
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Symmetry-breaking photoinduced charge transfer state in a near-IR absorbing meso-linked BODIPY dimer†
Fariyad
Ali
 a,
Elizabeth
Gehrmann
a,
Tianyi
Zhang
a,
Qasim Qayyum
Kashif
a,
Robbyn K.
Anand
a,
David Lee
Phillips
*ab and
Arthur H.
Winter
*a
a,
Elizabeth
Gehrmann
a,
Tianyi
Zhang
a,
Qasim Qayyum
Kashif
a,
Robbyn K.
Anand
a,
David Lee
Phillips
*ab and
Arthur H.
Winter
*a
aDepartment of Chemistry, Iowa State University, 1608 Gilman Hall, Ames, Iowa 50010, USA. E-mail: phillips@hku.hk; winter@iastate.edu
bDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China
First published on 29th April 2025
Abstract
The synthesis and characterization of a conformationally restrained near-IR absorbing homoleptic meso-linked BODIPY dye is reported. The photophysical properties have been investigated using steady-state and time-resolved femtosecond transient absorption spectroscopy. A combination of spectroscopies (steady-state absorption/emission, ultrafast transient absorption spectroscopy measurements), singlet oxygen generation quantum yields, and computational studies were conducted to evaluate the nature of the excited state of the dimer 3D compared to its monomer 3M as a control. The results are consistent with a symmetry-breaking intramolecular charge transfer (ICT) state for 3D being formed in polar solvents, but the formation of the ICT states competes with other photophysical channels (ISC, fluorescence) even in the most polar solvents tested, so the formation efficiency is less than the related “green” light absorbing BODIPY dimer 2 previously reported. The ICT state formation is attributed to the direct meso coupling of the BODIPY dimer, which stymies nonradiative deactivation through both a conformational restraint of the individual BODIPY subunits and a blocking the rotation of the bond connecting the two BODIPY multimers, preventing subunit electronic communication in the excited state and subsequent charge recombination. Consistent with some fraction of intramolecular charge transfer (ICT) states being formed upon photoexcitation (∼1 ps in MeOH and 500 fs in DMF), the lifetime, emission intensity, and absorbance are attenuated in polar solvents for the dimer than the monomer. Further, for 3D diminished ISC is observed in more polar solvents, which is attributed to an increase in the fraction of excited states undergoing the ICT pathway in polar solvents, while for the monomer 3M there is no effect of solvent polarity on ISC. Overall, this study provides insights into the delicate balance between different photophysical channels in symmetric dimers. The exceptional optical properties of this dimer make this chromophore a promising scaffold for initiating charge separation using wavelengths in the challenging-to-access far-red/near-IR region of the optical spectrum, while suggesting further improvements could be made to increase the fraction of excited states undergoing ICT.
Introduction
Photo-induced charge separation (CS) converts light energy into electrochemical potential, a process of significance across chemistry, physics, biology, and the emerging realm of molecular electronics and energy storage.1–8 Within molecular systems comprising identical and symmetric subunits, often termed multimers, CS between chromophores occurs through photoexcitation via excited-state symmetry breaking (SB).9,10 SB-CS frequently manifests within multimeric systems, with a notable illustration found in the photosynthetic reaction center.11–15In its essence, SB-CS is an excited-state mechanism wherein a symmetrical excited state undergoes electron transfer between two electronically degenerate states, leading to the formation of an asymmetric CS state.16 The pivotal factors influencing the rate of SB-CS include the electronic coupling between the chromophores and the surrounding solvent environment.17,18 Under scenarios characterized by weak coupling, electronic excitation localizes within one of the monomers within the multimer. This situation can theoretically be defined as symmetry-broken local-excited (LE) states, ultimately giving rise to a CS state.19 In contrast, strong coupling results in electronic excitation becoming delocalized. Consequently, Franck–Condon states transform into excitonic states, resulting in partial CS that culminates in a charge-transfer (CT) state. Notably, a polar solvent environment stabilizes the asymmetric SB-CS state, a phenomenon particularly relevant in biological systems.14
