
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-dependent ultrafast photodynamics of p-hydroxyphenacyl: deprotonation accelerates the photo-uncaging reaction†
Yannik
Pfeifer
 a,
Till
Stensitzki
a,
Jakub
Dostál
b,
Evgenii
Titov
c,
Miroslav
Kloz
b,
Peter
Saalfrank
c and
Henrike M.
Müller-Werkmeister
*a
a,
Till
Stensitzki
a,
Jakub
Dostál
b,
Evgenii
Titov
c,
Miroslav
Kloz
b,
Peter
Saalfrank
c and
Henrike M.
Müller-Werkmeister
*a
aInstitut für Chemie – Physikalische Chemie, Universität Potsdam, Karl-Liebknecht-Strasse 24-25, Potsdam, 14476, Germany. E-mail: henrike.mueller-werkmeister@uni-potsdam.de
bELI Beamlines Facility Extreme Light Infrastructure ERIC, Dolni Brezany, 25241, Czech Republic
cInstitut für Chemie – Theoretische Chemie, Universität Potsdam, Karl-Liebknecht-Strasse 24-25, Potsdam, 14476, Germany
First published on 23rd May 2025
Abstract
Photolabile protecting groups (PPGs) possess broad application potential as they allow spatially and temporally controlled release of the protected group. While the photochemistry of many PPGs has been studied in detail, data under aqueous conditions or depending on the pH are extremely rare. However, for applications under biological conditions in water, knowledge about the photochemistry under these is critical. Here, we studied the pH-dependent reaction dynamics of para-hydroxyphenacyl (pHP), one prominent example of PPGs, by ultrafast transient absorption spectroscopy and quantum chemical simulation. At neutral pH, the para-hydroxyl group of pHP is protonated and photoproduct formation occurs within less than 1 ns from a triplet state. At basic pH, the main scaffold gets deprotonated leading to a bathochromic shift of the characteristic absorption. Upon deprotonation the substrate release occurs directly from a singlet state, shortcutting the rate determining step of intersystem crossing (ISC) in the neutral case, resulting in an acceleration of the photochemical reaction with photoproduct formation observed at ∼1 ps.
Introduction
Photocaged compounds or photolabile protecting groups (PPGs) are a promising tool with applications ranging from chemical synthesis to biophysics as they allow direct, light-induced spatiotemporal control of substance release.1 In particular, experimental studies of otherwise non-light sensitive biological processes and dynamics can be enabled by PPGs: light activation of substance release is applied as the trigger for dynamics in microscopy, time-resolved spectroscopy or recently time-resolved serial crystallography.2–6Depending on the experimental requirements, different PPGs are used based on the required quantum yield, solubility, excitation wavelength and reaction dynamics or release times.5,7 In general, biophysical studies benefit from water-soluble PPGs with absorption bands that do not overlap significantly with protein (or DNA) absorption in the UV at ∼280 nm.
While the reaction dynamics of several PPGs have been studied in detail by ultrafast transient spectroscopies (UV-vis, Raman and IR) and theoretical approaches,1 the ultrafast photochemistry of PPGs is investigated rarely in aqueous solutions but rather in organic solvents. With increased demand for PPGs compatible with biophysical applications in water and at different pH values, spectroscopic data on the influence of these solvent conditions on the photochemistry and reaction dynamics of PPGs and hence their applicability under biological conditions are required.5–7
para-Hydroxyphenacyl (pHP) is a prototypical and broadly applied PPG, due to the ease of its functionalization when attached to the leaving group and its water-soluble properties combined with a favourable quantum yield.8–11
The photochemistry of pHP (with a variety of leaving groups) has been studied in detail for the protonated form, in organic solvents and with traces of water present. Scheme 1 shows a simplified reaction scheme of the irreversible uncaging reaction of the neutral form of pHP.9
 | ||
| Scheme 1 Reaction scheme for uncaging of para-hydroxyphenacyl-fluoroacetate (pHP-FAc) to para-hydroxyphenyl acetic acid (HPAA) and fluoroacetate (FAc) via a photo-Favorskii rearrangement by optical excitation in the UV.8,12 The reaction mechanism is known to require traces of H2O.9 An extended reaction scheme can be found in the ESI.† | ||
Experimental studies include ultrafast transient absorption and Raman spectroscopy, complemented by theoretical calculations.1,8,9,11,14–18 The reaction mechanism for the uncaging of protonated pHP upon exciting the S0 → S2 transition occurs via a photo-Favorskii rearrangement as proposed by Givens and coworkers.9 The initial photoexcitation is followed by rapid internal conversion to the S1-state. Then, intersystem crossing into the triplet manifold takes place. The triplet releases the substrate in a water-assisted reaction,9,19 forming a biradical-state that was initially spectrally illusive, but later found with improved experimental methods.9,20 The biradical state further decays on a similar timescale, giving rise to the final products in less than 1 ns. Recently, Van Wilderen and coworkers19 were able to directly observe the cleavage of leaving groups upon intersystem crossing from the S1 state using ultrafast UV-pump IR-probe spectroscopy.
