
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting the NMR chemical shifts of hydrides in SABRE-active Ir complexes by relativistic DFT†‡
Beatrice Bernadette
Mascitti
 a,
Giordano
Zanoni
a,
Federico
de Biasi
b,
Federico
Rastrelli
*a and
Giacomo
Saielli
*ac
a,
Giordano
Zanoni
a,
Federico
de Biasi
b,
Federico
Rastrelli
*a and
Giacomo
Saielli
*ac
aDipartimento di Scienze Chimiche, Università degli Studi di Padova via Marzolo, 1 – 35131, Padova, Italy. E-mail: federico.rastrelli@unipd.it
bInstitut des Sciences et Ingenierie Chimiques, École Polytechnique Fedérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
cIstituto per la Tecnologia delle Membrane del CNR, Sede di Padova, via Marzolo, 1 – 35131, Padova, Italy. E-mail: giacomo.saielli@unipd.it
First published on 11th June 2025
Abstract
Signal amplification by reversible exchange (SABRE) uses metal-mediated reversible binding of parahydrogen to transfer polarization via spin–spin couplings in transient complexes, particularly iridium complexes with N-heterocyclic carbenes (NHC). The magnetic parameters of such complexes, notably chemical shifts and coupling constant networks, are useful for elucidating their structure and assessing their effectiveness as spin catalysts. Moreover, Ir-bound hydrides have distinctive chemical shifts (−10 to −30 ppm) that provide valuable information about the complex geometry and, upon detailed analysis, about its substituents. However, interpreting the corresponding experimental NMR spectra becomes significantly more challenging than in simple organic molecules due to the presence of the heavy metal center which affects the hydrides resonance by the spin–orbit HALA (heavy atom on light atom) effect. In this work we show that density functional theory (DFT) can address these challenges by incorporating proper relativistic corrections, thus aiding the structural analysis of these systems.
Introduction
Among the various techniques that can achieve nuclear spin polarization in magnetic resonance spectroscopy, parahydrogen-induced polarization (PHIP) delivers dramatic signal enhancement with a relatively simple experimental setup.1 The methodologies for achieving PHIP are manifold. One approach, known as hydrogenative PHIP, involves the catalytic hydrogenation of unsaturated molecular moieties in a homogeneous solution. In this process, parahydrogen molecules irreversibly react with some unsaturated compounds, resulting in the hyperpolarization of the products.An alternate approach to hydrogenative PHIP is the non-hydrogenative technique known as signal amplification by reversible exchange (SABRE), which was originally developed by Duckett and collaborators.2 The enhancement of NMR signals in SABRE originates from the non-thermal spin order of parahydrogen, which is exploited through the mediation of a metal center that reversibly coordinates both the parahydrogen and the substrate to be polarized, enabling their exchange with free forms in solution. Polarization transfer from para-hydrogen (in the form of coordinated hydrides) to the nuclear spins of the substrate within the transient complex occurs essentially through spin–spin couplings, provided that the spin system is allowed to reach the conditions for an efficient spin-level anticrossing (LAC).3
In the context of SABRE, the most important class of compounds capable of satisfying the aforementioned requirements is based on iridium complexes that contain N-heterocyclic carbenes (NHC) such as 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes).
These “spin catalysts” are generally added in solution together with the substrate to be polarized, in the form of COD-protected Ir(I) NHC precursors. When molecular hydrogen (H2) is introduced in solution, it promotes an oxidative addition whereby Ir(I) is converted to Ir(III). This transformation results in the formation of two iridium-bound hydrides, while the substrate occupies the remaining coordination sites. Depending on the adopted conditions, this reaction can give rise to many different species which can be extremely difficult to track by just looking at the 1H resonances of the ligands. In contrast, the Ir-bound hydrides exhibit distinctive chemical shifts (typically spanning a range from −10 to −30 ppm) which can provide significant information on the geometry of the complex and, with more detailed analysis, on the nature of its substituents.
However, the chemical shifts of species involving heavy atoms are not easy to interpret due to the limited experimental data available and to the complex electronic environments inherent to the heavy atoms. In these systems, in fact, the well-known heavy atom on light atom (HALA) effect introduces an additional shielding contribution that primarily comes from the relativistic spin–orbit term in the Hamiltonian4 and influences the NMR resonances of the light atom bonded to the heavy atom, as in the case of hydrides and iridium.
In this context, the calculation of NMR properties by means of density functional theory (DFT) computational protocols is a valuable tool for experimentalists involved in the structural determination of unknown organic natural substances,5,6 organometallic systems containing heavy atoms,7–9 and complex fluid matrices.10 As mentioned above, when heavy atoms are present, relativistic effects need to be included in the calculation, since they strongly affect the chemical shift mainly through spin–orbit coupling.8,11–15
In this work, we investigate the application of DFT across various levels of theory, ranging from non-relativistic and ZORA scalar relativistic approaches -employed here only for comparative purposes- to more advanced two-component spin–orbit ZORA and four-component methodologies. Our primary objectives are twofold: first, to benchmark a selected set of DFT functionals against a series of well-characterized SABRE compounds, and second, to predict the chemical shifts of hydrides in previously uncharacterized reaction products arising from the interaction of the iridium catalyst with phenanthroline, thereby facilitating their structural elucidation. Spin–spin J-coupling constants are also computed and discussed in some relevant pyruvate complexes.
Computational details
The DFT calculations were run using the software packages Gaussian16,16 ADF201917–19 and ReSpect2019.20A first set of geometry optimizations, accounting also for dispersive interactions, were run at the ω-B97XD(PCM)/triple-ζ level of theory,21 where the basis set was LANL2TZ for iridium22 and 6-311G(d,p) for the remaining atoms. All geometries at this level were checked to be true minima by a frequency calculation. The long-range solvent effects were included in the optimization step by using the polarizable continuum model (PCM).23 The software used at this step was Gaussian 16.
NMR calculations, using the optimized geometries, were then run at four different relativistic levels of theory, either ZORA (zero-order regular approximation)24 or fully relativistic 4-component level, in all cases with the hybrid PBE0 functional: (i) ZORA scalar (PBE0-ZSC); (ii): ZORA spin–orbit (PBE0-ZSO); (iii) ZORA spin–orbit with inclusion of long-range solvent effects through the COSMO solvation model25 (PBE0(solv)-ZSO); (iv) four-component level (4c-PBE0). For the calculation of the chemical shifts, the basis set was TZ2P-ae for all atoms at the ZORA levels and dyall-vtz for Ir and upcS-1 for the other atoms at the 4c level. ADF was used for levels (i)–(iii) and ReSpect for level (iv). Additionally, at the PBE0-ZSO level, some relevant coupling constants involving the hydride atoms were calculated using the TZ2P-J basis set available in ADF and specifically optimized for spin–spin coupling constant calculations.
To test the effect of the geometry optimization, we also reoptimized all structures at the ZORA scalar relativistic level of theory using the B3LYP functional26–29 with Grimme330 correction to account for dispersive interactions and with inclusion of the long range solvent dielectric response using the COSMO method25 as implemented in ADF (B3LYPG3(solv)-ZSC). The basis set was all-electron TZ2P for all the atoms.
