
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ab initio study of silver–titanium interfaces in gas-phase and surface-supported clusters†
El yakout
El koraychy
* and
Riccardo
Ferrando
 *
*
Dipartimento di Fisica, Università di Genova, Via Dodecaneso 33, 16146 Genova, Italy. E-mail: elyakout.elkoraychy@edu.unige.it; riccardo.ferrando@unige.it
First published on 5th June 2025
Abstract
The TiAg interface is studied via density-functional theory calculations by considering different systems, ranging from free gas-phase clusters to small clusters supported on Ti(0001) and to an Ag monolayer on the same surface. Our results reveal that Ag impurities preferentially occupy surface sites over bulk or subsurface ones in Ti clusters and on the Ti(0001) surface, with the exception of icosahedral geometries where they are centrally incorporated due to local stress relief. For the gas-phase clusters, Ti@Ag core@shell arrangements are significantly more stable than their Ag@Ti counterparts, supporting the formation of chemically ordered arrangements. For surface-supported systems, small Ag clusters (up to tetramers) exhibit non-trivial adsorption behaviors. Ag clusters favor fcc over hcp stacking on Ti(0001); however, bulk-like stacking is obtained for complete monolayer adsorption. We further demonstrate that the adhesion between Ag and Ti is strong, with an adhesion energy of 2.4 J m−2, significantly stronger than that of Ag on Ti oxides. The charge analyses show that Ag adatoms gain charges from the Ti(0001) surface, and as the Ag cluster size increases, the net charge per Ag atom becomes progressively less negative. Our findings reveal that Ag and Ti can form sharp and well-defined interfaces with limited intermixing. These insights are relevant in the design of functional Ag coatings on Ti-based nanostructures for biomedical and electronic applications, where both mechanical robustness and interfacial stability are crucial.
1. Introduction
Titanium (Ti) has gained popularity in several fields due to its notable attributes, including biocompatibility, corrosion resistance in different environments, strength, elasticity, and excellent thermal and chemical stability.1 These characteristics have made Ti an ideal candidate for biomedical applications, particularly in prostheses due to its harmlessness, low reactivity, and good osteointegration.2,3Pure Ti does not exhibit antibacterial properties, and after implantation in the human body, therefore, a large number of bacteria can easily accumulate and eventually form a biofilm. The construction of an antibacterial coating on the surface of pure Ti is an effective way to reduce the infection and bacterial growth. Previous studies have shown that silver (Ag) can be a good coating material due to its effective antimicrobial characteristics that prevent the formation of bacterial biofilms.4–7
Beyond biomedical contexts, Ag is of particular interest as an interconnection material in a large variety of electronic devices because of its low electrical resistivity. However, Ag films can easily agglomerate during annealing due to poor adhesion of the Ag thin film on the SiO2/Si or glass substrate.8–10 These limitations render Ag films ineffective as interconnection materials. Hence, the fabrication of Ag films with improved thermal stability and low resistivity is significantly important. Previous works have shown that the thermal and mechanical stability of Ag films can be improved by introducing another metal as an additive to Ag.8,9,11–14 It has been determined that agglomeration of Ag films is related to the adhesion of the film to the substrate, and therefore the suppression of agglomeration can be improved by inserting an appropriate interfacial layer. In particular, a Ti interface has been proved to be a good candidate for enhancing the adhesion strength between an Ag film and a silica substrate.13
In this context, a microscopic understanding of the atomic-scale interactions between silver and titanium metal is essential. Previously, there have been theoretical studies focused on the interfacial effect between silver and titanium dioxide thin films.15–19 In ref. 19, Schvval et al. theoretically investigated the growth mode of Ag4 nanoclusters deposited on the stoichiometric surfaces of rutile (110) and anatase (101) TiO2. On both surfaces, they found that the Ag4 nanocluster prefers the 3D (tetrahedral shape of the tetramer) configuration, as experiments have indicated.20 This means that the adhesion energy of Ag on the TiO2 surface is very weak. This behavior has been confirmed theoretically using density functional theory,17 where it was found that the adhesion energy of Ag layers is very weak when they are deposited on a TiO2 surface as compared to other oxide surfaces. With respect to the large number of theoretical studies dedicated to the adhesion of silver on TiO2 surfaces,15–19,21 comparatively fewer results have been reported on the direct metal–metal interface between Ag and Ti.
To our knowledge, the fundamental understanding of atomic-scale interactions, bonding preferences, and charge transfer behavior in Ti–Ag systems remains incomplete. Investigating these systems through theoretical approaches provides valuable insights into their structural stability, electronic properties and interfacial behaviour, thereby enabling the rational design of advanced bimetallic materials for technological applications. To fill this gap, we tackle this issue in this work by means of density functional theory (DFT) calculations. To this end, we will consider a few different systems: Ag impurities in gas-phase Ti clusters; core@shell TiAg gas-phase clusters; small Agn (n = 2–4) clusters on the regular Ti(0001) surface; and an Ag monolayer on the same Ti(0001) surface.
