, is shifting beyond conventional Faradaic efficiency (FE) assessments to incorporate voltage efficiency (VE) and overall energy efficiency (EE).11,12 While FE has traditionally been used to evaluate CO2RR performance, EE provides a more comprehensive metric by considering the actual electrical energy input relative to the theoretical minimum energy required. However, one significant gap in current research is the inconsistent application of theoretical potential calculations, leading to inaccurate energy efficiency estimates. Moreover, conventional Pourbaix diagrams and theoretical potential calculations are typically constructed under standard conditions (25 °C).13,14 This method fails to accurately represent the practical operating conditions in experimental and industrial scenarios, where electrochemical systems typically operate at elevated temperatures. Since reaction enthalpy (ΔH) and Gibbs free energy (ΔG) vary with temperature, ignoring temperature dependence can lead to misinterpretations of energy efficiency values, further complicating the optimization of CO2 conversion technologies.
This study extends thermodynamic Pourbaix diagram analysis to address these gaps by incorporating temperature-dependent electrochemical modeling (25–100 °C) for key organic compounds, including hydrocarbons, alcohols, and organic acids. Additionally, theoretical potentials (ETN and ERE) for CO2RR and AWE are systematically calculated over a broad temperature range (0–1000 °C). This enables a quantitative evaluation of how the CO2 phase and operating conditions impact electrochemical performance. By providing a rigorous thermodynamic framework, this study quantifies the impact of CO2 phase transitions on reaction thermodynamics, evaluates temperature-dependent shifts in equilibrium potentials across CO2RR and AWE, and establishes a systematic approach for optimizing reaction conditions in practical electrochemical systems. Ultimately, this research contributes to the development of more efficient and selective electrochemical CO2 conversion technologies by refining thermodynamic analyses, optimizing reaction conditions, and advancing practical energy efficiency strategies for sustainable energy production.
Results and discussion
Thermodynamic modelling
Thermodynamic modeling and Pourbaix diagram calculations were performed using the FactSage software package. The FactPS database was employed to determine the theoretical potential of various electrochemical reactions and to assess the formation of potential byproducts associated with electrochemical energy conversion in aqueous environments (Table S1, ESI†). This study focused on energy conversion reactions occurring in aqueous conditions, particularly CO2RR and assisted water electrolysis using organic compounds. Pourbaix diagrams were generated over a temperature range from 25 °C (room temperature) to 100 °C (the boiling point of H2O at 1 atm) to analyze the thermodynamic feasibility of these processes under different operational conditions. To improve the clarity of the Pourbaix diagrams, inevitable byproducts with limited relevance or stability under the investigated conditions were excluded from the calculations.Calculation formulas
Electrochemical reactions involving charge transfer between reactants are crucial in energy conversion applications. The feasibility of these reactions is governed by thermodynamic parameters such as ΔG, ΔH, and theoretical equilibrium voltage. This study establishes a thermodynamic framework to evaluate the feasibility of CO2RR and AWE using organic compounds under varying temperatures and pH conditions. A comprehensive understanding of these thermodynamic constraints is essential for optimizing electrochemical CO2 utilization in practical applications. A general electrochemical reaction involving multiple reactants and products is expressed as:| αA + βB → γC + δD | (1) |
The thermodynamic feasibility of a reaction is determined by its Gibbs free energy change (ΔrG°), calculated using the Gibbs free energy of formation (ΔfG°) of reactants and products under standard conditions. The equation is given by:
 | (2) |
If ΔrG° is negative, the reaction is thermodynamically favorable and can proceed spontaneously. The maximum electrical work extractable from the reaction is given by:
| ΔrG° = −nFE° | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1).
Both ΔH and ΔG are temperature-dependent. Kirchhoff's law describes the temperature dependence of reaction enthalpy, and its equation is expressed as:15
 | (4) |
If the heat capacity change ( ) is assumed to be constant, this simplifies to:
) is assumed to be constant, this simplifies to:
 | (5) |
Similarly, the Gibbs free energy at any temperature can be determined using:
 | (6) |
For CO2RR, the phase of CO2 (gas and aqueous) significantly affects reaction feasibility due to pH-dependent speciation. In aqueous systems, CO2 undergoes equilibrium with bicarbonate (HCO3−) and carbonate (CO32−), altering the effective reduction potential. The acid–base equilibrium is given by:
 | (7) |
 | (8) |
The van’t Hoff equation describes how equilibrium constants (K) shift with temperature, particularly relevant for reactions involving solubility equilibria, which is given by:15
 | (9) |
The thermodynamic reversible voltage (ERE) defines the minimum potential required for an electrochemical reaction and is calculated as:
 | (10) |
The ΔG change at 25 °C for water electrolysis is 237.1 kJ mol−1, yielding ERE = 1.23 V.
The thermoneutral voltage (ETN) accounts for total enthalpy change (ΔH°), including both electrical and thermal energy:
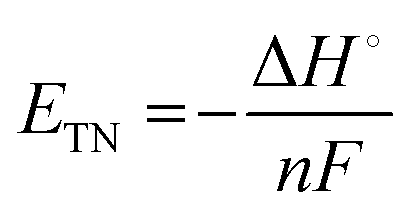 | (11) |
For water electrolysis, ΔH° = 286.0 kJ mol−1, resulting in ETN = 1.48 V. In practical applications, the actual operating voltage lies between ERE (1.23 V) and ETN (1.48 V), influenced by kinetic overpotentials and system inefficiencies.
To determine the feasibility of the reaction under non-standard conditions, the Nernst equation is applied:
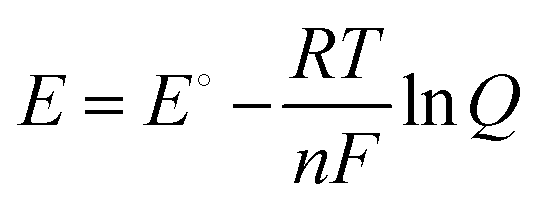 | (12) |
Q term becomes more significant, impacting equilibrium potential and selectivity.
This section presents the fundamental thermodynamic equations for evaluating electrochemical CO2 conversion and AWE under varying temperature and pH conditions. A comprehensive thermodynamic framework is developed by integrating Gibbs free energy, enthalpy, equilibrium potential, and reaction equilibria. These calculations form the foundation for the Pourbaix diagrams and theoretical potential analyses in the subsequent sections of this study.
CO2 reduction reactions to CO and hydrocarbons
The electrochemical CO2RR presents a promising pathway for converting CO2 into valuable organic compounds under sustainable conditions.16–19 The efficiency and selectivity of CO2RR are highly dependent on thermodynamic feasibility, electrolyte composition, CO2 phase (gaseous and aqueous), and operating temperature. To comprehensively assess these factors, this study constructs Pourbaix diagrams. It calculates thermodynamic voltages (ETN and ERE) for various CO2-derived products, including CO, hydrocarbons (CH4, C2H4, C2H6), organic acids (formic acid and acetic acid), and alcohols (CH3OH, CH3CH2OH, and C2H6O2).The feasibility of CO2RR is fundamentally dictated by the standard reduction potentials of key electrochemical half-reactions, which determine the minimum voltage required for CO2 conversion. A critical distinction arises between gaseous and aqueous CO2 reduction systems due to their differing equilibrium behaviors. In gaseous-phase CO2 systems, the standard reduction potentials remain independent of pH, as CO2 does not undergo phase transformation in solution. Conversely, in aqueous-phase systems, CO2 speciation shifts dynamically with pH, resulting in changes in equilibrium potential that impact reaction efficiency.
