
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NCI orbital decomposition and critical comparison to local correlation schemes†
Xaiza Aniban‡
 a,
Maxime Ferrer‡§
c,
M. Merced Montero-Campillod,
Ricardo A. Mata*a,
Julia Contreras-García*b and
Martí Gimferrer*a
a,
Maxime Ferrer‡§
c,
M. Merced Montero-Campillod,
Ricardo A. Mata*a,
Julia Contreras-García*b and
Martí Gimferrer*a
aInstitut für Physikalische Chemie, Tammannstrasse 6, Göttingen 37077, Germany. E-mail: rmata@gwdg.de; marti.gimferrerandres@uni-goettingen.de
bLaboratoire de Chimie Théorique (LCT), CNRS, Sorbonne Université, Paris 75005, France. E-mail: julia.contreras_garcia@sorbonne-universite.fr
cInstituto de Química Médica (CSIC), Juan de la Cierva 3, Madrid 28006, Spain
dDepartamento de Química, Universidad Autónoma de Madrid, Campus de Excelencia UAM-CSIC – Calle Tomás y Valiente s/n, Madrid 28049, Spain
First published on 6th June 2025
Abstract
In this study, we introduce novel orbital decomposition approaches for analyzing non-covalent interactions (NCIs) and dispersion interaction densities (DID), termed o-NCI and o-DID, respectively. Orbital pair analyses offers an opportunity to analyse in-depth NCIs in four model dimer systems to which dispersion forces contribute to different extents: argon, methane, water and benzene–acetylene dimers. The comparative calculations reveal that intuitive interpretations based solely on nearby σ- and π-orbital interactions may overlook substantial contributions from more distant orbitals. For instance, in the benzene–acetylene dimer, interactions between π-orbitals significantly contribute to the overall dispersion energy, rivaling traditional σ bond contributions. Overall, this work establishes a comprehensive framework for understanding NCIs through the lens of orbital contributions and highlights that our interpretations must account for the intricate interplay between different interaction types. It also underlines the differences between NCI and local correlation energy decomposition, paving the way for advancements in the design and analysis of molecular systems based on NCIs.
1 Introduction
The interplay of noncovalent interactions (NCIs) is essential for understanding numerous chemical and biological processes. According to Kollman's definition,1 NCIs are characterized by two key features: (1) the electrons in reactants and products remain paired, and (2) there is no alternation in the type of chemical bonding between reactants and products. NCIs encompass different types of chemical interactions, which depend on the nature of the elements (or synthons) involved in the interactions. These include hydrogen-, halogen-, chalcogen- and pnictogen-bonding, as well as π-effects, agostic, anagostic, and London dispersion forces, to name a few. Although these forces are significantly weaker than covalent interactions – by 1 or 2 orders of magnitude in small molecular systems – their cumulative effect can be substantial, and even dominant, in larger systems.2 For instance, NCIs play a key role in processes such as protein folding,3 protein–substrate (host–guest) interactions,4 packing of molecular crystals,5 self-assembly of nanomaterials,6,7 as well as in chemical reactions.8Given their significance, the field of NCIs has experienced significant growth over the years, with great efforts invested in developing accurate methods for their characterization.2,9 However, describing NCIs is not a straightforward task. In solution, the combination of multiple NCIs and complex solvation effects makes it challenging to pinpoint the characteristics of any isolated interaction. From quantum chemical calculations, its description is also complicated due to the ubiquitous presence of London dispersion forces, which are not properly accounted for by some commonly used methods. In systems where dispersion is dominant, it is a non-trivial task to find the right balance.10–12 Hence, an accurate description of these forces using ab initio methods is required but also a challenge in quantum chemistry.
Several theoretical approaches are available for describing NCIs. For instance, considering a system composed of two fragments (monomers), some methods decompose the interaction between monomers into chemically meaningful terms, such as the symmetry adapted perturbation theory (SAPT)13,14 and the energy decomposition analysis (EDA, in its many variants).15–19 From the latter, the Absolutely localized molecular orbital EDA (ALMO-EDA) has been particularly popular.20 Alternatively, real-space energy partitioning schemes, known as interacting quantum atoms (IQA) approaches,21,22 allow the decomposition of the system's energy into one- and two-center terms, leading to a natural emergence of NCIs from grouping inter-fragment energy contributions.23–25 In the real-space framework, other approaches based on a chemically grounded scalar or vector field exist, with the most popular ones being the non-covalent interaction (NCI) index,26 the restricted space partitioning,27 the quantum chemical topology (QCT)28,29 based on the quantum theory of atoms in molecules (QTAIM)30,31 and the electron localization function (ELF).32 Among the outlined methodologies, the NCI index is the most widely used tool for assessing NCIs due to its ability to spatially (in the real-space) localize the interactions while providing a rough idea of the atoms involved at a low computational cost (for further details, see Section 2.1). It complements QCT analysis by depicting interactions produced in regions where the density gradient approaches zero – a common signature of weak interactions.
Beyond the computational cost, computing weak interactions also suffers from basis set superposition error (BSSE).33 A feasible strategy for mitigating this issue is to apply BSSE corrections, such as the counterpoise (CP) method.34 However, its computational cost rapidly increases with the basis set size, and it has been proved that it may overestimate (overcorrect) the BSSE when using insufficiently large basis.35–37 Alternatively, one can rely on using local correlation (LC) methods, developed over the years to reduce the computational cost of well-established wavefunction methods, with minimal accuracy penalty. In this regard, pioneering work by Pulay and Sæbø,38–40 together with extensive efforts from several research groups,41–50 contributed to the development of the first generation LC methods based on projected atomic orbitals (PAOs). The latter, by construction, have the property of removing some of the BSSE, even intramolecular.51 The most recent LC methods are built upon pair natural orbitals (PNOs), numerically approaching the canonical results while being computationally much more efficient, but with little impact on the BSSE.52,53
LC schemes rely on using localized molecular orbitals (LMOs), obtained by transforming the canonical molecular orbitals. The localization procedure is not unique, with three of the most common schemes being the Förster–Boys,54,55 Edmiston–Ruedenberg56 and Pipek–Mezey,57 with the intrinsic bond orbitals (IBOs) providing a more recent and popular alternative.58 The use of LMOs bridges the gap between electronic structure and chemical intuition, linking quantum mechanical findings to “classical” chemical concepts.
