
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection and quantification of hydroperoxides and radicals within stabilized cool flames by means of SVUV-PEPICO spectroscopy†
Sébastien
Batut
 a,
Laure
Pillier
a,
Jérémy
Bourgalais
b,
Thomas
Panaget
a,
Pascal
Demaux
a,
Olivier
Herbinet
c,
Frédérique
Battin-Leclerc
c,
Gustavo A.
Garcia
d,
Laurent
Nahon
d and
Guillaume
Vanhove
*a
a,
Laure
Pillier
a,
Jérémy
Bourgalais
b,
Thomas
Panaget
a,
Pascal
Demaux
a,
Olivier
Herbinet
c,
Frédérique
Battin-Leclerc
c,
Gustavo A.
Garcia
d,
Laurent
Nahon
d and
Guillaume
Vanhove
*a
aUniversité de Lille, CNRS, PC2A, F-59000 Lille, France. E-mail: guillaume.vanhove@univ-lille.fr
bUniversité de Rennes, CNRS, IPR, F-, 35000, Rennes, France
cUniversité de Lorraine, CNRS, LRGP, F-54000 Nancy, France
dSynchrotron SOLEIL, L'Orme des Merisiers, Départementale 128, 91190 St Aubin, France
First published on 26th June 2025
Abstract
A novel coupling of a stagnation plate burner and the SAPHIRS instrument located at synchrotron SOLEIL has been performed to enable the observation of labile species and radicals within stabilized cool flames. Gases were extracted from a dimethyl ether/O2/O3/N2 cool flame with a capillary probe and expanded through two consecutive skimmers to reach the synchrotron vacuum ultraviolet-photoelectron photoion coincidence spectrometer. threshold photoelectron spectra (TPES) and total ion yields (TIY) were recorded as a function of the photon energy, and led to the identification of species that have so far never been observed in cool flames, including the only expected keto- or aldo-hydroperoxide (hydroperoxymethylformate), hydrogen peroxide, methyl and hydroperoxyl radicals. The quantification of the detected species has been performed and is compared with the predictions of a kinetic model, demonstrating the validity of the experimental approach and suggesting possible ways for improvement.
Introduction
Ever since their discovery by Humphry Davy in the 19th century, cool flames have garnered periodic interest due to their practical applications in coal mine safety,1 the formulation of fuels for internal combustion engines,2,3 and more recently in plasma-assisted ignition,4 constant volume chamber combustion5 and fire propagation under microgravity conditions.6,7 Early pioneering work performed in the 1960s at the Shell Thornton laboratory2,8 hypothesized that the ignition of cool flames and the first-stage ignition phenomenon in adiabatic reactors are caused by the formation of hydroperoxides (ROOH), following the addition of radicals to oxygen. Most notably, keto- and aldohydroperoxides were expected. These hydroperoxides decompose to yield radicals such as OH, causing indirect radical-chain branching9 which leads to autoignition. While this theory was consistent with indirect observations, such as the detection of cyclic ethers spawning from hydroperoxide decomposition, the high reactivity of hydroperoxides made their experimental observation a challenge for the following decades.Experimental proof for the formation of such indirect chain-branching agents was finally obtained in a motored engine and flow reactor using n-heptane and n-dodecane as the fuels, where Sahetchian et al.10,11 employed cold trapping, thin-layer chromatography and offline mass spectrometric analysis. This was later confirmed by Battin-Leclerc et al.,12 who used online synchrotron-based photoionization mass spectrometry (PIMS) coupled with a jet-stirred reactor (JSR) setup. By exposing gases extracted from the JSR to tuneable VUV radiation, soft ionization was achieved, consistent with the expected ionization energies of the ketohydroperoxides (KHP) formed by n-butane, although the branching fraction of the formed ketohydroperoxide isomers could not be accessed from these results. The identification of individual ketohydroperoxide isomers was then made possible through advanced techniques such as double imaging photoelectron photoion coincidence (i2-PEPICO) spectroscopy.13–16
In the past decade, burner-stabilized cool flames have emerged as a promising configuration to study ignition–extinction regimes,7 low-temperature oxidation kinetics17–20 and laminar burning velocities of cool flames.21,22 These studies have mostly focused on counterflow - in the premixed23 and diffusion6 flame regimes – and stagnation plate17,24 configurations. Detailed chemical information17–19 has been garnered in non-isothermal conditions relevant to the ignition-extinction regimes. Moreover, burner-stabilized cool flames offer low-dilution conditions conducive to detecting chain-branching agents and radicals. Sensitivity analysis has shown the importance of different reaction pathways in cool flames compared to jet-stirred/flow reactors, revealing their untapped potential for validating chemical kinetic models. However, despite these advances, and because of the numerous associated challenges, the coupling of burner-stabilized cool flames with synchrotron-based techniques, such as PIMS or i2-PEPICO, had not been attempted until now.
The present work presents the first coupling of a cool flame configuration with synchrotron-based techniques, to detect ketohydroperoxides and radicals in ozone (O3)-seeded cool flames of dimethyl ether (DME, CH3–O–CH3). DME is a model oxygenated fuel candidate for renewable energy systems, as it can be derived from biomass or captured CO2 and renewable electricity.25,26 Its symmetrical structure, small molecular size (molar mass 46 g mol−1), and high reactivity in the low-temperature combustion regime lead to the formation of a single aldohydroperoxide, hydroperoxymethylformate (HPMF, HOO–CH2–O–CHO, molar mass 92 g mol−1). This makes DME an ideal reference compound for studying low-temperature oxidation chemistry and ignition processes.