In this work, we address the photochemistry and reaction dynamics of pHP in its deprotonated form, present at basic pH. The work is motivated by a recent time-resolved serial crystallography study of the non-photosensitive enzyme fluoroacetate dehalogenase. Here, the substrate fluoroacetate was protected by pHP and its light-induced release was used to trigger and synchronize the protein dynamics of interest.3,12 The investigated enzyme can only be catalytically active under basic pH conditions due to the necessity of deprotonated side chains in the conserved catalytic triad.21
The requirement of basic pH conditions for the light-induced substrate release from para-hydroxyphenacyl-fluoroacetate (pHP-FAc) drastically changes the photophysical properties of pHP. Upon deprotonation (∼pKa 8),7,8,16,20 the absorption band of pHP shifts up to 328 nm (see Fig. 1 for the spectra of pHP-FAc), which prevents the spectral overlap with aromatic amino acid side chains and is beneficial in biophysical experiments such as the aforementioned crystallographic study. Earlier spectroscopic studies of pHP at high pH showed that the dissociation yield decreases upon deprotonation to ∼0.0916 or even down to 0.02–0.08,16 in case of substitutions on the phenol ring. Still, when accounting for the loss before application of pHP in biophysical experiments, the remaining yield can be sufficient.
 | ||
| Fig. 1 UV-vis absorption spectra of the studied compound pHP-FAc (para-hydroxyphenacyl-fluoroacetate) at pH 6.8 (0.2 mmol L−1) and pH 9.0 (0.115 mmol L−1), recorded in a cell with a 1 mm path length. Additionally experimental spectra of the pump pulses at 266 nm and 345 nm used in the reported ultrafast experiments are shown. Calculated transitions are illustrated as sticks and normalized spectra with a linewidth of 1500 cm−1 as dotted lines, with data reported in Table 1. Both, a calculation of the deprotonated molecule (blue) and with explicit water molecules (red) are shown. | ||
While there are proposed models for the reaction mechanism of photoproduct formation and release of the leaving group from the deprotonated scaffold,8 actual experimental data are sparse. Two studies have reported upon the photochemistry of the simplified parental scaffold without a leaving group, p-hydroxyacetophenone, including some pH-dependent data.18,20 Therefore, a detailed time-resolved spectroscopic study of the pH-dependent reaction dynamics of pHP with leaving groups is still missing. Aside from shifted absorption bands, further changes in the photochemistry and reaction dynamics of the deprotonated form must be expected, but a prediction of the effect of pH on the reaction mechanism or the altered timescales is difficult, i.e. it is not possible to directly know whether the deprotonated form of pHP will also show photoproduct formation (and hence release of the leaving group) below 1 ns or at longer times. We apply here a combination of ultrafast transient absorption spectroscopy and quantum chemical calculations to resolve the relevant timescales for the photochemistry of pHP at different pH values, using pHP-FAc as the model system.
Results and discussion
To investigate the photochemistry of the pHP-FAc uncaging reaction at basic pH, i.e. for the deprotonated structure of pHP, we performed ultrafast transient absorption spectroscopy. Notably, the experiments cover the relevant UV-region below 350 nm, in order to compare the dynamics of both, protonated pHP-FAc and deprotonated pHP-FAc (deprotonation at the para-hydroxyl group), in an aqueous buffer. Complementary quantum chemical calculations using time-dependent density functional theory (TD-DFT) were performed to support the analysis and provide guidance for assignment of the observed intermediate states (for details on the experiment and computation see the Methods section).Steady-state spectroscopy and quantum chemical calculations
The UV-vis absorption spectra of pHP-FAc at pH 6.8 and pH 9.0 are shown in Fig. 1. We used femtosecond pulses centred at 266 nm for exciting the neutral form of pHP-FAc at pH 6.8 and centred at 345 nm for exciting the deprotonated form of pHP-FAc at pH 9.0. The spectral profiles of these used pump pulses are also shown in Fig. 1. The corresponding excitation wavelengths and oscillator strengths for the lowest two singlet transitions for both cases, obtained from TD-DFT calculations, are listed in Table 1. The calculated structures of pHP-FAc are reported in Fig. 2, both for the protonated and the deprotonated form.| Reactant type | Transition | λ calc. [nm] (osc. strength) |
|---|---|---|
| Neutral | S0 → S1 | 303 (0.00) |
| S0 → S2 | 271 (0.44) | |
| Deprotonated | S0 → S1 | 315 (0.00) |
| S0 → S2 | 314 (0.62) | |
| Deprotonated with 3 explicit H2O | S0 → S1 | 318 (0.64) |
| S0 → S2 | 308 (0.00) |
 | ||
| Fig. 2 Optimized geometries of pHP-FAc for the protonated case (as present at pH 6.8, top row) and deprotonated case (as present at pH 9.0, bottom row). Calculations were carried out at the B3LYP/def2-TZVP/PCM(water) level. | ||
The bright transition observed for the neutral case can be assigned to the S0 → S2 transition, in good agreement with the experimental value of 277 nm and the calculated transition at 271 nm. The S0 → S1 transition does not yield the relevant oscillator strength in the calculation and is not observed experimentally as a significant feature. For the deprotonated case (calculated without and with explicit H2O), the experimentally observed transition is red-shifted to 329 nm. The calculations predict the S0 → S1 and S0 → S2 transitions to be very close in energy with the order depending on the inclusion of explicit water molecules. For the solvated deprotonated form of pHP-FAc the bright transition can be assigned to the S0 → S1 transition, in contrast to the protonated case. Importantly, the experimentally observed red-shift to 329 nm is well reproduced by the calculations, with the calculated transition wavelengths at ∼310–320 nm.
In addition, we analysed the nature of the excited states using natural transition orbital (NTO) analysis, and the resulting hole–particle pairs are reported in Table S1 of the ESI.† The key result is that the bright transitions are of the ππ* type, whereas the dark transitions are of the nπ* type.
Ultrafast transient spectroscopy of protonated pHP-FAc at pH 6.8
The transient absorption spectra obtained for pHP-FAc at neutral pH, i.e. for the protonated scaffold of pHP as shown in Fig. 2, are reported in Fig. 3 together with the plotted transient absorption change (traces) at selected wavelengths. The data were analysed from 300 fs onwards, as the early times are dominated by a coherent artifact (intense positive feature around time zero), manifested as a large broad positive signal. | ||
| Fig. 3 Top: Colormap of the ultrafast transient absorption spectra of (protonated) pHP-FAc in PBS buffer at pH 6.8, after excitation at 266 nm. Black contours are separated by a 0.4 mOD. Triangles indicate the position of the traces shown in the bottom plot. Labels mark features of the different transient states (Prod1 and Prod2: product absorption, S1: the singlet state of pHP-FAc, and T1 and T1*: the triplet state and radical triplet state). Data are corrected for group velocity dispersion (GVD) and for probe fluctuations. | ||
The first real signal can be observed at ∼300 fs, and we assign this positive feature, centred at 310 nm, to the excited state absorption (ESA) of the S1 state, which is formed after rapid internal conversion (IC) from the originally excited S2 state. We cannot clearly assign a corresponding bleach signature or stimulated emission, as this is expected outside the observable probe window below 280 nm and additionally masked by the coherent artifact.