2. Computational details
2.1. DFT framework
Our study was performed using the Quantum Espresso software.22 The Kohn–Sham equations were solved using a plane-wave basis set and periodic boundary conditions. The electron–ion interactions were described by projector-augmented wave (PAW) pseudo-potentials.23,24 The Perdew–Burke–Ernzerhof (PBE)25 version of the generalized gradient approximation (GGA) approach is used to treat the exchange–correlation interactions. Geometry optimization was performed using the Davidson scheme.26 For electronic relaxation, we applied the Fermi-level smearing approach of Marzari–Vanderbilt–DeVita–Payne with a smearing width of 0.02 Ry. Cutoffs for the wave function and charge density were set as suggested by the pseudopotentials that we used. A 3s23p64s23d2 valence configuration was used for titanium and a 5s1.55p04d9.5 valence configuration was used for silver. For all total energy optimizations, the equilibrium geometry was obtained until the convergence thresholds for energy, atomic forces and electronic calculations reached 10−4 Ry, 10−3 Ry a.u.−1, and 10−6 Ry, respectively.2.2. Gas-phase cluster modeling
For the free monometallic Ti and bimetallic TiAg nanoparticles, all the degrees of freedom of the metallic atoms were relaxed during the geometric optimizations. In order to avoid spurious interactions between cluster images, large cubic supercells of dimension 30 × 30 × 30 Å3 were considered, so the Brillouin zones were sampled using the gamma point approach.2.3. Supported cluster configurations
The Ti(0001) surface was modeled using two-dimensional periodic slabs, cleaved from an optimized hexagonal Ti bulk using the lattice parameters shown in Table S1 in the ESI.† A sufficient vacuum space (of about 20 Å) between two periodic images along the z direction was incorporated to avoid any spurious interaction between images (see Fig. 1). The Brillouin zone was sampled using the Monkhorst–Pack scheme27 with a 4 × 4 × 1 k-point. The chosen k-points used in our calculations were proven to be reasonable after convergence tests (see the ESI†). | ||
| Fig. 1 Bulk and surface slab model of the hcp Ti structure. (a) α-Ti unit cell of the space group P63/mmc. (b) Side view of the Ti(0001) surface slab model and (c) top view of the bidimensional periodic slab. Gray and pale turquoise spheres represent the surface and subsurface atoms of the Ti slab, respectively. | ||
All surface calculations were performed using the surface model of a 4 × 4 supercell with a thickness of four Ti(0001) atomic layers (corresponding to 64 Ti atoms in total). This slab size was selected after performing convergence tests (further details are provided in the ESI†). For all adsorption calculations, the last two layers of Ti atoms were kept fixed to mimic the bulk constraints and the top layers of the surface together with the cluster were relaxed. Adsorption of the Ti (or Ag) adatom, small clusters (up to 4 atoms) and monolayers was investigated. Various adsorption sites (top, bridge, hcp and fcc) were tested to identify the most favorable configurations.
2.4. Total energy and stability calculations
After DFT relaxation, we extracted the total energies and the atom coordinates for all systems. For gas-phase clusters, we calculated the cohesive energy Ecoh and the mixing energy Emix to identify the stable configuration. These energetic descriptors are defined for Ecoh as follows:| Ecoh = (55 − m)EatomTi + mEatomAg − E(Ti55−mAgm) | (1) |
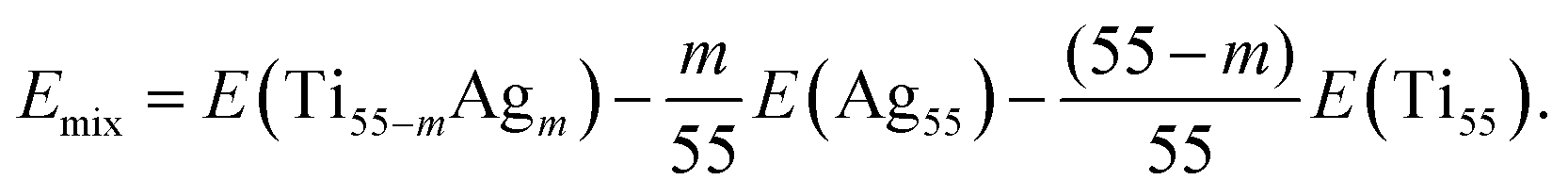 | (2) |
For supported nanoclusters and monolayers on Ti(0001), we computed the adhesion energy, interlayer spacing, and atom–surface distance to evaluate the adhesion strength and structural deformations. The adhesion energy Eadh is calculated using the total energy obtained from DFT calculations:
| Eadh = Ecluster + Esubstrate − Etotal | (3) |
2.5. Atomic charge analysis
Charge transfer was performed using the Bader method as implemented using the Henkelman code.30 The charge transfer trends were evaluated for Ag atoms on the Ti(0001) surface to understand the electronic environments. A negative Bader charge indicates a charge gain from the surrounding environment, whereas a positive value indicates the opposite.Charge density difference maps were computed using the following formula:
| Δρ = ρtotal − ρcluster − ρsubstrate | (4) |
3. Results and discussion
3.1. Free nanoparticles
In this section, we focus our attention on the stability of isolated nanoparticles. Clusters of size 55 atoms are considered. Size 55 is the geometrical magic size for the fcc cuboctahedron (Cubo) and Mackay icosahedron (Ih).32,33 Our DFT calculations indicate that the icosahedral structure is indeed the lowest in energy for Ti55, which is in agreement with the results in ref. 34. Indeed, strained Ih nanoparticles35 are often the most stable in small size ranges (N ∼ 100, where N is the number of atoms in the nanoparticle) due to their compact shape and surface.36The sites where the impurity is placed are shown in Fig. 2 and the energies calculated via DFT relaxations are given in the corresponding tables together with their cohesive and mixing energies. We mention that all energy calculations, including energy difference, cohesive energy and mixing energy, exhibit the same trend for predicting the best placement of the Ag atom in the Ti clusters. All the results show that the site preference of a single Ag atom embedded in Ti54 clusters depends strongly on the cluster geometry due to differences in local strain distribution and coordination environments. In the case of the icosahedron, the best site for the Ag atom is at the center. The central site of the icosahedron is known to be highly compressed and fully coordinated, so that the placement of a smaller atom such as Ag in that position can be beneficial to stress release,35 leading to a net energy gain. In contrast, in the cuboctahedral cluster, where the internal strain is absent, the Ag atom preferentially occupies low-coordination surface vertex sites rather than the central position as in the icosahedral geometry. The surface preference of the Ag atom in the cuboctahedron is driven by its weak bonding character, low cohesive energy (2.128 eV per atom for Ag vs. 4.225 eV per atom for Ti) and tendency for surface segregation (due to its lower surface energy compared to Ti).