While such SB-CS states offer the potential as light-harvesting initiators, the vast majority of SB-CS forming compounds absorb light in the ultraviolet region of the spectrum, and in these cases the CT state is frequently non-emissive. However, Thompson and co-workers15 reported that the donor/accepter-based BODIPY dimers 1 and 2 (Fig. 1) feature solvent-dependent intramolecular charge transfer (ICT) after photoexcitation to form emissive ICT states. Photo-induced SB in the excited states plays a key role in deciding the relaxation dynamics of the excited BODIPY conformers 1 and 2. The greater conformational flexibility of 1 compared to 2 promotes nonradiative relaxation pathways in the system and charge recombination.15,20 The directly meso-coupled 2 offered a longer-lived emissive CT state compared to 1, attributed to stymied electron recombination by the hindered bond rotation. Electronic communication between the individual meso-substituted BODIPY units is invoked as a major pathway for nonradiative deactivation.13,15,19,21 The exciting feature of the BODIPY systems 1 and 2 developed by Thompson and coworkers are that these dimers form an emissive CT state in a chromophore that absorbs intensely in the visible, which provides a facile way to initially separate charge carriers upon visible light irradiation with potential applications in photovoltaic devices. Similar to Thompson's work, we find evidence for a ICT state in the dimer in highly polar solvents, although the fraction of ICT states per excitation appears to be less than Thompson's structures due to competition with other photophysical channels. Nevertheless, we find that the emissive nature of the ICT state of 3D, its long lifetime (3.9–5.4 ns depending on solvent), and the exceptional molar extinction coefficient (up to 3.2 × 105 M−1 cm−1 in toluene) opens up the potential for derivatives of this structure to be exploited as a platform for light harvesting materials in photovoltaic and electronic devices in the NIR region.15
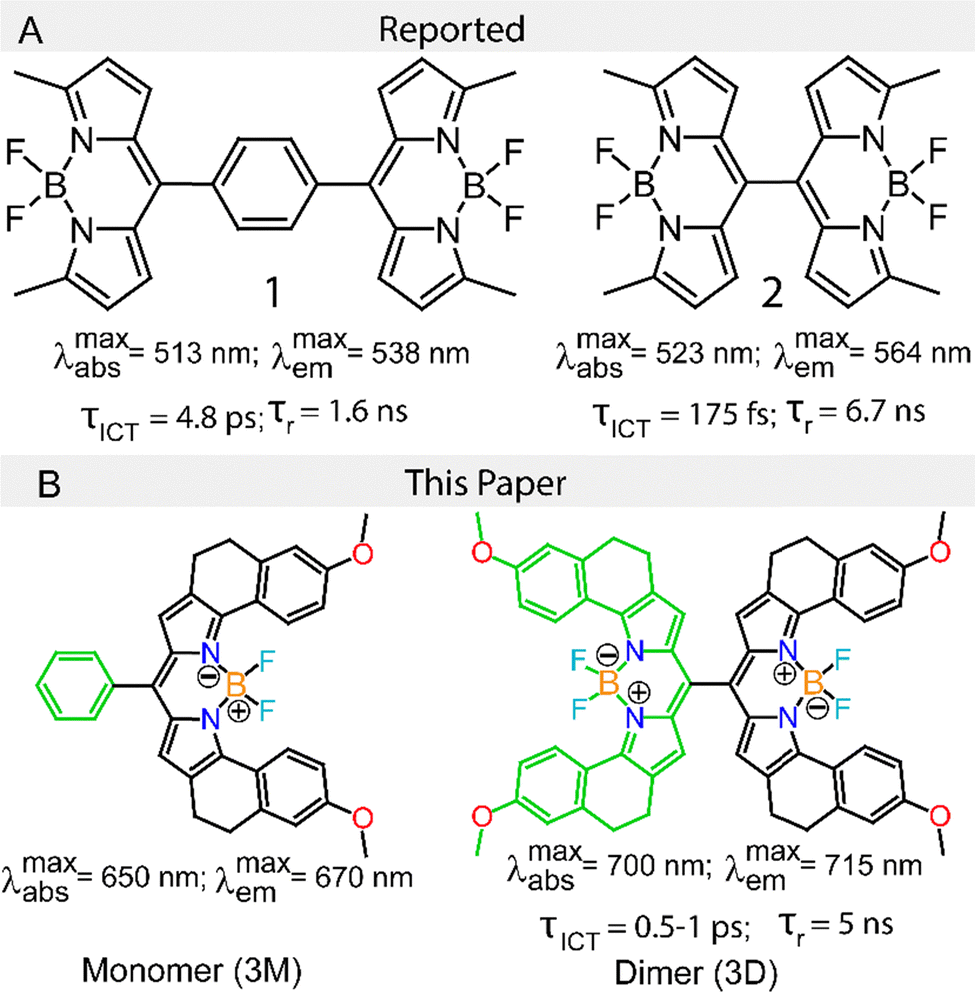 | ||
| Fig. 1 (A) Molecular structures of BODIPY dimers 1 and 2 as reported by Thompson and co-workers. Dimer 1 exhibits an ICT lifetime τICT of 4.8 ps in acetonitrile (ACN) and 18 ps in dichloromethane (DCM), with no observable ICT in toluene. Dimer 1 relaxes to the ground state with a time constant of 34 ps in ACN and 1.6 ns in DCM. In contrast, dimer 2 shows τICT ≤ 175 fs in ACN, 490 fs in DCM, and 4.5 ps in toluene. Compared to dimer 1, dimer 2 has a longer relaxation time constant of 650 ps in ACN and 6.7 ns in DCM. The free rotation of both monomers around the single bond in dimer 1 is responsible for the fast relaxation of the CT state, leading to nonradiative relaxation. In dimer 2, restricted rotation promotes rigidity in the system, resulting in a longer relaxation time for the CT state. (B) Molecular structures of the BODIPY monomer (3M) and dimer (3D). | ||
While the BODIPY dimers of Thompson and coworkers absorb green light, we anticipated that dimers with extended conjugation could enable a similar ICT state to be formed using longer wavelengths of light in the challenging to access far-red/near-IR region of the optical spectrum. With this motivation, a meso-substituted BODIPY monomer 3M and dimer 3D (Fig. 2) were synthesized to explore the role of the ICT process in solvents with varying polarity. A critical design feature of the dimer is that it is directly linked without a spacer in the hope that, similar to 2, the dimer would adopt a near-orthogonal geometry and that twisting about the connecting C–C bond in the ground and excited states would be sterically repressed in order to promote a long-lived CT state. This idea is given support by the results of computations described later. Furthermore, the individual BODIPY subunits are themselves conformationally restrained by bridging rings to prevent loose-bolt internal conversion effects of the individual BODIPY dyes and extend the excited state lifetime.
 | ||
| Fig. 2 Steady-state absorption and fluorescence emission spectra of BODIPY 3M (A) and 3D (B) in different solvents. The inset shows the molecular structures of 3M and 3D. The concentration was used 20 μM. | ||
Here, 3M and 3D were synthesized as new compounds (Fig. S1–S3, ESI†). The characteristic properties for 3M and 3D have been analyzed using steady state and time resolved spectroscopic techniques. The present study builds upon the vast body of literature that exist on the charge transfer dynamics with the substitution and solvents.14,15,17,19–23 Similar to Thompson's work, a solvent dependent ICT state is formed in the dimer. We find that the emissive nature of the ICT state of 3D, its long lifetime (3.9–5.4 ns depending on solvent), and the exceptional molar extinction coefficient (up to 3.2 × 105 M−1 cm−1 in toluene) opens up the potential for this structure to be exploited as a light harvesting material in photovoltaic and electronic devices in the NIR region.