Although some studies have investigated DME oxidation, particularly aiming at the quantification of KHPs, these efforts remain mostly limited to JSR and flow reactors coupled with SVUV-PIMS. Moshammer et al.27,28 detected, and reported significant fragmentation of HPMF near its adiabatic ionization energy (AIE ∼ 10 eV), primarily resulting in the loss of a CO molecule to produce a fragment at m/z 64 (HOO–CH2OH) with a much higher signal intensity than the parent KHP at m/z 92. Later, Couch et al.29 confirmed m/z 64 as the main fragment of HPMF but showed that this holds only at IEs below 10.5 eV. At higher energies, the additional loss of an OOH group produces a previously unreported fragment at m/z 31 (CH2OH), with partial photoionization cross sections (PICS) comparable to those of m/z 64 around 11 eV. Couch et al.25 also derived a total PICS for HPMF that was experimentally twice as low as the theoretical value explicitly calculated by Moshammer et al.23,24
Simultaneously, Liao et al.30 investigated the effect of ozone addition—known as a combustion promoter—on the low-temperature oxidation of DME in a JSR with SVUV-PIMS. They corroborated the identification of m/z 64 as the primary fragment of HPMF but observed differences in the total ion yield (TIY) curves compared to those reported by Couch et al. and Moshammer et al. Moreover, like Moshammer et al., they did not identify m/z 31 as a major fragment.
Thus, while this reactive system represents a promising tool for studying low-temperature oxidation processes, some ambiguities remain, particularly concerning the fragmentation patterns and photoionization behavior of HPMF. This makes it an excellent reference system for ongoing experimental development and refinement, building on previous studies of stabilized cool flames under similar conditions.17,21 These prior works have contributed to the creation of a well-validated kinetic mechanism tailored to these systems.
Experimental, numerical, and theoretical methods
Experimental method
The stagnation plate burner configuration developed in PC2A17 has been duplicated and adapted to enable the coupling at the undulator-based DESIRS31 VUV beamline of synchrotron SOLEIL, using the SAPHIRS end-station,32 equipped with the double-imaging photoelectron/photoion (i2PEPICO) spectrometer DELICIOUS III.33 Only the details relevant to this coupling are provided here, with a full description of the burner given in Panaget et al.17 As depicted in Fig. 1, the burner assembly is kept within a vessel, the pressure of which is fixed at 96.3 kPa (i.e. 0.95 atm) by means of both the pumping of the exhaust gases and a regulated flow of N2. This flow also helps flush the vessel and protect electrical connections from the burnt gases. | ||
| Fig. 1 Experimental setup used in this study. | ||
The composition of the fresh DME/O2/O3/N2 mixture entering the burner is given by the mole fractions x in Table 1, corresponding to a fuel-lean premixed flame with an equivalence ratio φ (i.e., the molar ratio of fuel to oxidizer with regards to stoichiometry) of 0.161. It exits the 10 mm diameter central nozzle, which is maintained at a temperature of 283 K, and flows toward a stagnation plate heated to 650 K, with the burner-plate distance set to 12 mm. A capillary probe with a 150 μm inner diameter and 220 μm outer diameter protrudes 800 μm from the stagnation plate into the flame gases, the total length of the probe being approximately 3 mm. An uncertainty of ±200 μm is assumed for the reported position, based on relevant studies of the spatial resolution of sampling probes in flames.34 This probe emerges on the other side of the stagnation plate within a 45° half-angle conical section, which allows the expansion of the sampled gases into the jet chamber, inside which the vessel is held and whose pressure is about 10−3 mbar.
| x DME | x O2 | x O3 | x N2 | Q burner/SL.min−1 | φ | Geometric strain rate α/s−1 |
|---|---|---|---|---|---|---|
| 0.043 | 0.759 | 0.026 | 0.173 | 2.32 | 0.161 | 41 |
The sampled gases further expand across two consecutive, 2 mm diameter skimmers, to first reach the differential chamber where the pressure is about 10−5 mbar before arriving to the spectrometer chamber whose pressure was around 10−7 mbar. Such expansion conditions facilitate a reduced collision environment, freezing the ongoing reactions and enabling the observation of transient molecular species and radicals. In the ionization chamber the gases are exposed to the VUV beam of DESIRS. Prior to entering the ionization chamber, the light supplied by DESIRS passes through a monochromator, equipped for this experiment with a 200 grooves per mm grating, resulting in a flux of ca. 1013 photons per second with a spectral resolution of ca. 25 meV at 10 eV. A gas filter located upstream of the monochromator, filled with Kr, effectively removes high harmonics from the undulator, providing a high spectral purity across the 9.5–12.7 eV range used during the experiments. The photon flux is recorded as a function of photon energy using a dedicated photodiode (AXUV from Opto Diode) and used to correct the spectra, while the photon energy scale was calibrated using the well-known ionization energy of the methyl radical35.
The molecular beam crosses the VUV synchrotron radiation at the centre of the i2PEPICO spectrometer DELICIOUS III.33 The coincident electrons and ions resulting from the ionization process are accelerated in opposite directions by a 88 V cm−1 continuous electric field and analysed by a velocity map imaging (VMI) spectrometer and a modified Wiley-McLaren time-of-flight (TOF) imaging spectrometer, respectively. The coincidence operation generates mass-filtered photoelectron images simultaneously for any peak in the TOF mass spectrum which are further treated by Abel inversion36 to yield mass-selected photoelectron spectra as a function of photon energy. The threshold photoelectron spectra (TPES) are obtained from these data according to the method outlined by Poully et al.37 and Briant et al.,38 here with an electron energy resolution of 17 meV. The TPES show resonant transitions from the neutral ground state toward vibronic states of the cation and serve as the vibronic fingerprint of the ionic species.14 These were used for the identification of the structures observed at different mass-to-charge ratios. Whenever possible, the TPES were preferred for identification because of their more distinctive spectral features to decipher isomers. However, when the signal-to-noise ratio does not allow an electron kinetic energy analysis for a specific mass-to-charge channel, the identification was based on the analysis of the respective total ion yield (TIY) curve obtained through integration over all electron energies. The signals as a function of photon energy were normalized by the photon flux, as measured offline with a dedicated AXUV photodiode.
