
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen migration reactions via low internal energy pathways in aminobenzoic acid dications†
Onni
Veteläinen
 *a,
Morsal
Babayan
a,
Lassi
Pihlava
b,
Abdul Rahman
Abid
a,
Antti
Kivimäki
c,
Edwin
Kukk
b,
Noelle
Walsh
c,
Samuli
Urpelainen
a,
Olle
Björneholm
d,
Marko
Huttula
a,
Matti
Alatalo
a,
Minna
Patanen
a and
Sergio
Díaz-Tendero
*efg
*a,
Morsal
Babayan
a,
Lassi
Pihlava
b,
Abdul Rahman
Abid
a,
Antti
Kivimäki
c,
Edwin
Kukk
b,
Noelle
Walsh
c,
Samuli
Urpelainen
a,
Olle
Björneholm
d,
Marko
Huttula
a,
Matti
Alatalo
a,
Minna
Patanen
a and
Sergio
Díaz-Tendero
*efg
aNano and Molecular Systems Research Unit, Faculty of Science, P.O. Box 3000, 90014 University of Oulu, Finland. E-mail: onni.vetelainen@oulu.fi
bDepartment of Physics and Astronomy, 20014 University of Turku, Finland
cMAX IV Laboratory, Lund University, 22100 Lund, Sweden
dDepartment of Physics and Astronomy, Uppsala University, Box 516, 751 20 Uppsala, Sweden
eDepartamento de Química, Módulo 13, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: sergio.diaztendero@uam.es
fCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain
gInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 22nd April 2025
Abstract
Hydrogen migration is a ubiquitous phenomenon upon dissociation of organic molecules. Here we investigate the formation of a H3O+ fragment after core-level photoionization and Auger decay in aminobenzoic acid molecules – a process that requires the migration of at least two hydrogen atoms. Using photoelectron–photoion coincidence spectroscopy, the formation of a H3O+ fragment is observed to be more probable in ortho-aminobenzoic acid than in meta- and para-aminobenzoic acid. Energy-resolved Auger electron–photoion coincidences are measured for the ortho-isomer to investigate the internal energy dependence of the fragmentation channels, most notably of those producing H3O+. The corresponding fragmentation channels and their mechanisms are investigated by exploring the potential energy surface with ab initio quantum chemistry methods and molecular dynamics simulations. Excited-state modeling of dicationic ortho-aminobenzoic acid is used to interpret features in the Auger spectra and identify the electronic states contributing to the signals in the Auger electron photoion coincidence map. We show that populating low-energy excited states of the dication is sufficient to trigger hydrogen migration and produce H3O+ efficiently.
Introduction
Aminobenzoic acid (ABA) is an aromatic molecule with carboxyl (–COOH) and amino (–NH2) functional groups. It has three isomers, ortho-, meta-, and para-ABA (oABA, mABA, and pABA) depending on the positioning of the functional groups. In addition, oABA and mABA have two rotational conformers based on the rotation of the carboxyl group (we refer to the lowest energy rotamers as oABA1 and mABA1, see ESI† for more discussion on the rotational configurations). The richness of isomers and the importance of the involved functional groups in various biochemical reactions and syntheses make ABAs an interesting set of molecules to study the structural dependency of radiation-induced intramolecular reactions.1–3Our previous photoelectron–photoion coincidence (PEPICO) study of oABA and mABA found that the fragmentation channels following core ionization and Auger decay are sensitive to the positioning of the functional groups, notably H3O+ and H2O production rates are higher in the ortho-isomer.4 The molecular geometry of aminobenzoic acid requires at least two hydrogen migration processes to form the H3O+ fragment. Hydrogen migration is a ubiquitous process in various chemical reactions and fragmentation processes following radiation damage.5–8 The isomer dependency of H3O+ production in ABAs offers an excellent case study to understand such processes better. Being the protonated form of water, H3O+ itself is a common molecular ion in the universe, playing an important role in both terrestrial systems and atmospheric processes, objects in the solar system and even in interstellar media.9–16 H3O+ is often formed from water and organic molecules and clusters following photoionization, for example by cosmic rays in the upper atmosphere. Signs of complex organic molecules, including amino acids and their precursors, have recently been reported to be found in asteroids17,18 and meteorites,19 and their formation and degradation in interstellar medium, like in ice grains, have gained a lot of interest.20–22
The formation mechanisms of H3O+ have been studied using various experimental techniques such as time-of-flight (TOF) mass spectroscopy as well as ab initio computational methods.5,23–30 For example, in a pump–probe study of gas phase ethanol, Kling et al.5 observed H3O+ formation from dicationic ethanol. Based on molecular dynamics simulations they proposed a double hydrogen migration mechanism, with the second hydrogen migration causing the C–O bond cleavage and H3O+ release within a timescale of several hundred femtoseconds. Shirota et al.23 also presented a double hydrogen mechanism for the formation of H3O+ from the C2H5O+ ion (a metastable intermediate in the photodissociation reactions of alcohols and ethers) based on ab initio calculations. Similar results for ethanol and C2H5O+ were obtained with density functional theory calculations by Ma et al.,26 who observed H3O+ formation in ethanol after electron beam irradiation. Hydrogen migration was also observed in ionized molecules of biological relevance, such as amino acids,6,31,32 nucleosides,33 sugar molecules,34 and clusters of amino acids.35