The aim of this work is twofold. First, we introduce an orbital decomposition of the NCI index (henceforth termed o-NCI) for the first time. This is complemented by a spatial analysis of LC methods aimed at reconciling the NCI index with the (localized) orbital picture (Section 2). By achieving an orbital representation of these interactions, we facilitate a direct comparison between schemes. Hence, we build a comparison between the two formulations utilizing four simple and well-established dimer systems. Our main focus will be on dispersion interactions for three reasons. First of all, it is the one interaction type that can be more strictly defined across different NCI analysis schemes. Secondly, given the importance of electron correlation in its description, it is the computationally most challenging interaction. Finally, it becomes a dominating force for large molecular systems, significantly impacting the PES topology.59,60
2 Methods
2.1 Non-covalent interactions (NCI) analysis
The non-covalent interaction index, known usually as NCI, is a visualization index based on the electronic density and its reduced density gradient.26 The base of this analysis is the empiric observation that non-covalent interactions can be associated with regions of low electronic density of the space and small values for the reduced density gradient, s, given by:
 | (1) |
In the NCI context, s(r) is plotted against the electron density, being the interactions represented as troughs. Fig. 1 (right side) shows one example, where the region of low density and low s can be associated with the non-covalent interaction in methane dimer. Representing in 3D the points in the peak (s < 0.5) leads to Fig. 1 (left side), where the region of interaction is revealed. For further details, we guide the reader to ref. 61–64.
 | ||
| Fig. 1 NCI surface of the methane dimer (left), and its associated s(ρ) plot (right). | ||
It should be noted that such an analysis does not set dispersion apart from other van der Waals (VdW) interactions, albeit it does contain it. It is described somewhat close in spirit to the Feynman interpretation of dispersion, as resulting from a shift in density.65 The reduced density gradient, s(r), is an adimensional measure that accounts for the inhomogeneity of the density at a given point of the real-space r, since s(r) measures the ratio at which the density changes with respect to the uniform electron gas (UEG), and was originally conceived as a correction for semi-local density functional approximations.66 In real molecular systems, this quantity can also be interpreted when the electron density importantly differ from the UEG.
As mentioned above, this procedure allows revealing different NCIs by changing focus from low- to high-density regions. However, the immediate chemical insight for the interaction in orbital terms is lost. In this section, we propose a new development of the NCI index, which enables to extract the pair-wise orbital contributions to non-covalent interactions. The so-called orbital non-covalent interaction (o-NCI) method enables one to clearly associate (localized) molecular orbitals with real-space interactions.
The reduced density gradient can be related to the relative bosonic kinetic energy density, tbose:67
 | (2) |
 | (3) |
Without loss of generality, we can assume real space orbitals, leading to
 | (4) |
Monocentric and bicentric terms along with the total s2 are shown for H2 with R = 1 Bohr in Fig. 2. s2 (blue line) is minimal and equal to zero at the nuclei and at the bond. When the monocentric and bicentric terms are analyzed we can see that the monocentric terms,  (red line) are responsible for the peaks at the hydrogen nuclei, which, in that case, is obtained by summing at each point of the grid the value of
(red line) are responsible for the peaks at the hydrogen nuclei, which, in that case, is obtained by summing at each point of the grid the value of  and
and  , whereas the bicentric term,
, whereas the bicentric term,  , is responsible for the bonding peak. This is the “contragradient region”.69,70 Note that the monocentric term leads to positive values in the bonding region, whereas
, is responsible for the bonding peak. This is the “contragradient region”.69,70 Note that the monocentric term leads to positive values in the bonding region, whereas  is negative values, thus leading to s2 = 0 at the bond mid-point.
is negative values, thus leading to s2 = 0 at the bond mid-point.
 | ||
| Fig. 2 1D NCI of H2 in the singlet spin state. Geometry and wavefunction evaluated at the B3LYP-D3(BJ)/aug-cc-pVTZ level of theory. | ||
However, as the number of shells increases, these contragradient regions also appear in the core. Hence, if we want to focus on intermolecular interactions, it is interesting to separate intra- and intermolecular bicentric contributions. If we define two molecular fragments, A and B, the  contribution can be separated into inter- and intramolecular terms:
contribution can be separated into inter- and intramolecular terms:
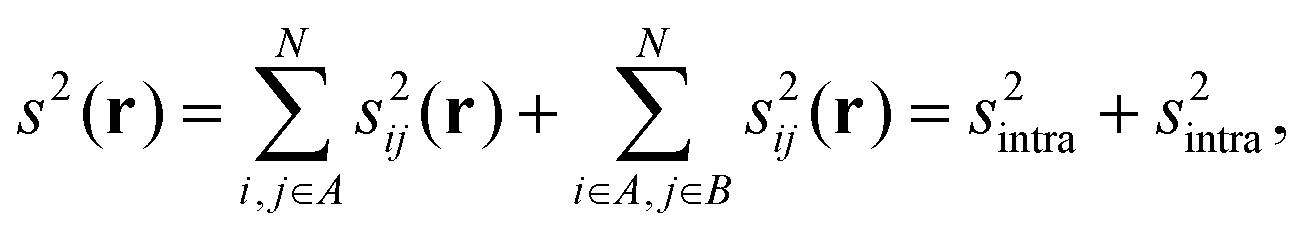 | (5) |
Two different options appear, local and global. It is possible to check the value of  at the minimum, as shown in Fig. 2; or to integrate
at the minimum, as shown in Fig. 2; or to integrate  within the NCI volume. Both options will be analyzed in the text.
within the NCI volume. Both options will be analyzed in the text.