Quantification of the relevant species was attempted from the fixed photon energy mass spectra obtained at selected quantification photon energies Eq above their respective ionization energies. The selected quantification energies, as well as the source for the used total and partial PICS are provided in Table 2. In the permanent regime, the acquired signal for species i at mass-to-charge ratio m can be calculated from eqn (1):
| Smi(E) = A.Dm.σmi(E).ni | (1) |
 | (2) |
| m/z | Formula | Species | E q/eV | PICS(Eq)/Mb | Ref. |
|---|---|---|---|---|---|
| 15 | CH3 | Methyl | 11.0 | 6.5 | 39 |
| 18 | H2O | Water | 12.7 | 5.9 | 40 |
| 30 | CH2O | Formaldehyde | 11.0 | 9.6 | 41 |
| 31 | C2H4O4 | HPMF fragment | 11.0 | 0.93 | 29 |
| 32 | CH3OH | Methanol | 11.0 | 4.4 ± 0.9 | 41 |
| 33 | HO2 | Hydroperoxyl | 11.5 | 1.5 ± 0.3 | 41 |
| 34 | H2O2 | Hydrogen peroxide | 11.0 | 0.37 ± 0.09 | 42 |
| 46 | C2H6O | DME | 11.0 | 11.4 ± 2.3 | 29 |
| 11.5 | 11.0 ± 2.2 | 29 | |||
| 12.7 | 13.2 ± 2.6 | 29,43 | |||
| 48 | O3 | Ozone | 12.7 | 3.2 | 44 |
| 60 | C2H4O2 | Methyl formate | 11.0 | 2.78 | 29 |
| 64 | C2H4O4 | HPMF fragment + hydroperoxymethanol | Non quantified | 29 | |
The mass discrimination factors of the SAPHIRS instrument have been measured previously,39 and have been used here. Given the uncertainty of the used PICS and the indirect measurement, an estimated overall uncertainty on ni of ±50% is assumed. In the case of the m/z = 31 fragment of HPMF, the contribution of 13CH2O was subtracted on the basis of the isotopic ratio, and assuming an identical PICS for 12CH2O and 13CH2O.
Kinetic modelling
The investigated flame was modelled using the stagnation plate burner model implemented in Cantera,45 following the method described in Panaget et al.17 Following the results of a recent characterization of the flow field in the same configuration via particle imaging velocimetry,21 the computational domain was restricted to a length of 6.5 mm relative from the stagnation plate. This length was chosen as the best compromise between achieving an accurate description of the axial velocity field and minimizing the constraint on the occurrence (or absence) of a cool flame. Simulations were performed using the DME/O3 model developed at PC2A,17,21 which has been extensively validated with cool flame speciation and laminar burning velocities results. Correlations derived from previous PIV data21 were used to calculate the velocity at the centre of the burner exit nozzle. Finally, in the absence of flow field data for the horizontal burner configuration, the flame spread rate in the modelling was adjusted to best fit the measured reactants and major products mole fractions at the measurement point, thereby allowing for a comparison of the branching between cool flame products.Theoretical quantum calculations and vibronic spectrum simulations
To facilitate the identification of the observed m/z channels, the spectra for hydroperoxymethanol, dimethyl ether, dioxirane and methyl formate were calculated. The simulations began with calculating the lowest-energy conformer for each isomer using density functional theory (DFT) with the M06-2X-D3 functional.46 This DFT functional was paired with the aug-cc-pVTZ Dunning's correlation-consistent basis set.47 Additionally, the zero-damped D3 dispersion correction developed by Grimme et al.48 was integrated into M06-2X. The lowest-energy structure was then used as the starting point for the cation optimization. All these calculations were performed within the harmonic approximation.Next, the adiabatic ionization energies of each isomer, both in its neutral and ionized forms, were calculated using the CBS-QB3 composite method49,50 with an accuracy of less than 100 meV. The PES were then simulated at 0 K using the time-independent adiabatic Hessian Franck–Condon (TI-AH|FC) model.51 The resulting stick spectrum was combined with a Gaussian profile featuring a 200 cm−1 bandwidth to match the experimental resolution. The 0–0 transition of the computed stick spectra was shifted to align with the experimental spectra, within the uncertainty of the energy calculations.
Results and discussion
Identification
A representative time-of-flight mass spectrum acquired at a PI energy of 11.5 eV with the proposed identification is shown in Fig. 2. A list of all the detected and quantified species is also provided in Table 2, but the detailed identification process will only be provided for selected m/z ratios. For the most abundant species, for which the identification has already been reported elsewhere, a comparison of the experimental and literature spectra is nevertheless provided in the ESI.† The relatively low electric field in the extraction region allows distinction of peaks originating from fragmentation events since those present a broader peak shape than the ones from parent ions due to the larger translational energies of the former. | ||
| Fig. 2 Typical TOF-MS recorded by synchrotron photoionization of sampled gas from the DME/O2/O3/N2 cool flame, acquired at a fixed 11.5 eV photon energy during 2 hours for the flame conditions reported in Table 1, at a distance of 800 ± 200 μm of the stagnation plate. The proposed identification is based on the following analysis. | ||
 | ||
| Fig. 3 Comparison of the TPE spectrum recorded at m/z = 15 (black symbols and lines) and the one reported for the methyl radical by Cunha de Miranda et al.35 | ||
 | ||
| Fig. 4 Comparison of the TIY spectrum recorded at m/z = 33 (black symbols and lines) and the one reported for the hydroperoxyl radical by Tang et al.52 | ||
 | ||
| Fig. 5 Comparison of the TPE spectrum recorded at m/z = 34 (black symbols and lines) and the one reported for hydrogen peroxide by Schio et al.53 | ||

In Couch et al.'s work, the m/z 31 and 64 channels were reported to yield significantly more signal than the parent ion and other fragmentation products (m/z =92, 91, 59) at all investigated photon energies. In agreement with this previous work, no signal was observed at m/z 92, 91 and 59 under our conditions.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hydroperoxymethanol signal branching ratio of 1:0.30. It should be noted that the TIY at m/z 64 is similar for the lower part to the one reported by Liao et al.30 in ozone-seeded conditions, which is also plotted in Fig. 6. The divergence in the higher energy part (i.e., above 10.5 eV) can be attributed to a lower HPMF:hydroperoxymethanol ratio in the current work, consistent with a much higher ozone loading (2.6% O3 in the mixture). On the opposite, the data acquired by Couch et al. at this m/z in the absence of ozone does not feature the lower energy contribution associated with hydroperoxymethanol. In comparison to the conditions of the present work and of the work of Liao et al., the results of Couch et al. were obtained in highly-diluted conditions to target the primary reactivity of DME. It is therefore likely that the formation of hydroperoxymethanol is caused by the reaction of secondary products of DME. Given that the mole fractions of HO2, H2O2 and CH2O are high in the current conditions, one can suggest that it originates from reactions between CH2O and H2O2, or between CH3O and HO2.