Our previous study established a difference in the H3O+ formation between oABA and mABA. In this study, we confirm the isomer dependency of H3O+ production for all the ABA isomers by measuring the PEPICO spectra following carbon 1s ionization and subsequent fragmentation of dicationic oABA and pABA in the gas phase. Furthermore, to investigate the fragmentation dynamics of oABA in more detail we measure the energy-resolved Auger electron photoion coincidence (AEPICO) spectra. The highest possible Auger electron energy is reached when the energy difference between the initial and final states of the Auger process is maximal, that is when the final state is the dicationic ground state. When the final state is an excited state of the dication, the Auger electron energy is necessarily lower. Therefore, higher Auger electron energies imply lower energy final states for the Auger process and thus lower internal energies available for the fragmentation processes. The energy-resolved AEPICO spectrum thus shows us the internal energies required for specific fragmentation processes, based on the Auger electron energy at which the cation signals appear. Similar internal energy – Auger electron energy mapping has been utilised e.g. in a study of dissociation timescales of the diiodothiophene dication.36 We observe that the H3O+ signal appears in a narrow energy range right after the stable dication signal in the AEPICO map, i.e. the fragmentation process occurs with low internal energies of the dication and the corresponding Auger final state is some low-lying excited state of the dication. By modeling the excited states with ab initio quantum chemistry methods we can identify the final dicationic electronic states contributing to the H3O+ and other signals. We then explore the potential energy surface to identify multiple potential pathways of H3O+ formation, with activation energies that match the experiment. Ab initio molecular dynamics were also used to simulate the fragmentation processes.
Methods
Experimental details
The experiments were performed at the gas-phase endstation37 of the FinEstBeAMS beamline38,39 at the MAX IV Laboratory in Lund, Sweden. Powder-form samples of aminobenzoic acid (C7H7NO2, molar mass 137.136 g mol−1) were placed in a resistively heated oven in a glass crucible. Both studied isomers, ortho- and para-aminobenzoic acid (Sigma-Aldrich, Merck Group, St. Louis, US) had higher than 98% purity and were used without any further purification. Effusive beams of gas-phase molecules were created by heating the samples to ∼50–60 °C (oABA) and ∼100 °C (pABA). Non-coincidence Auger electron spectra were measured with a Scienta R4000 (Scienta Omicron GmbH) electron energy analyser. The photon energy of 350 eV was selected with an SX-700 type plane-grating monochromator (FMB Feinwerk-und Messtechnik GmbH, Berlin, Germany) with a 600-lines-mm−1 grating. The electron energy analyser was operated with a 50 eV pass energy and 0.8 mm curved entrance slit, yielding 100 meV kinetic energy resolution which is close to the lifetime broadening of C 1s core hole states in carbon compounds.40 The spectrum was energy calibrated using the C 1s → 3σ−2 1Σg Auger transition in CO2 at 251.9 ± 0.1 eV41 as a reference. The polarization vector of linearly polarized synchrotron radiation was parallel to the electron detection axis of the electron energy analyser, but to the first approximation, the Auger intensities should not depend on the photoionization anisotropy parameter β. However, since the β value of the atomic C 1s orbital is 2 (and only slightly lower in a molecular environment), this configuration typically maximises the photoelectron counts when recording C 1s PEPICO spectra. This is important for higher purity of PEPICO data as the electron analyser has a much smaller acceptance angle than the ion analyser.The Scienta R4000 analyser is equipped with a fast position-sensitive detector (Quantar Inc., Model 3395A) to allow electron–ion coincidence measurements. The coincidence data was collected during two different experimental campaigns, and both times, the C 1s PEPICO spectrum of oABA was collected to confirm reproducibility. In coincidence measurements, performed also with 350 eV photon energy, the electron analyser was operated in a constant kinetic energy mode with a pass energy of 200 eV. When an electron within the kinetic energy range of interest arrived at the detector, an electric field was ramped up to extract positive ions present in the interaction region towards a modified Wiley–McLaren type multi-hit-capable ion time-of-flight (TOF) spectrometer, which is equipped with a position-sensitive delay line detector (HEX80, RoentDek Handels GmbH). Details regarding the TOF settings can be found in the ESI.† A pulse generator operating at approximately 60 Hz was also used to trigger the ion extraction field. Since these pulses are not correlated with electron triggers, they lead only to “false” ion collection. These can then be used to subtract the false coincidence ion background from the electron-triggered data.37,42 The amount of false coincidences was kept low by limiting the electron count rate to 10–20 counts s−1 by closing the exit slit of the monochromator to 25 μm. The data analysis was carried out using Igor Pro software (Wavemetrics, Inc. USA), augmented with curve fitting43,44 and coincidence data handling macro packages by E. Kukk.