2.2 Dispersion interactions analysis with local correlation
The interpretation of dispersion in the context of local correlation is quite straightforward. Given that the excitations are spatially resolved, one can distinguish London dispersion energy terms from other correlation contributions. The former should arise from an excitation pair whereby both monomers are involved but the electron “ownership” is untouched. In other words, an {ij} → {ab} excitation with i,a located in one monomer and j,b in a different monomer. We apply the common nomenclature of using i,j as occupied orbital indices and a,b for virtuals. This provides the definition of each orbital pair contribution (in the restricted closed–shell formalism) to the dispersion energy as
 | (6) |
The monomers need to be defined but the approach is flexible enough to also handle intramolecular energy terms. One only needs to define grouping criteria for the different orbitals.71,72
The pair dispersion energy terms edispij can in turn be used to build a visual representation of dispersion interactions. How this is achieved is again not unequivocally defined. In previous works, and a definition which has been used by other groups as well,73–75 we have weighted the respective orbital densities as
 | (7) |
| Piμν = CμiCνi, | (8) |
The DID definition is significantly different from that obtained in the NCI framework. It is close to the London dispersion picture, where electron clouds for the most part keep their form and interact over a distance. Although it is not possible to reach the same type of analysis, the DID and NCI pictures are comparable when defining zones of weak interaction between monomers. We propose to provide such zones of interaction by displaying regions of space where the DID of different monomers overlap. This brings us to the following expression
 | (9) |
The main issue is that one is depending on the individual orbital densities ρi which commonly contain orthogonalization tails, irrespective of the basis set size used. In order to have a stricter spatial location of these overlap zones, we opted to strictly orthogonalize the localized orbitals, setting MO coefficients from AOs centered in other monomers to zero. This leads to the strictly orthogonalized individual orbital densities
 | (10) |
These orthogonalized density matrices ![[D with combining tilde]](https://www.rsc.org/images/entities/i_char_0044_0303.gif) iμν can then be used in eqn (9) to build the respective ρi(r) values, and provide the basis for the plots later shown in the manuscript labeled as o-DID.
iμν can then be used in eqn (9) to build the respective ρi(r) values, and provide the basis for the plots later shown in the manuscript labeled as o-DID.
3 Computational details
Geometry optimizations for all systems were performed with the Molpro 2021.2 package76 at the SCS-MP2 level together with the aug-cc-pVTZ basis set for all atoms except of H, where the cc-pVTZ basis was employed. To confirm the nature of the stationary points (minima), vibrational frequency calculations at the same level of theory were performed.Evaluation of the DID surfaces, as well as the quantification of its orbital interactions, was realized at the SCS-LMP2 level with the developers’ version of the Molpro2018.1 code where the o-DID algorithm was implemented. The NCI surfaces and its orbital decomposition (o-NCI) were obtained with a developers version of the NCIPLOT version 4.063 software using the same set of localized orbitals as in the SCS-LMP2 calculations. Numerical integrations are carried out following the integration scheme already implemented in NCIPLOT on a grid 0.05, 0.05, 0.05 a.u. along x, y, z.63 For the SCS-LMP2 calculations, and the o-DID and o-NCI analysis, Pipek–Mezey localized orbitals were employed.
The energy decomposition analysis (EDA) results were obtained with the Su and Li formulation of the Kitaura–Morokuma (KM) analysis.77 In particular, this method decomposes the interaction energy (ΔEInt) between molecular fragments (in our case monomers) into electrostatic (ΔEElec), exchange (ΔEExch), repulsion (ΔERep) and polarization (ΔEPol) energy terms, solely based on the reference Hartree–Fock calculation
| ΔEInt = ΔEElec + ΔEExch + ΔERep + ΔEPol. | (11) |
The correlation contribution was decomposed with the PNO-SCS-LMP2 scheme, which combines the simplicity of the spin-scaled MP2 method and the accuracy of higher-level methods, into dispersion (ΔEDisp) and ionic (ΔEIonic) contributions. Both decompositions were performed with the Molpro 2018.1 developer's version, using the aug-cc-pVTZ (cc-pVTZ for H) basis set.
Visualization and representation of the molecular systems, dispersion interaction densities, NCI surfaces, and orbital contributions (o-DID and o-NCI) were performed with the VMD 1.9.4a38 software.78 All figures in this paper were generated using Matplotlib.79
4 Results and discussion
To compare the two methodologies presented, we investigated four dimer systems (see Fig. 3) where the interaction between monomers is rather well-known. These include two dispersion-dominated systems (argon and methane dimers), the water dimer – whose interaction is characterized by its strong electrostatic and polarization components – and the benzene–acetylene dimer, whose interaction lies in between the two regimes, with both dispersion and polarization playing significant roles. The comparison is structured as follows: first, we analyze the contact surfaces from both the NCI and DID perspectives, as well as their respective orbital decompositions (Sections 4.1 and 4.2). Second, we examine the behavior of these systems along their dissociation profiles, comparing them with the Su and Li formulation of the Kitaura–Morokuma energy decomposition analysis. The latter is applied (Section 4.3) to complement the local correlation analysis with an energetic decomposition. It should be noted that both the NCI and DID analyses, together with their corresponding orbital decompositions, are not restricted to dimeric systems. These methods are very versatile and applicable to any number of monomers, as reported in ref. 71 for the DID case. | ||
| Fig. 3 Selected model systems for evaluation. | ||
4.1 Contact surfaces
Estimating non-covalent interactions can be challenging. However, their role in many important biological and materials processes highlights the need to identify the main atoms involved. This identification is a crucial step towards inverse design. In the present study, one of the noteworthy points is to perform a comparative assessment of the contact surface between the molecular fragments using the aforementioned methodologies.We begin our analysis with dispersion forces, which are very effectively isolated through the LC approach.51 In local correlation, where electrons are localized in specific orbitals, a more intuitive picture of orbital information is readily available. Using o-DID, the productive interaction of each monomer in local orbital representation is highlighted. Visual inspection of the contact surfaces from o-DID (Fig. 4) reveals that their shape and volume are highly dependent on the interacting system studied.