Under standard conditions, the reduction of gaseous CO2 proceeds through multiple proton–electron transfer steps, resulting in the formation of CO, CH4, C2H4, and C2H6. The equilibrium potentials for these reactions remain independent of pH, as CO2 in the gas phase does not undergo speciation. The corresponding half-reactions at standard conditions (25 °C, 1 atm) are expressed as follows:20
| CO2(g) + 2H+(aq) + 2e− ⇌ CO(g) + H2O(l), E° = −0.104 V vs. SHE | (13) |
| CO2(g) + 8H+(aq) + 8e− ⇌ CH4(g) + 2H2O(l), E° = 0.169 V vs. SHE | (14) |
| 2CO2(g) + 12H+(aq) + 12e− ⇌ C2H4(g) + 4H2O(l), E° = 0.079 V vs. SHE | (15) |
| 2CO2(g) + 14H+(aq) + 14e− ⇌ C2H6(g) + 4H2O(l), E° = 0.143 V vs. SHE | (16) |
These reactions illustrate that the electrochemical reduction of gaseous CO2 requires an external potential input to drive product formation under standard conditions. The overall energy demand is dictated by the number of protons and electrons involved, with methane production requiring the highest charge transfer. As shown in Fig. S1 (ESI†), the stability of equilibrium potentials across varying pH levels makes gaseous CO2 advantageous for maintaining consistent reaction selectivity. However, these theoretical potentials should be applied exclusively to gas-phase reactions, as aqueous systems require consideration of CO2 phase transitions and related speciation effects.
In aqueous-phase systems, CO2 undergoes pH-dependent speciation, leading to the formation of HCO3− and CO32−, which significantly impact the thermodynamics of the reduction reaction. The half-reactions for aqueous-phase CO2 reduction under standard conditions are as follows:
| CO2(aq) + 2H+(aq) + 2e− ⇌ CO(aq) + H2O(l), E° = −0.150 V vs. SHE | (17) |
| CO2(aq) + 8H+(aq) + 8e− ⇌ CH4(aq) + 2H2O(l), E° = −0.159 V vs. SHE | (18) |
| 2CO2(aq) + 12H+(aq) + 12e− ⇌ C2H4(aq) + 4H2O(l), E° = 0.083 V vs. SHE | (19) |
| 2CO2(aq) + 14H+(aq) + 14e− ⇌ C2H6(aq) + 4H2O(l), E° = 0.144 V vs. SHE | (20) |
Fig. 1 presents Pourbaix diagrams of CO and hydrocarbons (CH4, C2H4, and C2H6) across various temperatures (25–100 °C), providing insights into the pH-dependent stability of CO2 reduction products in aqueous conditions and the electrochemical potential required for selective hydrocarbon formation. The equilibrium potential for CO2 reduction in aqueous systems is strongly influenced by pH, particularly under alkaline conditions, where CO2 undergoes speciation into HCO3− and CO32−. This phase-dependent shift increases the required overpotential for CO2RR, making reduction reactions less favorable in basic environments.
 | ||
| Fig. 1 Pourbaix diagrams of hydrocarbons including (a) CO, (b) CH4, (c) C2H4, and (d) C2H6 using CO2(aq) as a function of temperature from 25 to 100 °C. | ||
In contrast, gaseous CO2 maintains stable equilibrium potentials across different pH values, as it does not undergo speciation changes (Fig. S1, ESI†). This stability enhances reaction selectivity and minimizes pH-induced fluctuations that could hinder efficiency. To account for these phase differences, Pourbaix diagrams were constructed for both gas-phase and aqueous-phase CO2 systems. The graphs illustrate key phase boundaries, showing that the pKa values for CO2(aq) to HCO3− are 6.381, 6.288, and 6.205, while those for HCO3− to CO32− are 10.329, 10.194, and 10.243 at 25 °C, 50 °C, and 100 °C, respectively. These values indicate a strong pH dependency in aqueous systems, resulting in significant shifts in equilibrium potential with increasing pH.
A comparison of gaseous and aqueous CO2 reduction underscores the importance of selecting optimal phase and electrolyte conditions to maximize conversion efficiency. While gaseous CO2 enables stable equilibrium potentials, aqueous-phase CO2 introduces pH-dependent shifts that influence reduction and oxidation pathways, affecting reaction efficiency and selectivity. The reduction potentials for CO2-to-CO conversion are −0.150 V and −1.312 V vs. SHE at pH 0 and pH 14, respectively, at 25 °C (Fig. 1(a)). As temperature increases to 100 °C, these potentials decrease further to −0.176 V and −1.640 V vs. SHE at pH 0 and pH 14, respectively. For CO2-to-CH4 reduction, the equilibrium potentials are 0.159 V at pH 0 and −0.753 V at pH 14 at 25 °C (Fig. 1(b)). These values decrease to 0.122 V and −1.021 V vs. SHE at 100 °C. Similarly, for CO2-to-C2H4 reduction, the equilibrium potentials at 25 °C are 0.083 V at pH 0 and −0.857 V at pH 14 (Fig. 1(c)), shifting further down to 0.044 V and −1.135 V at 100 °C. Lastly, for CO2-to-C2H6 reduction, the equilibrium potentials are 0.144 V at pH 0 and −0.780 V at pH 14 at 25 °C (Fig. 1(d)). These values decrease to 0.103 V and −1.056 V at 100 °C. The trends of the CO2-to-CO, CH4, C2H4, and C2H6 reactions indicate that under aqueous conditions, the theoretical reduction potentials become more hostile as pH increases after CO2 transitions to the HCO3− and CO32− phases, leading to higher overpotentials for CO2RR in neutral and alkaline environments. This increased overpotential negatively impacts the efficiency of CO2 reduction, making CO2RR less favorable under basic conditions.
Fig. 2 compares the potential gap (ECO2RR − EHER) between aqueous CO2RR and hydrogen evolution reaction (HER) across various pH and temperature conditions. The results highlight that while it thermodynamically hinders CO2-to-CO conversion under all conditions, CO2-to-CH4, –C2H4, and –C2H6 exhibit potential windows where HER competition is minimized, suggesting an optimal pH range for selective hydrocarbon formation. The CO2-to-CO reaction is unable to avoid HER competition at any pH or temperature (Fig. 2(a)). The potential gaps between CO2-to-CO and HER are −0.150 V and −0.484 V, respectively, at pH 0 and pH 14, at 25 °C. As the temperature increases to 100 °C, these potential gaps further decrease to −0.176 V and −0.604 V at pH 0 and pH 14, respectively.