Computational details
Two computational approaches were used to study the fragmentation: by calculating potential energy landscapes of the dication (carried out only for oABA1) and using molecular dynamics (for all isomers). The ground state potential energy surface exploration, including geometry optimizations for minima and transition states, intrinsic reaction coordinate calculations, frequency calculations and zero-point corrections, was performed using second-order Møller–Plesset perturbation theory45 with the correlation consistent valence double-zeta basis set cc-pVDZ.46 The singlet and triplet excited state energies of dicationic oABA1 were calculated using the equation of motion coupled cluster method with single and double excitations (EOM-CCSD)47,48 with the cc-pVTZ basis set, using the MP2/cc-pVDZ optimized geometry of the neutral molecule. The molecular dynamics were performed using the atom centered density matrix propagation model (ADMP).49–51 ADMP belongs to the extended Lagrangian family of molecular dynamics methods, in which the electronic degrees of freedom are propagated by introducing a fictitious term in the Lagrangian. Here, all isomers of ABA (including the rotamers) were considered. Applying the Franck–Condon approximation, the dynamics of the dicationic systems were propagated starting from the optimized coordinates of the neutral molecules. Both the geometry optimizations and the molecular dynamics were done at the B3LYP52,53/6-31G(d,p)54 level of density functional theory. At the beginning of each trajectory, the internal energy was assumed to be randomly distributed along the nuclear degrees of freedom in the dicationic ground state. Three internal energy estimates were used: 10 eV, 20 eV and 30 eV. For each molecule and internal energy estimate 1024 trajectories were computed. The propagation time for each trajectory was 250 fs, with a time step of 0.1 fs. All calculations were performed using the Gaussian1655 computational chemistry package.Results and discussion
Fig. 1 shows a schematic of the processes studied in oABA and the structural formula of its lower energy rotational conformer oABA1. Fig. 1(a) indicates the involved energy levels. The oABA molecule in its neutral ground state in the gas-phase is core-ionised from the C 1s level using 350 eV photons. The core-ionised states are well separated in energy, and their identification and binding energies were determined using the results of our previous study.4 The core-ionised states decay via Auger electron emission to double-ionised states. Due to a short core-hole lifetime (fs timescale) we can assume that dicationic excited states of the canonical structure of the neutral molecule are populated. The lowest DIP can be reached after a significant rearrangement in the dicationic final state, with a redistribution of the internal energy, as will be discussed later. The Auger spectrum (Fig. 1(b)) is a typical C 1s Auger electron spectrum of a multiatomic organic compound, a rather featureless broad asymmetric distribution of electrons from ∼280 eV to ∼215 eV. Compared to the overall broadness of the spectrum, we have used a very small step size of 50 meV to record the spectrum in order to resolve two small peaks, marked with an asterisk, at 267.9 and 269.1 eV at the high-energy onset of the spectrum. Fig. 1(c) shows four TOF spectra of coincident ions (false coincidence subtracted) related to C 1s photoelectrons (black trace) and Auger electrons with low, medium, and high kinetic energies (red, green, and blue traces, respectively). Note that there is a double peak around m/z = 19 in the blue spectrum. This is a signature of the high kinetic energy of the detected ions where we lose those which are emitted perpendicular to the spectrometer axis. | ||
| Fig. 1 A schematic of the processes and energy levels involved in this study with corresponding electron and ion spectra. (a) Energy level diagram (not to scale). The neutral ground state (GS) is set as the 0 of the energy scale. 350 eV photons initiate the process by photoionising the molecule from the C 1s levels. The binding energies (BEs) of chemically distinct carbon atoms are indicated next to the energy levels and taken from ref. 4. Core-ionised states decay via Auger decay to double-ionised (DI) states. The electronic transitions take place in the fs time scale, and thus the vertical double ionization potential (DIP) (calculated value) defines the onset of the experimental Auger electron spectrum. The state of the lowest DIP (calculated value) requires a significant restructuring of the molecule and is reached long after the Auger decay. (b) Molecular structure and the C 1s Auger electron spectrum of oABA. The asterisk marks two peaks which are discussed in the text. The kinetic energy regions I–III are depicted with blue, green, and red, respectively, to the right part of the graph. (c) TOF spectra in coincidence with all C 1s photoelectrons (labelled as total, black line) and three Auger regions: high kinetic energy region I (blue), medium kinetic energy region II (green), and low kinetic energy region III (red). Stable dications and H3O+ are indicated in the spectrum. The structural formula of oABA1 is shown. | ||