 | ||
| Fig. 4 o-DID contact surfaces calculated at the SCS-LMP2/aug-cc-pVTZ, H = cc-pVTZ level. Isocontour values are enclosed in parenthesis for the following molecular systems: (a) argon dimer (0.20), (b) methane dimer (0.11), (c) benzene–acetylene (7.00), and (d) water dimer (50.00). | ||
For the case dispersion-dominated systems, such as the argon and methane dimers (Fig. 4(a) and (b)), the overlap density is symmetric due to the dimer conformation. The density profile for the argon dimer adopts a prolate ellipsoidal shape, evidently indicating an equal contribution from both constituent atoms. Similarly, in the methane dimer, the overlap density underscores the C–H interactions of one monomer and the other, resulting in a uniform density distribution from all the C–H arms oriented towards one another.
In the case of the benzene–acetylene dimer (Fig. 4(c)), the overlap density exhibits a distinctive shape, akin to a pear-shaped form. Notably, the semi-planar base of this density distribution is situated near the benzene ring, providing an intuitive indication of the uniform contribution stemming from multiple π-orbitals. Conversely, the density progressively narrows as it extends toward the acetylene moiety, implying that the source of dispersion contribution from this region is associated with orbitals that are spatially close.
Finally, the overlap density from the water dimer (Fig. 4(d)) adopts a distinctive configuration reminiscent of a lone pair from the oxygen atom of the left water molecule. This density distribution conveys that the orbitals responsible for dispersion interactions predominantly involve the O–H orbital of the water molecule on the right, interacting with the p-orbitals of the oxygen atom in the left water monomer. Notably, the volume of density associated with the actively interacting O–H orbital on the right water molecule is considerably larger compared to the non-contribution of benzene in the benzene–acetylene dimer.
Moving forward to analyze non-covalent interactions, the NCI method allows for the visual identification of weakly interacting regions of the space and their nature (repulsive or attractive), as indicated by coloring (red for repulsive and blue for attractive), with color intensity correlating to interaction strength. However, the surface shape provides less immediate chemical bonding information (Fig. 5).
 | ||
| Fig. 5 NCI surfaces evaluated using the pair orbitals obtained at the SCS-LMP2/aug-cc-pVTZ, H = cc-pVTZ level. Surfaces represented using s = 0.35 and a color scale of −0.05 < ρ < 0.05. | ||
NCI isosurfaces enable the distinction between pairwise and many-body based on the isosurface shape.80,81 Two-body interactions typically appear as disc-shaped surfaces between the involved atoms, as observed in Ar2 and water dimer shown in Fig. 5. In the argon dimer, the disc appears in between the two Ar atoms, whereas in the water dimer, it shows up in between the expected position of the lone pair and the hydrogen in the electron-accepting molecule (the right water molecule). In contrast, many-body interactions present more extended surfaces that cover the atoms involved. This phenomenon is evident in the methane dimer and the benzene–acetylene dimer. For the former, the surface appears in-between the H atoms pointing towards the other molecule, while in the latter, a cone shape typical of T-shape interactions with benzene rings appears. These highlight the interaction with the benzene π structure.82 These observations have been derived from the analysis of many structures over the years, providing an ad hoc link between orbitals and NCI. However, a proper mathematical link was missing. This also hampers more subtle analyses. For example, it is not possible to know from the NCI picture to which extent further away entities are contributing to the interaction. For example in the case of benzene–acetylene, is only the C–H bond contributing to the interaction with benzene, or also the triple bond have a contribution? Although the analysis of both surfaces (DID and NCI) provides valuable information, to gain further insight we propose to make use of their (localized) orbital decomposition introduced in Section 2.
4.2 Individual orbital interactions
Fig. 6 provides a graphical representation of the interacting LMOs for each studied system, complemented by a bar graph illustrating the contributions (in percentage) of each pair of LMOs involved in dispersion interactions. For the methane dimer (Fig. 6(a)), the preeminent orbital interactions arise from the face-to-face alignment of the C–H bonds. The cumulative energy stemming from these σC–H interactions accounts for approximately 71% of the total dispersion interaction energy. The remaining 29% is attributed to the peripheral C–H bonds interacting with the LMOs of the other monomer.
 | ||
| Fig. 6 Pair orbital analysis of (a) methane dimer, (b) benzene–acetylene, and (c) water dimer. In benzene–acetylene, the contributions below 1% were omitted for clarity. For ease of visualization, orbital densities are chosen as representation and the individual orbital pair labels were removed. The same figures have been included in the ESI,† but including the orbital pairs labels. Isocontour values of 0.1 were used for isosurface representations except for benzene–acetylene, where a value of 0.025 was employed. | ||
The utility of pair orbital analysis becomes particularly evident when investigating complex interactions that may not lend themselves to immediate chemical interpretations. This can be illustrated in the case of the benzene–acetylene dimer (Fig. 6(b)). Without the orbital analysis, one might intuitively attribute the density overlap observed in Fig. 4(c) to the interaction between σC2H2 orbital and the π orbitals of benzene (πC6H6). This is a valid interpretation, but the latter only accounts for 28% of the dispersion contribution. The pair orbital analysis reveals additional interactions that may elude immediate chemical intuition. Specifically, the interaction between each π orbital of C2H2 and each π orbital of C6H6 contributes, on average, approximately 4.5% to the overall dispersion interaction. This cumulative interaction represents a substantial 27% of the total dispersion contribution rivaling the σC2H2–πC6H6 interaction in significance. The substantial πC2H2–πC6H6 interaction explains the DID shape; not positioned midway between the two molecules, but rather skewed toward C2H2. Altogether, these two categories of interactions account for 55% of the overall dispersion interaction. Albeit not as predominant as the previously discussed types of interactions, it is worth noting that the σC2H2–σC6H6 still constitutes a notable fraction, contributing approximately 18% to the overall dispersion interaction. The remaining interactions are of minor magnitude and are distributed among numerous orbital pairs, rendering them less chemically meaningful in the context of the interaction analysis.