Starting at ∼1 ps the transient spectra are dominated by a long-lived positive signal, centred at 400 nm, which is very broad from ∼350 nm to 450 nm and decays after ∼100 ps. We assign this signal to the T1 state formed from the S1 state by ISC.
At 285 nm, we observe a corresponding, long-lived negative signal, with a similar decay time at ∼100 ps. We assign this signal to an overlap of the initial bleach of the S0 state (before 1 ps, not fully resolved), followed by the broad signature of the T1 state, which is populated from 1 ps to ∼100 ps. After 100 ps, the appearance of an intermediate triplet species absorbing near 285 nm can be observed as a positive rise in the time-trace (Fig. 3 bottom). The intermediate disappears again within ∼400 ps. We observe two further positive signals, centred at 340 nm and 390 nm, respectively, increasing on a timescale larger than 1000 ps. These can be assigned to the formation of a photo product from the triplet state. However, due to the strong overlap with the dominant ESA signature of the T1 state, the sequential order of product formation cannot be fully observed in the transient absorption spectra.
Our experimental data for the reaction dynamics of pHP-FAc at neutral pH in H2O replicate earlier studies very well:14,18,22,23 initial excitation into the S2-state is followed by rapid internal conversion (IC) to the S1-state within less than 200 fs. The S1-state decays by intersystem crossing with a time-constant of around 2 ps. The main marker of the triplet-state is a positive band at 400 nm, assigned to a triplet-to-triplet transition. According to our calculations, this band is dominated by the T1 to T10 transition (see the ESI,† Table S2).
To further quantify the data, the time-dependent data were analysed using a global fit analysis after 450 fs, with three exponentials and a constant component, assuming a sequential model. The resulting EADS (evolutionary associated decay spectra) are reported in Fig. 4, and the associated DAS (decay associated spectra) can be found in the ESI† Fig. S6.
 | ||
| Fig. 4 Spectral components assuming a sequential model for the photochemistry of pHP-FAc in PBS at pH 6.8, upon excitation at 266 nm (see the ESI,† Fig. S9 for a reaction scheme). As only data after 450 fs are used for analysis, the ultrafast transition of the initially excited S2 to the S1 is not covered by the model (the S1 state is reached after fast internal conversion). The colour code is identical for the reaction scheme and the spectral components. | ||
At neutral pH, pHP-FAc forms the S1 state after initial photoexcitation into S2 and subsequent IC within 200–300 fs. The corresponding spectrum for S1 (EADS, blue, Fig. 4) shows a negative signature at 410 nm in addition to the ESA signal at 310 nm, which is observed directly in the experimental data. We assign this negative signal at 410 nm to the stimulated emission from S1 to S0, despite the lack of observable oscillator strength for the S0 → S1 transition in the absorption spectra, which we assign to the stimulated emission (SE) of the S1 → S0 transition.
However, an earlier ultrafast fluorescence study by Ma and co-workers14 reported S1 fluorescence of pHP at this wavelength. Hence, there must be some structural rearrangement in the excited state that partly allowed this transition.
The triplet with its characteristic absorption at 400 nm, which we assigned to a T1 → T10 transition (see the ESI,† Table S2 for calculations), is then formed with a time-constant of 2.4 ps via ISC, in full agreement with earlier studies.23
This initial triplet state is assigned to a neutral triplet ground state and decays within 213 ps to another state. This T1* state cannot be directly observed in these transient spectra due to masking by the bleach at 285 nm and is only clearly detected in the global analysis. We assign this state to an intermediate photoproduct after the release of the leaving group with a subsequent structural rearrangement to the short lived triplet radical intermediate (spiroketone) as proposed in previous studies by Slanina23 and Park.24 Our data do not allow to directly resolve the release of the leaving group, which was recently reported to occur directly upon T1 formation within few ps.19 Within 0.9 ns, the formation of the final photoproducts is observed matching timescales reported earlier for the neutral case.1,11,19,22 Based on the generally accepted reaction mechanism (see the ESI,† Fig. S9), we assign photoproduct 1 to para-hydroxybenzyl alcohol and photoproduct 2 to para-hydroxy phenylacetic acid (HPPA).