 | ||
| Fig. 2 Site preference of Ag replacing one Ti in the Ti55 icosahedral (first row) and cuboctahedral (second row) clusters. Blue and gray spheres represent silver and titanium atoms, respectively. Tables show the relative energy differences ΔE together with cohesive Ecoh and mixing Ecoh energies. The zero of ΔE corresponds to the best position for the Ag impurity in the Ti structure. | ||
On the other hand, our findings indicate that subsurface sites of the Ag atom are energetically costly for both geometries. The most probable explanation for these behaviours lies on the interplay of several factors, in particular, the local structural relaxation. The interatomic distances of the 12 nearest neighbours of the Ag central atom are 2.87 Å in the cuboctahedron and 2.79 Å in the icosahedron, showing a shortening of the bonds of nearly 3% in the latter compared to the former. In both the cuboctahedron and the icosahedron, the 12 nearest neighbours around the central impurity are placed at the same distance, preserving the symmetry of the structures upon relaxation. In contrast, when the Ag impurity is placed in the subsurface position, there is some distortion of the structures around it. The twelve nearest neighbours are placed in a relatively large range of distances, i.e. between 2.59 and 2.89 Å in the cuboctahedron and between 2.66 and 2.84 Å in the icosahedron. These distortions reflect unfavourable local strain and poor bonding compatibility, which correlates with higher total energies and lower site preference. In summary, sites that are highly symmetric and with short bond lengths (as in the icosahedral center) or lower coordination (as in surface vertices), which minimize the bonding demands, are preferred by Ag.
To reinforce these findings, we evaluated the ECN and dav values29,37 (see Fig. S2 and S3 in the ESI†). The results provide compelling quantitative evidence supporting our previous energetic findings. In the icosahedral cluster, the central site exhibits a maximized coordination environment and minimal local strain, as reflected by the high ECN and the shortest average bond lengths for the Ag atom, highlighting its strong preference for this symmetric and compact geometry. In contrast, the cuboctahedral configuration exhibits a broader range of ECN and dav values, with Ag favoring surface positions where local coordination is lower and bonding is more flexible. This difference emphasizes the geometry dependence of Ag incorporation, driven by the interplay between symmetry, coordination and local relaxation effects.
These findings indicate that the bonding environment in the cuboctahedral motif is less favorable for Ag incorporation. This is further corroborated by the cohesive energy values, where the Ti54Ag icosahedral cluster exhibits a slightly higher cohesive energy compared to its cuboctahedral counterpart.
For the composition Ti53Ag2, all possible placements of Ag atoms in the icosahedron have been considered. They can be both inside the cluster (one in the center and the other on the subsurface or both in the subsurface sites), both on the surface of the cluster (occupying vertices or edges) or one inside and another in the surface vertex site or edge site. The optimized structures and their relative energies are shown in Fig. 3, going from the best configuration (left image) to the worst one (right image). The best structure corresponds to that in which one Ag atom occupies the innermost site and the other occupies the surface vertex site (see the first left image in Fig. 3). This specific arrangement is energetically preferred due to the complementary role played by each Ag atom in distinct environments. The central Ag atom relieves compressive stress and stabilizes the core of the icosahedron. On the other hand, the vertex surface Ag atom minimizes bonding strain and takes advantage of Ag's surface-seeking behavior. However, the very worst configuration is shown in the last right image of Fig. 3, which corresponds to the structure with two Ag atoms on the subsurface. These arrangements introduce significant local distortions in the Ti lattice, as reflected by irregular bond lengths and non-uniform atomic displacements. This shows that the Ti icosahedron core cannot accommodate more than one silver atom.
 | ||
| Fig. 3 Configurations (a)–(g) of Ti53Ag2 in the icosahedral structure, arranged from best (a) to worst (g) in terms of chemical ordering. Titanium atoms are shown in gray and silver atoms in blue. Relative energies of DFT calculations are given. Zero energy corresponds to the lowest placement of Ag atoms in the Ti icosahedron. | ||
Now, we will discuss the stability of TiAg icosahedral clusters at two different compositions, Ti42Ag13 and Ti13Ag42, which give the possibility of forming perfect core@shell arrangements. For both compositions, we considered two more patterns besides core@shell, i.e. random mixing and ordered mixing “vertex arrangement”. The latter pattern is formed by placing the minority atoms in the surface vertices and at the central site of the Ih structure. All these configurations are illustrated in Fig. 4 with their corresponding relative energies.
 | ||
| Fig. 4 Optimized homotops of Ti42Ag13 (upper panel, (a)–(c)) and Ti13Ag42 (lower panel, (d)–(h)). Values represent relative energies of each homotop. Titanium atoms are shown in gray and silver atoms in blue. | ||
For Ti42Ag13, the best atomic arrangement corresponds to the configuration in which one Ag atom is at the central site and 12 Ag atoms at each of the surface vertex sites. This configuration represents a three-shell-like structure with an ordered mixing surface “Ag vertices arrangement”. This chemical ordering balances the need for strain relief in a highly coordinated, compressed environment while preserving surface segregation of the remaining Ag atoms. However, the configuration with all Ag atoms in the core forming a highly symmetric Ag@Ti core@shell arrangement is significantly higher in energy, by 16.4 eV. Placing the lower cohesive energy element (Ag atoms) in the highly coordinated environment (nanoparticle core) and the high surface energy element (Ti atoms) on the surface reduces the cluster stability and increases its total energy. Finally, for Ti13Ag42, the Ti@Ag core@shell chemical arrangement is energetically the most favorable over all the considered configurations. This preserves the natural chemical ordering tendency of AgTi systems observed in bulk and nanoclusters: Ti prefers bulk, and Ag prefers the surface.