The same levels of theory used for the NMR calculation of the systems optimized at the ω-B97XD(PCM) where then used also for this second set of geometries, with the addition of a non-relativistic level (NR). Moreover, still using this set of geometries, we also used the SSB-D functional,31 at the ZSO/TZ2P level, for the NMR calculation. A summary of all levels of theory used can be found in Table 1.
| Entry | Geometry opt level | NMR level |
|---|---|---|
| (i) | ω-B97XD(PMC)/Triple-ζ | PBE0-ZSC/TZ2P |
| (ii) | ω-B97XD(PMC)/Triple-ζ | PBE0-ZSO/TZ2P |
| (iii) | ω-B97XD(PMC)/Triple-ζ | PBE0-ZSO(COSMO)/TZ2P |
| (iv) | ω-B97XD(PMC)/Triple-ζ | 4c-PBE0/(vtz,upcS-1) |
| (v) | B3LYPG3-COSMO-ZSC/TZ2P | NR-PBE0/TZ2P |
| (vi) | B3LYPG3-COSMO-ZSC/TZ2P | PBE0-ZSC/TZ2P |
| (vii) | B3LYPG3-COSMO-ZSC/TZ2P | PBE0-ZSO/TZ2P |
| (viii) | B3LYPG3-COSMO-ZSC/TZ2P | PBE0-ZSO(COSMO)/TZ2P |
| (ix) | B3LYPG3-COSMO-ZSC/TZ2P | 4c-PBE0/(vtz,upcS-1) |
| (x) | B3LYPG3-COSMO-ZSC/TZ2P | SSDB-ZSO/TZ2P |
The 1H chemical shifts, δ, are calculated as δ = σref − σ, where σref is the shielding constant of 1H in tetramethylsilane, calculated at the same level of theory.
The structural formula of the iridium hydrides used for the benchmark calculations with the computational protocols of Table 1 are shown in Chart 1 and Chart 2. The solvent used for the experimental determination of the chemical shift is methanol, except for [Ir(H)2(IMes)(NH3)3]+ which was measured in dichloromethane. The corresponding dielectric constant of 32.613 and 8.93 for methanol and dichloromethane, respectively, was used in the treatment of long-range solvent effects with continuum models, that one of dichloromethane only for the case of complex [Ir(H)2(IMes)(NH3)3]+.
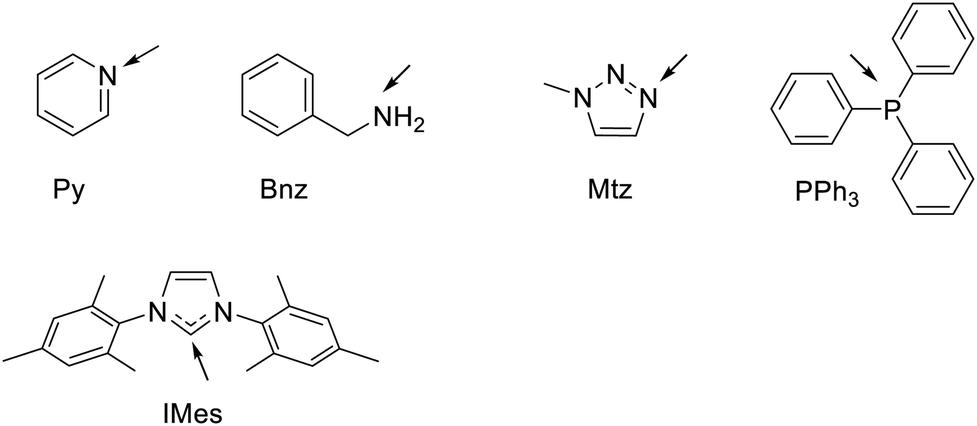 | ||
| Chart 1 Structural formula and abbreviations of the ligands relevant to this work. The arrows indicate the Ir binding atom. Py = pyridine, Bnz = benzylamine, Mtz = 1-methyl-1H-1,2,3-triazole, PPh3 = triphenyl phosphine, IMes = 1,3-Bis(2,4,6-trimethylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene. | ||
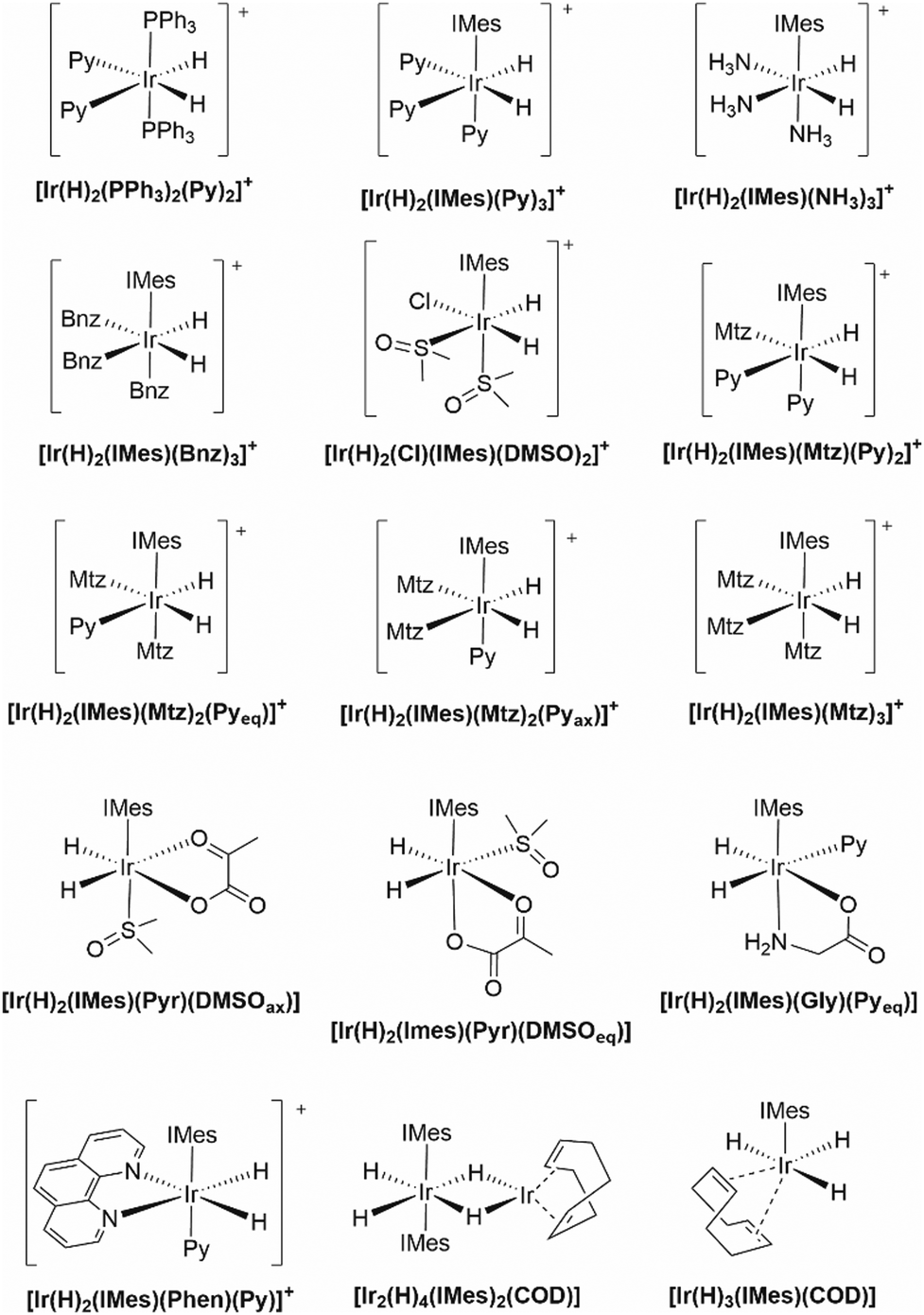 | ||
| Chart 2 Structural formula and nomenclature of iridium benchmark complexes investigated in this work. Pyr = pyruvate anion, Gly = glycinate anion, COD = cycloocta-1,5-diene. | ||
For each computational protocol listed in Table 1 we have calculated the linear fit of the correlation between the calculated and the experimental chemical shifts of the hydrides
| δcalc = aδexp + b, | (1) |
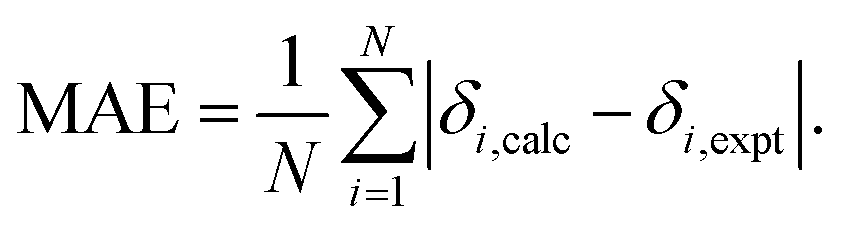 | (2) |
Results and discussion
Benchmark calculations
We begin the analysis of the results by first looking at the overall performance of the various levels of theory employed. Statistical parameters for all protocols are reported in Table 2 while their graphical representation can be found in ESI,‡ Fig. S7–S11. For the geometries minimized at the ω-B97XD level, the graphs of the correlations of calculated (levels (i)–(iv)) vs. experimental chemical shifts are also shown in Fig. 1.| Level | a | b/ppm | R 2 | MAE/ppm | Max Err/ppm |