Results and discussion
Steady-state and time resolved fluorescence
Steady-state absorption and emission spectra of BODIPY conjugates monomer (3M) and dimer (3D) were recorded at room temperature in solvents of varying polarity (Fig. 2). 3M shows absorption bands at 600 nm and 650 nm (Fig. 2(A) and Fig. S4A, ESI†) while 3D shows a sharp absorption band at ∼700 nm along with a shoulder band 640 nm (Fig. 2(B) and Fig. S4C, ESI†). In the emission spectra, 3M shows a prominent peak at 665 nm with a shoulder band at ∼725 nm after excitation (λex) at 600 nm (Fig. 2(A) and Fig. S4B, ESI†) while 3D (λex = 640 nm) shows emission maxima at ∼715 nm with two shoulder bands at ∼640 nm and 780 nm (Fig. 2(B) and Fig. S4D, ESI†). The absorption maxima (λmaxabs) and emission maxima (λmaxem) of both the conjugates are shown in Table 1. A small Stokes shift is seen for both 3M and 3D with varying the polarity of the solvents. The trend in the shift of λmaxabs and λmaxem with the solvent polarity and the absorption features are very similar for both the conjugates which is well corroborated with the earlier report, although smaller in magnitude.15 The λmaxabs for 3D is ∼50 nm redshifted compare to that of 3M. This is consistent with a prior report from our group where substitution leads to a redshift the absorption maxima when the conjugation has been extended.24–26 This is not the usual case for BODIPY systems where typically there is no significant shift in the wavelength for meso-substitution.14,22 The fluorescence quantum yield (ϕf) has been recorded in the solvents for both 3M and 3D (Table 1). The lower ϕf for 3D in polar solvents compared to 3M is consistent with the formation of a less emissive charge transfer state.15 The observation of small Stokes shifts and similar ϕf for 3M indicate a similar dipole in the excited state, while the longer emission wavelength for 3D in polar solvents suggests a more polar excited state for 3D.27| Solvent | E T(30) | Monomer (3M) | Dimer (3D) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λ maxabs (nm) | λ maxem (nm) | ε (M−1 cm−1) × 105 | ϕ f | τ (ns) | λ maxabs (nm) | λ maxem (nm) | ε (M−1 cm−1) × 105 | ϕ f | τ (ns) | ||
| Cyclohexane | 30.9 | 648 | 662 | 2.13 | 0.39 | 6.4 | 697 | 705 | 1.9 | 0.24 | 5.2 |
| Toluene | 33.9 | 656 | 668 | 1.80 | 0.48 | 5.5 | 707 | 718 | 3.2 | 0.23 | 5.1 |
| DCM | 45.6 | 652 | 669 | 1.94 | 0.41 | 6.4 | 705 | 715 | 1.5 | 0.22 | 5.4 |
| CAN | 55.4 | 653 | 672 | 1.09 | 0.43 | 6.3 | 699 | 716 | 2.4 | 0.22 | 4.9 |
| MeOH | 55.4 | 645 | 662 | 1.94 | 0.39 | 5.9 | 697 | 711 | 1.5 | 0.17 | 3.9 |
| DMF | 43.3 | 646 | 666 | 2.33 | 0.44 | 5.6 | 708 | 724 | 2.3 | 0.19 | 4.1 |
Time-resolved fluorescence decays for 3M and 3D have been recorded in different solvents (Fig. 3). A single exponential decay is observed in all of the solvents examined and the decay is faster with increasing solvent polarity. The extracted fluorescence lifetimes from the decays are shown in Table 1. The negligible change in the fluorescence lifetime for 3M and a significant change and faster decay was observed for 3D. The observation of the redshift in the steady-state and the fast decay kinetics from the time-resolved fluorescence decay data can be the evidence of the formation of the charge transfer state.15 However, the magnitude of the red-shift and the lifetime change is less than the systems reported by Thompson, suggesting that potentially a smaller fraction of excited 3D molecules form the ICT state even in the most polar solvents we tested.
 | ||
| Fig. 3 Time-resolved fluorescence decay of 3M (A) and 3D (B) in different solvents with varying polarity. λex = 360 nm and λem = λmaxem. | ||
Spectroelectrochemistry has been a successful technique recently used to confirm the presence of a CT State in BODIPY dimer systems for showing the double reduction potential which is not always true for the system that has a very small energy gap between the S1 and CT state.10 Both 3M and 3D have been investigated (Fig. S5, ESI†) and observed a single reversible reduction potential for 3M and 3D. 3D shows a little more negative reduction potential than 3M, which suggests the presence of a CT state but delocalized with the S1 state.10
Singlet oxygen generation
ISC was investigated using trapping experiments. The singlet oxygen trap, DPBF (1,3-diphenylisobenzofuran), was used to monitor the generation of singlet oxygen (1O2) using 3M and 3D as a photosensitizer (PS) (Fig. 4).10,28–31 The DPBF absorption was monitored at 412 nm as a function of irradiation time (LED centered at 625 nm) containing 50 μM of DPBF with 20 μM of 3M and 3D in ACN. The absorption band of DBPF almost disappears within 30 seconds in the presence of 3M (Fig. 4(A)) but the absorption decay is three times slower in the presence of 3D (Fig. 4(B)). This can be clearly seen when the decay of the DPBF absorption has been compared in the absence and presence of the PSs (Fig. 4(C)). A control experiment monitoring the absorption of DPBF without any PSs showed a negligible change in the absorbance of DPBF (Fig. 4(C), black circles). The singlet oxygen quantum yield (ϕΔ) has been calculated using methylene blue (MB, ϕΔ = 0.6) as the standard reference for both PSs (see the ESI† for methods) because the ϕΔ is related to the triplet state quantum yield of the compound.10,31 The ϕΔ for 3M was 1.4% and for 3D it was 0.5%. The ϕΔ for 3D is almost three times less compared to 3M which is a similar trend that is observed for the decay of the DPBF absorption for 3M and 3D (Fig. 4(A) and (B)). | ||
| Fig. 4 Absorption spectra of DPBF with 3M (A), 3D (B). (C) Change in the absorption of DPBF (black circle) without photosensitizer. Green and red circles represent the change in the absorption of DPBF in the presence of 3M and 3D photosensitizer, respectively. (D, E) Schematic representation for excited electron relaxation in the presence of 3M (D), 3D (E), of both the PSs. The concentration of DPBF was 50 μM, while 20 μM was used for both 3M and 3D photosensitizers (PSs). (D) For 3M, a fast component (70–174 ps) corresponds to vibrational cooling (i), while a 5 ns component represents the fluorescence lifetime of the S1 state (ii). Additionally, a long-lived component (>8 ns), which exceeds the fs-TA instrument time limit, is assigned to the triplet state lifetime, varying with solvent polarity. (E) Similar to 3M, 3D exhibits vibrational relaxation (53–94 ps) (i) and an S1 state fluorescence lifetime of approximately 5 ns (ii). In both toluene and ACN, no CT state was observed (iv); however, intersystem crossing (ISC) occurs with time constants of 378 ps in toluene and 570 ps in ACN (iii). In contrast, for MeOH and DMF, the CT state is created with a lifetime of 0.5–1 ps, followed by charge recombination (CR) occurring within ∼4–5 ns. A long component (>8 ns) is assigned to the triplet state lifetime. | ||
The 1O2 capability of both PSs was evaluated in the less polar solvent toluene (Fig. S6A, ESI†), showing a similar trend as in ACN (Fig. 4(C)). For 3M, negligible differences in the DPBF absorption decay were observed between ACN and toluene (Fig. S6B, ESI†), but 3D exhibited a significant difference (Fig. S6C, ESI†), indicating that 3M produces more triplet states. 3M undergoes vibrational relaxation (i), S1 state relaxation to the ground state (ii), and ISC (iii) to populate the triplet state (Fig. 4(D)). In contrast, 3D involves charge transfer (CT) (iv), creating an additional relaxation pathway. Consequently, excited electrons in 3D have to compete between relaxing to the CT state or to the triplet state via ISC (Fig. 3(E)), while in 3M, the electrons either undergo ISC or relax to the ground state (Fig. 4(D)). This explains why 3D shows less ISC and triplet state formation, serving as evidence for its CT state.