:hydroperoxymethanol signal branching ratio of 1:0.30. It should be noted that the TIY at m/z 64 is similar for the lower part to the one reported by Liao et al.30 in ozone-seeded conditions, which is also plotted in Fig. 6. The divergence in the higher energy part (i.e., above 10.5 eV) can be attributed to a lower HPMF:hydroperoxymethanol ratio in the current work, consistent with a much higher ozone loading (2.6% O3 in the mixture). On the opposite, the data acquired by Couch et al. at this m/z in the absence of ozone does not feature the lower energy contribution associated with hydroperoxymethanol. In comparison to the conditions of the present work and of the work of Liao et al., the results of Couch et al. were obtained in highly-diluted conditions to target the primary reactivity of DME. It is therefore likely that the formation of hydroperoxymethanol is caused by the reaction of secondary products of DME. Given that the mole fractions of HO2, H2O2 and CH2O are high in the current conditions, one can suggest that it originates from reactions between CH2O and H2O2, or between CH3O and HO2.
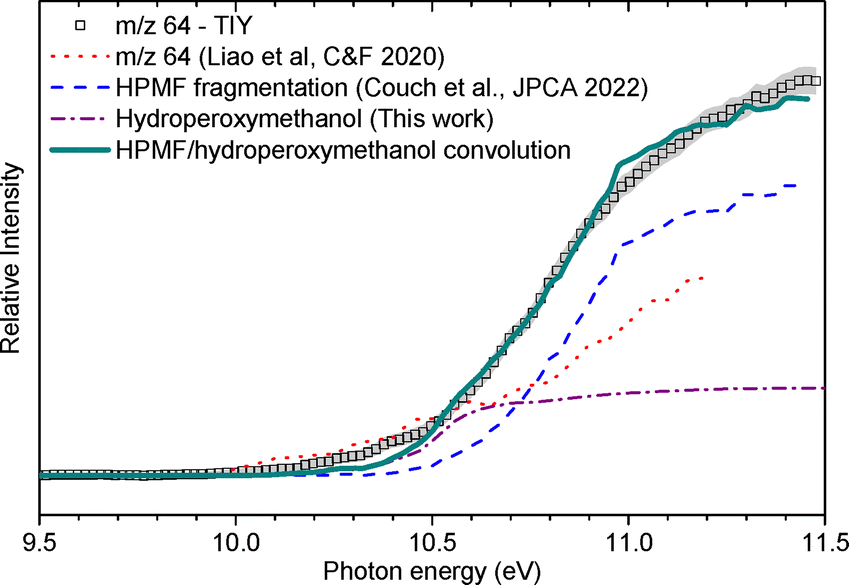 :13CH2O:CH3O signal branching ratio of 1:0.20:0.08. The signal of m/z 31 is dominated by the fragment of HPMF, which is consistent with the fact that it is the main fragment of the KHP along with m/z 64. This is also confirmed by the broad shape of the TOF peak which is correlated to dissociative ionization generating fragments with larger kinetic energy release distributions. The contribution from 13CH2O is also confirmed by the distinctive peak at 10.9 eV visible in the TPE spectrum shown in the SM. A contribution of the methoxy radical to the TPES cannot be ruled out, in accordance with the convolution presented in Fig. 7. The fragmentation of methanol could also contribute to the observed signal. This, however, corresponds to an appearance energy of around 11.5 eV based on Cool et al.,29 which is at the edge of our current energy range and cannot be properly quantified.
:13CH2O:CH3O signal branching ratio of 1:0.20:0.08. The signal of m/z 31 is dominated by the fragment of HPMF, which is consistent with the fact that it is the main fragment of the KHP along with m/z 64. This is also confirmed by the broad shape of the TOF peak which is correlated to dissociative ionization generating fragments with larger kinetic energy release distributions. The contribution from 13CH2O is also confirmed by the distinctive peak at 10.9 eV visible in the TPE spectrum shown in the SM. A contribution of the methoxy radical to the TPES cannot be ruled out, in accordance with the convolution presented in Fig. 7. The fragmentation of methanol could also contribute to the observed signal. This, however, corresponds to an appearance energy of around 11.5 eV based on Cool et al.,29 which is at the edge of our current energy range and cannot be properly quantified.