|---|---|---|---|---|---|
| a At the 4-component level (protocols (iv) and (ix)) the NMR calculation for the dimeric compound [Ir2(H)4(IMes)2(COD)] was unfeasible with our computational facilities. Therefore, we used a model system where all methyl groups of IMes were replaced by hydrogen atoms keeping the rest of the structures as in the original optimized geometry. | |||||
| (i) | 0.289 | −1.66 | 0.5697 | 12.21 | 17.54 |
| (ii) | 0.846 | 0.71 | 0.9518 | 3.88 | 6.51 |
| (iii) | 0.860 | 0.50 | 0.9607 | 3.39 | 4.97 |
| (iv)a | 0.927 | 0.14 | 0.9446 | 1.90 | 4.01 |
| (v) | 0.257 | −1.36 | 0.5470 | 14.04 | 18.96 |
| (vi) | 0.284 | −1.57 | 0.5078 | 13.40 | 17.58 |
| (vii) | 0.811 | 0.43 | 0.9292 | 4.32 | 7.08 |
| (viii) | 0.826 | 0.19 | 0.9444 | 3.79 | 5.48 |
| (ix)a | 0.888 | −0.26 | 0.9244 | 2.52 | 4.64 |
| (x) | 0.691 | 1.21 | 0.9176 | 7.59 | 11.26 |
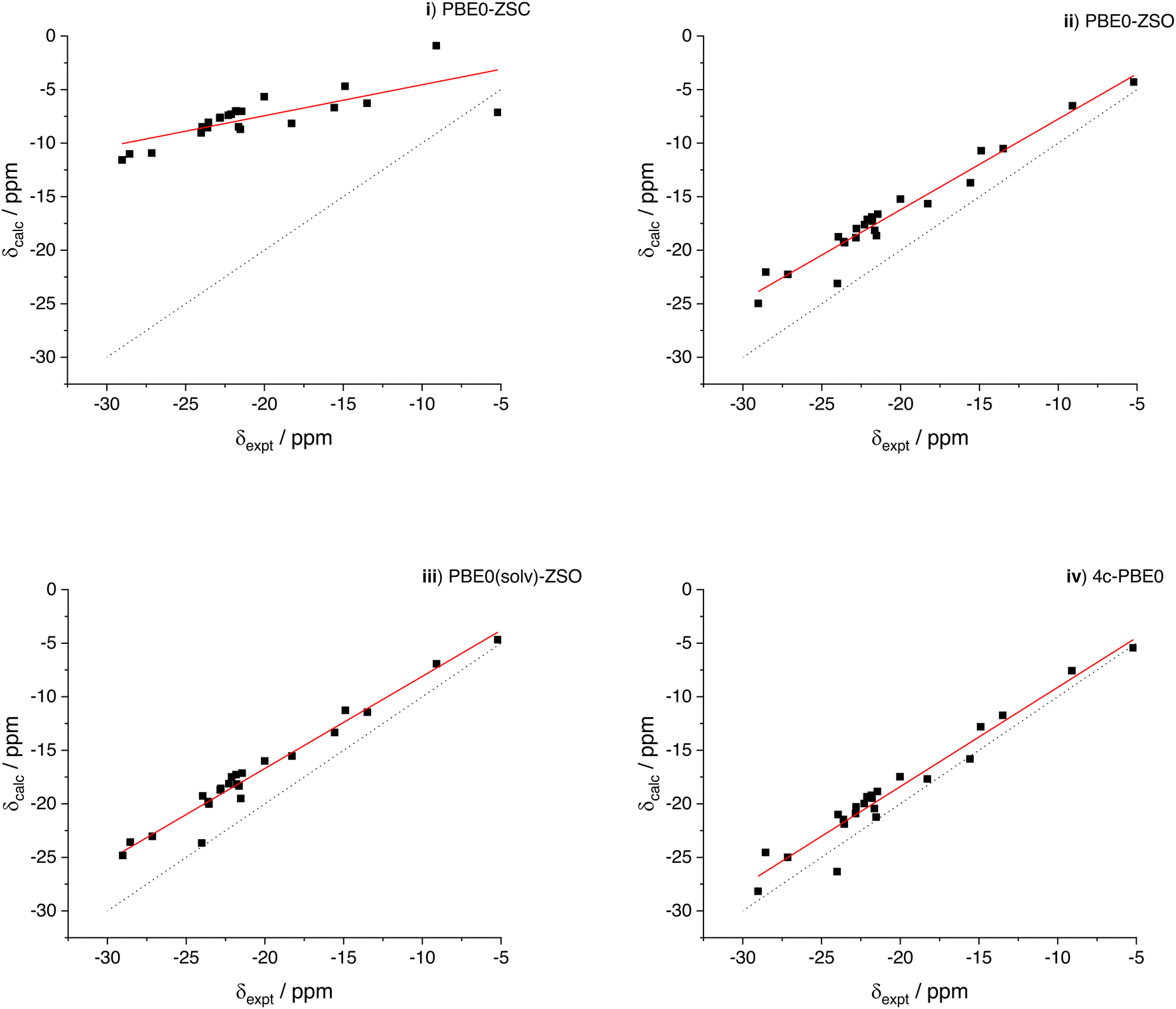 | ||
| Fig. 1 Correlation between experimental and calculated 1H chemical shifts of the iridium hydrides of Chart 2. The levels of theory (i)–(iv) are reported in Table 1. (Filled squares) data points; (black dot line) unitary line as a guide for the eye; (solid red line) linear fitting, with the relevant parameters in Table 2. See also the footnote of Table 2. From the statistical analysis of method PBE0-ZSO (see below) the width of the confidence band with 95% probability around −20 ppm is 1 ppm. | ||
First we note that the inclusion of spin–orbit coupling in the calculation of the chemical shifts of the hydrides directly bonded to the heavy iridium atom is mandatory, as a consequence of the well-known HALA effect (heavy atom on light atom).4 The ZORA scalar-relativistic level, (i), and the analogous spin–orbit level, (ii), have a very different performance. Despite the overall correlation even at level (i) has the correct trend, the absolute values are largely underestimated due to the neglect of the SO contribution. Therefore, the slope of the fitting line is much smaller than unity and the MAE and MAX Err are above 10 ppm. A significant improvement is instead obtained after inclusion of the spin–orbit term in the NMR calculation, still at the ZORA level of theory, as can be observed at the level of theory (ii). Here the correlation improves significantly and also the MAE and MAX Err drop down to 3.88 and 6.51 ppm, respectively. Inclusion of the solvent in the NMR calculation also slightly improves the results (level (iii)) while a more significant improvement, especially in terms of MAE and MAX Err, is obtained at the 4-component level (level (iv)), where we have 1.90 and 4.01 ppm for MAE and MAX Err, respectively.