3.2. Nanoclusters on Ti(0001)
First, we will search for the best placement of one Ti or Ag atom on the Ti(0001) slab. To this end, we construct different initial configurations as presented in Fig. 5. In the top row, a titanium or a silver atom (orange sphere) is located on the surface of the Ti(0001) slab as an adatom at different adsorption sites such as top (a), bridge (b), and fcc and hcp (c). In the second row, the silver atom (blue sphere) is placed on the Ti(0001) surface (d) and in the subsurface (e) sites, respectively. | ||
| Fig. 5 Top views of the Ti(0001) surface with an adsorbed adatom at different positions. The Top row represents surface adsorption of a Ti or Ag adatom (orange sphere) on four high-symmetry adsorption sites of the Ti(0001) surface: Top (a), Bridge (b), and hcp (right inset image) and fcc (left inset image) (c). The bottom row corresponds to the Ag atom (blue ball) replacing the Ti surface (d) and the subsurface atom (e) of the Ti(0001) substrate with the Ti adatom (black ball) on the hcp surface site. The white balls represent the first nearest neighbor Ti surface atoms of the adatom, and the gray balls are its second nearest neighbor Ti surface atoms. | ||
In Table 1, we show the energy differences ΔE and adhesion energies Eadh of single Ti or Ag atom at different sites of the Ti(0001) substrate. In all cases, there is a correlation between ΔE and Eadh and they reflect the true interaction strength of the atoms with the surface. A small ΔE (stable adsorbed state) corresponds to a high Eadh (strong adsorption) and vice versa. For Ti adsorption, our results indicate that an isolated Ti adatom prefers to occupy the top site over the hollow ones of the Ti(0001) surface. Therefore, the best adsorption site of a single atom does not correspond to the bulk stacking of Ti layers, which would indeed indicate a preference for the hcp site. This behavior bears some resemblance to what has previously been found for self-adsorption of iridium atoms on Ir(111),38 where an isolated iridium atom was found to preferentially occupy surface hcp sites rather than the normal bulk fcc ones.
| Atom type | Initial site | ΔE (eV) | E adh (eV) | Final site |
|---|---|---|---|---|
| Ti atom | Top | 0.000 | 4.542 | Top |
| Bridge | 0.066 | 4.476 | Bridge | |
| fcc | 0.095 | 4.447 | fcc | |
| hcp | 0.130 | 4.412 | hcp | |
| Ag atom | Top | 0.358 | 2.627 | Top |
| Bridge | 0.000 | 2.986 | fcc | |
| fcc | 0.000 | 2.986 | fcc | |
| hcp | 0.033 | 2.952 | hcp | |
| Surf | 0.564 | 2.422 | Surf | |
| Subsurf | 1.136 | 1.850 | Subsurf |
For an Ag atom, we find that the configurations on the surface of Ti(0001) are more stable than those in which Ag is incorporated in the surface and subsurface layers (surf and subsurf configurations of Fig. 5, respectively). This result is consistent with previous results in the literature, obtained using DFT calculations and semi-empirical models.39,40 This means that the exchange mechanism between an Ag atom and Ti surface or subsurface atoms is not energetically favorable during the deposition of Ag atoms onto this Ti surface. Therefore, the Ag atom prefers to stay on the surface occupying fcc or hcp sites, whose energy difference is quite small, less than 0.04 eV.
Now we examine the adsorption of small Ti and Ag nanoclusters on the Ti(0001) substrate, for sizes up to 4 atoms. Cluster shapes and placements are illustrated in Fig. 6, whereas the energies of the different configurations are reported in Table 2.
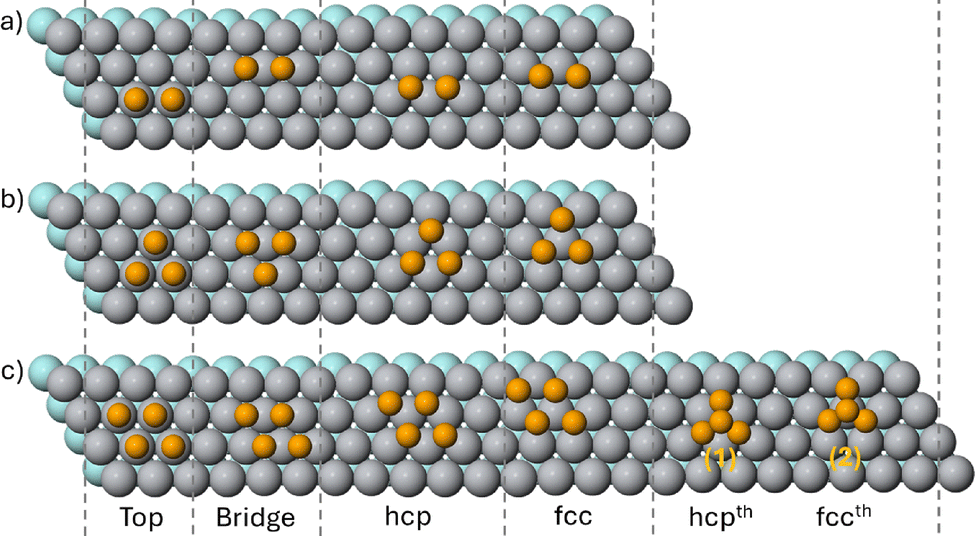 | ||
| Fig. 6 Top view of clusters on the Ti(0001) surface at different positions. Rows show the cluster sizes from dimers to tetramers. The orange balls represent the atoms of the cluster. The gray balls show the outermost layer atoms of the substrate. Atoms of the second layer are illustrated by pale turquoise spheres. | ||
| Cluster | Type | Initial site | δE (eV) | E adh | Final site |
|---|---|---|---|---|---|
| Dimer | Ti | Top | 0.271 | 3.038 | Top |
| Bridge | 0.000 | 3.309 | Bridge | ||
| fcc | 0.001 | 3.307 | Bridge | ||
| hcp | 0.002 | 3.307 | Bridge | ||
| Ag | Top | 0.347 | 1.757 | Top | |
| Bridge | 0.042 | 2.062 | bridge | ||
| fcc | 0.000 | 2.104 | fcc | ||
| hcp | 0.022 | 2.082 | hcp | ||
| Trimer | Ti | Top | 0.396 | 2.638 | Top |
| Bridge | 0.000 | 3.034 | hcp | ||
| fcc | 0.046 | 2.988 | fcc | ||
| hcp | 0.000 | 3.034 | hcp | ||
| Ag | Top | 0.351 | 1.712 | Top | |
| Bridge | 0.035 | 2.027 | Bridge | ||
| fcc | 0.000 | 2.063 | fcc | ||
| hcp | 0.007 | 2.056 | hcp | ||
| Tetramer | Ti | fcc | 0.000 | 2.704 | fcc |
| hcp | 0.039 | 2.665 | hcp | ||
| fccth | 0.513 | 2.145 | fccth | ||
| hcpth | 0.556 | 2.102 | hcpth | ||
| Ag | fcc | 0.000 | 1.778 | fcc | |
| hcp | 0.011 | 1.766 | hcp | ||
| fccth | 0.294 | 1.723 | fccth | ||