Quantification and kinetic modelling
The determined mole fractions at the sampling probe location are compared with simulation results in Fig. 8. While measurements were only performed at one position of the flame, one can observe the mole fractions of the major species (left) lie within the experimental uncertainty. Notably, the determined mole fraction of HPMF is in fair agreement with the predictions of the kinetic model, and further validates this experimental approach for the observation and quantification of organic hydroperoxides. One can observe higher discrepancies for ozone and water, for which the quantification was performed at a photon energy of 12.7 eV. This could be due to the use of DME as an internal standard, while other species than DME could contribute to the m/z = 46 signal at this photon energy. Methanol is underpredicted by about a factor of two, which is consistent with the performance of the kinetic model in previous work on DME cool flames by means of gas chromatographic analysis,17 and could be correlated with the underestimation of the mole fraction of the methyl radical by three orders of magnitude (see Table 3). The discrepancy between the experimental and modelled mole fractions of the methyl radical can also partly originate from the contribution of the fragmentation of heavier species to the TIY signal at the quantification photon energy. Further work on the methanol submodel will therefore be needed to improve the DME kinetic model. Finally, as visible from Table 3, the agreement is however not as satisfying in the case of HO2 and H2O2, which could suggest that losses could still take place, for example through reactivity in the capillary probe, and requires further experimental development. | ||
| Fig. 8 Simulated (lines) and measured mole fraction at the probe position in the investigated flame. | ||
| Species | Measured mole fraction | Simulated mole fraction |
|---|---|---|
| HPMF | 4 ± 2 × 10−4 | 7 × 10−4 |
| Methyl formate | 1.8 ± 0.9 × 10−3 | 5 × 10−3 |
| Ozone | 0.017 ± 0.008 | 0.009 |
| Dimethyl ether | 0.03 ± 0.02 | 0.02 |
| Hydrogen peroxide | 3 ± 2 × 10−4 | 2 × 10−3 |
| Hydroperoxyl | 7 ± 4 × 10−5 | 3 × 10−4 |
| Methanol | 6 ± 3 × 10−4 | 1 × 10−4 |
| Formaldehyde | 3 ± 2 × 103 | 2 × 10−3 |
| Water | 0.05 ± 0.03 | 0.03 |
| Methyl | 3 ± 2 × 10−5 | 1 × 10−8 |
Conclusions
This study provides the first experimental evidence of the presence of keto- or aldo-hydroperoxides within cool flames, by means of a novel coupling of a stagnation plate cool flame burner with the i2-PEPICO SAPHIRS instrument at the DESIRS beamline of synchrotron SOLEIL. Gases extracted upstream from a plate-stabilized DME/O2/O3/N2 cool flame by means of a capillary probe were expanded into the spectrometer, leading to the following conclusions: Unambiguous proof of the formation of hydroperoxymethylformate, the only expected aldohydroperoxide from DME low-temperature gas phase oxidation, has been obtained through the detection of its fragments at mass-to-charge ratios of 31 and 64, as previously reported in flow-reactor conditions in the literature. Through the analysis of the signal collected for m/z = 64, and in accordance with data reported by others, the existence of an ozone-dependent pathway yielding hydroperoxymethanol is suggested. Additional detection of the methyl, methoxy and hydroperoxyl radicals, as well as hydrogen peroxide was also possible. The quantification of most of these species is attempted here for the first time in a cool flame configuration. Kinetic modelling of the studied configuration shows good agreement on the major species, as well as hydroperoxymethyl formate, the aldohydroperoxide responsible for the initiation of the cool flame. The measured mole fractions for HO2 and H2O2 are however lower than those predicted by the kinetic model, suggesting that losses could take place during the sampling. In the case of methanol and the methyl radical, the opposite is true, suggesting that improvement in the model could be needed. Further work will aim at investigating more complex fuels, that yield several KHP isomers, as well as reaching better jet conditions in order to enable molecular beam conditions prone to quantitative measurement of radicals.Author contributions
Sébastien Batut: conceptualization, investigation, methodology. Laure Pillier: conceptualization, investigation, methodology, supervision, writing – review & editing. Jérémy Bourgalais: formal analysis, investigation, validation, writing – review & editing. Thomas Panaget: investigation, writing – review & editing. Pascal Demaux: investigation. Olivier Herbinet: investigation, writing – review & editing. Frédérique Battin-Leclerc: writing – review & editing. Gustavo A. Garcia: formal analysis, investigation, validation, writing – review & editing. Laurent Nahon: resources, writing – review & editing. Guillaume Vanhove: conceptualization, formal analysis, funding acquisition, investigation, methodology, supervision, validation, writing – original draft.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We acknowledge SOLEIL for provision of synchrotron radiation under project 20220879 and we are grateful to Jean-François Gil for his technical help around the SAPHIRS set-up. The authors thank the Région Hauts-de-France, the Ministère de l’Enseignement Supérieur et de la Recherche and the European Fund for Regional Economic Development for their financial support to the CPER ECRIN program. The CaPPA project (Chemical and Physical Properties of the Atmosphere) is funded by the French National Research Agency (ANR) through the PIA (Programme d’Investissement d’Avenir) under contract “ANR-11-LABX-0005-01” and by the Regional Council “Hauts-de-France” and the “European Funds for Regional Economic Development” (FEDER). Theoretical calculations in this work were performed using HPC resources from the EXPLOR centre hosted by the University of Lorraine (Project: 2021EXTXX2356).References
- H. Davy, VIII. Some New Experiments and Observations on the Combustion of Gaseous Mixtures, with an Account of a Method of Preserving a Continued Light in Mixtures of Inflammable Gases and Air without Flame, Philos. Trans. R. Soc. London, 1997, 107, 77–85, DOI:10.1098/rstl.1817.0009.
- A. Fish, The Cool Flames of Hydrocarbons, Angew. Chem., Int. Ed. Engl., 1968, 7(1), 45–60, DOI:10.1002/anie.196800451.
- M. Carlier, C. Corre, R. Minetti, J. F. Pauwels, M. Ribaucour and L. R. Sochet, Autoignition of Butane: A Burner and a Rapid Compression Machine Study, Symp. Int. Combust., 1991, 23(1), 1753–1758, DOI:10.1016/S0082-0784(06)80453-4.
- G. Vanhove, M.-A. Boumehdi, S. Shcherbanev, Y. Fenard, P. Desgroux and S. M. Starikovskaia, A Comparative Experimental Kinetic Study of Spontaneous and Plasma-Assisted Cool Flames in a Rapid Compression Machine, Proc. Combust. Inst., 2017, 36(3), 4137–4143, DOI:10.1016/j.proci.2016.09.007.