The pair orbital analysis also provides valuable insights into the dispersion interaction within the water dimer (Fig. 6(c)). Conventionally, one might interpret the dispersion interaction as arising primarily from the interaction between the σOH orbital and the p-orbitals of the oxygen atom in the other water molecule. Indeed, this orbital interaction contributes significantly, accounting for 42% of the overall dispersion interaction energy. However, another form of interaction is uncovered. Specifically, the σOH orbital of the upper water molecule interacting with the two σOH LOs of the lower water molecule collectively contributes with 20% of the dispersion energy. This nuanced depiction provided by pair orbital analysis underscores the multifaceted nature of dispersion interactions within the water dimer, extending beyond the conventional σOH – p-orbital interaction. Overall, it shows how dispersion is a sum of small contacts, with (almost) every bit of density contributing.
 | ||
| Fig. 7 o-NCI decomposition of the most relevant o-DID terms for (a) methane dimer, (b) benzene–acetylene, and (c) water dimer. o-NCI terms normalized for the sum of all intermolecular contributions (including the not represented here, adding to 100%). | ||
On the other extreme, the methane dimer interaction, depicted in Fig. 7(a), reveals contributions from three C–H bonds on each molecule, resulting in a highly multiatomic interaction characterized by 6 main interactions from the closer C–H pairs and 3 minor ones from the opposite C–H bonds, collectively yielding 81–66% of the interaction. This finding explains the big surface observed in Fig. 5(b).
The multiatomic contribution from the benzene ring is also easily identifiable in Fig. 7(b). Here, we observe that the NCI surface's T-shape is determined by the contribution from the three σC–H(acetylene)–πC–H(benzene) interactions, which comprise the main contributors (61–71%). In contrast, contributions from the intermolecular π–π interactions are negligeable, comprising only 6–2%.
Both local and global analyses yield similar qualitative results regarding the relative relevance of orbital pairs. However, different quantitative relative weights appear depending on the bonding type involved. To provide guidance on the use of s2 local or global approaches, we will compare them with o-DID in the following section.
From the o-NCI perspective, it generally appears that spatially closer orbitals have greater importance than those identified by o-DID. A clear example is the benzene–acetylene dimer, where all NCI contributions can be attributed to the σC–H(acetylene)–πC–H(benzene) interaction, while o-DID reveals a large cumulative πC–H(acetylene)–πC–H(benzene) contribution. A similar scenario is observed in the water dimer, where contributions also stem primarily from the electron-receiving σO–H orbitals.
Notably, the s2-global approach exhibits close alignment with o-DID in the methane dimer, where dispersive interactions prevail (see inset in Fig. 7(a)). Conversely, in the benzene–acetylene and water dimers, in which the bonding exhibits a greater electrostatic component, the contributions from the main orbital pairs are substantially larger for both s2 approaches.
The smaller contribution from the further (2nd, 3rd, etc.) orbital pairs can be attributed to the fact that NCI predominantly accounts for the “local” contact between densities, giving a lower weight to long-distance interaction between densities. As far as the first group of orbital pairs, the bigger differences between o-NCI and o-DID obtained for water dimer and benzene–acetylene could be related to differing aims behind each method: DID is specifically designed to reveal dispersion, while potentially overlooking other energetic components (e.g., polarization) that are shorter in range.
To further investigate this, in the next section we will analyze the behavior of o-DID and o-NCI alongside an energetic partitioning along the dissociation profiles of the studied systems.
4.3 Potential energy surface behaviour
In order to look beyond dispersion (and in general electron correlation effects) we make use of the Su and Li formulation of the Kitaura–Morokuma analysis.77 The latter decompose the interaction energy between molecular fragments (in our case monomers) into electrostatic, exchange, repulsion and polarization energy terms, solely on the basis of the reference Hartree–Fock calculation. For the electron correlation part, we decompose this contribution with the PNO-SCS-LMP2 scheme into dispersion and ionic contributions. With this, we aim to compare the behaviour of the energy values from DID and NCI with the associated energy component from KM-EDA upon dissociation.In order to verify the connection between NCI and dispersion, we have analyzed the evolution of o-NCI in the Ar dimer. Given the fact that the o-NCI minimum is divided by the electron density, it is only suitable for equilibrium distances, so we have focused on s2. Results are presented in Fig. 8 (top-left). The long-distance behavior (marked with black dots) has been fitted, showing a r5.5 trend, which is close enough to the expected rate. A similar analysis has been carried out for the dimers in our study, forcing an increasing distance and relaxing the rest of the coordinates. Results for methane dimer (Fig. 8 (top-right)), where the dispersion component is very important, also lead to an exponent close to six (6.14). Instead, it is clear that shorter-range components play an important role in water dimer and benzene–acetylene (Fig. 8 bottom), where the exponents are 10.37 and 12.06, respectively. An exponent close to six (5.64) is only recovered at even larger distances. Hence, it is expected that greater deviations are found between o-DID and o-NCI for this case. As such, in line with our comparative discussion of LC and NCI index results, there is a strong agreement between the two approaches in dispersion-dominated systems. Given that the NCI index (both in its original and in the form of o-NCI) also includes other interactions, and with a different resolution than that of LC decompositions, the comparison becomes increasingly difficult. This is further confirmed by the wave function energy decomposition on the four systems, provided in S8.