Fig. 2 compares the TOF mass spectra of all three isomers recorded in coincidence with C 1s photoelectrons (hν = 350 eV). The m/z ranges for the main detected ions are marked in Fig. 2, and the sharp peaks originating from doubly charged species with very low or no kinetic energy are indicated. The low m/z range contains CHi+ (i = 0,…,4), NHj+ (j = 0,…,4), and OHk+ (k = 0,…,3) fragments, followed by a broad region with ions for example C2Hn+, CNHn+, and COHn+ (n = 0,…,2, m/z ∼ 26–30). The m/z range covering 37–41 is a typical range for benzene ring fragments which can here also include an amino group. COOH+ has m/z of 45, and this signature is overlapped with two sharp peaks at ∼44.6 and ∼45.6, originating from doubly charged ions with masses of 89 and 91 amu, respectively. These doubly charged ions can be, for example, a parent ion which has lost a COOH-group and some additional hydrogens. The following m/z range 49–55 is again a typical range for benzene ring fragments, and the next doubly charged ion, especially strong for pABA, is at m/z = 54.5. This is interpreted as a parent ion which has lost 28 amu, which indicates the emission of a CO or CNH2 fragment. In oABA, there is a sharp peak at m/z = 59.6, which would be a doubly charged parent which has lost a neutral fragment with a mass of 18 amu, the plausible interpretation being emission of neutral H2O. Interestingly, in mABA and pABA a sharp peak is observed at m/z = 60, which would correspond to a fragment arising from the fragmentation of a doubly charged parent with a loss of 17 amu, i.e. neutral OH or NH3. After m/z = 60, there is a region of ring fragments up to a doubly charged parent ion signature at m/z = 68.6, especially visible for oABA and mABA, but very small in pABA. In all isomers, ring fragments around m/z = 74 are visible, as well as some signatures of [M–COOH]+ and additional H-loss ions at m/z = 90–92. Notice that in Fig. 2 the m/z peaks 16, 17, and 19 have flat tops in the TOF spectra recorded for oABA and mABA, while in pABA, the lower field setting resulted in the loss of some ions emitted perpendicular to the spectrometer axis, leading to double peak structures.
 | ||
| Fig. 2 TOF spectra of all aminobenzoic acid isomers, measured in coincidence with C 1s photoelectrons. The data in the spectra of oABA and mABA are taken from ref. 4, and they have been measured with a higher extraction field in the TOF spectrometer, and thus the TOF axis differs compared to pABA. The m/z ratio is depicted as an indicative axis on the top. | ||
Fig. 3 shows the energy-resolved AEPICO coincidence map in the high energy region of the Auger electron, corresponding to KE region I in Fig. 1. The AEPICO map is constructed by presenting the Auger electron – ion events as a contour plot where the colour scale of the map refers to the number of coincidence events. The kinetic energy of the events Auger electron defines the position on the x-axis, while the TOF of the detected ion defines the placement with respect to the y-axis. The most important fragments discussed here are labelled in the projected TOF spectrum on the right-hand panel of the coincidence map. Some selected profiles are shown in the ESI† (see Fig. S1). The first, well-isolated intense signal from the right at 269.1 eV is due to the stable dication, and it matches the first peak labelled with an asterisk in Fig. 1(b). The H3O+ and [M–H3O]+ signals appear at 1.2 eV lower Auger kinetic energy than the stable dication, and these signals form the second peak marked with an asterisk in Fig. 1(b). They peak in a very narrow energy range, but H3O+ is produced in small amounts at lower Auger kinetic energies as well. Due to broadness of the lower field TOF peaks, there might also be a small contribution from the H2O+ fragment in the H3O+ signal. However, other channels such as [M–H2O]2+ and [C6H5N]2+ dominate the fragmentation dynamics below 267 eV Auger kinetic energies (higher internal energies).