Ultrafast transient spectroscopy of deprotonated pHP-FAc at pH 9
Transient absorption spectra obtained for pHP-FAc at a basic pH of 9.0 in Tris buffer, i.e. for the deprotonated PPG scaffold are reported in Fig. 5. At this pH, the absorption of the chromophore is red-shifted (see Fig. 1). The lower panel in Fig. 5 shows selected time traces. Similar to the neutral case, data are dominated by a coherent artifact for early times and only analysed from 300 fs onwards.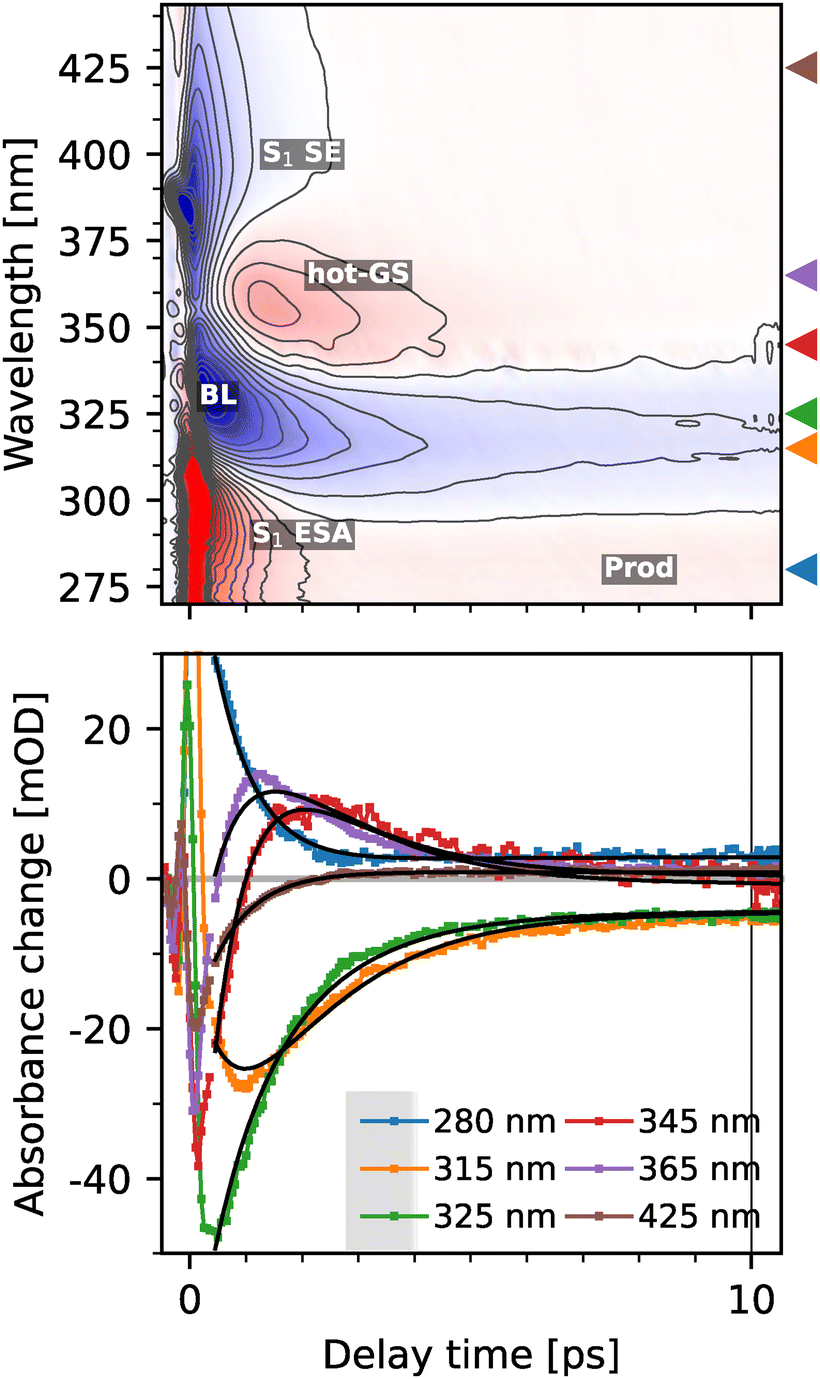 | ||
| Fig. 5 Top: Colormap of the transient spectra of (deprotonated) pHP-FAc in TRIS at pH 9.0 upon excitation at 345 nm; black contours are separated by 5 mOD. Triangles indicate the position of the traces shown in the bottom plot. Positions of stimulated emission (SE), excited state absorption (ESA), bleach (BL), all signatures of S1 of pHP-FAc, product absorption (Prod) and the hot ground state (hot-GS) of pHP-FAc are labelled. Black lines show a global two exponential fit of the data. | ||
Transient spectra were recorded also for long time delays using two synchronized laser sources (see the Experimental section) as we expected different photochemistry for the deprotonated case in contrast to the protonated case, but could not make an a priori prediction about whether the reaction dynamics would be accelerated or slowed down. Additional data for long time scales are reported in ESI† Fig. S3, clearly indicating that the reaction dynamics for the photo-uncaging reaction of pHP-FAc at pH 9 are completed on a timescale well below 10 ps.
The transient UV-vis spectra of deprotonated pHP-FAc show three key features below 1 ps. First, a strong negative signal at 330–340 nm, which can be assigned as the ground state bleach of the S0 → S1 transition (BL); second, a very broad, negative feature stretching from ∼380 to 440 nm, which can only be attributed as the red-shifted stimulated emission (SE) from S1 back to S0. The third characteristic signal at early times is a positive signal centred at 290 nm. We assign this signal to the ESA of the S1 state. This assignment is supported by the similar time constant we observe for the decay of the BL and ESA signal around ∼1 ps, comparing the spectral traces at 280 nm (ESA without influence from the BL) and 325 nm (BL). While the early times up to ∼2 ps of the positive signal at 290 nm can thus be assigned to the ESA signal from the S1 state, we observe a long-lived positive signal, which can be assigned to the photoproduct in the same wavelength region (see the ESI,† Fig. S2 for a more detailed analysis). Within the first ps, the S1-state undergoes IC to the vibrationally excited (‘hot’) ground state S0 at 365 nm, as described by the red-shifted replica of the early absorption band. This hot state relaxes into the vibrational ground state by vibrational cooling within 2 ps. The remaining signal thus can be assigned to the photoproducts of the reaction, since no further signal changes are observed up to hundreds of nanoseconds.