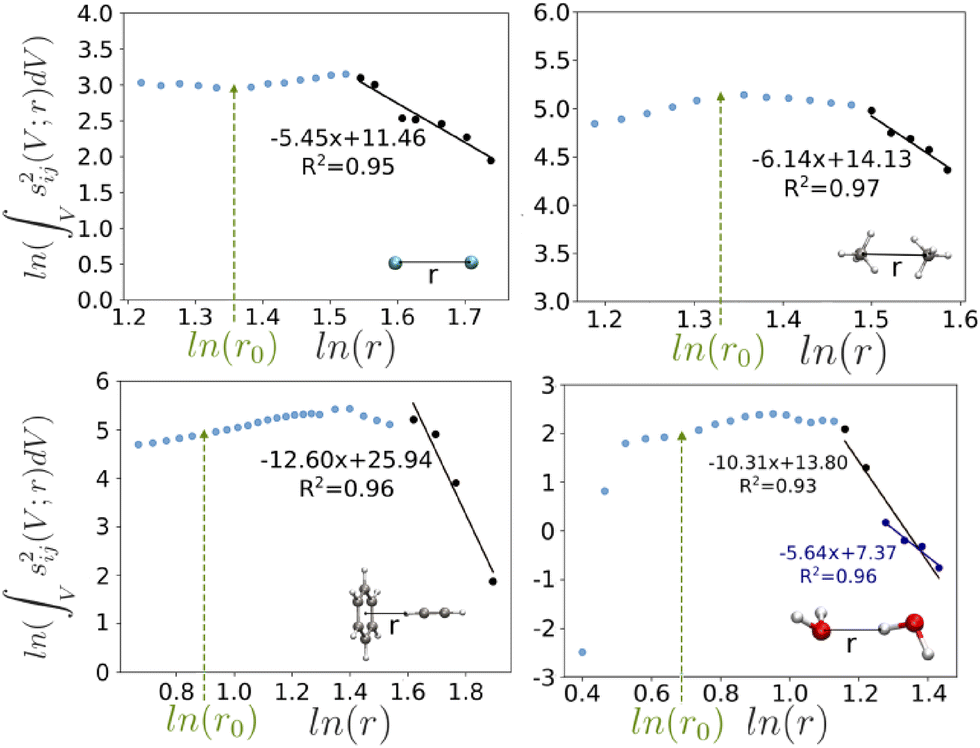 | ||
| Fig. 8 Long-range s2 integrals behavior for the studied systems at different intermolecular distances. Fits for long-range are added in black, and equilibrium geometries are marked with a green line. For the water dimer, a second fit at very long distances has been added in dark blue, while the long-range fit in black contains both black and dark blue data points. | ||
5 Conclusions
In this study, we have introduced the orbital decomposition of the non-covalent interactions (NCI) and the dispersion interactions densities methods. Also, we have conducted a comparative analysis of NCIs through the application of orbital decomposition approaches. By examining four distinct dimer systems – argon and methane dimers, the water dimer, and the benzene–acetylene dimer – we have demonstrated the effectiveness of these methodologies in elucidating the nature and contributions of dispersion interactions. Visual representations of contact surfaces highlighted distinct differences among the studied systems, with variations in overlap density providing insight into the contributions from specific orbital interactions. For example, in the methane dimer, face-to-face C–H interactions dominated the dispersion energy, while in the benzene–acetylene dimer, a significant contribution came from the interaction between π orbitals in addition to σ bond interactions from acetylene.The pair orbital analysis further clarified the complexities of these interactions, illustrating how intuitive interpretations based solely on σ and π orbital interactions close in space may overlook substantial contributions from other orbital pairs to dispersion. In the benzene–acetylene dimer, interactions between π orbitals accounted for a notable fraction of the dispersion energy, rivaling traditional σ bond contributions. The insights gained from the pair orbital analysis and o-NCI indicate that NCIs comprise a summation of multiple small contributions, underscoring the importance of a detailed breakdown of interactions in orbital pairs.
Our analysis revealed that both approaches (NCI and DID) can provide consistent results when dispersion is the dominating factor (argon and methane dimers). The analysis becomes much more complicated when other intermolecular interactions play a strong role. On the side of wave function theory, there is not a single unique definition, while in NCI these different forces cannot be disentangled. At the very least, we can confirm that in the NCI index range where dispersion forces are analyzed, other interactions are also included (see Fig. S7, ESI†). This is the first time that an orbital decomposition has been introduced within the NCI framework, which can be extremely useful in building bridges with other theories.
Overall, this work not only establishes a comprehensive framework for understanding non-covalent interactions through the lens of orbital contributions but also highlights the interplay between various interaction types, paving the way for further advancements in the design and analysis of molecular systems based on non-covalent interactions.
Data availability
The code used for o-NCI calculations is available at https://github.com/juliacontrerasgarcia/o-NCI. The code for LC analysis is provided in the Molpro program package.76Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The development of the o-NCI approach was initially tested from monocentric contributions. Although it did not lead to the intermolecular analysis we hereby highlight, it inspired the birth of the o-NCI approach. Hence, we would like to thank I. Iribarren, and O. Eisenstein, who were involved in a seminal approach. Funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 217133147/SFB 1073, project C03, and 389479699/GRK2455 is gratefully acknowledged. M. M. Montero-Campillo is grateful to the Ministerio de Ciencia, Innovación y Universidades for her José Castillejo International Mobility Grant (CAS19/00070) at the Sorbonne Université. J. Contreras-García thanks ANR TcPredictor S22JRAR036, ANR Fisciency S23JRAR060 and Emergence-SU H2Ox S23JR31014 for funding and GENCI for computational time AP010810189.Notes and references
- P. A. Kollman, Acc. Chem. Res., 1977, 10, 365–371 CrossRef CAS.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 1999, 100, 143–168 CrossRef PubMed.
- S. Fiedler, J. Broecker and S. Keller, Cell. Mol. Life Sci., 2010, 67, 1779–1798 CrossRef CAS PubMed.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- B. Silvi, Phys. Rev. Lett., 1994, 73, 842–845 CrossRef CAS PubMed.
- H. Fenniri, P. Mathivanan, K. L. Vidale, D. M. Sherman, K. Hallenga, K. V. Wood and J. G. Stowell, J. Am. Chem. Soc., 2001, 123, 3854–3855 CrossRef CAS PubMed.
- P. Kruse, E. R. Johnson, G. A. DiLabio and R. A. Wolkow, Nano Lett., 2002, 2, 807–810 CrossRef CAS.