The various contributions to the chemical shift can be appreciated more quantitatively by computing the difference in the calculated chemical shifts (which corresponds to the difference in shielding constants with an opposite sign) at given levels of theory. The results are shown in Fig. 2 and they highlight a more significant SO contribution to the shielding (more negative calculated chemical shift) as the experimental resonance becomes more and more shielded. The effect of the ZORA approximation is also not negligible since the 4-c results are systematically 2–3 ppm more shielded and the effect is larger as the chemical shift of the hydrides becomes more negative. This result is qualitatively very similar to what reported few years ago for a large dataset of proton and carbon chemical shifts of halogenated marine natural substances.32 Finally, the solvent effect in the NMR calculation amounts to about 1 ppm contribution, still as a shielding effect. Unfortunately, it is not possible to run NMR calculations including the solvent effect at the 4-c level with the ReSpect software, but we may expect a further decrease of the MAE down to about 1 ppm for a hypothetical 4-c level with long range solvent effects included.
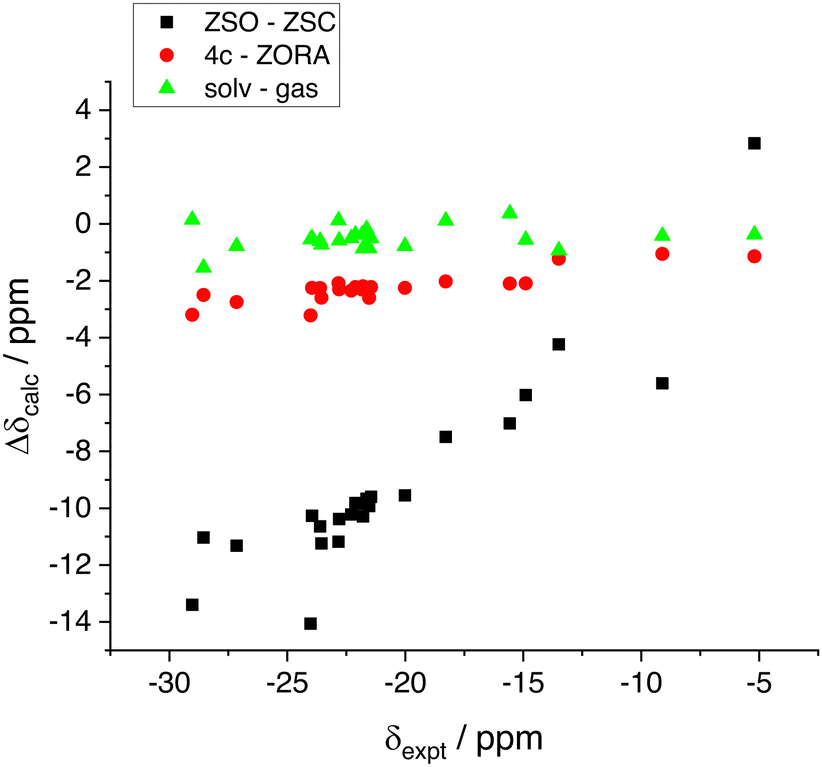 | ||
| Fig. 2 The various contributions (4-c vs. ZORA, spin–orbit and solvent effect) to the shielding constant of the hydrides calculated as a difference between two levels of theory. (ZSO–ZSC) is obtained as δ(ii)–δ(i); (4c–ZORA) is obtained as δ(iv)–δ(ii); (solv–gas) is obtained as δ(iii)–δ(ii). | ||
Concerning the correlations shown in Fig. 1, we finally note a couple of minor issues. First, the chemical shift of bridge hydrogens in the dimeric compound [Ir2(H)4(IMes)2(COD)], although the Hs are bonded to two iridium atoms, are not significantly shielded (expt. value of −5.20 ppm). The scalar result, level (i), appears quite off the correlation line, but the agreement is nicely recovered at the spin–orbit levels. Interestingly, however, the SO contribution appears as a deshielding effect in this case, contrary to all other compounds investigated. The second point to be noted is the calculated result for the hydride in trans position with respect to the carbonyl oxygen in [Ir(H)2(IMes)(Pyr)(DMSOeq)], (expt. value −24.01 ppm). The calculated values when including the SO correction appear somewhat off the linear correlation (although in better agreement with the experiment) with all levels (ii)–(iv). Curiously, at the scalar level, although largely wrong compared to the experiment, the calculated result for this resonance is almost perfectly aligned with the linear fitting line of the level (i). Therefore, it is the calculated SO contribution that, for this specific geometry, is affected by a larger error compared to the other compounds. The effect is not related with explicit coordination of the pyruvate by a methanol molecule, as it might be argued since methanol is the solvent. We calculated the chemical shift also using a geometry with an explicit methanol molecule obtaining an almost identical result (changes of the order of 10−2 ppm in the calculated values).
The results obtained with the second set of geometries optimized at the ZORA scalar relativistic level (see Computational details section) are qualitatively very similar, though generally exhibiting slightly worse correlation parameters when compared with the NMR results obtained with the ω-B97XD geometries (with the NMR calculations at the same level of theory, that is comparing (i) with (vi), (ii) with (vii), (iii) with (viii) and (iv) with (ix). In addition, we note that the quality of the NMR results at the non-relativistic level (v) is quite similar to that one at the ZORA scalar level indicating that only including the scalar relativistic corrections does not bring in any notable improvement. Finally, as we can see in Table 2, the SSB-D functional does not show any better correlation compared to the PBE0 functional for this set of compounds, although it was found to have a good performance when tested at the non-relativistic level with a set of small organic and inorganic molecules containing only light atoms.33
Since this study focuses on the structural elucidation of the many reaction products that can result from hydrogenation of the SABRE catalyst (see below), we deemed it not essential to extensively and systematically investigate the performance of other functionals. Instead, we are satisfied with the relatively good performance of PBE0 shown above and we will now proceed with the discussion of some experimental results. Moreover, the PBE0 functional has been found to perform rather well for the prediction of NMR properties of organometallic systems of iridium.34
The case of phenanthroline
1,10-Phenanthroline (Phen) is a bidentate ligand that strongly binds to the Ir atom in SABRE-active complexes. In 2015 Duckett and coworkers proposed an interesting use of 2,2′-bipyridine and 1,10-phenanthroline for the rapid deactivation of the SABRE catalyst following the hyperpolarization of a selected target. In fact, it was observed that the introduction of these chelating ligands can significantly extend the relaxation times of polarized targets, thereby enhancing the potential of SABRE for diagnostic MRI applications.35 The same work reports the experimental chemical shifts for the [Ir(H)2(IMes)(Phen)(Py)]Cl complex, which has been incorporated into the set of benchmark molecules used to test the different levels of theory (see the Computational results section above). From a general standpoint, phenanthroline is of particular interest due to its rigid bidentate structure, which, upon proper modification, enables the anchoring of the Ir catalyst onto auxiliary substrates.In this study, we examine the fate of phenanthroline in the absence of pre-formed SABRE-active complexes (Experimental details are reported in ESI,‡ Fig. S1–S6). After mixing 10 mM 1,10-phenanthroline with 1 mM [IrCl(COD)(IMes)] in CD3OD and bubbling H2 for several minutes, the hydride region of the 1H NMR spectrum displays two singlets at −20.31 and −20.91 ppm (For reference, the CHD2OD residual signal in the same spectrum appears at 3.33 ppm). One of these signals (−20.31 ppm) exhibits large amplification after bubbling with p-H2, while the other one does not (see ESI,‡ Fig. S5). Selective NOESY experiments conducted with a 0.2 s mixing time reveal that the protons responsible for the signal at −20.31 ppm undergo chemical exchange with H2 in solution, whereas those resonating at −20.91 ppm exhibit no exchange. It is worth noting that Duckett has reported a similar lack of exchange for the hydrides of [Ir(H)2(IMes)(Bpy)(L1)]+, where Bpy is 2,2′-bipyridine and L1 is nicotinamide.35
The portion of the spectrum relative to phenanthroline shows three distinct spin systems, one of which pertains to free phenanthroline. The other two spin systems, whose signals can be isolated by chemical shift selective filter (CSSF) TOCSY (see ESI‡), are associated with phenanthroline bound to Ir (Fig. 3). Only one of these two spin systems provides polarized signals under SABRE conditions (3 bar p-H2 and 60 Gauss external field).