- C. Runnoo, B. Boust, M. Bellenoue and Q. Michalski, Characterization of Re-Ignition Events in Constant Volume Combustion, In AIAA SCITECH 2025 Forum; AIAA SciTech Forum; American Institute of Aeronautics and Astronautics, 2025 DOI:10.2514/6.2025-1180.
- S. H. Won, B. Jiang, P. Diévart, C. H. Sohn and Y. Ju, Self-Sustaining n-Heptane Cool Diffusion Flames Activated by Ozone, Proc. Combust. Inst., 2015, 35, 881–888 CrossRef CAS.
- Y. Ju, C. B. Reuter, O. R. Yehia, T. I. Farouk and S. H. Won, Dynamics of Cool Flames, Prog. Energy Combust. Sci., 2019, 75, 100787, DOI:10.1016/j.pecs.2019.100787.
- A. Fish, I. A. Read, W. S. Affleck and W. W. Haskell, The Controlling Role of Cool Flames in Two-Stage Ignition, Combust. Flame, 1969, 13(1), 39–49, DOI:10.1016/0010-2180(69)90026-1.
- Z. Wang, O. Herbinet, N. Hansen and F. Battin-Leclerc, Exploring Hydroperoxides in Combustion: History, Recent Advances and Perspectives, Prog. Energy Combust. Sci., 2019, 73, 132–181, DOI:10.1016/j.pecs.2019.02.003.
- K. A. Sahetchian, R. Rigny and S. Circan, Identification of the Hydroperoxide Formed by Isomerization Reactions during the Oxidation of n-Heptane in a Reactor and CFR Engine, Combust. Flame, 1991, 85(3), 511–514, DOI:10.1016/0010-2180(91)90153-3.
- N. Blin-Simiand, F. Jorand, K. Sahetchian, M. Brun, L. Kerhoas, C. Malosse and J. Einhorn, Hydroperoxides with Zero, One, Two or More Carbonyl Groups Formed during the Oxidation of n-Dodecane, Combust. Flame, 2001, 126(1–2), 1524–1532 CrossRef CAS.
- F. Battin-Leclerc, O. Herbinet, P.-A. Glaude, R. Fournet, Z. Zhou, L. Deng, H. Guo, M. Xie and F. Qi, Experimental Confirmation of the Low-Temperature Oxidation Scheme of Alkanes, Angew. Chem., Int. Ed., 2010, 49(18), 3169–3172, DOI:10.1002/anie.200906850.
- F. Battin-Leclerc, J. Bourgalais, Z. Gouid, O. Herbinet, G. Garcia, P. Arnoux, Z. Wang, L.-S. Tran, G. Vanhove, L. Nahon and M. Hochlaf, Chemistry Deriving from OOQOOH Radicals in Alkane Low-Temperature Oxidation: A First Combined Theoretical and Electron–Ion Coincidence Mass Spectrometry Study, Proc. Combust. Inst., 2021, 38, 309–319 CrossRef CAS.
- J. Bourgalais, H.-H. Carstensen, O. Herbinet, G. A. Garcia, P. Arnoux, L.-S. Tran, G. Vanhove, L. Nahon, M. Hochlaf and F. Battin-Leclerc, Product Identification in the Low-Temperature Oxidation of Cyclohexane Using a Jet-Stirred Reactor in Combination with SVUV-PEPICO Analysis and Theoretical Quantum Calculations, J. Phys. Chem. A, 2022, 126(34), 5784–5799, DOI:10.1021/acs.jpca.2c04490.
- J. Bourgalais, C. Smith Lewin, O. Herbinet, G. A. Garcia, P. Arnoux, L.-S. Tran, G. Vanhove, L. Nahon and F. Battin-Leclerc, Refining the Chain-Branching Process in the Low-Temperature Oxidation of 1-Hexene with Synchrotron-Based PEPICO Spectroscopy, Combust. Flame, 2023, 258, 113065, DOI:10.1016/j.combustflame.2023.113065.
- N. Belhadj, M. Lailliau, R. Benoit and P. Dagaut, Experimental and Kinetic Modeling Study of n-Hexane Oxidation. Detection of Complex Low-Temperature Products Using High-Resolution Mass Spectrometry, Combust. Flame, 2021, 233, 111581, DOI:10.1016/j.combustflame.2021.111581.
- T. Panaget, N. Mokrani, S. Batut, A. Lahccen, Y. Fenard, L. Pillier and G. Vanhove, Insight into the Ozone-Assisted Low-Temperature Combustion of Dimethyl Ether by Means of Stabilized Cool Flames, J. Phys. Chem. A, 2021, 125, 9167–9179 CrossRef CAS PubMed.
- T. Panaget, K. Potier, S. Batut, A. Lahccen, Y. Fenard, L. Pillier and G. Vanhove, How Ozone Affects the Product Distribution inside Cool Flames of Diethyl Ether, Proc. Combust. Inst., 2023, 39, 325–333 CrossRef.
- K. De Ras, T. Panaget, Y. Fenard, J. Aerssens, L. Pillier, J. W. Thybaut, G. Vanhove and K. M. Van Geem, An Experimental and Kinetic Modeling Study on the Low-Temperature Oxidation of Oxymethylene Ether-2 (OME-2) by Means of Stabilized Cool Flames, Combust. Flame, 2023, 253, 112792, DOI:10.1016/j.combustflame.2023.112792.
- M. Zhou, M. Lee, Y. Ju and Y. Suzuki, Spatial Distribution and Temporal Evolution of Wall-Stabilized DME/O2 Premixed Cool Flames, Combust. Flame, 2025, 271, 113814, DOI:10.1016/j.combustflame.2024.113814.
- T. Panaget, P. Bragança, B. Lecordier, A. Lahccen, C. Cuvier, S. Batut, Y. Fenard, G. Vanhove and L. Pillier, Deriving Cool Flame Propagation Speeds by Means of an Ozone-Seeded, Stagnation Plate Burner Configuration, Fuel, 2024, 362, 130766, DOI:10.1016/j.fuel.2023.130766.