Varying solvent polarity was also explored for 3D to assess its impact on the CT state (Fig. S6C, ESI†). A faster DPBF absorption decay was observed in toluene compared to ACN, indicating a higher ISC yield in the less polar solvent.28 This is due to two possible relaxation pathways (CT and ISC). Polar solvents favor CT (iv), reducing ISC (iii), unlike 3M. Thus, 3D shows a faster DPBF absorption decay in less polar solvents. The distinct behaviour of 3D compared to 3M and their different 1O2 rates in solvents with varying polarity confirm that ISC occurs from the S1 state, not from the CT state.
Femtosecond transient absorption (fs-TA)
fs-TA spectroscopy was used to investigate the ultrafast dynamics and photoinduced ICT dynamics in the BODIPY systems. Both 3M and 3D were excited using λpump = 600 nm and 640 nm, respectively. The fs-TA spectrum was recorded from 0 to 7 ns delay between the pump and probe pulses for 3M (Fig. S7A–C, ESI†) and 3D (Fig. 5(A), (B) and Fig. S8, ESI†) systems in different solvents. The TA features from 400–600 nm for both of the systems is assigned to the excited state absorption (ESA) which is broad for 3M but separated into two features for 3D. A sharp and prominent negative absorption is observed at 650 nm for 3M (Fig. S7A–C, ESI†) and 710 nm for 3D (Fig. 5(A), (B) and Fig. S8, ESI†), and is assigned to ground state bleach (GSB) because the negative absorption matches with the ground state absorption spectra (Fig. 1(A) and (B)).16,32–34 The fs-TA spectrum for 3D has been investigated at the initial delays to see the effect of polarity on the CT state (Fig. 5(A) and Fig. S8A, C, E, ESI†). Apart from that, for 3D, a very distinct and red shifted negative band, not observed for 3M, was observed which increases in intensity with the delay time. The occurrence possibility and dynamics of this feature is discussed later using global analysis vide and infra. Time-resolved decay kinetics have been recorded at ESA for 3M (Fig. S9, ESI,†λprobe = 445 nm), 3D (Fig. 5(C), λprobe = 454 nm) in the different solvents with varying polarity.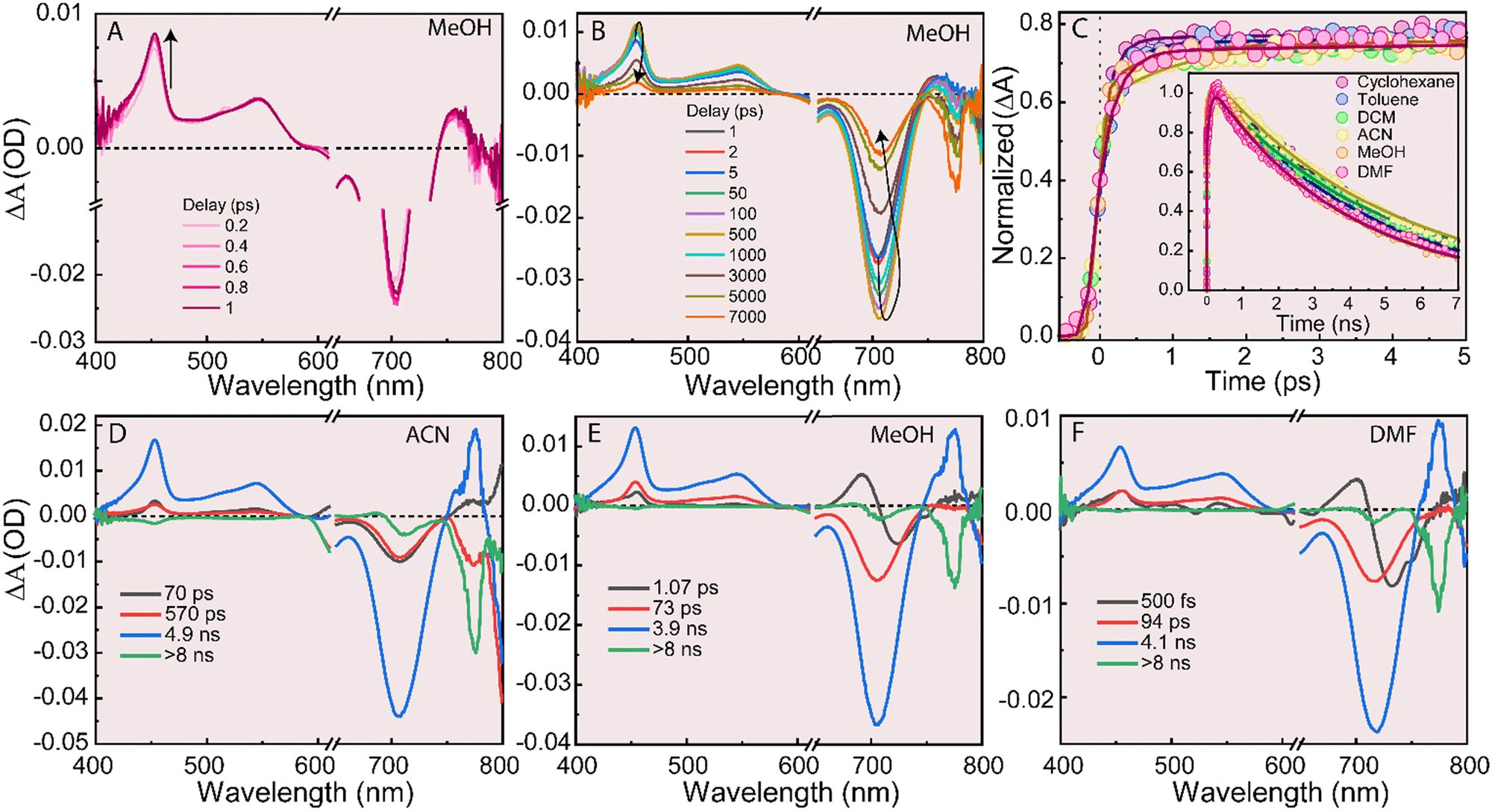 | ||
| Fig. 5 (A), (B) fs-TA spectrum of 3D in MeOH with delay (0.2–1 ps, A) and (1 ps–7 ns, B). (C) Time-resolved decay kinetics of 3D at λprobe = 454 nm with delay (0–7 ns) time in the different solvents with varying the polarity. (D)–(F) Decay associated difference spectra (DADS) of 3D in ACN (D), MeOH (E) and DMF (F) using global analysis. For all the solvents λpump = 640 nm has been used to excite the 3D. | ||
As a contro, the lifetime was extracted from the decay at ESA for 3M. The growth times changed from 60–120 ps (Table S1, ESI†), varied in the different solvents along with a long decay component ∼5 ns (Table S1, ESI†). The 5 ns component is similar to the fluorescence lifetime obtained from the TCSPC (Fig. 3(A) and Table 1) and assigned as the lifetime of the S1 state but the growth time (60–120 ps) has been assigned to the vibrational cooling based on some previous reports.35–41 The observation of the two lifetime components for 3M confirms that the excited species relaxes to the ground state without passing through a charge transfer state. The GSB is not completely recovered within the available delay range (∼7 ns), iindicating the possibility of a long-lived species (triplet state) which is confirmed by observing singlet oxygen generation (discussed previously, Fig. 4(A) and (C)).