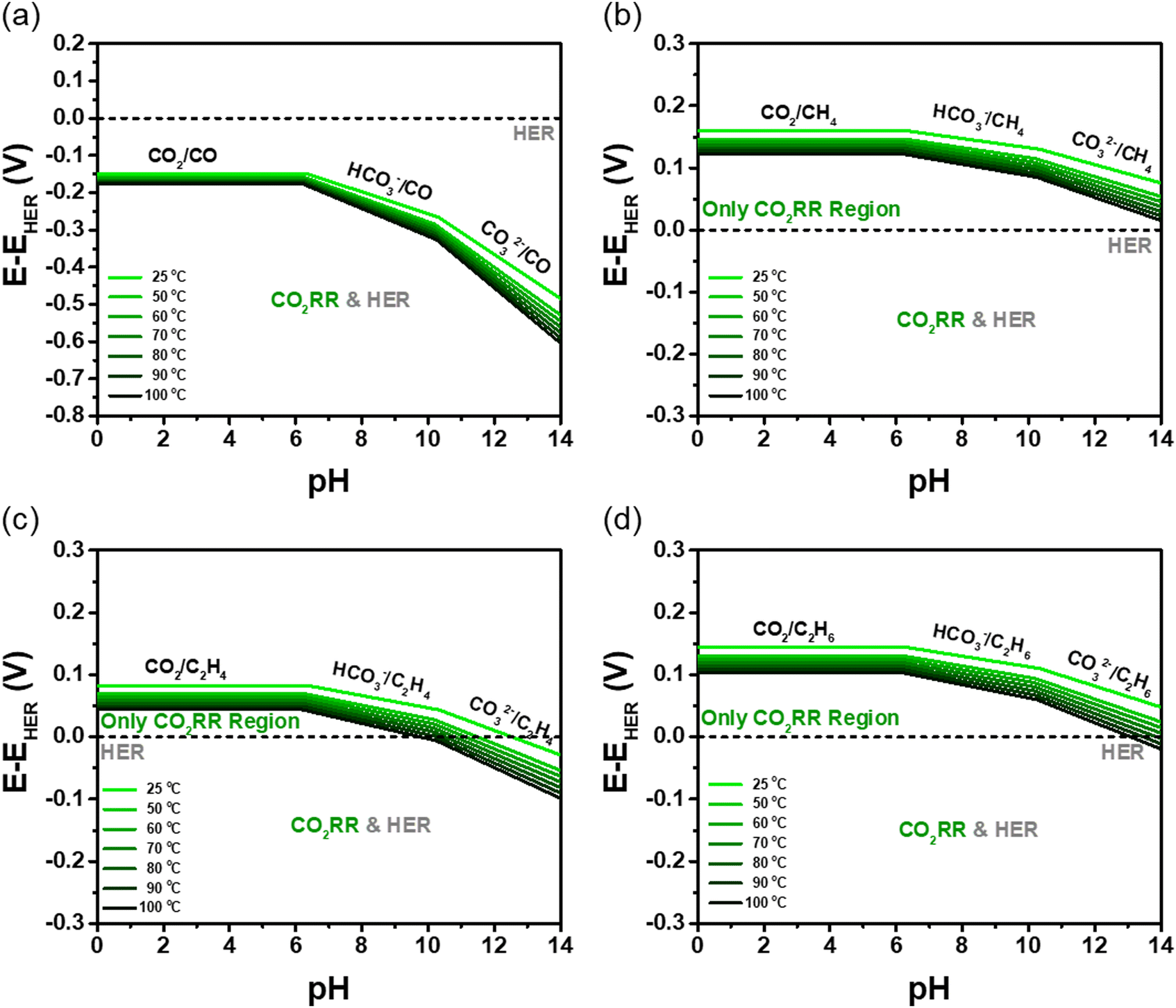 | ||
| Fig. 2 Potential gap between CO2RR and HER for the formation of (a) CO, (b) CH4, (c) C2H4, and (d) C2H6 as a function of temperature (25–100 °C). | ||
In contrast, the CO2-to-CH4 reaction exhibits a CO2RR-dominant region across all pH levels within the 25–100 °C temperature range (Fig. 2(b)). The potential gaps between CO2-to-CH4 and HER are 0.159 V and 0.076 V, respectively, at pH 0 and pH 14, at 25 °C. As the temperature increases to 100 °C, these potential gaps further decrease to 0.122 V and 0.016 V at pH 0 and pH 14, respectively. In contrast, the CO2-to-CH4 reaction exhibits a CO2RR-dominant region across all pH levels within the 25–100 °C temperature range (Fig. 2(b)). The potential gaps between CO2-to-CH4 and HER are 0.159 V and 0.076 V, respectively, at pH 0 and pH 14, at 25 °C. As the temperature increases to 100 °C, these potential gaps further decrease to 0.122 V and 0.016 V at pH 0 and pH 14, respectively. Notably, within the pH range of 0 to 6.381 at 25 °C, the largest potential gap (0.159 V) between CO2RR and HER is observed. These conditions provide a wide operating window where HER can be suppressed, thereby improving the selectivity and efficiency of CO2RR.
Similarly, the CO2-to-C2H4 reaction predominantly occurs within the CO2RR region, except under strongly basic conditions (Fig. 2(c)). As the temperature increases, the exclusive CO2RR region gradually diminishes. The potential gaps between CO2-to-C2H4 and HER are 0.083 V and −0.028 V, respectively, at pH 0 and pH 14, at 25 °C. At 100 °C, these potential gaps decrease to 0.044 V and −0.099 V at pH 0 and 14, respectively. The CO2-to-C2H6 reaction follows a trend similar to that of CO2-to-CH4, maintaining a large CO2RR-dominant region (Fig. 2(d)). However, at temperatures exceeding 80 °C, HER becomes unavoidable under strongly basic conditions. The potential gaps between CO2-to-C2H6 and HER are 0.144 V and 0.048 V, respectively, at pH 0 and pH 14, at 25 °C. As the temperature increases to 100 °C, these potential gaps further decrease to 0.103 V and −0.019 V at pH 0 and pH 14, respectively. These thermodynamic analyses provide deeper insights into reaction mechanisms, enabling improved efficiency and guiding the selection of optimal operating temperatures and pH conditions for practical applications.
Fig. 3 presents the theoretical potential trends for CO2 reduction to CO and hydrocarbons over a temperature range of 0–1000 °C. The VRE and VTN define the minimum and total energy requirements, respectively, under ideal conditions. For CO2-to-CO conversion, below 100 °C, both VTN and VRE decrease as temperature increases (Fig. 3(a)). The calculated VRE for CO2-to-CO remains higher than that of HER/OER. At the same time, VTN is comparable to HER/OER, indicating unavoidable HER competition during CO2RR for CO formation. However, beyond 100 °C, VRE for CO2-to-CO begins to decrease, eventually crossing below the VRE of HER/OER at approximately 800 °C, suggesting a shift in thermodynamic favorability. Additionally, the Fischer–Tropsch process, which involves the conversion of CO to hydrocarbons, was analyzed over a temperature range of 0–1000 °C (Fig. S2, ESI†). This process is highly temperature-dependent, with temperature control critical in product distribution and reaction efficiency. Fig. 3(b) compares the VTN and VRE values for CO2-to-hydrocarbon conversion, including CH4, C2H4, and C2H6, with those of HER/OER over a broad temperature range (0–1000 °C). At 25 °C, the VTN values for CH4/OER (1.153 V), C2H4/OER (1.219 V), and C2H6/OER (1.155 V) are significantly lower than that of HER/OER (1.481 V). Similarly, the VRE values at 25 °C for CH4/OER (1.060 V), C2H4/OER (1.150 V), and C2H6/OER (1.086 V) are also considerably lower than that of HER/OER (1.229 V). These lower theoretical potentials for CO2-to-hydrocarbon conversion indicate a competitive advantage over hydrogen evolution, as the reduction of CO2 to hydrocarbons occurs at a lower potential than HER/OER. This suggests that hydrocarbon formation at 25 °C could be selectively favored over hydrogen production, depending on the applied electrochemical conditions.