- M. Hajilou, T. Ombrello, S. H. Won and E. Belmont, Experimental and Numerical Characterization of Freely Propagating Ozone-Activated Dimethyl Ether Cool Flames, Combust. Flame, 2017, 176, 326–333, DOI:10.1016/j.combustflame.2016.11.005.
- C. B. Reuter, S. H. Won and Y. Ju, Experimental Study of the Dynamics and Structure of Self-Sustaining Premixed Cool Flames Using a Counterflow Burner, Combust. Flame, 2016, 166, 125–132, DOI:10.1016/j.combustflame.2016.01.008.
- M. Lee, Y. Fan, C. B. Reuter, Y. Ju and Y. Suzuki, DME/Oxygen Wall-Stabilized Premixed Cool Flame, Proc. Combust. Inst., 2019, 37(2), 1749–1756, DOI:10.1016/j.proci.2018.05.059.
- U. Lee, J. Han, M. Wang, J. Ward, E. Hicks, D. Goodwin, R. Boudreaux, P. Hanarp, H. Salsing, P. Desai, E. Varenne, P. Klintbom, W. Willems, S. L. Winkler, H. Maas, R. De Kleine, J. Hansen, T. Shim and E. Furusjö, Well-to-Wheels Emissions of Greenhouse Gases and Air Pollutants of Dimethyl Ether from Natural Gas and Renewable Feedstocks in Comparison with Petroleum Gasoline and Diesel in the United States and Europe, SAE Int. J. Fuels Lubr., 2016, 9(3), 546–557 CrossRef CAS.
- Y. Fenard and G. Vanhove, A Mini-Review on the Advances in the Kinetic Understanding of the Combustion of Linear and Cyclic Oxymethylene Ethers, Energy Fuels, 2021, 35(18), 14325–14342, DOI:10.1021/acs.energyfuels.1c01924.
- K. Moshammer, A. W. Jasper, D. M. Popolan-Vaida, Z. Wang, V. S. Bhavani Shankar, L. Ruwe, C. A. Taatjes, P. Dagaut and N. Hansen, Quantification of the Keto-Hydroperoxide (HOOCH2OCHO) and Other Elusive Intermediates during Low-Temperature Oxidation of Dimethyl Ether, J. Phys. Chem. A, 2016, 120(40), 7890–7901, DOI:10.1021/acs.jpca.6b06634.
- K. Moshammer, A. W. Jasper, D. M. Popolan-Vaida, A. Lucassen, P. Diévart, H. Selim, A. J. Eskola, C. A. Taatjes, S. R. Leone, S. M. Sarathy, Y. Ju, P. Dagaut, K. Kohse-Höinghaus and N. Hansen, Detection and Identification of the Keto-Hydroperoxide (HOOCH2OCHO) and Other Intermediates during Low-Temperature Oxidation of Dimethyl Ether, J. Phys. Chem. A, 2015, 119(28), 7361–7374, DOI:10.1021/acs.jpca.5b00101.
- D. E. Couch, C. R. Mulvihill, R. Sivaramakrishnan, K. Au, C. A. Taatjes and L. Sheps, Quantification of Key Peroxy and Hydroperoxide Intermediates in the Low-Temperature Oxidation of Dimethyl Ether, J. Phys. Chem. A, 2022, 126(50), 9497–9509, DOI:10.1021/acs.jpca.2c06959.
- H. Liao, S. Kang, N. Hansen, F. Zhang and B. Yang, Influence of Ozone Addition on the Low-Temperature Oxidation of Dimethyl Ether in a Jet-Stirred Reactor, Combust. Flame, 2020, 214, 277–286, DOI:10.1016/j.combustflame.2019.12.036.
- L. Nahon, N. de Oliveira, G. A. Garcia, J.-F. Gil, B. Pilette, O. Marcouillé, B. Lagarde and F. Polack, DESIRS: A State-of-the-Art VUV Beamline Featuring High Resolution and Variable Polarization for Spectroscopy and Dichroism at SOLEIL, J. Synchrotron Radiat., 2012, 19(4), 508–520, DOI:10.1107/S0909049512010588.
- X. Tang, G. A. Garcia, J.-F. Gil and L. Nahon, Vacuum Upgrade and Enhanced Performances of the Double Imaging Electron/Ion Coincidence End-Station at the Vacuum Ultraviolet Beamline DESIRS, Rev. Sci. Instrum., 2015, 86(12), 123108, DOI:10.1063/1.4937624.
- G. A. Garcia, B. K. Cunha de Miranda, M. Tia, S. Daly and L. Nahon, DELICIOUS III: A Multipurpose Double Imaging Particle Coincidence Spectrometer for Gas Phase Vacuum Ultraviolet Photodynamics Studies, Rev. Sci. Instrum., 2013, 84(5), 053112, DOI:10.1063/1.4807751.
- N. Lamoureux and P. Desgroux, In Situ Laser-Induced Fluorescence and Ex Situ Cavity Ring-Down Spectroscopy Applied to NO Measurement in Flames: Microprobe Perturbation and Absolute Quantification, Energy Fuels, 2021, 35(9), 7107–7120, DOI:10.1021/acs.energyfuels.0c03806.
- B. K. Cunha de Miranda, C. Alcaraz, M. Elhanine, B. Noller, P. Hemberger, I. Fischer, G. A. Garcia, H. Soldi-Lose, B. Gans, L. A. Vieira Mendes, S. Boyé-Péronne, S. Douin, J. Zabka and P. Botschwina, Threshold Photoelectron Spectroscopy of the Methyl Radical Isotopomers, CH3, CH2D, CHD2 and CD3: Synergy between VUV Synchrotron Radiation Experiments and Explicitly Correlated Coupled Cluster Calculations, J. Phys. Chem. A, 2010, 114(14), 4818–4830, DOI:10.1021/jp909422q.