- S. S. Sheiko, F. C. Sun, A. Randall, D. Shirvanyants, M. Rubinstein, H. il Lee and K. Matyjaszewski, Nature, 2006, 440, 191–194 CrossRef CAS PubMed.
- P. Hobza and J. Řezáč, Chem. Rev., 2016, 116, 4911–4912 CrossRef CAS PubMed.
- H. C. Gottschalk, A. Poblotzki, M. A. Suhm, M. M. Al-Mogren, J. Antony, A. A. Auer, L. Baptista, D. M. Benoit, G. Bistoni, F. Bohle, R. Dahmani, D. Firaha, S. Grimme, A. Hansen, M. E. Harding, M. Hochlaf, C. Holzer, G. Jansen, W. Klopper, W. A. Kopp, L. C. Kröger, K. Leonhard, H. Mouhib, F. Neese, M. N. Pereira, I. S. Ulusoy, A. Wuttke and R. A. Mata, J. Chem. Phys., 2018, 148, 014301 CrossRef PubMed.
- H. C. Gottschalk, A. Poblotzki, M. Fatima, D. A. Obenchain, C. Pérez, J. Antony, A. A. Auer, L. Baptista, D. M. Benoit, G. Bistoni, F. Bohle, R. Dahmani, D. Firaha, S. Grimme, A. Hansen, M. E. Harding, M. Hochlaf, C. Holzer, G. Jansen, W. Klopper, W. A. Kopp, M. Krasowska, L. C. Kröger, K. Leonhard, M. Mogren Al-Mogren, H. Mouhib, F. Neese, M. N. Pereira, M. Prakash, I. S. Ulusoy, R. A. Mata, M. A. Suhm and M. Schnell, J. Chem. Phys., 2020, 152, 164303 CrossRef CAS PubMed.
- T. L. Fischer, M. Bödecker, S. M. Schweer, J. Dupont, V. Lepère, A. Zehnacker-Rentien, M. A. Suhm, B. Schröder, T. Henkes, D. M. Andrada, R. M. Balabin, H. K. Singh, H. P. Bhattacharyya, M. Sarma, S. Käser, K. Töpfer, L. I. Vazquez-Salazar, E. D. Boittier, M. Meuwly, G. Mandelli, C. Lanzi, R. Conte, M. Ceotto, F. Dietrich, V. Cisternas, R. Gnanasekaran, M. Hippler, M. Jarraya, M. Hochlaf, N. Viswanathan, T. Nevolianis, G. Rath, W. A. Kopp, K. Leonhard and R. A. Mata, Phys. Chem. Chem. Phys., 2023, 25, 22089–22102 RSC.
- K. Szalewicz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 254–272 Search PubMed.
- E. G. Hohenstein and C. D. Sherrill, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 304–326 Search PubMed.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- G. K. Schenter and E. D. Glendening, J. Phys. Chem., 1996, 100, 17152–17156 CrossRef CAS.
- R. Z. Khaliullin, M. Head-Gordon and A. T. Bell, J. Chem. Phys., 2006, 124, 8753–8765 CrossRef PubMed.
- P. R. Horn and M. Head-Gordon, J. Chem. Phys., 2015, 143, 114111 CrossRef PubMed.
- M. Mandado and J. M. Hermida-Ramón, J. Chem. Theory Comput., 2011, 7, 633–641 CrossRef CAS PubMed.
- P. R. Horn, Y. Mao and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067–23079 RSC.
- P. Salvador, M. Duran and I. Mayer, J. Chem. Phys., 2001, 115, 1153–1157 CrossRef CAS.
- M. A. Blanco, A. M. Pendás and E. Francisco, J. Chem. Theory Comput., 2005, 1, 1096–1109 CrossRef CAS PubMed.
- D. Suárez, N. Díaz, E. Francisco and A. MartínPendás, ChemPhysChem, 2018, 19, 973–987 CrossRef PubMed.
- A. F. Silva, L. J. Duarte and P. L. A. Popelier, Struct. Chem., 2020, 31, 507–519 CrossRef CAS.
- J. M. Guevara-Vela, E. Francisco, T. Rocha-Rinza and Á. M. Pendás, Molecules, 2020, 25, 4028 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- M. Kohout, Int. J. Quantum Chem., 2003, 97, 651–658 CrossRef.
- P. L. A. Popelier, The Chemical Bond II, Springer International Publishing, 2016, pp. 71–117 Search PubMed.
- P. L. A. Popelier, J. Mol. Model., 2022, 28, 1–41 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- N. R. Kestner, J. Chem. Phys., 1968, 48, 252–257 CrossRef CAS.
- S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- P. Salvador and M. Duran, J. Chem. Phys., 1999, 111, 4460–4465 CrossRef CAS.
- P. Salvador, B. Paizs, M. Duran and S. Suhai, J. Comput. Chem., 2001, 22, 765–786 CrossRef CAS.
- M. Mentel and E. J. Baerends, J. Chem. Theory Comput., 2013, 10, 252–267 CrossRef PubMed.
- S. Sæbø and P. Pulay, Chem. Phys. Lett., 1985, 113, 13–18 CrossRef.
- P. Pulay and S. Sæbø, Theor. Chim. Acta, 1986, 69, 357–368 CrossRef CAS.
- S. Sæbø and P. Pulay, J. Chem. Phys., 1988, 88, 1884–1890 CrossRef.
- P. Maslen and M. Head-Gordon, Chem. Phys. Lett., 1998, 283, 102–108 CrossRef CAS.
- G. Hetzer, P. Pulay and H.-J. Werner, Chem. Phys. Lett., 1998, 290, 143–149 CrossRef CAS.
- G. E. Scuseria and P. Y. Ayala, J. Chem. Phys., 1999, 111, 8330–8343 CrossRef CAS.
- M. Schütz, Phys. Chem. Chem. Phys., 2002, 4, 3941–3947 RSC.
- H.-J. Werner, F. R. Manby and P. J. Knowles, J. Chem. Phys., 2003, 118, 8149–8160 CrossRef CAS.