The final observable photoproduct is characterized by a small positive band near 280 nm, and in addition, the remaining bleaching signal; additionally, a small constant background is observed. Following the previous work from Givens and coworkers,9 we assign the signal to p-quinone methide, which has its absorption maximum at 276 nm. The p-quinone methide (PQM) is assumed to be the product of decarbonylation of the spiroketone intermediate. This assignment is further supported by TD-DFT calculations, which show a bright absorption transition at 284 nm (see Table S3 for details, ESI†). In addition, p-hydroxybenzyl alcohol or its deprotonated form may also be a by-product of the reaction.9 While both forms do not absorb in the probed region, their existence would explain the weaker strength of the product band relative to the bleaching band (see Fig. S2, ESI†). The latter is surprising, since according to our calculations both the reactant and PQM have similar oscillator strengths for their lowest allowed transitions.
To quantify the photoreaction in more detail, we modelled the data with an exponential model. Here, at least two exponentials and a constant are necessary to describe the data. The raw decay associated spectra are shown in Fig. S8 (ESI†). We opted to model the data using a branching model (see Fig. 6), in which the decay of the initially excited S1 state either leads to the generation of the hot ground state or to the formation of the products. The branching ratio was determined by assuming equal bleaching signals of the hot GS and product at 310 nm, resulting in a yield of 12%. A similar yield can be estimated by comparing the signal amplitude of the bleaching directly after excitation with its final value.
 | ||
| Fig. 6 Species associated difference spectra (SADS) of pHP-FAc in an aqueous TRIS buffer at pH 9 after excitation at 345 nm, assuming the kinetic model shown at the bottom right. The resulting time constants and yields are shown at the bottom right. Components have the same colours in the kinetic scheme and the spectra. The calculation of the yield is described in the main text. | ||
However, due to the overlap of the S1 ESA with the final product absorption, we cannot determine the rise of the product band directly. Still, we can rule out product formation from the hot-GS, since the signal at 280 nm remains constant after the initial decay of the S1 state within 750 fs (see also the ESI,† Fig. S1). The 750 fs are therefore an upper limit for the cleavage of the leaving group.
A rapid cleavage of the leaving group after excitation is also supported by calculations. Attempts to optimize the excited-state geometry of the S1 state have failed; however, the final structure of the run already shows breakage of the bond between the leaving group and the scaffold. Furthermore, the energy gap between S0 and S1 is only ∼0.1–0.2 eV (see the ESI,† Fig. S10), indicating the presence of a conical intersection between the two states.
Therefore, we propose the following reaction scheme for pHP-FAc in its deprotonated form at pH 9.0 (see Fig. 7): after the initial excitation to the S1 state of pHP-FAc (structure 1), a conical intersection is reached within 750 fs. Most of the excited population relaxes back to the ground state by vibrational cooling on a time scale of 1.6 ps, while the remaining population continues along a reaction coordinate resulting in photo cleavage. The concomitant release of the leaving group from the main scaffold is illustrated by structure 2, followed by the subsequent rearrangement to the initial photoproduct, the spectroscopically elusive spiroketone, illustrated as intermediate structure 3. Finally, the product state is reached.
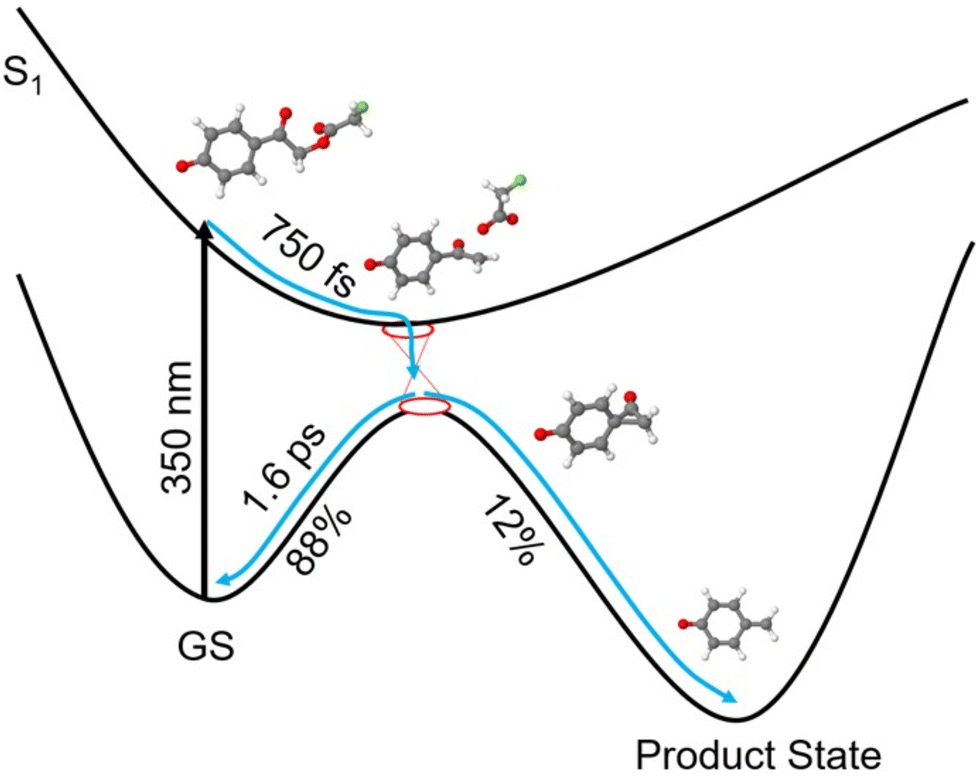 | ||
| Fig. 7 Schematic proposed energy landscape for the uncaging reaction of para-hydroxyphenacyl-fluoroacetate at basic pH, for the deprotonated case (detailed results from calculations are shown in the ESI,† Fig. S10). After the initial excitation into the S1-state, a conical intersection is reached within 750 fs. The majority of the excited population relaxes back into the ground state by vibrational cooling on a timescale of 1.6 ps, while the remaining population proceeds to the cleavage reaction. First, the release of the leaving group fluoroacetate occurs, then the spiroketone intermediate is formed as an initial, short-lived photoproduct, followed by the formation of the PQM as the product state. | ||
As the cleavage reaction crucially depends on the availability of solvent waters, we measured the photoreaction for deprotonated pHP-FAc as the control in an ACN/Tris 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture at pH 9 (results are shown in the ESI,† Fig. S4, S5 and S8). The solvent change only slows down the reaction slightly, the S1-lifetime increases to 1 ps and vibrational cooling occurs within 3 ps instead of 1.6 ps. The yield (0.14) is identical within the errors; therefore, we conclude that sufficient water molecules are located near the cleavage site of pHP-FAc as the yield is not negatively affected. A small reduction in the yield could be caused by the increased availability of hydrogen-bonding partners, resulting in a shorter lifetime of the reactive complex.