 | ||
| Fig. 3 Theoretical potentials (VTN and VRE) of (a) CO2RR to CO and (b) hydrocarbons, as a function of temperature from 0 to 1000 °C. | ||
As the temperature increases to 100 °C, the theoretical potentials for all reactions decrease. However, beyond 100 °C, the theoretical potentials gradually increase with temperature, except for VRE of C2H4/OER and HER/OER, which continue to decline. At temperatures above 600 °C, the VRE of CO2-to-hydrocarbon conversion exceeds that of HER/OER, indicating that hydrogen evolution becomes thermodynamically unavoidable in this temperature range. However, from the perspective of VTN, CO2-to-hydrocarbon conversion still maintains a lower potential than HER/OER, suggesting that despite the inherent competition with HER at high temperatures, hydrocarbon formation remains thermodynamically viable. These thermodynamic understandings provide a fundamental understanding of the temperature-dependent feasibility of CO2RR and hydrocarbon synthesis, offering guidance for optimizing electrocatalytic CO2 conversion under practical operating conditions.
CO2 reduction reactions to organic acids
Electrochemical CO2 reduction can produce hydrocarbons and organic acids, which are significant in various industrial applications.21,22 Understanding the thermodynamic feasibility of these reactions is essential for optimizing selectivity and efficiency in CO2 conversion systems. Fig. 4 presents a thermodynamic analysis of CO2 reduction to organic acids, specifically formic acid (HCOOH) and acetic acid (CH3COOH), as a function of pH and temperature. The Pourbaix diagrams illustrate variations in equilibrium potential, while the potential gap analysis highlights the competition between CO2RR and HER. Given the similar trends observed in CO2-to-hydrocarbon conversions, except in certain regions due to product phase changes, understanding these thermodynamic constraints is crucial for optimizing CO2RR selectivity under varying electrochemical conditions. Under standard conditions, the reduction of gaseous CO2 proceeds via multiple proton–electron transfer steps, forming HCOOH and CH3COOH. Since gaseous CO2 does not undergo pH-dependent speciation, its equilibrium potential remains stable across all pH conditions, providing a consistent reaction environment. However, in practical electrochemical CO2RR systems, CO2 is typically dissolved in aqueous electrolytes, where it undergoes acid–base equilibria, forming HCO3− and CO32− species. This conversion has a significant impact on electrochemical behavior, altering the equilibrium potential as a function of pH. The half-reactions for organic acid formation from gaseous and aqueous CO2 are as follows:23| CO2(g) + 2H+(aq) + 2e− ⇌ HCOOH(aq), E° = −0.114 V vs. SHE | (21) |
| 2CO2(g) + 8H+(aq) + 8e− ⇌ CH3COOH(aq) + H2O(l), E° = −0.106 V vs. SHE | (22) |
| CO2(aq) + 2H+(aq) + 2e− ⇌ HCOOH(aq), E° = −0.071 V vs. SHE | (23) |
| 2CO2(aq) + 8H+(aq) + 8e− ⇌ CH3COOH(aq) + H2O(l), E° = 0.128 V vs. SHE | (24) |
 | ||
| Fig. 4 Pourbaix diagrams of organic acids, including (a) formic acid and (b) acetic acid, using CO2(aq) as a function of temperature from 25 to 100 °C. Potential gap between CO2RR and HER for the formation of (c) HCOOH and (d) CH3COOH as a function of temperature (25–100 °C). | ||
These results indicate that aqueous-phase CO2RR exhibits a slight increase in equilibrium potential compared to its gaseous counterpart, potentially lowering the overpotential requirements for organic acid production. However, at higher pH levels, the dominance of HCO3− and CO32− leads to increased overpotential requirements, reducing conversion efficiency.
As shown in Fig. 4(a), the Pourbaix diagram for CO2-to-HCOOH conversion exhibits temperature dependence and significant variations across intermediate pH values due to CO2 speciation (CO2(aq) ⇌ HCO3− ⇌ CO32−) and the acid–base equilibrium of HCOOH. The dissociation of HCOOH into formate (HCOO−) follows a pKa of 3.7153, 3.7357, and 3.8693 at 25, 50, and 100 °C, respectively, introducing a noticeable inflection point in this pH range, which further alters the thermodynamic favorability of HCOOH formation.
A similar trend is observed in Fig. 4(b) for CO2-to-CH3COOH conversion, where the equilibrium potential exhibits significant variations at intermediate pH due to CO2 speciation effects and the acid–base equilibrium of CH3COOH. The dissociation of CH3COOH into acetate (CH3COO−) follows pKa values of 4.7347, 4.7655, and 4.944 at 25, 50 °C, and 100 °C, leading to a distinct inflection point within this pH range, influencing the reaction energetics.
Compared to CO2-to-hydrocarbon conversions, these organic acid formation reactions involve an additional acid–base equilibrium step, resulting in more pronounced variations in equilibrium potential across different pH regions. This increased complexity introduces multiple transition points, making the thermodynamic landscape of CO2-to-organic acid conversion more complex than that of hydrocarbon formation. Therefore, a more precise thermodynamic analysis is required for accurate predictions.
While the Pourbaix diagrams provide valuable insights into equilibrium, they do not account for HER competition under practical conditions. Fig. 4(c) and (d) compare the potential gaps between CO2RR and HER, identifying selective reaction regions where CO2RR is thermodynamically favored. As shown in Fig. 4(c), the equilibrium potential of HER is more favorable than that of CO2-to-HCOOH, making HER the thermodynamically favored reaction under most conditions. Consequently, HER dominates across a wide pH range, significantly lowering the faradaic efficiency of HCOOH production. However, within a moderate pH range (∼6–11) at temperatures below 50 °C, the equilibrium potential for CO2-to-HCOOH, which is effectively governed by the HCO3−/HCOO− equilibrium, shifts more positive than that of HER, partially suppressing HER and enabling a selective CO2RR region. As temperature increases, this selective window narrows, reducing the effectiveness of CO2RR. In contrast, Fig. 4(d) shows that CO2-to-CH3COOH conversion occurs predominantly at more positive equilibrium potentials, making it less susceptible to HER competition except under highly basic and high-temperature conditions. Since CH3COOH formation requires multiple proton–electron transfer steps, it exhibits a larger overpotential window relative to HER, reducing unwanted H2 evolution. As a result, CH3COOH production remains selective across a broader pH range. However, at temperatures exceeding 60 °C, a slight downward shift in equilibrium potential occurs under highly basic conditions (pH > 13–14), leading to partial HER overlap. Despite this, CH3COOH formation remains unaffected mainly by HER, maintaining a high selectivity window for CO2RR.