In contrast to the simple fits for 3M, 3D decay kinetics have been fitted with a triexponential fitting at λprobe = 454 nm. Two growth components (the first vary from 0.2–1 ps and the second vary from 61–158 ps, Table 2) and one decay component has been observed in the solvents examined (Fig. 5(C)). The long growth component is similar to the observed growth component for 3M and assigned for the vibrational relaxation/cooling. The long decay component (∼5 ns) is similar to the fluorescence lifetime observed using TCSPC and assigned the lifetime of S1 state.15 The very fast component assigned to the formation of ICT states which are faster compared to the earlier reports based on the meso-linked BODIPY dimers.13,15,19 The credibility of assigning the fast component to form the CT state has been analyzed using the global analysis of both 3M and 3D in different solvents which is discussed further.
| Solvent | λ probe = 454 nm | τ CT (ps) | τ ISC = 1/kISC (ps) | |||||
|---|---|---|---|---|---|---|---|---|
| τ 1 (fs) | a 1 | τ 2 (fs) | a 2 | τ 3 (fs) | a 3 | |||
| m: not measured, h: not observed | ||||||||
| Cyclohexane | <0.2 | −0.52 | 101 | −0.48 | ≤5 | 1 | m | m |
| Toluene | 0.23 | −0.65 | 89 | −0.35 | ≤5 | 1 | h | 378 |
| DCM | 0.25 | −0.46 | 61 | −0.54 | ≤5 | 1 | m | m |
| ACN | 0.97 | −0.3 | 158 | −0.7 | ≤5 | 1 | h | 570 |
| MeOH | 0.46 | −0.26 | 69 | −0.74 | ≤5 | 1 | 1.07 | h |
| DMF | 0.23 | −0.6 | 100 | −0.4 | ≤5 | 1 | 0.5 | h |
From the fs-TA spectra and kinetics, 3M shows only two components (growth and decay) and a long-lived species. 3M has been analyzed using global analysis (see ESI† for more details). The global spectra of 3M are almost the same with varying the polarity of the solvents (Fig. S10, ESI†). The first component changing (70–174 ps) with changing the solvents and is similar to the growth component observed by fitting the fs-TA kinetics (Fig. S7, ESI†) and again is assigned as the vibrational cooling time and the long component (∼5 ns) is similar to the fluorescence lifetime of the S1 state (Fig. 4(D)).42 The corresponding spectral features of both of the components are very similar to each other and confirms the same electronic state which is populated through the ps component and decays with the ns component. But for 3D, the global analysis has been done by considering the presence of a CT state.
The global analysis of 3D shows the presence of the three components along with considering a long component (Fig. 5(D)–(F) and Fig. S11, ESI†). For the less polar solvents (Toluene and ACN), the shortest component (53 ps for toluene and 70 ps for ACN) and the long component (∼5 ns) have same spectral features and are present over the spectra range (Fig. 4(E), 5(D) and Fig. S11, ESI†). These two components are similar to the global components present in the monomer and can be assigned similarly as vibrational cooling or relaxed S1 state and the fluorescence lifetime of the S1 state respectively.42 These two components are present in more polar solvents (73 ps for MeOH and 94 ps for DMF) as well (Fig. 4(E) and 5(E), (F)). A short component (τISC = 378 ps for Toluene and τISC = 570 ps for ACN, Fig. 5(D), Fig. S10, ESI† and Table 2) which have a similar spectral features like the other two components discussed above except for a negative band at ∼775 nm that matches with the long component (>8 ns) spectral feature. This component was observed only for the less polar solvents and appears to be absent for MeOH and DMF. As it is confirmed from the 1O2 generation experiment, 3D shows more triplet state in less polar solvents (Fig. S4C, ESI†), and more CT state in the more polar solvents which is discussed later in this paragraph. Based on these result discussed here, this component is assigned for the ISC rate happening from the S1 state.42,43 The low ϕΔ for 3D and disappeared the ISC component with solvent polarity confirm that the ISC process occurs from the S1 state, not from the CT state which is discussed in the previous section. Due to high efficiency for the formation of CT state, ISC process was not efficient in the more polar solvent.42 A long-lived component which is more than the instrument resolution (>8 ns) is present in all of the solvents and is assigned to the lifetime of a triplet state and can be resolved using nanosecond TA spectroscopy.42 A component with a lifetime ∼1 ps (absent in the less polar solvents) is observed in MeOH (Fig. 5(E) and Table 2). The spectral features correlate well with the fs-TA spectrum observed at the initial delays and have a positive absorption in the red end of the spectrum (Fig. 5(A)). The results discussed in this article suggest that this component can be assigned as the lifetime of the CT state (τCT) which is similar to the lifetime observed for a meso-linked BODIPY dimer along with some reported BODIPY conjugates.15,16,32–34,44 This component becomes faster with the polarity (τCT ∼ 500 fs for DMF) which is an evidence for a CT state (Fig. 5(F) and Table 2).42,443D does not show the separate relaxation pathways for the CT state along with the S1 relaxation to the ground state. For such a kind of system, the CT state is delocalized with the S1 state,10 this is the potential reason which is further supported by calculating the charge density distribution discussed later.
Molecular modelling to understand geometrical properties and how structural changes impact the transition energies associated with charge transfer (CT) in the BODIPY dimer. To assess the molecular geometry of 3D we optimized the geometry of the S0 state for 3D. Upon optimization using a semi-empirical method (AM1), a near-orthogonal geometry was obtained (a dihedral angle of 84°, as depicted in Fig. S12, ESI†). A relaxed dihedral scan indicates substantial barriers for rotation due to steric hindrance. Furthermore, the vertical energies obtained from TD-DFT along the ground state IRC indicate a similarly substantial excited state S1 barrier for rotation. This barrier for twisting in the excited state was previously suggested by Thompson to be critical for the long lifetime of the charge transfer state and could rationalize the long-lived CT state for 3D.