- T. S. Chwee, A. B. Szilva, R. Lindh and E. A. Carter, J. Chem. Phys., 2008, 128, 224106 CrossRef PubMed.
- A. Venkatnathan, A. B. Szilva, D. Walter, R. J. Gdanitz and E. A. Carter, J. Chem. Phys., 2004, 120, 1693–1704 CrossRef CAS PubMed.
- M. S. Lee, P. E. Maslen and M. Head-Gordon, J. Chem. Phys., 2000, 112, 3592–3601 CrossRef CAS.
- N. J. Russ and T. D. Crawford, J. Chem. Phys., 2004, 121, 691–696 CrossRef CAS PubMed.
- T. Crawford and R. A. King, Chem. Phys. Lett., 2002, 366, 611–622 CrossRef CAS.
- M. Schütz, G. Rauhut and H.-J. Werner, J. Phys. Chem. A, 1998, 102, 5997–6003 CrossRef.
- H.-J. Werner, G. Knizia, C. Krause, M. Schwilk and M. Dornbach, J. Chem. Theory Comput., 2015, 11, 484–507 CrossRef CAS PubMed.
- Q. Ma and H.-J. Werner, J. Chem. Theory Comput., 2015, 11, 5291–5304 CrossRef CAS PubMed.
- S. F. Boys, Rev. Mod. Phys., 1960, 32, 296–299 CrossRef CAS.
- J. M. Foster and S. F. Boys, Rev. Mod. Phys., 1960, 32, 300–302 CrossRef CAS.
- C. Edmiston and K. Ruedenberg, Rev. Mod. Phys., 1963, 35, 457–464 CrossRef CAS.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- S. Suárez-Pantiga, P. Redero, X. Aniban, M. Simon, C. Golz, R. A. Mata and M. Alcarazo, Chem. – Eur. J., 2021, 27, 13358–13366 CrossRef PubMed.
- Y. S. Al-Hamdani, P. R. Nagy, A. Zen, D. Barton, M. Kállay, J. G. Brandenburg and A. Tkatchenko, Nat. Commun., 2021, 12, 3927 CrossRef CAS PubMed.
- J. Contreras-García, W. Yang and E. R. Johnson, J. Phys. Chem. A, 2011, 115, 12983–12990 CrossRef PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- R. A. Boto, F. Peccati, R. Laplaza, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-García, J. Chem. Theory Comput., 2020, 16, 4150–4158 CrossRef CAS PubMed.
- R. Laplaza, F. Peccati, R. A. Boto, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-García, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 11, e1497 Search PubMed.
- R. P. Feynman, Phys. Rev., 1939, 56, 340–343 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. T. Roberto, A. Boto, J. Contreras-García and J.-P. Piquemal, Mol. Phys., 2016, 114, 1406–1414 CrossRef.
- A. Savin, O. Jepsen, J. Flad, O. K. Andersen, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1992, 31, 187–188 CrossRef.
- C. Woodrow Wilson and W. A. Goddard, Chem. Phys. Lett., 1970, 5, 45–49 CrossRef.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- A. Wuttke and R. A. Mata, J. Comput. Chem., 2016, 38, 15–23 CrossRef PubMed.
- S. Riedel, P. Pyykkö, R. A. Mata and H.-J. Werner, Chem. Phys. Lett., 2005, 405, 148–152 CrossRef CAS.
- K. Dzialkowski, A. Gehlhaar, C. Wölper, A. A. Auer and S. Schulz, Organometallics, 2019, 38, 2927–2942 CrossRef CAS.
- G. Bistoni, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 10, e1442 Search PubMed.
- A. Altun, F. Neese and G. Bistoni, J. Chem. Theory Comput., 2018, 15, 215–228 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, P. Celani, W. Györffy, A. Hesselmann, D. Kats, G. Knizia, A. Köhn, T. Korona, D. Kreplin, R. Lindh, Q. Ma, F. R. Manby, A. Mitrushenkov, G. Rauhut, M. Schütz, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, J. A. Black, P. J. Bygrave, R. Cimiraglia, D. L. Cooper, D. Coughtrie, M. J. O. Deegan, A. J. Dobbyn, K. Doll, M. Dornbach, F. Eckert, S. Erfort, E. Goll, C. Hampel, G. Hetzer, J. G. Hill, M. Hodges, T. Hrenar, G. Jansen, C. Köppl, C. Kollmar, S. J. R. Lee, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, B. Mussard, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. A. Peterson, K. Pflüger, R. Pitzer, I. Polyak, M. Reiher, J. O. Richardson, J. B. Robinson, B. Schröder, M. Schwilk, T. Shiozaki, M. Sibaev, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, J. Toulouse, M. Wang, M. Welborn and B. Ziegler, MOLPRO, 2021.2, a package of ab initio programs, see https://www.molpro.net Search PubMed.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- R. Chaudret, B. de Courcy, J. Contreras-García, E. Gloaguen, A. Zehnacker-Rentien, M. Mons and J.-P. Piquemal, Phys. Chem. Chem. Phys., 2014, 16, 9876–9891 RSC.
- R. A. Boto, J.-P. Piquemal and J. Contreras-García, Theor. Chem. Acc., 2017, 136, 139 Search PubMed.
- A. Armstrong, R. A. Boto, P. Dingwall, J. Contreras-García, M. J. Harvey, N. J. Mason and H. S. Rzepa, Chem. Sci., 2014, 5, 2057–2071 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Orbital density plots, pair orbital analysis in o-DID, and wave function energy decomposition analysis (PDF), together with xyz coordinates of all systems (XYZ). See DOI: https://doi.org/10.1039/d5cp01057h |
| ‡ These authors contributed equally to this work. |
| § Current address: Ecole Nationale Supérieure de Chimie de Paris, Université PSL, CNRS, Institute of Chemistry for Life and Health Sciences, Paris 75005, France. |
| This journal is © the Owner Societies 2025 |