These results highlight the substantial influence of pH and temperature on CO2RR selectivity. The analysis indicates that HCOOH formation is highly susceptible to HER competition due to its equilibrium potential being close to that of HER, significantly reducing its faradaic efficiency. In contrast, CH3COOH demonstrates a broader CO2RR-selective window, making it a more viable candidate for practical electrochemical CO2 conversion applications. Therefore, precise optimization of temperature and pH conditions is essential to enhance CO2RR selectivity while minimizing HER interference.
The calculated VRE and VTN for CO2RR to organic acids are compared in Fig. 5, providing insights into the competition between CO2RR and HER under varying temperature conditions (0–1000 °C). Evaluating the thermodynamic feasibility of CO2RR in relation to water electrolysis is crucial for optimizing reaction selectivity and energy efficiency in electrochemical CO2 conversion. At 25 °C, the VRE values for HCOOH/OER (1.410 V) and CH3COOH/OER (1.132 V) are both lower than that of HER/OER (1.229 V). However, the VRE of HCOOH/OER is higher than that of HER/OER, indicating unavoidable competition with HER during formic acid formation. Conversely, the VTN values at 25 °C for HCOOH/OER (1.329 V) and CH3COOH/OER (1.133 V) are significantly lower than that of HER/OER (1.481 V). This suggests that CO2-to-organic acid conversion occurs at a lower thermodynamic threshold than HER/OER, reducing direct competition with HER and improving selectivity under practical electrochemical conditions. The VTN of the CO2RR for organic acid is lower than the VRE of the reactions, unlike the trends of HER/OER.
 | ||
| Fig. 5 Theoretical potentials (VTN and VRE) of CO2RR to organic acids, as a function of temperature from 0 to 1000 °C. | ||
As the temperature increases to 100 °C, both VRE and VTN for CO2-to-organic acid conversion remain lower than those of HER/OER, respectively, indicating a thermodynamically favorable pathway. However, beyond 100 °C, the VTN of HER/OER becomes lower than that of CO2-to-CHOOH, demonstrating that HER is unavoidable during CO2RR for CHOOH formation. Additionally, the VRE of CO2-to-CH3COOH exceeds that of HER/OER from approximately 160 °C, further emphasizing the thermodynamic limitations of organic acid formation under high-temperature conditions.
The potential window for CO2 reduction before HER competition is depicted, revealing that a larger VTN gap between HER/OER and CO2RR/OER favors CO2RR in terms of thermodynamic feasibility. These results highlight that HER competition is less dominant in these regions, underscoring the crucial role of enthalpy in governing reaction selectivity. Thus, the thermodynamic analysis of CO2RR, integrating Pourbaix diagrams and theoretical voltage calculations, provides critical insights into product stability, reaction feasibility, and temperature-dependent behavior. Extending the analysis beyond standard conditions (25 °C), this study establishes a practical framework for designing high-efficiency CO2 conversion systems, optimizing hydrocarbon and organic acid production under industrially relevant conditions.
CO2 reduction reactions to alcohols
The electrochemical reduction of CO2 to alcohols, including methanol (CH3OH), ethanol (C2H5OH), and ethylene glycol (C2H6O2), has also been widely studied.24 These reactions follow a multi-electron, multi-proton transfer pathway, with equilibrium potential shifts primarily governed by pH and temperature. Like hydrocarbon formation, these reactions are influenced by CO2 speciation (CO2(aq) ⇌ HCO3− ⇌ CO32−), which dictates the thermodynamic feasibility of alcohol production under varying electrochemical conditions. The half-reactions for alcohol formation from gaseous and aqueous CO2 are as follows:23,25| CO2(g) + 6H+ + 6e− ⇌ CH3OH(aq) + H2O(l), E° = 0.032 V vs. SHE | (25) |
| 2CO2(g) + 2H+ + 12e− ⇌ CH3CH2OH(aq) + 3H2O(l), E° = 0.0902 V vs. SHE | (26) |
| 2CO2(g) + 10H+ + 10e− ⇌ C2H6O2(l) + H2O(l), E° = 0.009 V vs. SHE | (27) |
| CO2(aq) + 6H+ + 6e− ⇌ CH3OH(aq) + H2O(l), E° = 0.046 V vs. SHE | (28) |
| 2CO2(aq) + 2H+ + 12e− ⇌ CH3CH2OH(aq) + 3H2O(l), E° = 0.105 V vs. SHE | (29) |
| 2CO2(aq) + 10H+ + 10e− ⇌ C2H6O2(l) + H2O(l), E° = 0.030 V vs. SHE | (30) |
As shown in Fig. 6(a)–(c), the Pourbaix diagrams illustrate the variations in equilibrium potential for the formation of methanol, ethanol, and ethylene glycol as a function of pH and temperature. The formation of alcohols requires the stepwise addition of protons and electrons to CO2-derived intermediates, with equilibrium potentials strongly influenced by electrolyte pH. As pH increases, CO2 undergoes a phase transition to HCO3− and CO32−, shifting the equilibrium potential more negative and increasing the overpotential required for alcohol production.
 | ||
| Fig. 6 Pourbaix diagrams of (a) methanol, (b) ethanol, and (c) ethylene glycol as a function of temperature from 25 to 100 °C. The potential gap between CO2RR and HER for the formation of (d) CH3OH, (e) CH3CH2OH, and (f) C2H6O2 as a function of temperature (25–100 °C). | ||
The equilibrium potentials for CO2-to-CH3OH conversion are 0.046 V and −0.894 V vs. SHE at pH 0 and pH 14, respectively, at 25 °C (Fig. 6(a)). As temperature increases to 100 °C, these potentials decrease further to 0.007 V and −1.172 V vs. SHE at pH 0 and pH 14, respectively. For the reduction of CO2 to CH3CH2OH, the equilibrium potentials at 25 °C are 0.105 V at pH 0 and −0.835 V vs. SHE at pH 14 (Fig. 6(b)). These values further decrease to 0.063 V and −1.116 V vs. SHE at 100 °C. Similarly, for CO2-to-C2H6O2 reduction, the equilibrium potentials at 25 °C are 0.030 V at pH 0 and −0.909 V vs. SHE at pH 14 (Fig. 6(c)), shifting further down to −0.009 V and −1.188 V vs. SHE at 100 °C. This trend is consistent with CO2-to-hydrocarbon reduction, where lower pH conditions favor product formation due to CO2 speciation. The stability regions of methanol, ethanol, and ethylene glycol shift toward lower pH values, indicating that their formation is thermodynamically more favorable under acidic conditions. In alkaline environments, the equilibrium potential becomes increasingly negative, making alcohol formation less efficient due to the higher energy input required for reduction.
Fig. 6(d)–(f) compares the potential gaps between CO2RR for alcohol formation and HER, revealing selective electrochemical windows where alcohol production can be favored over HER. Like hydrocarbons, the formation of alcohol follows a comparable thermodynamic trend, reinforcing the close relationship between CO2-to-alcohol and CO2-to-hydrocarbon conversion pathways. Unlike CO2-to-organic acid conversions, which exhibit distinct transition points due to additional acid–base equilibria of products, alcohol formation is primarily governed by CO2 speciation and electrolyte pH, resulting in a more continuous thermodynamic shift.