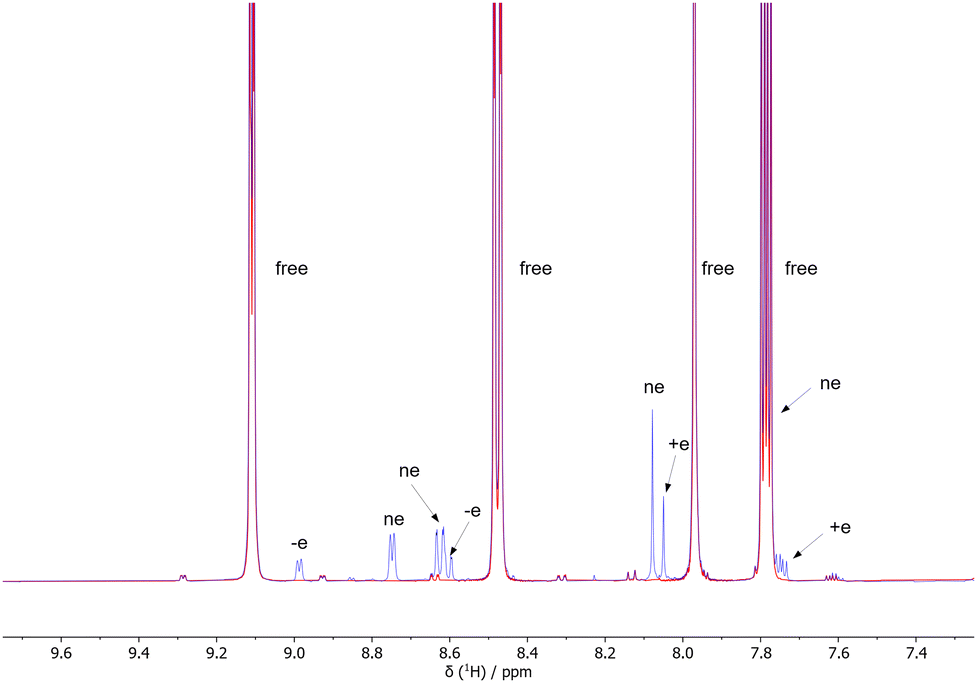 | ||
| Fig. 3 500 MHz 1H NMR subspectrum of 10 mM 1,10-phenanthroline and 1 mM [IrCl(COD)(IMes)] in CD3OD before (red) end after (blue) H2 pressurization (3 bar). The letters indicate the signals of phenanthroline bound to the Ir complex that provide positively enhanced (+e), negatively enhanced (−e) or non-enhanced (ne) signals under SABRE conditions. The label free indicates free phenanthroline in solution. | ||
Combining all the above information we conclude that the two major complexes observed in solution should be symmetric, with phenanthroline bound to Ir in the equatorial position.
On this basis, we propose a selection of possible candidate structures as responsible for the two main resonances observed experimentally, see Chart 3. The choice is done considering the species that could possibly occupy the axial position, namely MeOH, H2O, Cl−, along with an additional phenanthroline molecule (assuming fast exchange with free phenanthroline).
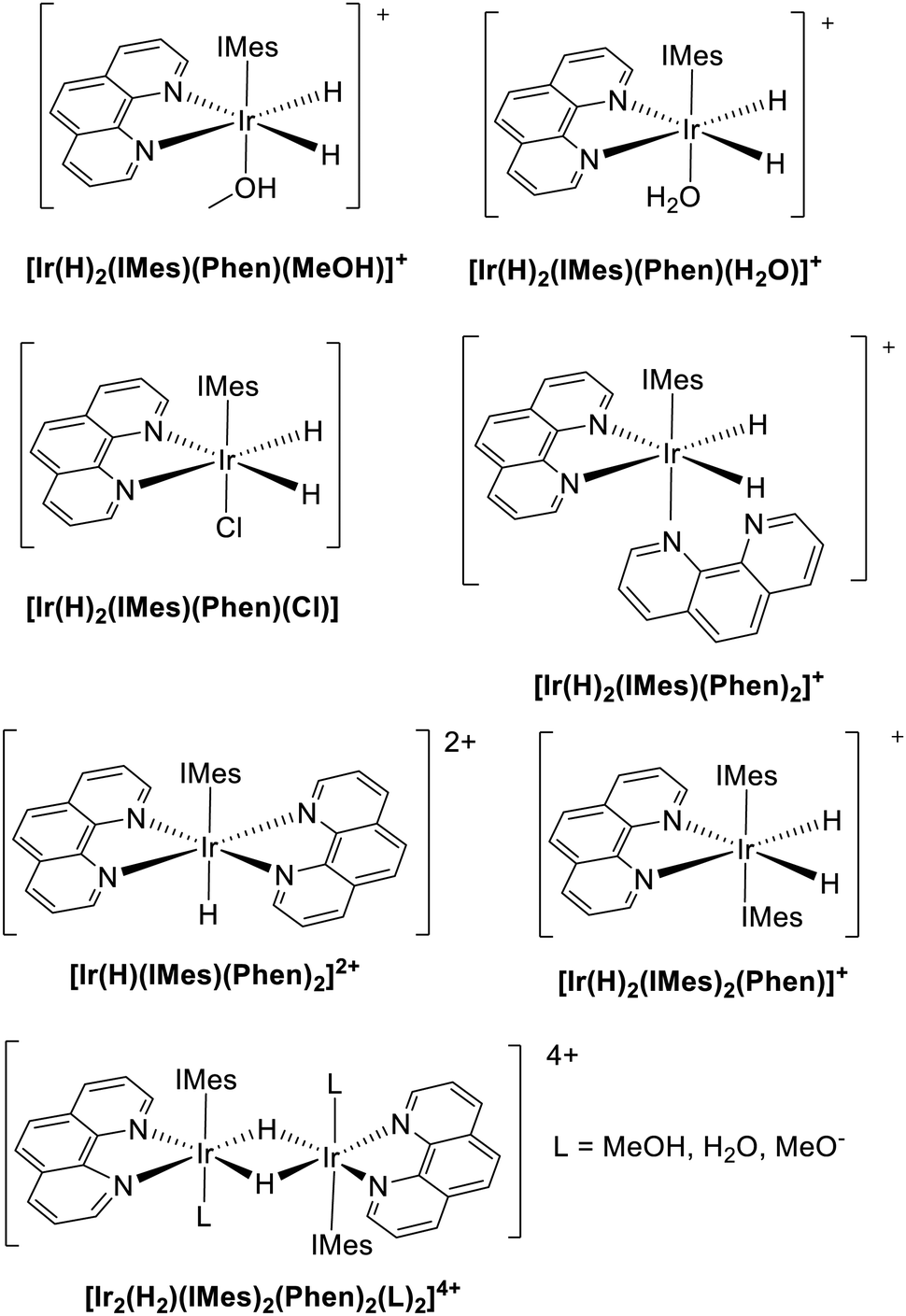 | ||
| Chart 3 Structural formulas of hypothetical structures responsible for the experimental hydrides’ resonances observed at −20.31 and −20.91 ppm and computationally tested using level of theory (ii). Note that, in the experimental practice, deuterated methanol and water are employed, whereas their non-deuterated counterparts are used in the calculations. Since vibrational effects are not included in our computational protocol, the final DFT results are identical. | ||
Moreover, given that dimers with bridging hydrides have also been reported (see e.g.[Ir2(H)4(IMes)2(COD)]) our investigation is here extended to the centrosymmetric species [Ir2(H)2(IMes)2(Phen)2(L)2]4+, see Chart 3. For the sake of completeness, the results of the calculations for some other hypothetical species are included in the ESI,‡ see Table S1, even if, in such cases, simple chemical and/or symmetry arguments can be used to rule them out immediately.