:50 mixture at pH 9 (results are shown in the ESI,† Fig. S4, S5 and S8). The solvent change only slows down the reaction slightly, the S1-lifetime increases to 1 ps and vibrational cooling occurs within 3 ps instead of 1.6 ps. The yield (0.14) is identical within the errors; therefore, we conclude that sufficient water molecules are located near the cleavage site of pHP-FAc as the yield is not negatively affected. A small reduction in the yield could be caused by the increased availability of hydrogen-bonding partners, resulting in a shorter lifetime of the reactive complex.
Conclusion
The presented experimental results on the photochemically induced reaction dynamics for the uncaging reaction of the photocaged compound para-hydroxyphenacyl-fluoroacetate (pHP-FAc) constitute a systematic ultrafast transient absorption study focusing on the role of pH in modulating reaction mechanisms and timescales for this class of molecules.While we originally expected an entirely altered release mechanism with possibly much longer lifetimes for intermediates and the release of the leaving group, the spectroscopic data reveal the opposite: for the neutral pH case the photochemistry of pHP, irrespective whether studied in water or organic solvents, is already among the fastest reported for photocaged compounds. We were able to confirm previously reported reaction times for the neutral case of pHP and show that also for fluoroacetate as the leaving group, photoproduct formation faster than 1 ns can be observed. The rate limiting step during the photochemistry is the formation of a triplet state T1via intersystem crossing (ISC) within 2.4 ps and the subsequent rearrangement to an intermediate photoproduct within 200 ps and the final photoproduct formation at 0.9 ns. While we cannot monitor the release of the leaving group directly in this study, upper timescales for the release are in good agreement with previous studies.
In contrast, the deprotonated form of pHP-FAc undergoes excitation to the S1 state followed by either fast relaxation to a hot ground state (main population) or direct formation of the photoproduct from the vibrationally cooled S1 state via a conical intersection, providing a direct explanation for the previously observed low quantum yields at high pH.
We interpret the instantaneous product formation from S1 within 750 fs in the absence of further spectroscopic signatures of other intermediates, (neither at ultrashort nor long timescales), such, that the vibrationally cooled S1 state of the deprotonated pHP species is electronically (and structurally) closely related to the product-forming state T1* of the neutral case. In short, the absence of a triplet state for deprotonated pHP shortcuts the reaction mechanism for pHP at basic pH, thereby giving direct access to a product-forming structure within less than 1 ps. This reflects common strategies used in photochemistry, aiming to stabilize short-lived singlet states to increase reaction rates. The presented transient absorption spectra do not yield direct information on the nature of the transient structure and further, structure sensitive, experiments are necessary to fully unravel the reaction intermediates of this extremely fast photo-uncaging reaction.
Generally, the results demonstrate the urgent necessity to study the reaction dynamics of photolabile protecting groups or photocaged compounds applied in biophysical studies under aqueous conditions and at relevant pH as these can be altered in a non-predictable way.
Experimental
Sample preparation
The synthesis of the compound para-hydroxyphenacyl-fluoroacetate (pHP-FAc) was described in detail previously,12 and further details (NMR results) are reported in the ESI.† For all spectroscopic experiments, samples were prepared in PBS buffer at pH 6.8 for the neutral conditions and in TRIS buffer at pH 9 for the alkaline environment while the pH was adjusted with either HCl or NaOH. Reference experiments in a buffer/ACN (50:50) mixture are reported in the ESI.† The concentration of the samples was adjusted to 0.2 mmol L−1 in PBS and 0.115 mmol L−1 for the conjugate base yielding an OD of 0.5 at their excitation wavelengths in the 2 mm flow cuvette. The pH was checked after each experiment to ensure that the triggered uncaging reaction did not alter the dominant form (protonated or deprotonated pHP) depending on the experimental conditions.