The CO2-to-CH3OH reaction avoids HER competition under acidic and neutral conditions, particularly at low temperatures (Fig. 6(d)). The potential gaps (ECO2RR − EHER) between CO2-to-CH3OH and HER are 0.046 V and −0.065 V at pH 0 and pH 14, respectively, at 25 °C. As the temperature increases to 100 °C, these potential gaps decrease to 0.007 V and −0.136 V at pH 0 and pH 14, respectively, indicating a reduced thermodynamic driving force for CH3OH formation at elevated temperatures. The CO2-to-CH3CH2OH reaction exhibits a broader CO2RR-dominant region than CO2-to-CH3OH (Fig. 6(e)). At 25 °C, the potential gaps between CO2-to-CH3CH2OH and HER are 0.105 V at pH 0 and −0.007 V at pH. As the temperature increases to 100 °C, these values decrease to 0.063 V and −0.080 V at pH 0 and pH 14, respectively. These conditions provide a wider operating window, which minimizes HER competition and thereby improves both the selectivity and efficiency of CO2RR. Among alcohol formation pathways, the CO2-to-C2H6O2 reaction exhibits the narrowest CO2RR-dominant region (Fig. 6(f)). As temperature and pH increase, this exclusive CO2RR region gradually diminishes. The potential gaps between CO2-to-C2H6O2 and HER are 0.030 V at pH 0 and −0.081 V at pH 14 at 25 °C, which further decrease to −0.009 V and −0.151 V at 100 °C, respectively. These results highlight the increasing thermodynamic limitations on ethylene glycol formation as temperature and pH rise.
The VTN and VRE for CO2-to-alcohol conversion were calculated using enthalpy change and Gibbs free energy. Fig. 7 compares the VTN and VRE values for methanol (MeOH), ethanol (EtOH), and ethylene glycol (EgOH) formation with those of HER/OER over a broad temperature range (0–1000 °C). At 25 °C, the VRE values for MeOH/OER (1.213 V), EtOH/OER (1.145 V), and EgOH/OER (1.220 V) are all lower than that of HER/OER (1.229 V). Among them, the VRE of MeOH/OER and EgOH/OER are particularly close to HER/OER, indicating inevitable competition with HER. Conversely, the VTN values at 25 °C for MeOH/OER (1.255 V), EtOH/OER (1.181 V), and EgOH/OER (1.233 V) are significantly lower than that of HER/OER (1.481 V). From the VTN perspective, CO2-to-alcohol conversion exhibits a lower potential than HER/OER, suggesting that alcohol formation is more thermodynamically favorable than HER at 25 °C, despite the inherent competition with HER. These lower theoretical potentials for CO2-to-alcohol conversion indicate a competitive advantage over hydrogen evolution, as CO2 reduction to alcohols occurs at a lower potential than HER/OER. This suggests that alcohol formation at 25 °C could be selectively favored over hydrogen production, depending on the applied electrochemical conditions.
 | ||
| Fig. 7 Theoretical potentials (VTN and VRE) of CO2-to-alcohol reactions as a function of temperature from 0 to 1000 °C. | ||
As the temperature increases to 100 °C, the theoretical potentials for all reactions decrease. Notably, the VRE of HER/OER decreases rapidly, surpassing the VRE of MeOH/OER and EtOH/OER. Meanwhile, the VRE of EtOH/OER remains consistently lower than that of HER/OER from 0 to 100 °C, suggesting a more favorable electrochemical pathway. Beyond 100 °C, the theoretical potentials gradually increase with temperature, except for VRE of HER/OER, which continues to decline. At temperatures above 350 °C, the VRE of all CO2-to-alcohol conversions exceeds that of HER/OER, indicating that hydrogen evolution becomes thermodynamically unavoidable in this temperature range. However, from the VTN perspective, CO2-to-alcohol conversion still maintains a lower potential than HER/OER, except for EgOH/OER, suggesting that despite the inherent competition with HER at high temperatures, alcohol formation remains thermodynamically viable. The findings elucidate the temperature-dependent viability of CO2RR and alcohol synthesis, providing valuable guidelines for tailoring reaction conditions and enhancing the efficiency of electrocatalytic CO2 conversion in practical applications.
Assisted water electrolysis using organic compounds
Water electrolysis is a promising method for hydrogen production; however, its widespread application is constrained by the high energy demand of the OER, which is the kinetically sluggish half-reaction. AWE has been explored as an alternative approach to address this challenge, wherein organic oxidation reactions replace OER, thereby reducing the overall cell voltage.26 The oxidation of organic molecules, such as methanol (MOR), ethanol (EOR), and ethylene glycol (EGOR), has been demonstrated as a more energy-efficient alternative to conventional water splitting.27–29Fig. 6(a)–(c) presents the Pourbaix diagrams of methanol, ethanol, and ethylene glycol, depicting the equilibrium potentials for both CO2-to-alcohol formation and oxidation pathways. Since reduction and oxidation reactions share the same equilibrium potentials, these diagrams provide insights into the feasibility of both processes under various electrochemical conditions.The feasibility of AWE depends on the electrochemical stability and oxidation potential of organic compounds, both of which vary with pH and temperature. To further analyze the oxidation behavior, Fig. 8 illustrates the calculated oxidation potentials of methanol, ethanol, and ethylene glycol, comparing them with those of conventional water electrolysis. The potential gap between conventional water electrolysis and AWE highlights the energy advantage of organic oxidation. Additionally, the inflection points at pH 6.38 and pH 10.33 correspond to phase transitions of CO2 species, indicating changes in oxidation behavior as a function of pH. Depending on the applied potential and pH, methanol undergoes electrochemical oxidation to CO2, HCO3−, or CO32− (Fig. 8(a)). The data indicate that methanol remains stable under acidic conditions. However, it becomes increasingly susceptible to oxidation at lower potentials in neutral pH environments. At 25 °C, the required potentials for HER/MOR are 0.046 V at pH 0 and −0.065 V at pH 14. As the temperature increases to 100 °C, these values decrease to 0.007 V at pH 0 and −0.136 V at pH 14. In strongly alkaline conditions, the potential gap between water electrolysis and methanol oxidation widens, enabling more efficient oxidation. Notably, methanol oxidation to CO32− becomes more favorable in alkaline environments.
 | ||
| Fig. 8 Potential difference between water electrolysis and assisted water electrolysis using (a) methanol, (b) ethanol, and (c) ethylene glycol as a function of temperature from 25 to 100 °C. (d) VRE and VTN of water electrolysis and assisted water electrolysis using alcohols as a function of temperature from (0 to 1000 °C). | ||
Similarly, the ethanol oxidation reaction follows trends comparable to HER but requires an additional potential input (Fig. 8(b)). At 25 °C, the required potentials for HER/EOR are 0.105 V at pH 0 and −0.007 V at pH 14. As the temperature increases to 100 °C, these values decrease to 0.063 V at pH 0 and −0.080 V at pH 14. The oxidation of ethylene glycol follows a similar trend, requiring the lowest energy input among alcohol oxidation reactions (Fig. 8(c)). The oxidation potentials shift with pH, and the potential gap relative to water electrolysis increases significantly in strongly alkaline environments. At 25 °C, the required potentials for HER/EGOR are 0.030 V at pH 0 and −0.081 V at pH 14. As the temperature increases to 100 °C, these values decrease to −0.009 V at pH 0 and −0.151 V at pH 14. This significant potential difference between water electrolysis and AWE using alcohols highlights the potential for energy-saving strategies in electrochemical hydrogen production. While VRE and VTN are widely used to evaluate water electrolysis efficiency, their application in the AWE field remains limited.