The level of theory (ii), that is PBE0-ZSO/TZ2P//ω-B97XD/triple-ζ, was chosen for the prediction of the chemical shift of the hydrides of the hypothetical structures conceived above and shown in Chart 3.
Although this level is not the most accurate in terms of direct comparison between calculated and experimental chemical shifts, it has a relatively high predictive power (the correlation coefficient is even slightly higher than level (iv) and it has a computational cost lower than level (iv). Using this level of theory, once the chemical shifts of the hydrides are obtained for a putative structure, they are rescaled using the fitting parameters of level (ii) as
| δest = (δcalc − b)/a, | (3) |
| Structure | σ calc | δ calc | δ est |
|---|---|---|---|
| [Ir(H)(IMes)(Phen)2]2+ | 41.94 | −10.44 | −13.18 |
| [Ir(H)2(IMes)(Phen)2]+ | 46.28 | −14.78 | −18.31 |
| [Ir(H)2(IMes)(Phen)(MeOH)]+ | 46.54 | −15.04 | −18.61 |
| [Ir(H)2(IMes)(Phen)(H2O)]+ | 46.87 | −15.37 | −19.00 |
| [Ir(H)2(IMes)(Phen)(Cl)] | 47.23 | −15.73 | −19.43 |
| [Ir(H)2(IMes)2(Phen)]+ | 48.26 | −16.76 | −20.64 |
| [Ir2(H)2(IMes)2(Phen)2 (MeOH)2]4+ | 51.95 | −20.45 | −25.01 |
| [Ir2(H)2(IMes)2(Phen)2(MeO)2]2+ | 53.70 | −22.20 | −27.07 |
| [Ir2(H)2(IMes)2(Phen)2(Cl)2]2+ | 54.37 | −22.87 | −27.86 |
First of all, based on the comparison of the estimated chemical shifts and the experimental values (−20.31 and −20.91 ppm) we can exclude the possibility of a dimer formed in solution, since the most likely ones reported in Table 3 have an estimated chemical shift significantly lower than the observed resonances (between −25 and −27 ppm). On the other hand, the monohydride compound [Ir(H)(IMes)(Phen)2]2+ is predicted to be much less shielded and therefore can be safely excluded.
Although discarding the many structures that do not match the experimental values has narrowed down the set of likely candidates, still there remain several species with an estimated chemical shift of around −20 ppm, all of them compatible with the experimental data. The computational protocol, however, exhibits a relatively high degree of error, as evidenced by the correlation coefficient of 0.9518 and the data dispersion observed in Fig. 1. Consequently, this level of uncertainty prevents the unambiguous assignment of any of the remaining proposed structures shown in Chart 3.
Reverting to the experimental procedure, we then prepared a sample containing 1 mM (in place of 10 mM) 1,10-phenanthroline and 1 mM [IrCl(COD)(IMes)]. Hydrogenation of this sample under 3 bar H2 resulted in a substantial reduction of the signal at −20.91 ppm relative to the signal at −20.31 ppm (see Fig. S6 of ESI‡). The signal at −20.91 ppm also remained unchanged upon re-hydrogenation of the sample. This observation suggests that the excess of phenanthroline indeed plays a role in the formation of the complex having hydride resonances at −20.91 ppm and, based on this evidence, we assign this signal to the [Ir(H)2(IMes)(Phen)2]+ complex.
The hydrides resonating at −20.31 ppm, in contrast, display signal amplification upon p-H2 bubbling, indicating an efficient exchange with dissolved p-H2. Additionally, under SABRE conditions, the phenanthroline ligand trans to these hydrides undergoes 1H polarization. In order to clarify whether this complex involves Cl− as a ligand, we prepared a sample containing 1 mM [IrCl(COD)(IMes)], 2 mM 1,10-phenanthroline and 5 mM tetrabuthyl (TBU) ammonium chloride. Hydrogenation of this sample under 3 bar H2 led to a significant reduction in the signal at −20.91 ppm and to the emergence of a signal at −20.76 and −20.79 ppm, the former of which tended to disappear over time (Fig. S6, ESI‡). Notably, the signal at −20.79 ppm was also detected at trace levels in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 sample of 1,10-phenanthroline and [IrCl(COD)(IMes)] with no TBU Cl, suggesting that this signal may be referred to the species [Ir(H)2(Phen)(IMes)(Cl)].
:1 sample of 1,10-phenanthroline and [IrCl(COD)(IMes)] with no TBU Cl, suggesting that this signal may be referred to the species [Ir(H)2(Phen)(IMes)(Cl)].
Based on this evidence, we assign the hydrides resonating at −20.31 ppm to the complex with MeOD in the axial position [Ir(H)2(IMes)(Phen)(MeOH)]+ (see Chart 3). Indeed, equatorial coordination of methanol to the Ir center has already been reported in the context of catalyst deactivation.36
The pyruvate spin system
The estimate of the optimal field for achieving an effective homonuclear or heteronuclear SABRE is guided by an analysis of the conditions required for level anticrossing (LAC). This analysis necessitates a precise knowledge of the coupling constants J between the various nuclei at play in the transfer of spin order from p-H2 to exchangeable substrate molecules. It is therefore interesting to assess whether the same computational approach used for predicting hydrides chemical shifts can also yield accurate predictions of the coupling constants networks in SABRE-active spin systems. In this context, pyruvate emerges as a good candidate molecule because it has been extensively investigated due to its potential to provide largely enhanced signals.The protocols for SABRE hyperpolarization of pyruvate were introduced and subsequently refined by Duckett's group. However, it is only recently that the small 1H–13C J couplings of pyruvate bound to the IMes-Ir catalyst have been experimentally determined using 1H–13C PASADENA spectra.37 Measuring the J couplings in these complexes poses in fact significant challenges due to their intrinsically small values and to the line broadening effects resulting from chemical exchange processes. Indeed, simulations of 13C spectra of pyruvate as a function of the polarization field have highlighted the importance of employing accurate J coupling values, including those of small magnitude.38 Therefore, we have calculated the coupling constants and carbon chemical shifts for the relevant pyruvate systems, namely the [Ir(H)2(IMes)(Pyr)(DMSOax/eq)] complexes. For the case of the equatorial DMSO coordination, we also considered the possibility of pyruvate coordinating with two possible orientations. The results are shown in Table 4.