| hcpth | 0.337 | 1.681 | hcpth |
For the Ti dimer, the best placement is with both atoms on the Bridge site. The Top site is much higher in energy, and both fcc and hcp sites are unstable. The best position of the Ti trimer is on the hcp stacking, but it is quasi-degenerate with the fcc stacking. The Bridge position becomes unstable, and the Top position is high in energy. For the tetramer, we consider two shapes, the flat rhombus and the three-dimensional tetrahedron. The former is much lower in energy than the latter for all placements on the surface. For both the rhombus and the tetrahedron, the fcc stacking is slightly lower in energy than the hcp stacking, showing that the bulk-like continuation has not yet been accomplished for this cluster size.
For Ag clusters, the best placement is always on the fcc stacking, but with very small energy differences with respect to the hcp stacking. Bridge sites are slightly higher in energy, while Top sites are much higher in energy. For tetramers, the planar configuration is much more favourable than the tetrahedron. This result is at variance with those for tetramers on titanium oxides. In fact, in ref. 19, Schvval et al. theoretically investigated Ag4 nanoclusters deposited on the stoichiometric surfaces of rutile (110) and anatase (101) TiO2. For both surfaces, they found that Ag4 nanoclusters prefer the tetrahedral shape configuration, in agreement with experiments.20 If we compare these results with ours, we find a clear indication that the adhesion energy of Ag on the TiO2 surface is much smaller than that for Ag on pure Ti.
The results of adhesion energy reveal a clear size-dependent trend in the adhesion strength of clusters on the Ti(0001) surface. Specifically, the adhesion energy per cluster atom decreases progressively with the adsorbed cluster size, from ∼3 eV for adatoms to ∼1 eV in the case of tetramers. This behavior is attributed to the reduced number of cluster–substrate bonds per atom in larger clusters, as the intra-cluster interactions compete with direct surface bonding.
In Table 3, we list the optimized structural parameters of adatoms and clusters on the Ti(0001) surface. For each cluster size, our calculations show that dad(Ag)–surf(Ti) is somewhat higher than dad(Ti)–surf(Ti). For instance, the Ag adatom at the Top site is about 2.173 Å above the surface Ti layer, whereas it is only 1.799 Å for the Ti adatom. For each type, the value becomes fairly higher with the increasing cluster size. These suggest that the chemical bonds between Ag and surface Ti atoms are stronger than Ti–Ti and are reinforced with increasing Ag adatoms. This behavior may be due to the high electronegativity of silver, which enhances the longitudinal interaction between Ag and Ti atoms.
 , where d1,2 is the average atomic layer spacing between atomic layers 1 and 2 after adsorption and d0 is the bulk layer spacing of Ti. Δ represents the buckling which is the vertical displacement of the Ti atoms nearest to adatoms in the first Ti layer. The inset image shows buckling Δ and vertical distances between the adsorbates and the top surface Ti layer dad–surf
, where d1,2 is the average atomic layer spacing between atomic layers 1 and 2 after adsorption and d0 is the bulk layer spacing of Ti. Δ represents the buckling which is the vertical displacement of the Ti atoms nearest to adatoms in the first Ti layer. The inset image shows buckling Δ and vertical distances between the adsorbates and the top surface Ti layer dad–surf
|
|
|||||
|---|---|---|---|---|---|
| Cluster | Type | Adsorption site | d ad–surf (Å) | Δd1,2 (%) | Δ (Å) |
| Adatom | Ti | Top | 1.799 | −5.4487 | 0.432 |
| Ag | Top | 2.173 | −5.7271 | 0.369 | |
| Dimer | Ti | Bridge | 1.969 | −4.8057 | 0.160 |
| Ag | Bridge | 2.139 | −5.5056 | 0.065 | |
| Trimer | Ti | fcc | 2.077 | −4.4388 | 0.127 |
| hcp | 2.039 | −4.0248 | 0.150 | ||
| Ag | fcc | 2.192 | −5.2729 | 0.025 | |
| hcp | 2.190 | −5.2125 | 0.055 | ||
| Tetramer | Ti | fcc | 2.076 | −4.0415 | 0.079 |
| hcp | 2.105 | −4.0197 | 0.129 | ||
| Ag | fcc | 2.171 | −5.0724 | 0.084 | |
| hcp | 2.190 | −4.9305 | 0.076 | ||

After the cluster adsorption, the distance between the two topmost layers of the Ti(0001) substrate was obviously changed, with respect to the bulk spacing value of 2.321 Å. The relaxation of the topmost interlayer of the Ti(0001) slab in the presence of adatoms is illustrated in the fifth column of Table 3 for different adsorbed clusters. The adsorption of Ti and Ag adatoms results in a remarkable contraction in the separation between the first and second layers of the Ti atom slab in comparison with the clean Ti(0001) one (−6.388%). Moreover, the magnitude of contraction steadily increases with the increasing cluster size and it is stronger for Ti adsorbates than for Ag ones.