In particular, temperature-dependent theoretical potentials for AWE have not been thoroughly investigated. Fig. 8(d) compares the VTN and VRE values of MOR, EOR, and EGOR with those of OER across a broad temperature range (0–1000 °C). The results indicate that the VTN values at 25 °C for MOR (0.226 V), EOR (0.301 V), and EGOR (0.248 V) are significantly lower than that of OER (1.481 V), highlighting the energetic advantage of organic oxidation in reducing the required electrolysis voltage. Similarly, the VRE values at 25 °C for MOR (0.016 V), EOR (0.084 V), and EGOR (0.009 V) are also considerably lower than that of OER (1.229 V). As temperature increases, VRE decreases, reducing the energy demand for assisted electrolysis, whereas VTN increases, indicating that enthalpic benefits remain unchanged at elevated temperatures. Given that the oxidation potential of organic compounds is substantially lower than that of OER, organic oxidation can be selectively promoted while simultaneously suppressing OER as a competing side reaction.
The simulated results confirm that AWE using organic compounds effectively reduces the energy input required for hydrogen production. Replacing OER with organic oxidation can significantly reduce the total cell voltage, thereby improving overall energy efficiency and lowering operational costs for large-scale electrolysis applications. Furthermore, since many industrial processes generate waste alcohols and other organic compounds, AWE offers a sustainable approach to utilizing these byproducts for hydrogen production. This thermodynamic framework, which integrates Pourbaix diagrams and theoretical voltage calculations, provides a systematic approach for selecting optimal organic compounds and operating conditions for AWE.
Conclusion
This study presents a comprehensive thermodynamic framework for understanding electrochemical CO2 conversion, encompassing CO2RR and AWE under varying temperature (0–1000 °C) and pH conditions. By integrating Pourbaix diagrams and theoretical potentials (VTN and VRE), this work systematically evaluates the thermodynamic feasibility, equilibrium potential shifts, and reaction selectivity across different electrochemical environments. The results highlight the critical role of CO2 phase transitions in governing reaction selectivity and energy efficiency.Based on the comprehensive thermodynamic analysis presented in this work, we summarize the optimal conditions for the selective conversion of CO2 to various products. Hydrocarbon formation (e.g., CH4, C2H4) is thermodynamically favorable under low to moderate pH and temperature (<100 °C), exhibiting higher theoretical potentials than HER under these conditions. Organic acids, such as HCOOH and CH3COOH, exhibit specific thermodynamic selectivity windows, particularly in the pH range of 6–10, and are less susceptible to HER competition at low to intermediate temperatures. Alcohols (e.g., CH3OH, C2H5OH) exhibit higher sensitivity to temperature variations, with selective formation favored under mildly acidic and low-temperature conditions.
Furthermore, in addition to their thermodynamic advantages, acidic to neutral pH conditions also offer practical benefits for system operation. Under strongly alkaline conditions, the formation of alkali metal (bi)carbonate salts can cause precipitation, potentially clogging the flow channels of the electrolyzer and hindering long-term operational stability.6 These results provide guidance for identifying suitable operating conditions for CO2RR, enabling the rational design of reaction environments tailored for product-specific selectivity within a thermodynamic framework.
This thermodynamic understanding is not limited to CO2RR. Beyond CO2RR, this study highlights that phase-dependent shifts in equilibrium potential also play a critical role in AWE, where organic oxidation replaces the conventional OER. The VTN and VRE of organic oxidation reactions remain significantly lower than those of OER across a wide temperature range, reaffirming the energetic advantage of AWE. Furthermore, under strongly alkaline conditions, AWE becomes even more thermodynamically favorable due to the enhanced deprotonation of organic species and the shift of carbonate equilibria (CO2 ⇌ HCO3− ⇌ CO32−). This behavior contrasts with CO2RR, where increasing alkalinity leads to a decrease in thermodynamic favorability, reflecting the fundamentally opposite nature of oxidation and reduction reactions.
This study integrates temperature-dependent Pourbaix diagrams with equilibrium potential modeling to further strengthen the thermodynamic predictions and provide a unified perspective. These insights highlight the crucial role of thermodynamics in guiding pathway selection, mitigating overpotentials, and improving overall energy efficiency in electrochemical systems. These findings are particularly relevant for enhancing CO2RR selectivity, improving organic-assisted water electrolysis, and reducing energy demands in electrochemical hydrogen production.
Author contributions
J. C.: conceptualization, data curation, investigation, methodology, software, visualization, writing—original draft preparation, writing – review, and editing. H. L.: conceptualization, investigation, writing – original draft preparation, writing – review, and editing. S. S.: supervision, validation, visualization, writing – review, and editing. S. P.: resources, visualization, writing – review, and editing. J. S: resources, visualization, writing – review, and editing. G. H. J.: visualization, supervision, validation, writing – review, and editing. U. S.: conceptualization, investigation, visualization, supervision, funding acquisition, validation, writing – original draft preparation, writing – review, and editing.Data availability
The data that support the findings of this study are included in the published article and its ESI.† The raw data are also available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea grant funded by the Ministry of Science and the Korean Government (MSIT), Republic of Korea (NRF-2021M3H4A6A01045764) and Korea Institute of Energy Technology Evaluation and Planning (KETEP) granted financial resources from Ministry of Trade, Industry & Energy, Republic of Korea (No. 20224000000320). This work was also supported by the Henan Center for Outstanding Overseas Scientists (GZS2024020).References
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- C. Xu, P. Hong, Y. Dong, M. Robert, G. Shao and Y. Lei, Toward Complete CO2 Electroconversion: Status, Challenges, and Perspectives, Adv. Energy Mater., 2025, 2406146 Search PubMed.
- J. Li, Y. Ma, X. Mu, X. Wang, Y. Li, H. Ma and Z. Guo, Recent Advances and Perspectives on Coupled Water Electrolysis for Energy-Saving Hydrogen Production, Adv. Sci., 2025, 12, 2411964 Search PubMed.
- H. Choi, D.-K. Lee, M.-K. Han, G. Janani, S. Surendran, J. H. Kim, J. K. Kim, H. Cho and U. Sim, Review—Non-Noble Metal-Based Single-Atom Catalysts for Efficient Electrochemical CO2 Reduction Reaction, J. Electrochem. Soc., 2020, 167, 164503 CrossRef CAS.
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E.-S. Jeong, V. Subramanian, U. Sim and K. T. Nam, Achieving highly efficient CO2 to CO electroreduction exceeding 300 mA cm−2 with single-atom nickel electrocatalysts, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC.
- T. H. Ha, J. Kim, H. Choi and J. Oh, Selective Zero-Gap CO2 Reduction in Acid, ACS Energy Lett., 2024, 9, 4835–4842 CrossRef CAS.