The absorption spectra were also predicted by TD-DFT (Fig. 6(A) and (B)). The broad first excitonic peak of the absorption spectra for both of the fluorophores are blue shifted by 100 nm in comparison to the experimental results which is common in BODIPY conjugates, because TD-DFT omits double excitations which are important in stabilizing the BODIPY excited state.14,15,27,45 The calculations predict a 50 nm shift for the 3D than 3M which is in good agreement with the experimental results (Table 1 and Fig. 2(A), (B)). The localization of HOMO and LUMO throughout both 3M and 3D is shown in Fig. 6(C) and (D). For 3M, the HOMO is largely localized to the BODIPY subunit but more extended conjugation onto the linked phenyl rings is observed for the LUMO (Fig. 6(C)). The charge density is symmetrically distributed in both the HOMO and LUMO due to the strong coupling between the two BODIPY subunits of the 3D (Fig. 6(D)). This confirms the delocalization of electrons across both subunits, which prevents significant charge separation.10 As a result, the system does not exhibit noticeable charge-separation feature with changes in solvent polarity, as observed in the fs-TA measurement (Fig. 5(A) and (B)). In the excited state, the dihedral twisting from a purely orthogonal configuration leads to an electron density delocalization between the multimers in the S1 state, enabling electron transfer.14,46
 | ||
| Fig. 6 (A), (B) TD-DFT absorption spectra of 3M and 3D using CAM-B3LYP/6-311 G(d,p) level of theory. (C), (D) pictorial representation of HOMO and LUMO of 3M and 3D respectively. | ||
Conclusion
In conclusion, the BODIPY dimer 3D is a potent chromophore that shows spectroscopic patterns consistent with some polarity-dependent ICT, making it of interest as a light-harvesting material in the challenging-to-access near-IR region of the optical spectrum. On the other hand, the red-shift and fluorescence quenching in polar solvents is less than that observed by Thompson, suggesting that ICT is not an exclusive pathway in 3D and competes with other photophysical channels. ISC is a competitive channel as observed by the production of 1O2. Despite having a relatively low singlet oxygen generation quantum yield, ϕΔ ∼ 0.5–1.5%, rapid generation of 1O2 can be attributed to the intense absorption properties of 3D (ε ∼ 20 × 104 M−1 cm−1), which leads to a higher quantum efficiency (in contrast, the Q bands of porphyrins used in PDT usually have ε < 2 × 104 M−1 cm−1). Thus, derivatives with improved ISC may find use in PDT applications. The long excited state lifetime is attributed to the conformationally-restrained nature of the BODIPY units, and the direct meso attachment, which stymies electronic coupling between the charge separated monomers in the excited state. These novel BODIPY dimers with unique optical properties may pave the way for further exploration in areas such as molecular electronics, light-harvesting and photovoltaics, and biological sensing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
AHW thanks NSF (CHE-2055335) and PRF (62317-ND4) and the Leroy Edward Phillips Foundation for support. DLP thanks the Hong Kong Research Grants Council (GGRF 17316922) for support.References
- G. Feher, J. P. Allen, M. Y. Okamura and D. C. Rees, Structure and Function of Bacterial Photosynthetic Reaction Centres, Nature, 1989, 111–116, DOI:10.1038/339111a0.
- J. L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Molecular Understanding of Organic Solar Cells: The Challenges, Acc. Chem. Res., 2009, 42(11), 1691–1699, DOI:10.1021/ar900099h.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, Generation of Triplet Excited States via Photoinduced Electron Transfer in Meso-Anthra-BODIPY: Fluorogenic Response toward Singlet Oxygen in Solution and in Vitro, J. Am. Chem. Soc., 2017, 139(18), 6282–6285, DOI:10.1021/jacs.7b00551.
- M. R. Wasielewski, Photoinduced Electron Transfer in Supramolecular Systems for Artificial Photosynthesis, Chem. Rev., 1992, 92(3), 435–461, DOI:10.1021/cr00011a005.
- J. Yang, D. Wang, H. Han and C. Li, Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis, Acc. Chem. Res., 2013, 46(8), 1900–1909, DOI:10.1021/ar300227e.
- C. S. Ponseca, P. Chábera, J. Uhlig, P. Persson and V. Sundström, Ultrafast Electron Dynamics in Solar Energy Conversion, Chem. Rev., 2017, 117(16), 10940–11024, DOI:10.1021/acs.chemrev.6b00807.
- O. A. Bozdemir, R. Guliyev, O. Buyukcakir, S. Selcuk, S. Kolemen, G. Gulseren, T. Nalbantoglu, H. Boyaci and E. U. Akkaya, Selective Manipulation of ICT and PET Processes in Styryl-Bodipy Derivatives: Applications in Molecular Logic and Fluorescence Sensing of Metal Ions, J. Am. Chem. Soc., 2010, 132(23), 8029–8036, DOI:10.1021/ja1008163.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, Twisted Intramolecular Charge Transfer and Aggregation-Induced Emission of BODIPY Derivatives, J. Phys. Chem. C, 2009, 113(36), 15845–15853, DOI:10.1021/jp902962h.
- E. Vauthey, Photoinduced Symmetry-Breaking Charge Separation, ChemPhysChem, 2012, 2001–2011, DOI:10.1002/cphc.201200106.
- Y. Liu, J. Zhao, A. Iagatti, L. Bussotti, P. Foggi, E. Castellucci, M. Di Donato and K. L. Han, A Revisit to the Orthogonal Bodipy Dimers: Experimental Evidence for the Symmetry Breaking Charge Transfer-Induced Intersystem Crossing, J. Phys. Chem. C, 2018, 122(5), 2502–2511, DOI:10.1021/acs.jpcc.7b10213.
- E. Sebastian and M. Hariharan, Null Exciton-Coupled Chromophoric Dimer Exhibits Symmetry-Breaking Charge Separation, J. Am. Chem. Soc., 2021, 143(34), 13769–13781, DOI:10.1021/jacs.1c05793.
- J. Kong, W. Zhang, G. Li, D. Huo, Y. Guo, X. Niu, Y. Wan, B. Tang and A. Xia, Excited-State Symmetry-Breaking Charge Separation Dynamics in Multibranched Perylene Diimide Molecules, J. Phys. Chem. Lett., 2020, 11(24), 10329–10339, DOI:10.1021/acs.jpclett.0c03210.