The buckling Δ reported in the last column of Table 3 quantitatively indicates the extent of surface deformation, encompassing surface folds or flexures.41 For instance, when the adatom or clusters are adsorbed on any adsorption site of the Ti(0001) surface, the first layer of Ti atoms connected to the adatom or clusters moves down, while the first layer of Ti atoms not bonded to the adatom or clusters exhibits a downward displacement, which results in a large Ti surface layer buckling after the adsorption of a single atom. The bending morphology of the initial Ti layer becomes less significant with the increasing cluster size.
3.3. Ag and Ti monolayers on Ti(0001)
Our results on small clusters have shown that they do not preferentially adopt the hcp bulk-like stacking. Therefore, it is interesting to check whether the hcp stacking is recovered for a complete monolayer. This is indeed a consistency check for the calculations in the case of an added Ti monolayer, whereas it is a non-trivial point in the case of an Ag monolayer. We will use the 4 × 4 slab, so that our monolayer consists of n = 16 atoms. Our calculations, whose results are presented in Table 4, show that the hcp stacking is preferred for both Ti and Ag monolayers. These results indicate that as the number of Ti or Ag atoms in a cluster increases, binding of cluster atoms at hcp sites is expected to become more advantageous.| Type | Stacking | δE (eV) | d ad–surf (Å) | Δd1,2 (%) | W adhesion (J m−2) |
|---|---|---|---|---|---|
| Ti ML | fcc | 0.046 | 2.200 | +2.3905 | — |
| hcp | 0.000 | 2.180 | +3.0855 | — | |
| Ag ML | fcc | 0.021 | 2.347 | −3.5625 | 2.384 |
| hcp | 0.000 | 2.340 | −3.2055 | 2.429 |
In order to measure the bonding strength at the interface between Ag and Ti surfaces, we calculated its adhesion work using the following equation:17
 | (5) |
The work of adhesion is presented in Table 4 for two different stackings of the Ag monolayer. The obtained values show that the titanium surface demonstrates a high affinity with the silver monolayer, with Wadh of 2.384 and 2.429 J m−2 for fcc and hcp stackings of the Ag layer, respectively. These results confirmed that both Ag fcc and hcp stackings show a good adhesion on the Ti surface with a small strength variation of 0.045 J m−2.
Regarding the spacing between the first and second Ti layers (Δd1,2) after the adsorption of the Ti or Ag monolayer, it is noted that substrate relaxation in the Ti/Ti(0001) system is significantly larger than the Ag/Ti(0001) one at both fcc and hcp stackings.
To gain insight into the local electronic properties at the interface between Ag clusters and the Ti(0001) surface, we perform Bader charge analysis and examine charge density difference maps. The results are given only for the most stable adsorption configuration of each system, including the Ag adatom, dimer, trimer, tetramer, and monolayer on the Ti(0001) surface.
Bader charge analysis shows that Ag atoms are always negatively charged as illustrated in Table 5 regardless of the cluster size, meaning that Ag atoms consistently gain charges when supported on the Ti(0001) surface. This is expected due to the higher electronegativity of Ag with respect to Ti, leading to electron transfer from nearest Ti atoms to Ag. It is revealed that the net charge per Ag atom decreases in magnitude as the Ag cluster size increases, from −0.604 e for an adatom to −0.358 e per atom in the monolayer configuration. This reduction reflects that the charge transfer from Ti to Ag is the strongest for the Ag adatom or small clusters, where the Ag atoms are in direct contact with the reactive Ti surface. These atoms experience stronger local polarization and enhanced interactions with the Ti surface, resulting in more significant charge accumulation. As the cluster size increases, Ag–Ag interactions become more prominent, and the fraction of Ag atoms in direct contact with the Ti surface decreases. In the case of the monolayer, each Ag atom is coordinated not only with Ti but also with neighboring Ag atoms, which reduces the local charge transfer per atom.
| Ag cluster | Best adsorption site | Average charge | Total charge on Ag |
|---|---|---|---|
| Adatom | fcc | −0.604 | −0.604 |
| Dimer | fcc | −0.560 | −1.120 |
| Trimer | fcc | −0.525 | −1.575 |
| Tetramer | fcc | −0.509 | −2.035 |
| ML | hcp | −0.358 | −5.726 |
These results highlight that charge transfer at the Ag/Ti interface is highly sensitive to local coordination and interface density and further support the view that Ag atoms act as moderately electronegative acceptors when adsorbed on Ti surfaces. The trend also reflects a transition from a highly localized charge redistribution regime for small clusters to a more delocalized regime in large clusters and monolayers.
To support the Bader charge analysis, we also examined the charge density difference isosurfaces for the Ag adatom, clusters and monolayer on the Ti(0001) surface. These isosurfaces are visualized in Fig. 7, where the charge-depletion regions are displayed in cyan and the charge-accumulation regions are shown in yellow.