- G. A. Garcia, L. Nahon and I. Powis, Two-Dimensional Charged Particle Image Inversion Using a Polar Basis Function Expansion, Rev. Sci. Instrum., 2004, 75(11), 4989–4996, DOI:10.1063/1.1807578.
- J. C. Poully, J. P. Schermann, N. Nieuwjaer, F. Lecomte, G. Grégoire, C. Desfrançois, G. A. Garcia, L. Nahon, D. Nandi, L. Poisson and M. Hochlaf, Photoionization of 2-Pyridone and 2-Hydroxypyridine, Phys. Chem. Chem. Phys., 2010, 12(14), 3566–3572, 10.1039/B923630A.
- M. Briant, L. Poisson, M. Hochlaf, P. de Pujo, M.-A. Gaveau and B. Soep, Ar2 Photoelectron Spectroscopy Mediated by Autoionizing States, Phys. Rev. Lett., 2012, 109(19), 193401, DOI:10.1103/PhysRevLett.109.193401.
- B. Gans, L. A. V. Mendes, S. Boyé-Péronne, S. Douin, G. Garcia, H. Soldi-Lose, B. K. C. De Miranda, C. Alcaraz, N. Carrasco, P. Pernot and D. Gauyacq, Determination of the Absolute Photoionization Cross Sections of CH3 and I Produced from a Pyrolysis Source, by Combined Synchrotron and Vacuum Ultraviolet Laser Studies, J. Phys. Chem. A, 2010, 114(9), 3237–3246, DOI:10.1021/jp909414d.
- P. M. Dehmer and D. M. P. Holland, Photoionization of Rotationally Cooled H2O and D2O in the Region 650–990 Å, J. Chem. Phys., 1991, 94(5), 3302–3314, DOI:10.1063/1.459805.
- L. G. Dodson, L. Shen, J. D. Savee, N. C. Eddingsaas, O. Welz, C. A. Taatjes, D. L. Osborn, S. P. Sander and M. Okumura, VUV Photoionization Cross Sections of HO 2, H 2 O 2, and H 2 CO, J. Phys. Chem. A, 2015, 119(8), 1279–1291, DOI:10.1021/jp508942a.
- B. Dong, Z. Hu, Q. Xu, B. Liu, Q. Zhu, J. Guan, C. Liu, Y. Pan, L. Hu, J. Fang and Z. Wang, Improving Quantification of Hydrogen Peroxide by Synchrotron Vacuum Ultraviolet Photoionization Mass Spectrometry, Combust. Flame, 2022, 242, 112214, DOI:10.1016/j.combustflame.2022.112214.
- R. Botter, J. M. Pechine and H. M. Rosenstock, Photoionization of Dimethyl Ether and Diethyl Ether, Int. J. Mass Spectrom. Ion Phys., 1977, 25(1), 7–25, DOI:10.1016/0020-7381(77)80100-9.
- J. Berkowitz, Absolute Partial Photoionization Cross Sections of Ozone, Int. J. Mass Spectrom., 2008, 271(1), 8–14, DOI:10.1016/j.ijms.2007.11.005.
- D. G. Goodwin, H. K. Moffat and R. L. Speth, Cantera: An Object-Oriented Software Toolkit for Chemical Kinetics, Thermodynamics, and Transport Processes, 2018, https://www.cantera.org Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120(1), 215–241, DOI:10.1007/s00214-007-0310-x.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, Electron Affinities of the First-row Atoms Revisited. Systematic Basis Sets and Wave Functions, J. Chem. Phys., 1992, 96(9), 6796–6806, DOI:10.1063/1.462569.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, A Complete Basis Set Model Chemistry. VI. Use of Density Functional Geometries and Frequencies, J. Chem. Phys., 1999, 110(6), 2822–2827, DOI:10.1063/1.477924.
- J. A. Montgomery, Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, A Complete Basis Set Model Chemistry. VII. Use of the Minimum Population Localization Method, J. Chem. Phys., 2000, 112(15), 6532–6542, DOI:10.1063/1.481224.
- J. Bloino, A. Baiardi and M. Biczysko, Aiming at an Accurate Prediction of Vibrational and Electronic Spectra for Medium-to-Large Molecules: An Overview, Int. J. Quantum Chem., 2016, 116(21), 1543–1574, DOI:10.1002/qua.25188.
- X. Tang, X. Lin, G. A. Garcia, J.-C. Loison, C. Fittschen, A. Röder, D. Schleier, X. Gu, W. Zhang and L. Nahon, Threshold Photoelectron Spectroscopy of the HO2 Radical, J. Chem. Phys., 2020, 153(12), 124306, DOI:10.1063/5.0022410.
- L. Schio, M. Alagia, A. A. Dias, S. Falcinelli, V. Zhaunerchyk, E. P. F. Lee, D. K. W. Mok, J. M. Dyke and S. Stranges, A Study of H2O2 with Threshold Photoelectron Spectroscopy (TPES) and Electronic Structure Calculations: Redetermination of the First Adiabatic Ionization Energy (AIE), J. Phys. Chem. A, 2016, 120, 5220–5229 CrossRef CAS PubMed.
- X. Tang, X. Lin, G. A. Garcia, J.-C. Loison, C. Fittschen, X. Gu, W. Zhang and L. Nahon, Threshold Photoelectron Spectroscopy of the Methoxy Radical, J. Chem. Phys., 2020, 153(3), 031101, DOI:10.1063/5.0016146.
- T. A. Cool, J. Wang, K. Nakajima, C. A. Taatjes and A. Mcllroy, Photoionization Cross Sections for Reaction Intermediates in Hydrocarbon Combustion, Int. J. Mass Spectrom., 2005, 247(1), 18–27, DOI:10.1016/j.ijms.2005.08.018.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00887e |
| This journal is © the Owner Societies 2025 |