- L. Estergreen, A. R. Mencke, D. E. Cotton, N. V. Korovina, J. Michl, S. T. Roberts, M. E. Thompson and S. E. Bradforth, Controlling Symmetry Breaking Charge Transfer in BODIPY Pairs, Acc. Chem. Res., 2022, 55(11), 1561–1572, DOI:10.1021/acs.accounts.2c00044.
- L. Ma, Z. Kuang, Z. Wang, H. Zhao, Y. Wan, X. F. Zhang, Y. Li and A. Xia, Ultrafast Charge Separation Driven by Torsional Motion in Orthogonal Boron Dipyrromethene Dimer, J. Phys. Chem. Lett., 2023, 14(3), 702–708, DOI:10.1021/acs.jpclett.2c03581.
- M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Symmetry-Breaking Intramolecular Charge Transfer in the Excited State of Meso-Linked BODIPY Dyads, Chem. Commun., 2012, 48(2), 284–286, 10.1039/c1cc12260f.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, Spin-Orbit Charge-Transfer Intersystem Crossing (SOCT-ISC) in Bodipy-Phenoxazine Dyads: Effect of Chromophore Orientation and Conformation Restriction on the Photophysical Properties, J. Phys. Chem. C, 2019, 123(37), 22793–22811, DOI:10.1021/acs.jpcc.9b06170.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Tuning Symmetry Breaking Charge Separation in Perylene Bichromophores by Conformational Control, Chem. Sci., 2019, 10(45), 10629–10639, 10.1039/c9sc03913a.
- N. Mataga, H. Yao, T. Okada and W. Rettig, Charge-Transfer Rates in Symmetric and Symmetry-Disturbed Derivatives of 9,9′-Bianthryl, J. Phys. Chem., 1989, 93(9), 3383–3386, DOI:10.1021/j100346a004.
- J. H. Golden, L. Estergreen, T. Porter, A. C. Tadle, D. M. R. Sylvinson, J. W. Facendola, C. P. Kubiak, S. E. Bradforth and M. E. Thompson, Symmetry-Breaking Charge Transfer in Boron Dipyridylmethene (DIPYR) Dimers, ACS Appl. Energy Mater., 2018, 1(3), 1083–1095, DOI:10.1021/acsaem.7b00214.
- Z. Wang, L. Ma, H. Zhao, Y. Wan, X. F. Zhang, Y. Li, Z. Kuang and A. Xia, Spin-Orbit Charge-Transfer Intersystem Crossing in Heavy-Atom-Free Orthogonal Covalent Boron-Dipyrromethene Heterodimers, Phys. Chem. Chem. Phys., 2023, 25(36), 24386–24394, 10.1039/d3cp01934a.
- H. L. Kee, C. Kirmaiery, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheldt, R. R. Birge, J. S. Lindsey and D. Holten, Structural Control of the Photodynamics of Boron-Dipyrrin Complexes, J. Phys. Chem. B, 2005, 109(43), 20433–20443, DOI:10.1021/jp0525078.
- S. Y. Kim, Y. J. Cho, H. J. Son, D. W. Cho and S. O. Kang, Photoinduced Electron Transfer in a BODIPY- Ortho -Carborane Dyad Investigated by Time-Resolved Transient Absorption Spectroscopy, J. Phys. Chem. A, 2018, 122(13), 3391–3397, DOI:10.1021/acs.jpca.8b01539.
- R. Montero, V. Martínez-Martínez, A. Longarte, N. Epelde-Elezcano, E. Palao, I. Lamas, H. Manzano, A. R. Agarrabeitia, I. López Arbeloa, M. J. Ortiz and I. Garcia-Moreno, Singlet Fission Mediated Photophysics of BODIPY Dimers, J. Phys. Chem. Lett., 2018, 9(3), 641–646, DOI:10.1021/acs.jpclett.7b03074.
- P. Shrestha, K. C. Dissanayake, E. J. Gehrmann, C. S. Wijesooriya, A. Mukhopadhyay, E. A. Smith and A. H. Winter, Efficient Far-Red/Near-IR Absorbing BODIPY Photocages by Blocking Unproductive Conical Intersections, J. Am. Chem. Soc., 2020, 142(36), 15505–15512, DOI:10.1021/jacs.0c07139.
- P. Shrestha, A. Mukhopadhyay, K. C. Dissanayake and A. H. Winter, Efficiency of Functional Group Caging with Second-Generation Green- and Red-Light-Labile BODIPY Photoremovable Protecting Groups, J. Org. Chem., 2022, 87(21), 14334–14341, DOI:10.1021/acs.joc.2c01781.
- J. A. Peterson, D. Yuan and A. H. Winter, Multiwavelength Control of Mixtures Using Visible Light-Absorbing Photocages, J. Org. Chem., 2021, 86(14), 9781–9787, DOI:10.1021/acs.joc.1c00658.
- W. Miao, R. Gan, W. Liu, X. Guo, Q. Wu, Y. Wei, C. Yu, E. Hao and L. Jiao, Palladium Catalyzed, Annulative Multiple C–H Bond Activations of BODIPYs to Access π-Extended Polycyclic Heteroaromatic Molecules with Deep Red Emissions and Self-Aggregation Behaviors, Chem. – Eur. J., 2023, 29(35), e202300449, DOI:10.1002/chem.202300449.
- N. Epelde-Elezcano, E. Palao, H. Manzano, A. Prieto-Castañeda, A. R. Agarrabeitia, A. Tabero, A. Villanueva, S. de la Moya, Í. López-Arbeloa, V. Martínez-Martínez and M. J. Ortiz, Rational Design of Advanced Photosensitizers Based on Orthogonal BODIPY Dimers to Finely Modulate Singlet Oxygen Generation, Chem. – Eur. J., 2017, 23(20), 4837–4848, DOI:10.1002/chem.201605822.
- X. F. Zhang, X. Yang and B. Xu, PET-Based bisBODIPY Photosensitizers for Highly Efficient Excited Triplet State and Singlet Oxygen Generation: Tuning Photosensitizing Ability by Dihedral Angles, Phys. Chem. Chem. Phys., 2017, 19(36), 24792–24804, 10.1039/c7cp02645e.
- X. F. Zhang, BisBODIPY as PCT-Based Halogen Free Photosensitizers for Highly Efficient Excited Triplet State and Singlet Oxygen Formation: Tuning the Efficiency by Different Linking Positions, Dyes Pigm., 2017, 146, 491–501, DOI:10.1016/j.dyepig.2017.07.051.