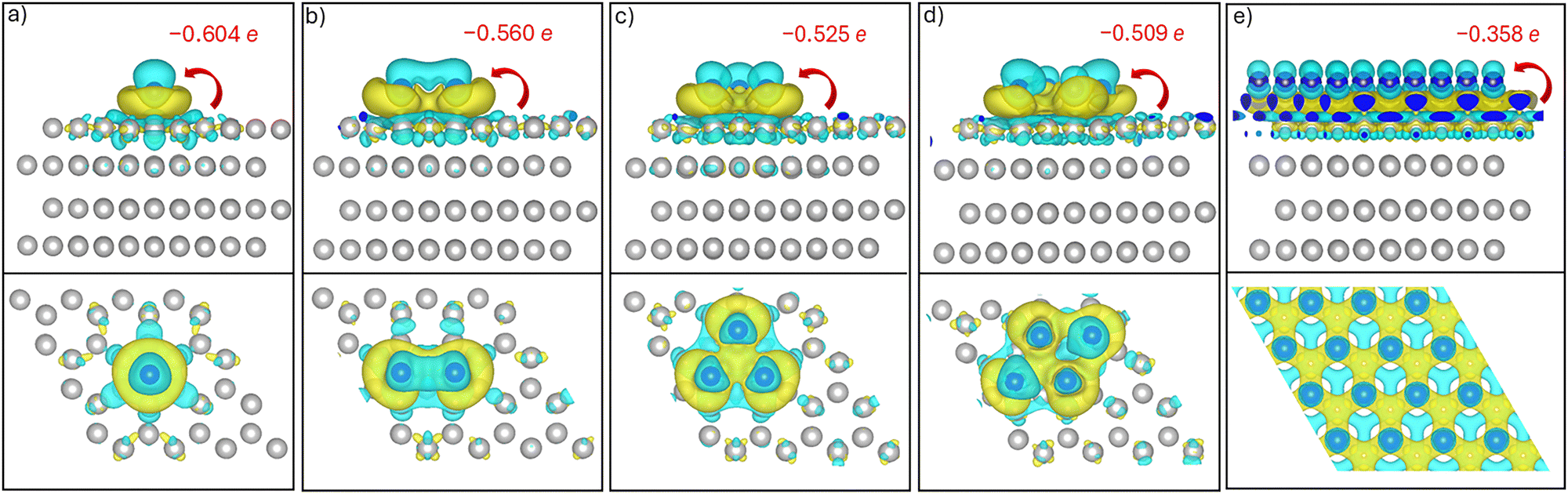 | ||
| Fig. 7 Side (top row) and top views (bottom row) of the charge density difference isosurfaces for Ag adatom (a), dimer (b), trimer (c), tetramer (d) and monolayer (e) on the Ti(0001) surface. Values represent the Bader charge gained by one Ag atom in the clusters and monolayer. Electron accumulation and depletion between Ag and Ti atoms are shown in yellow and cyan colors, respectively. | ||
The maps clearly show charge accumulation at the Ag–Ti bonding interface and depletion from the surface Ti atoms, indicating charge transfer from the substrate to the adatom. This interfacial polarization is consistent with Bader charge analysis. For small clusters, this polarization is strongly localized beneath the Ag atoms, forming distinct charge accumulation lobes that reflect direct Ag–Ti bonding. As the Ag cluster size increases, the charge density difference maps reveal a broadening of the charge redistribution zone. In the monolayer case, although individual Ag atoms show weaker localized accumulation, the overall interfacial polarization becomes more delocalized and uniform.
4. Conclusions
Our results of gas-phase and surface supported TiAg clusters help in establishing some clear tendencies. In general, the incorporation of Ag atoms in Ti clusters and surfaces preferentially takes place in surface sites, with the only exception being the central site in the icosahedron. Subsurface sites are always less favourable than those in the surface layer. Our results concerning adatoms on the Ti(0001) surface show that even the incorporation in the surface layer after an exchange causes a notable energy penalty. In addition, the formation of core@shell Ti@Ag gas-phase clusters seems to be energetically favourable. All these results concur in predicting a quite sharp interface between Ag and Ti, with a limited amount of interdiffusion, because there might be a significant bottleneck to full intermixing both in bulk samples and in clusters.The structure of that sharp interface is, however, non-trivial. In fact, when placing small Ag clusters on the Ti(0001) surface, one finds that the preferential stacking does not correspond to the hcp bulk-like continuation, but to the fcc stacking instead. Energy differences between these stackings are, however, quite small. The bulk-like stacking is recovered for a full Ag monolayer. A similar type of complex behaviour is also found for small Ti clusters on Ti(0001).
The interface between Ag and Ti is sharp but is characterized by quite a strong adhesion, which is much stronger than that of Ag on Ti oxide surfaces, as demonstrated by the comparison between the preferential shape of Ag tetramers, which is flat on Ti(0001) but tetrahedral on Ti oxides.
Charge analyses show that there is some charge transferred from the surface to the Ag clusters, which supports the strong bonding of these clusters to the Ti surface.
Author contributions
E. E. performed and analyzed calculations. R. F. assisted in the analysis of the simulations and supervised the work. Both authors contributed to the drafting of the paper.Data availability
Data for this article are available at Zenodo at https://zenodo.org/records/15017886 – input and output files from DFT calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded under the National Recovery and Resilience Plan (NRRP), Mission 4, Component 2, Investment 1.1, Call for tender No. 104 published on 2.2.2022 by the Italian Ministry of University and Research (MUR), funded by the European Union – NextGenerationEU – Project Title PINENUT – CUP D53D23002340006 – Grant Assignment Decree No. 957 adopted on 30/06/2023 by the Italian Ministry of University and Research (MUR). The authors acknowledge networking support from the IRN Nanoalloys of CNRS. The authors thank Cesare Roncaglia for useful discussions.Notes and references
- I. Gurappa, Mater. Charact., 2002, 49, 73–79 CrossRef CAS.