Spectroscopy
Transient absorption spectra were measured on a home-built setup at ELI beamlines/Dolny Brezany. The setup has been described in detail previously.25 The central laser system consists of two 1 kHz laser amplifiers independently seeded with one Ti:sapphire oscillator. The output of one amplifier (Femtopower, Spectra Physics, 800 nm, 20 fs, 4 mJ) was split into two parts. One part was frequency tripled and served as the 266 nm pump (1/e2-diameter in focus: 160 μm, 110 nJ per pulse). The remaining part was used to generate a white light supercontinuum in an argon-filled hollow-core fibre (Ultrafast Innovations) spanning 270–1000 nm, used as the probe beam (1/e2-diameter in focus: 70 μm). The 340 nm pump (1/e2-diameter in focus: 110 μm, 700 nJ per pulse) was generated using a commercial optical parametric amplifier (TOPAS, Light Conversion) seeded with the second laser amplifier (Solstice, Spectra Physics, 800 nm, 30 fs, 7 mJ). The timing between the pump and probe pulses was controlled using an optomechanical delay line covering the range of approximately 0–8 ns. Additionally, the delay between the probe pulse and the 340 nm pump could be tuned by up to <1 ms by adjusting the electronic synchronization of both laser amplifiers. The relative polarization of the pump and probe beams was set to magic angle. Both beams were chopped by a pair of optomechanical choppers [Thorlabs] allowing for fast detection of transient absorption signals, corrected by pump scattering and the detector dark noise on a shot-to-shot basis. The probe beam transmitted through the sample was spectrally dispersed in a home-built dual-channel prism spectrometer and detected using a CCD camera (Entwicklungsbüro Stresing). The second (reference) channel of the spectrometer was used for corrections from probe fluctuations using an approach described previously.26 The sample was kept moving during experiments in a 2 mm-thick optical flow-through cell comprised of quartz. It was constantly flown using a peristaltic pump to avoid baking of the sample to the windows. Data analysis of the transient spectra was performed with the skultrafast package,‡ incl. the GVD correction.Computational methods
Molecular structures were optimized in the electronic ground state (S0) using density functional theory (DFT) at the B3LYP27,28/def2-TZVP29 level of approximation. Solvent effects were accounted for with a polarizable continuum model (PCM)30,31 for water. In addition, for the deprotonated pHP-FAc, the complex with three explicit water molecules was considered and first optimized in the S0 state with dispersion-corrected DFT at the B3LYP+D3(BJ)32/def2-TZVP/PCM(water) level. Vertical electronic transitions were computed with the linear response time-dependent DFT (TD-DFT)33 at the TD-B3LYP/def2-TZVP/PCM(water) level of theory. The nature of the excited states was characterized by the natural transition orbital (NTO) analysis34 as reported in the ESI† (Fig. S1). The geometry optimizations in the T1 state were done using spin-unrestricted (U) DFT at the UB3LYP/def2-TZVP/PCM(water) level. The geometry optimizations in the S1 state were performed with TD-B3LYP/def2-TZVP/PCM(water). Linear response PCM formulation was used for the excited-state optimizations and calculations of the vertical transitions. Nonequilibrium solvation was used for the calculation of the vertical transitions, and the equilibrium solvation was used for the geometry optimizations. The calculations were performed using Gaussian 16.35Author contributions
Y. P. performed the ultrafast spectroscopy experiments, supported by J. D. and M. K.; Y. P. and T. S. performed the data analysis. Y. P. performed the synthesis of the samples. E. T. and P. S. conceptualized the calculations, done by E. T., and interpreted the results. Y. P., T. S., E. T. and H. M.-W. discussed the data and wrote the manuscript with support from all co-authors. H. M.-W. planned and conceptualized the study.Data availability
Data for this article, including the experimental data and the scripts used to analyze the data are available at Zenodo at https://doi.org/10.5281/zenodo.14944110.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge ELI Beamlines in Dolní Břežany, Czech Republic, for the provision of laser beamtime and would like to thank the instrument group and facility staff for their assistance. LM2017094, MEYS – Large research infrastructure project ELI Beamlines. The authors thank Dr Alessandra Picchiotti (U Hamburg, formerly ELI) for helpful discussions. H. M.-W. acknowledges the generous funding by the Tenure-Track program of Uni Potsdam and the State of Brandenburg, and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy (EXC 2008/1-390540038, UniSysCat). H. M.-W., E. T. and P. S. acknowledge the funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within SFB/CRC 1636, ID 510943930. E. T. is also grateful to the DFG for financial support via the project number 454020933. M. K. was financially supported by the Czech Science Foundation (project No. 21-09692M).References
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy, Chem. Rev., 2013, 113, 119–191 Search PubMed
.
- Y. Li, M. Wang, F. Wang, S. Lu and X. Chen, Recent progress in studies of photocages, Smart Mol., 2023, 1, e20220003 CrossRef
- P. Mehrabi, E. C. Schulz, R. Dsouza, H. M. Müller-Werkmeister, F. Tellkamp, R. J. D. Miller and E. F. Pai, Time-resolved crystallography reveals allosteric communication aligned with molecular breathing, Science, 2019, 365, 1167–1170 CrossRef CAS PubMed
- A. C. Kneuttinger, A guide to designing photocontrol in proteins: methods, strategies and applications, Biol. Chem., 2022, 403, 573–613 CrossRef CAS PubMed
- D. C. F. Monteiro, E. Amoah, C. Rogers and A. R. Pearson, Using photocaging for fast time-resolved structural biology studies, Acta Crystallogr., Sect. D:Struct. Biol., 2021, 77, 1218–1232 CrossRef CAS PubMed
- J. I. Zaitseva-Kinneberg, A. Puchert, Y. Pfeifer, H. Yan, B. A. Yorke, H. M. Müller-Werkmeister, C. Uetrecht, J. Rehbein, N. Huse, A. R. Pearson and M. Sans, Synthesis and characterisation of α-carboxynitrobenzyl photocaged L-aspartates for applications in time-resolved structural biology, RSC Adv., 2019, 9, 8695–8699 RSC
- Y. Pfeifer and H. M. Müller-Werkmeister, pH dependency of photolabile protection groups used for applications in dynamic structural biology, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-vchtp.