| |Jexpt|/Hz | J calc/Hz | δ expt(C)/ppm | δ calc(C)/ppm | |||
|---|---|---|---|---|---|---|
| [Ir(H)2(IMes)(Pyr)(DMSOax)] | Ha–Hb | 10.48 | −7.768 | |||
| C1–Ha | 0.55 | 0.286 | C1 (COO−) | 168.55 | 174.60 | |
| C1–Hb | 0.014 | −0.911 | ||||
| C2–Ha | 0.93 | −1.332 | C2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
206.41 | 217.82 | |
| C2–Hb | 0 | 0.451 | ||||
| [Ir(H)2(IMes)(Pyr)(DMSOeq)] | Ha–Hb | 6.55 | −4.869 | |||
| C1–Ha | 0.32 | 0.141 | C1 (COO−) | 166.40 | 173.51 | |
| C1–Hb | 0.25 | 0.23 | ||||
| C2–Ha | 0.41 | 0.509 | C2 (CO) |
196.50 | 220.90 | |
| C2–Hb | 2.69 | −1.421 | ||||
| [Ir(H)2(IMes)(Pyr)(DMSOeq)] (alternative Pyr coordination) | Ha–Hb | 6.55 | −4.793 | |||
| C1–Ha | 0.32 | 0.137 | C1 (COO−) | 166.40 | 172.07 | |
| C1–Hb | 0.25 | −0.977 | ||||
| C2–Ha | 0.41 | 0.691 | C2 (CO) |
196.50 | 226.91 | |
| C2–Hb | 2.69 | −0.622 |
First, we note that the 2J(1H,1H) couplings are systematically underestimated by a factor of about 0.7. Since these are accurately measured and assigned from the spectra, we take this scaling factor as a systematic error of the computational protocol. The remaining couplings are rather small and only qualitatively in agreement with the experimental values, that is both calculated and experimental values are generally below 1 Hz, but the correlation is poor for such small couplings. Indeed, using larger basis sets such as QZ4P-J could result in a much better agreement between calculated and experimental coupling constants. However, such calculations would be quite demanding for these relatively large systems.
Second, we highlight the possibility of an alternative binding mode for pyruvate in [Ir(H)2(IMes)(Pyr)(DMSOeq)], in which the positions of the carbonyl and carboxyl groups are interchanged with respect to the structure proposed in ref. 37, see Fig. 4. This complex, having an axial carbonyl group, exhibits a higher energy (approximately 2.8 kcal mol−1 in terms of ΔG from ref. 37 3.7 kcal mol−1 from this work using level (ii)) and provides a less favourable agreement between the calculated and experimental J couplings compared to the complex with an axial carboxyl group. In particular, the 2J(1H,13C) coupling between Hb and C2, see Fig. 4, which exhibits a relatively large experimental value of 2.69 Hz, is calculated to be significantly smaller than experimentally observed, even after considering the scaling factor of about 0.7, while the result obtained from the original structure is in better agreement.
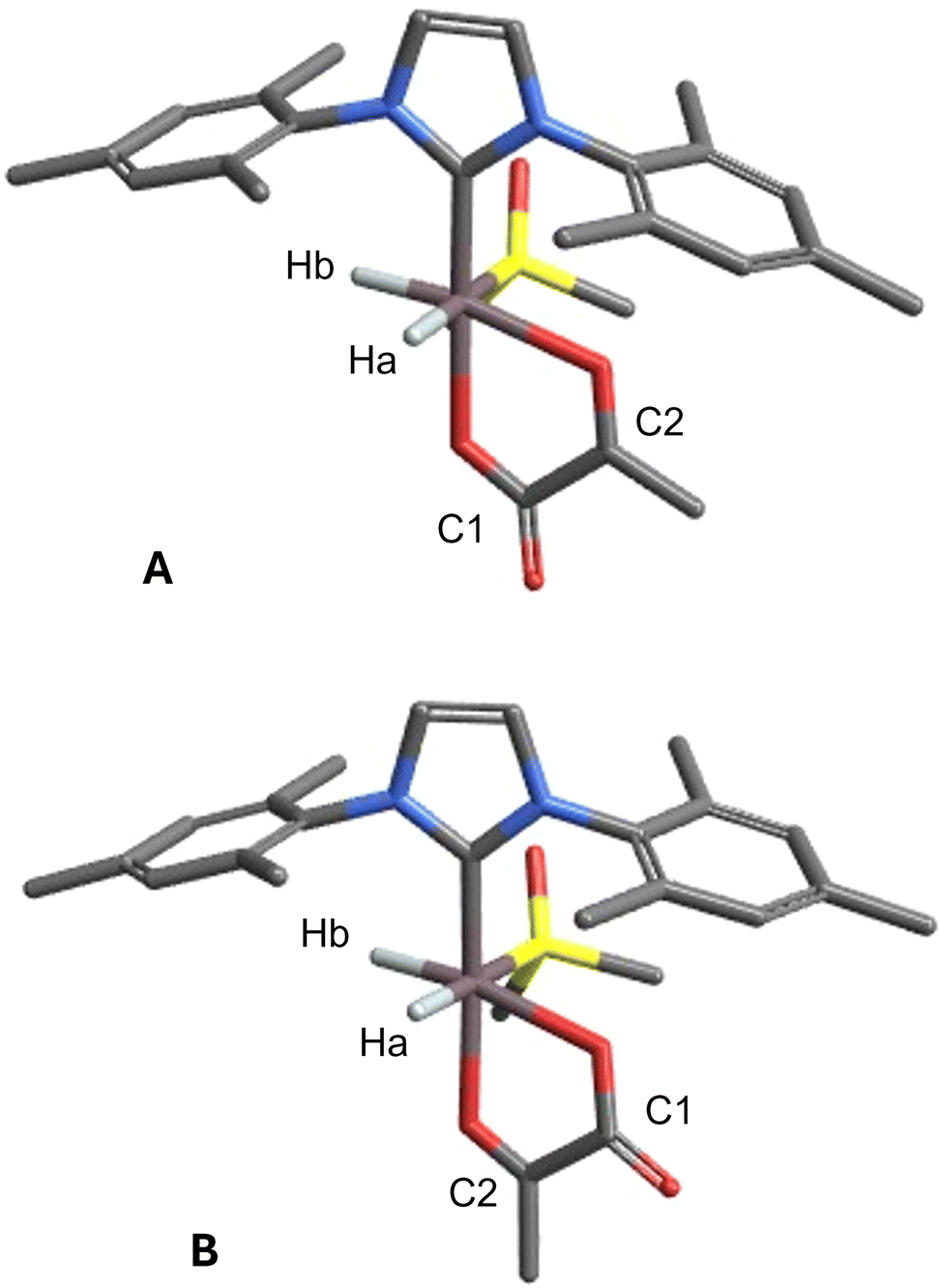 | ||
| Fig. 4 Alternative geometries of [Ir(H)2(IMes)(Pyr)(DMSOeq)] with atom labels. Geometry (A) has the lowest relative energy. Geometry (B) is discarded based on its higher relative energy and on the J-coupling pattern, see text and ref. 37. | ||
Overall, we find that the predicted carbon chemical shifts and J couplings are close to the experimental values and confirm the geometrical arrangement of pyruvate, in the case of the [Ir(H2)(IMes)(Pyr)(DMSOeq)] with the carboxylate oxygen in the axial position and the carbonyl oxygen in the equatorial position.
Conclusions
In this study, we employed relativistic density functional theory protocols to examine the chemical shift of several iridium hydride complexes which are of relevance in SABRE experiments.The validation of the computational protocols using a set of literature compounds with known structure and hydride chemical shifts allows to draw some general conclusions. First, the spin–orbit coupling is necessary to achieve an acceptable agreement between calculated and experimental results, since it is responsible for the large shielding of the hydride resonances (around −20 ppm) through the HALA mechanism.4 Then, although a systematic benchmarking of various functionals and basis sets was outside the scope of this study, we can say that the PBE0 functional with a triple-ζ quality basis set represents a very good compromise between accuracy and computational cost. Finally, long-range solvent effects also give a noticeable contribution, though quantitatively less important than the SO term, as well as the use of a fully relativistic 4-component level instead of a ZORA level.