- A. Etiemble, C. Lopes, G. I. Nkou Bouala, J. Borges, A. Malchère, C. Langlois, F. Vaz and P. Steyer, Surf. Coat. Technol., 2019, 358, 646–653 CrossRef CAS.
- A. Fonseca-García, J. Pérez-Alvarez, C. Barrera, J. Medina, A. Almaguer-Flores, R. B. Sánchez and S. E. Rodil, Mater. Sci. Eng. C, 2016, 66, 119–129 CrossRef PubMed.
- E. Unosson, D. Rodriguez, K. Welch and H. Engqvist, Acta Biomater., 2015, 11, 503–510 CrossRef CAS PubMed.
- C. Lopes, C. Gabor, D. Cristea, R. Costa, R. Domingues, M. Rodrigues, J. Borges, E. Alves, N. Barradas, D. Munteanu and F. Vaz, Appl. Surf. Sci., 2020, 505, 144617 CrossRef CAS.
- G. Benetti, E. Cavaliere, A. Canteri, G. Landini, G. M. Rossolini, L. Pallecchi, M. Chiodi, M. J. Van Bael, N. Winckelmans, S. Bals and L. Gavioli, APL Mater., 2017, 5, 036105 CrossRef.
- K. M. Elattar, A. A. Ghoniem, F. O. Al-Otibi, M. S. El-Hersh, Y. A. Helmy and W. I. A. Saber, Appl. Sci., 2023, 13, 10110 CrossRef CAS.
- M. Kawamura, Y. Inami, Y. Abe and K. Sasaki, Jpn. J. Appl. Phys., 2008, 47, 8917 CrossRef CAS.
- M. Kawamura, D. Fukuda, Y. Inami, Y. Abe and K. Sasaki, J. Vac. Sci. Technol., A, 2009, 27, 975–978 CrossRef CAS.
- H. C. Kim, N. D. Theodore and T. L. Alford, J. Appl. Phys., 2004, 95, 5180–5188 CrossRef CAS.
- K. Sugawara, M. Kawamura, Y. Abe and K. Sasaki, Microelectron. Eng., 2007, 84, 2476–2480 CrossRef CAS.
- Y. Minamide, M. Kawamura, Y. Abe and K. Sasaki, Vacuum, 2009, 84, 657–662 CrossRef CAS.
- M. Kawamura, Z. Zhang, R. Kiyono and Y. Abe, Vacuum, 2013, 87, 222–226 CrossRef CAS.
- E. B. Choi and J.-H. Lee, Appl. Surf. Sci., 2017, 415, 67–74 CrossRef CAS.
- Z. D. Davis and B. J. Tatarchuk, Appl. Surf. Sci., 2015, 353, 679–685 CrossRef CAS.
- B. Sarma and B. K. Sarma, Appl. Surf. Sci., 2017, 410, 557–565 CrossRef CAS.
- D. Cornil, H. Wiame, B. Lecomte, J. Cornil and D. Beljonne, ACS Appl. Mater. Interfaces, 2017, 9, 18346–18354 CrossRef CAS PubMed.
- A. S. Mazheika, V. E. Matulis and O. A. Ivashkevich, J. Mol. Struct. THEOCHEM, 2009, 909, 75–78 CrossRef CAS.
- A. B. Schvval, A. Juan and G. F. Cabeza, Appl. Surf. Sci., 2019, 490, 343–351 CrossRef CAS.
- E. Lira, J. O. Hansen, L. R. Merte, P. T. Sprunger, Z. Li, F. Besenbacher and S. Wendt, Top. Catal., 2013, 56, 1460–1476 CrossRef CAS.
- D. Cornil, N. Rivolta, V. Mercier, H. Wiame, D. Beljonne and J. Cornil, ACS Appl. Mater. Interfaces, 2020, 12, 40838–40849 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. R. Davidson, J. Comput. Phys., 1975, 17, 87–94 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- R. Hoppe, Z. Kristallogr. - Cryst. Mater., 1979, 150, 23–52 CrossRef CAS.
- J. L. F. Da Silva, J. Appl. Phys., 2011, 109, 023502 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- R. Ferrando, Structure and Properties of Nanoalloys, Elsevier, 2016 Search PubMed.
- N. Canestrari, D. Nelli and R. Ferrando, Nat. Commun., 2025, 16, 1655 CrossRef CAS PubMed.
- S.-Y. Wang, J.-Z. Yu, H. Mizuseki, J.-A. Yan, Y. Kawazoe and C.-Y. Wang, J. Chem. Phys., 2004, 120, 8463–8468 CrossRef CAS PubMed.
- D. Nelli, C. Roncaglia and C. Minnai, Adv. Phys.: X, 2023, 8, 2127330 Search PubMed.
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 77, 371–423 CrossRef CAS.
- M. J. Piotrowski, P. Piquini and J. L. F. Da Silva, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155446 CrossRef.
- S. C. Wang and G. Ehrlich, Phys. Rev. Lett., 1989, 62, 2297–2300 CrossRef CAS PubMed.
- Y. Zhou, R. Smith, S. D. Kenny and A. L. Lloyd, Nucl. Instrum. Methods Phys. Res., Sect. B, 2017, 393, 122–125 CrossRef CAS.
- Y. Zhou, A. L. Lloyd, R. Smith and S. D. Kenny, Surf. Sci., 2019, 679, 154–162 CrossRef CAS.
- J. Zhao, Y. Xu, S. Liu and X. Ding, Appl. Surf. Sci., 2022, 586, 152810 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01019e |
| This journal is © the Owner Societies 2025 |