- S. Bandyopadhyay, S. Kundu, A. Giri and A. Patra, A Smart Photosensitizer Based on a Red Emitting Solution Processable Porous Polymer: Generation of Reactive Oxygen Species, Chem. Commun., 2018, 54(66), 9123–9126, 10.1039/c8cc04328k.
- M. Hu, A. A. Sukhanov, X. Zhang, A. Elmali, J. Zhao, S. Ji, A. Karatay and V. K. Voronkova, Spiro Rhodamine-Perylene Compact Electron Donor-Acceptor Dyads: Conformation Restriction, Charge Separation, and Spin-Orbit Charge Transfer Intersystem Crossing, J. Phys. Chem. B, 2021, 125(16), 4187–4203, DOI:10.1021/acs.jpcb.1c02071.
- X. Zhang, A. Elmali, R. Duan, Q. Liu, W. Ji, J. Zhao, C. Li and A. Karatay, Charge Separation, Recombination and Intersystem Crossing of Directly Connected Perylenemonoimide-Carbazole Electron Donor/acceptor Dyads, Phys. Chem. Chem. Phys., 2020, 22(11), 6376–6390, 10.1039/c9cp06914c.
- X. Xiao, T. Mu, A. A. Sukhanov, Y. Zhou, P. Yu, F. Yu, A. Elmali, J. Zhao, A. Karatay and V. K. Voronkova, The Effect of Thionation of the Carbonyl Group on the Photophysics of Compact Spiro Rhodamine-Naphthalimide Electron Donor-Acceptor Dyads: Intersystem Crossing, Charge Separation, and Electron Spin Dynamics, Phys. Chem. Chem. Phys., 2023, 25(46), 31667–31682, 10.1039/d3cp04891h.
- H. Chen, N. An, Y. Wang, G. Wang, S. Mukherjee, H. Bian, J. Ma, J. Liu and Y. Fang, Tracking the Intramolecular Charge Transfer Process of 2,6-Substituted D-A BODIPY Derivatives, J. Phys. Chem. B, 2023, 127(9), 2044–2051, DOI:10.1021/acs.jpcb.3c00347.
- S. M. Aly, A. Usman, M. Alzayer, G. A. Hamdi, E. Alarousu and O. F. Mohammed, Solvent-Dependent Excited-State Hydrogen Transfer and Intersystem Crossing in 2-(2′-Hydroxyphenyl)-Benzothiazole, J. Phys. Chem. B, 2015, 119(6), 2596–2603, DOI:10.1021/jp508777h.
- W. Zhang, J. Kong, D. Hu, M. Tao, X. Niu, S. Vdović, D. Aumiler, Y. Ma and A. Xia, Solvation-Dependent Excited-State Dynamics of Donor-Acceptor Molecules with Hybridized Local and Charge Transfer Character, J. Phys. Chem. C, 2020, 124(10), 5574–5582, DOI:10.1021/acs.jpcc.0c00003.
- S. Cetindere, S. T. Clausing, M. Anjass, Y. Luo, S. Kupfer, B. Dietzek and C. Streb, Covalent Linkage of BODIPY-Photosensitizers to Anderson-Type Polyoxometalates Using CLICK Chemistry, Chem. – Eur. J., 2021, 27(68), 17181–17187, DOI:10.1002/chem.202102897.
- T. Suhina, S. Amirjalayer, S. Woutersen, D. Bonn and A. M. Brouwer, Ultrafast Dynamics and Solvent-Dependent Deactivation Kinetics of BODIPY Molecular Rotors, Phys. Chem. Chem. Phys., 2017, 19(30), 19998–20007, 10.1039/c7cp02037f.
- Y. Lee, S. Das, R. M. Malamakal, S. Meloni, D. M. Chenoweth and J. M. Anna, Ultrafast Solvation Dynamics and Vibrational Coherences of Halogenated Boron-Dipyrromethene Derivatives Revealed through Two-Dimensional Electronic Spectroscopy, J. Am. Chem. Soc., 2017, 139(41), 14733–14742, DOI:10.1021/jacs.7b08558.
- A. A. Cullen, K. Heintz, L. O’Reilly, C. Long, A. Heise, R. Murphy, J. Karlsson, E. Gibson, G. M. Greetham, M. Towrie and M. T. Pryce, A Time-Resolved Spectroscopic Investigation of a Novel BODIPY Copolymer and Its Potential Use as a Photosensitiser for Hydrogen Evolution, Front. Chem., 2020, 8(19), 584060, DOI:10.3389/fchem.2020.584060.
- Y. Dong, A. Elmali, J. Zhao, B. Dick and A. Karatay, Long-Lived Triplet Excited State Accessed with Spin–Orbit Charge Transfer Intersystem Crossing in Red Light-Absorbing Phenoxazine-Styryl BODIPY Electron Donor/Acceptor Dyads, ChemPhysChem, 2020, 21(13), 1388–1401, DOI:10.1002/cphc.202000300.
- K. Chen, I. V. Kurganskii, X. Zhang, A. Elmali, J. Zhao, A. Karatay and M. V. Fedin, Intersystem Crossing and Electron Spin Selectivity in Anthracene-Naphthalimide Compact Electron Donor-Acceptor Dyads Showing Different Geometry and Electronic Coupling Magnitudes, Chem. – Eur. J., 2021, 27(27), 7572–7587, DOI:10.1002/chem.202100611.
- X. Chen, N. Rehmat, I. V. Kurganskii, P. Maity, A. Elmali, J. Zhao, A. Karatay, O. F. Mohammed and M. V. Fedin, Efficient Spin–Orbit Charge-Transfer Intersystem Crossing and Slow Intramolecular Triplet–Triplet Energy Transfer in Bodipy–Perylenebisimide Compact Dyads and Triads, Chem. – Eur. J., 2023, 29(61), e202302137, DOI:10.1002/chem.202302137.
- M. R. Momeni and A. Brown, Why Do TD-DFT Excitation Energies of BODIPY/aza-BODIPY Families Largely Deviate from Experiment? Answers from Electron Correlated and Multireference Methods, J. Chem. Theory Comput., 2015, 11(6), 2619–2632, DOI:10.1021/ct500775r.
- M. Kellogg, A. Akil, D. S. Muthiah Ravinson, L. Estergreen, S. E. Bradforth and M. E. Thompson, Symmetry Breaking Charge Transfer as a Means to Study Electron Transfer with No Driving Force, Faraday Discuss., 2019, 216, 379–394, 10.1039/c8fd00201k.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00609k |
| This journal is © the Owner Societies 2025 |