- S. N. Senadheera, K. Stensrud, C. Perera, J. Wirz, P. G. Conrad, D. Heger, A. L. Yousef and R. S. Givens, p -Hydroxyphenacyl photoremovable protecting groups—Robust photochemistry despite substituent diversity, Can. J. Chem., 2011, 89, 364–384 CrossRef PubMed
- R. S. Givens, D. Heger, B. Hellrung, Y. Kamdzhilov, M. Mac, P. G. Conrad, E. Cope, J. I. Lee, J. F. Mata-Segreda, R. L. Schowen and J. Wirz, The photo-Favorskii reaction of p-hydroxyphenacyl compounds is initiated by water-assisted, adiabatic extrusion of a triplet biradical, J. Am. Chem. Soc., 2008, 130, 3307–3309 CrossRef CAS PubMed
- I. Bownik, P. Šebej, J. Literák, D. Heger, Z. Šimek, R. S. Givens and P. Klán, 4-Hydroxyphenacyl Ammonium Salts: A Photoremovable Protecting Group for Amines in Aqueous Solutions, J. Org. Chem., 2015, 80, 9713–9721 CrossRef CAS PubMed
- P. G. Conrad, R. S. Givens, B. Hellrung, C. S. Rajesh, M. Ramseier and J. Wirz,
p-Hydroxyphenacyl phototriggers: The reactive excited state of phosphate photorelease, J. Am. Chem. Soc., 2000, 122, 9346–9347 Search PubMed
- E. C. Schulz, P. Mehrabi, H. M. Müller-Werkmeister, F. Tellkamp, A. Jha, W. Stuart, E. Persch, R. De Gasparo, F. Diederich, E. F. Pai and R. J. D. Miller, The hit-and-return system enables efficient time-resolved serial synchrotron crystallography, Nat. Methods, 2018, 15, 901–904 CrossRef CAS PubMed
- T. Stensitzki, Skultrafast – a python package for time-resolved spectroscopy, Zenodo repository DOI:10.5281/zenodo.5713589.
- C. Ma, M. K. Wai, S. C. Wing, P. Zuo, J. T. W. Kan, P. H. Toy and D. L. Phillips, Ultrafast time-resolved study of photophysical processes involved in the photodeprotection of p-hydroxyphenacyl caged phototrigger compounds, J. Am. Chem. Soc., 2005, 127, 1463–1472 CrossRef CAS PubMed
- Q. Cao, X. Guan, M. W. George, D. L. Phillips, C. Ma, W. M. Kwok, M. Li, Y. Du, X.-Z. Sun and J. Xue, Ultrafast time-resolved transient infrared and resonance Raman spectroscopic study of the photo-deprotection and rearrangement reactions of p-hydroxyphenacyl caged phosphates, Faraday Discuss., 2010, 145, 171–183 RSC
- K. Stensrud, J. Noh, K. Kandler, J. Wirz, D. Heger and R. S. Givens, Competing Pathways in the Photo-Favorskii Rearrangement and Release of Esters: Studies on Fluorinated p -Hydroxyphenacyl-Caged GABA and Glutamate Phototriggers, J. Org. Chem., 2009, 74, 5219–5227 CrossRef CAS PubMed
- A. L. Houk, R. S. Givens and C. G. Elles, Two-Photon Activation of p-Hydroxyphenacyl Phototriggers: Toward Spatially Controlled Release of Diethyl Phosphate and ATP, J. Phys. Chem. B, 2016, 120, 3178–3186 CrossRef CAS PubMed
- P. Zuo, C. Ma, W. M. Kwok, W. S. Chan and D. L. Phillips, Time-Resolved Resonance Raman and Density Functional Theory Study of the Deprotonation Reaction of the Triplet State of p -Hydroxyacetophenone in Water Solution, J. Org. Chem., 2005, 70, 8661–8675 CrossRef CAS PubMed
- L. J. G. W. Van Wilderen, D. Kern-Michler, C. Neumann, M. Reinfelds, J. Von Cosel, M. Horz, I. Burghardt, A. Heckel and J. Bredenbeck, Choose your leaving group: selective photodeprotection in a mixture of p HP-caged compounds by VIPER excitation, Chem. Sci., 2023, 14, 2624–2630 RSC
- Ĺ. Klíčová, P. Šebej, T. Šolomek, B. Hellrung, P. Slavíček, P. Klán, D. Heger and J. Wirz, Adiabatic triplet state tautomerization of p-hydroxyacetophenone in aqueous solution, J. Phys. Chem. A, 2012, 116, 2935–2944 CrossRef PubMed
- P. W. Y. Chan, A. F. Yakunin, E. A. Edwards and E. F. Pai, Mapping the Reaction Coordinates of Enzymatic Defluorination, J. Am. Chem. Soc., 2011, 133, 7461–7468 CrossRef CAS PubMed
- C. Ma, W. M. Kwok, W. S. Chan, Y. Du, J. Tze, W. Kan, P. H. Toy and D. L. Phillips, Ultrafast Time-Resolved Transient Absorption and Resonance Raman Spectroscopy Study of the Photodeprotection and Rearrangement Reactions of p-Hydroxyphenacyl Caged Phosphates, J. Am. Chem. Soc., 2006, 128, 2558–2570, DOI:10.1021/ja0532032
- T. Slanina, P. Šebej, A. Heckel, R. S. Givens and P. Klán, Caged Fluoride: Photochemistry and Applications of 4-Hydroxyphenacyl Fluoride, Org. Lett., 2015, 17, 4814–4817 CrossRef CAS PubMed
- C. H. Park and R. S. Givens, New photoactivated protecting groups. 6. p-Hydroxyphenacyl: A phototrigger for chemical and biochemical probes, J. Am. Chem. Soc., 1997, 119, 2453–2463 CrossRef CAS
- S. Kaziannis, M. Broser, I. H. M. van Stokkum, J. Dostal, W. Busse, A. Munhoven, C. Bernardo, M. Kloz, P. Hegemann and J. T. M. Kennis, Multiple retinal isomerizations during the early phase of the bestrhodopsin photoreaction, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2318996121 Search PubMed
- Y. Feng, I. Vinogradov and N.-H. Ge, General noise suppression scheme with reference detection in heterodyne nonlinear spectroscopy, Opt. Express, 2017, 25, 26262 CrossRef CAS PubMed
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623–11627 Search PubMed
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
-
M. E. Casida, Recent Advances in Density Functional Methods, World Scientific, 1995, vol. 1, pp. 155–192 Search PubMed
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and analysis of pHP-FAc; supporting computational results: Tables S1–S3, Fig. S10; supporting experimental results: Fig. S1–S9. See DOI: https://doi.org/10.1039/d5cp01049g |
| ‡ https://zenodo.org/record/5713589.13 |
| This journal is © the Owner Societies 2025 |