However, it is noteworthy that the linear correlation at the ZSO level is already considerably strong and it can be used to predict the hydride chemical shifts of hypothetical structures within a range of approximately 1 ppm. It is clear that such accuracy is not sufficient to unambiguously assign experimental resonances that can be as close as few tenths of a ppm. However, compared to the range of chemical shifts potentially observable in iridium hydrides complexes (from about −10 to about −30 ppm) this interval is sufficiently narrow to allow discarding those structural proposals whose calculated chemical shift is very different from the experimental observation. Indeed, in some experiments involving phenanthroline as a ligand, this approach has been useful to discard several proposed structures that, in principle, could have accounted for two major resonances observed at −20.31 and −20.91 ppm. These resonances have been finally assigned to [Ir(H)2(IMes)(Phen)(MeOH)]+ and [Ir(H)2(IMes)(Phen)2]+, respectively.
Concerning spin–spin couplings, we have highlighted the potential of comparing calculated and experimental J values involving the hydrides to confirm or discard conformational isomers of the bidentate pyruvate ligand.
Finally, we expect this approach to have implications not only in the identification of unknown species but also in the rational design of novel catalysts, wherein the electronic environment of the metal center can be modulated through the strategic selection of appropriate ligands.
Author contributions
BBM: investigation, writing – review & editing. GZ and FDB: conceptualization, investigation. FR and GS: conceptualization, methodology, investigation, formal analysis, writing – original draft, review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.‡Acknowledgements
This work was funded by the Italian Association for Cancer Research (AIRC) under the Investigators Grants scheme (IG 25003). CloudVeneto Consortium and the Computational Chemistry Community of the Department of Chemical Sciences of the University of Padova (C3P) are acknowledged for the use of computing and storage facilities. F. R. acknowledges helpful discussions with Cristina Tubaro and Marco Tessari.Notes and references
- J. Eills, D. Budker, S. Cavagnero, E. Y. Chekmenev, S. J. Elliott, S. Jannin, A. Lesage, J. Matysik, T. Meersmann, T. Prisner, J. A. Reimer, H. Yang and I. V. Koptyug, Chem. Rev., 2023, 123, 1417–1551 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- D. A. Barskiy, S. Knecht, A. V. Yurkovskaya and K. L. Ivanov, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 33–70 CrossRef CAS PubMed.
- M. Kaupp, O. L. Malkina, V. G. Malkin and P. Pyykkö, Chem. – Eur. J., 1998, 4, 118–126 CrossRef CAS.
- M. O. Marcarino, M. M. Zanardi, S. Cicetti and A. M. Sarotti, Acc. Chem. Res., 2020, 53, 1922–1932 CrossRef CAS PubMed.
- A. Bagno and G. Saielli, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 228–240 CAS.
- J. Autschbach and S. Zheng, Annu. Rep. NMR Spectrosc., 2009, 67, 1–95 CrossRef CAS.
- Y. Xiao, W. Liu and J. Autschbach, in Relativistic Theories of NMR Shielding, ed. W. Liu, Springer, Berlin, Heidelberg, 2017, pp. 657–692 Search PubMed.
- J. Vicha, S. Komorovsky, M. Repisky, R. Marek and M. Straka, J. Chem. Theory Comput., 2018, 14, 3025–3039 CrossRef CAS.
- G. Saielli, Adv. Theory Simul., 2018, 1, 1800084 CrossRef.
- R. Pigliapochi, A. J. Pell, I. D. Seymour, C. P. Grey, D. Ceresoli and M. Kaupp, Phys. Rev. B, 2017, 95, 54412 CrossRef.
- J. Vicha, J. Novotný, S. Komorovsky, M. Straka, M. Kaupp and R. Marek, Chem. Rev., 2020, 120, 7065–7103 CrossRef CAS PubMed.
- I. L. Rusakova and L. B. Krivdin, Mendeleev Commun., 2018, 28, 1–13 CrossRef CAS.
- I. L. Rusakova and Y. Y. Rusakov, Magnetochemistry, 2023, 9, 24 CrossRef CAS.
- K. Kozioł, I. A. Aucar, K. Gaul, R. Berger and G. A. Aucar, J. Chem. Phys., 2024, 161, 64307 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B1, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde, E. J. Baerends, C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- E. J. Baerends, T. Ziegler, A. J. Atkins, J. Autschbach, D. Bashford, O. Baseggio, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerritger, L. Cavallo, C. Daul, D. P. Chong, D. V. Chulhai, L. Deng, R. M. Dickson, J. M. Dieterich, D. E. Ellis, M. van Faassen, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. Goez, A. W. Götz, S. Gusarov, F. E. Harris, P. van den Hoek, Z. Hu, C. R. Jacob, H. Jacobsen, L. Jensen, L. Joubert, J. W. Kaminski, G. van Kessel, C. König, F. Kootstra, A. Kovalenko, M. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, S. M. Morton, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, M. Pavanello, C. A. Peeples, P. H. T. Philipsen, D. Post, C. C. Pye, H. Ramanantoanina, P. Ramos, W. Ravenek, J. I. Rodríguez, P. Ros, R. Rüger, P. R. T. Schipper, D. Schlüns, H. van Schoot, G. Schreckenbach, J. S. Seldenthuis, M. Seth, J. G. Snijders, M. Solà, S. M. M. Swart, D. Swerhone, G. te Velde, V. Tognetti, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo and A. L. Yakovlev, ADF 2019.102, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2017, http://www.scm.com.
- M. Repisky, S. Komorovsky, M. Kadek, L. Konecny, U. Ekström, E. Malkin, M. Kaupp, K. Ruud, O. L. Malkina and V. G. Malkin, J. Chem. Phys., 2020, 152, 184101 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- A. Klamt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 699–709 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. Swart, M. Solà and F. M. Bickelhaupt, J. Chem. Phys., 2009, 131, 94103 CrossRef PubMed.
- G. Casella, A. Bagno, S. Komorovsky, M. Repisky and G. Saielli, Chem. – Eur. J., 2015, 21, 18834–18840 CrossRef CAS PubMed.
- L. Armangué, M. Solà and M. Swart, J. Phys. Chem. A, 2011, 115, 1250–1256 CrossRef PubMed.
- J. Vícha, M. Patzschke and R. Marek, Phys. Chem. Chem. Phys., 2013, 15, 7740–7754 RSC.
- R. E. Mewis, M. Fekete, G. G. R. Green, A. C. Whitwood and S. B. Duckett, Chem. Commun., 2015, 51, 9857–9859 RSC.
- B. J. A. van Weerdenburg, A. H. J. Engwerda, N. Eshuis, A. Longo, D. Banerjee, M. Tessari, C. F. Guerra, F. P. J. T. Rutjes, F. M. Bickelhaupt and M. C. Feiters, Chem. – Eur. J., 2015, 21, 10482–10489 CrossRef CAS PubMed.
- C. D. Assaf, X. Gui, A. A. Auer, S. B. Duckett, J.-B. Hövener and A. N. Pravdivtsev, J. Phys. Chem. Lett., 2024, 15, 1195–1203 CrossRef CAS PubMed.
- A. Browning, K. Macculloch, P. TomHon, I. Mandzhieva, E. Y. Chekmenev, B. M. Goodson, S. Lehmkuhl and T. Theis, Phys. Chem. Chem. Phys., 2023, 25, 16446–16458 RSC.
Footnotes |
| † In memory of Alessandro Bagno [1958–2015]. |
| ‡ Electronic supplementary information (ESI) available: NMR spectra of the phenanthroline system, additional correlation parameters, cartesian geometries of the molecules studied. See DOI: https://doi.org/10.1039/d5cp01214g |
| This journal is © the Owner Societies 2025 |