- Z. Zhu, W. Tang, J. Wang, L. Zhao, Y. Lin, Z. Li, X. Niu, J. S. Chen and R. Wu, Insights into Operating Conditions on Electrocatalytic CO2 Reduction, Adv. Energy Mater., 2025, 2405768 CrossRef CAS.
- Y. Wang, D. Zhang, B. Sun, X. Jia, L. Zhang, H. Cheng, J. Fan and H. Li, Divergent Activity Shifts of Tin-Based Catalysts for Electrochemical CO2 Reduction: pH-Dependent Behavior of Single-Atom Versus Polyatomic Structures, Angew. Chem., Int. Ed., 2025, 64, e202418228 Search PubMed.
- C. Jo, S. Surendran, M.-C. Kim, T.-Y. An, Y. Lim, H. Choi, G. Janani, S. Cyril Jesudass, D. Jun Moon, J. Kim, J. Young Kim, C. Hyuck Choi, M. Kim, J. Kyu Kim and U. Sim, Meticulous integration of N and C active sites in Ni2P electrocatalyst for sustainable ammonia oxidation and efficient hydrogen production, Chem. Eng. J., 2023, 463, 142314 Search PubMed.
- H. Choi, S. Surendran, D. Kim, Y. Lim, J. Lim, J. Park, J. K. Kim, M.-K. Han and U. Sim, Boosting eco-friendly hydrogen generation by urea-assisted water electrolysis using spinel M2GeO4 (M = Fe, Co) as an active electrocatalyst, Environ. Sci.: Nano, 2021, 8, 3110–3121 RSC.
- H. Choi, M. G. Ha, J. Suh, C. Lim, B. Kim, S. E. Wang, J. Y. Lee, H.-S. Oh and J. Oh, Effect of the nitrogen/carbon ratio in the organic ligand of a nickel single-atom catalyst on its electrochemical activity in CO2 reduction, Appl. Catal., B, 2024, 355, 124192 CrossRef CAS.
- Y. Li, H. Zhang, T. Chen, Y. Sun, F. Rosei and M. Yu, Dual-Interfacial Electrocatalyst Enriching Surface Bonded H for Energy-Efficient CO2-to-CH3OH Conversion, Adv. Funct. Mater., 2024, 34, 2312970 CrossRef CAS.
- J. Choi, H. Lim, S. Surendran, X. Lu, K. Jin, H. Park, H.-Y. Jung and U. Sim, Dichalcogenides as Emerging Electrocatalysts for Efficient Ammonia Synthesis: A Focus on Mechanisms and Theoretical Potentials, Adv. Funct. Mater., 2025, 2422585 CrossRef.
- M. König, J. Vaes, E. Klemm and D. Pant, Solvents and Supporting Electrolytes in the Electrocatalytic Reduction of CO2, iScience, 2019, 19, 135–160 Search PubMed.
- P. W. Atkins, J. De Paula and J. Keeler, Atkins’ physical chemistry, Oxford University Press, 2023 Search PubMed.
- J. Choi, S. Yoo, G. Janani, S. Surendran, S. Shanmugapriya, H. Choi, G. Kwon, K. Jin and U. Sim, Recent research trends in the rational designing of single atom catalysts for electrochemical CO2 reduction reaction to CO—a mini review, Carbon Lett., 2025, 35, 469–479 CrossRef CAS.
- H. Song, C. A. Fernández, H. Choi, P.-W. Huang, J. Oh and M. C. Hatzell, Integrated carbon capture and CO production from bicarbonates through bipolar membrane electrolysis, Energy Environ. Sci., 2024, 17, 3570–3579 RSC.
- B. Kim, Y. C. Tan, Y. Ryu, K. Jang, H. G. Abbas, T. Kang, H. Choi, K.-S. Lee, S. Park, W. Kim, P.-P. Choi, S. Ringe and J. Oh, Trace-Level Cobalt Dopants Enhance CO2 Electroreduction and Ethylene Formation on Copper, ACS Energy Lett., 2023, 8, 3356–3364 CrossRef CAS.
- Y. Kim, B. Kim, H. Choi, S. Kim, Y. Yun and J. Oh, Modulating the electronic structure of Au using a heterostructure for efficient electrochemical CO2 reduction, Chem. Eng. J., 2023, 461, 142126 CrossRef CAS.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Electrocatalytic reduction of CO2 and CO to multi-carbon compounds over Cu-based catalysts, Chem. Soc. Rev., 2021, 50, 12897–12914 Search PubMed.
- X. Zhang, W. Huang, L. Yu, M. García-Melchor, D. Wang, L. Zhi and H. Zhang, Enabling heterogeneous catalysis to achieve carbon neutrality: Directional catalytic conversion of CO2 into carboxylic acids, Carbon Energy, 2024, 6, e362 CrossRef CAS.
- H. Shen, H. Jin, H. Li, H. Wang, J. Duan, Y. Jiao and S.-Z. Qiao, Acidic CO2-to-HCOOH electrolysis with industrial-level current on phase engineered tin sulfide, Nat. Commun., 2023, 14, 2843 CrossRef CAS PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Carbon dioxide electroreduction to C2 products over copper–cuprous oxide derived from electrosynthesized copper complex, Nat. Commun., 2019, 10, 3851 CrossRef PubMed.
- S. Wang, H. D. Jung, H. Choi, J. Kim, S. Back and J. Oh, Delicate control of a gold-copper oxide tandem structure enables the efficient production of high-value chemicals by electrochemical carbon dioxide reduction, Nano Energy, 2024, 130, 110176 CrossRef CAS.
- C. Guo, Y. Guo, Y. Shi, X. Lan, Y. Wang, Y. Yu and B. Zhang, Electrocatalytic Reduction of CO2 to Ethanol at Close to Theoretical Potential via Engineering Abundant Electron-Donating Cu+ Species, Angew. Chem., Int. Ed., 2022, 61, e202205909 CrossRef CAS PubMed.
- F. Arshad, Tu Haq, I. Hussain and F. Sher, Recent Advances in Electrocatalysts toward Alcohol-Assisted, Energy-Saving Hydrogen Production, ACS Appl. Energy Mater., 2021, 4, 8685–8701 CrossRef CAS.
- F. Liu, T. Wang, J. Li, T. Wei, Z. Ye, D. Dong, B. Chen, Y. Ling and Z. Shao, Elevated-temperature bio-ethanol-assisted water electrolysis for efficient hydrogen production, Chem. Eng. J., 2022, 434, 134699 CrossRef CAS.
- Q. Liu, S. Du, T. Liu, L. Gong, Y. Wu, J. Lin, P. Yang, G. Huang, M. Li, Y. Wu, Y. Zhou, Y. Li, L. Tao and S. Wang, Efficient Low-temperature Hydrogen Production by Electrochemical-assisted Methanol Steam Reforming, Angew. Chem., Int. Ed., 2024, 63, e202315157 CrossRef CAS PubMed.
- Q. Mao, K. Deng, H. Yu, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, In Situ Reconstruction of Partially Hydroxylated Porous Rh Metallene for Ethylene Glycol-Assisted Seawater Splitting, Adv. Funct. Mater., 2022, 32, 2201081 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01408e |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2025 |