 | ||
| Fig. 3 Auger electron photoion coincidence map in the high Auger electron energy region (region I in Fig. 1). The color scale refers to the number of counts. The corresponding total Auger electron and TOF spectra are depicted in the top and right-hand panels respectively. | ||
Note that production of H3O+ in oABA is mainly observed in the high-kinetic-energy region of the Auger electron spectrum, which corresponds to a low internal excitation energy in the remaining dication. We have obtained deeper insight into the double ionization in this region, and the subsequent fragmentation mechanisms with quantum chemistry calculations. We focus on the rotamer 1 of oABA, which is expected to be the dominant configuration at the experimental temperature, based on the energy difference between the ortho-rotamers (∼120 meV).4 First, the high-kinetic-energy region of the Auger electron spectrum (see region I in Fig. 1(c) and 3) was investigated by excited states calculations of dicationic oABA using the geometry of the neutral molecule, computed at the MP2/cc-pVDZ level, i.e., assuming vertical ionization in the Franck–Condon region. The excited states were modelled using the equation of motion coupled cluster method with single and double excitations (EOM-CCSD).47,48 The results are presented in Table 1, and the energies are referred to the dicationic ground state. The vertical double ionization potential is the lowest possible energy final state in the Auger process assuming that nuclei do not have time to move in the core-ionised state (lifetime in order of a few fs), and thus corresponds to the highest possible Auger electron energy in the Auger electron spectrum. Lower Auger electron energies correspond to excited states as the Auger final states. We concentrate on the low-lying excited states as in that region the most interesting spectral features are observed, namely the peak at 269.1 eV due to stable dications and 267.9 eV where H3O+ production occurs. Using the results in Table 1 we may interpret the AEPICO map features in Fig. 3. A single Gaussian fit to the first signal from the right (i.e. the stable dication M2+ signal) in the AEPICO map depicted in Fig. 3 gives a full-width-at-half-maximum (FWHM) of 1.4 eV, which is considerably higher than the experimental broadening of 0.45 eV. This can be due to significant vibrational broadening, and/or two or more overlapping electronic states. A double Gaussian fit also gives a total FWHM of 1.4 eV and a peak separation of 0.65 eV, which matches well the computed first triplet excitation energy of 0.59 eV. We can conclude that both the dicationic ground state S0 and the first triplet state T1 can contribute to the M2+ signal. Fitting the H3O+ signal was much more challenging due to the low counts, but it is approximately as wide as the M2+ signal, and therefore likely composed of multiple electronic states as well. We propose that these are the T2, S1, S2 and T3 excited states which are grouped closely together in energy and whose average energy (1.56 eV) is 1.26 eV higher than the average energy of the S0 and T1 states, matching the experimental peak separation of 1.2 eV between the M2+ and H3O+ signals. The higher energy S3, S4 and T4 excited states then contribute to signals further to the left in the coincidence map, where multiple fragmentation channels such as the [M–H2O]2+ and C6H5N2+ channels start to overlap.
| Singlet states | S1 | S2 | S3 | S4 |
|---|---|---|---|---|
| Energy (eV) | 1.45 | 1.51 | 2.51 | 3.92 |
| Triplet states | T1 | T2 | T3 | T4 |
| Energy (eV) | 0.59 | 1.39 | 1.88 | 2.33 |
The fragmentation mechanisms of dicationic ABA were investigated through an exploration of the potential energy surface (PES) as well as ab initio molecular dynamics simulations. Fig. 4 presents the results of the PES exploration. We assume a very efficient energy redistribution from the electronic excited states populated in the Auger process, towards nuclear degrees of freedom in electronic ground state (electron–phonon coupling). Thus, the PES exploration is carried out in the electronic ground state. The minimum Min 1 (highlighted in red) is reached by relaxation following vertical ionization. Hydrogen migration (through transition state TS 1, highlighted in green) followed by H2O roaming connect Min 1 to a more stable configuration in the minimum Min 2 (highlighted in blue). The reconfiguration to this more stable structure is a common step in the calculated pathways.
 | ||
| Fig. 4 Results of the potential energy surface (PES) exploration for dicationic oABA with singlet spin multiplicity at the MP2/cc-pVDZ level. Energies include zero-point corrections and are referred to the neutral ground state. (a) A straightforward pathway to H3O+ emission through hydrogen migration and water roaming. The energy needed to release neutral water is also depicted. (b) A more complex series of hydrogen migrations to produce H3O+. (c) H3O+ is produced after the roaming water abstracts a proton from the NH2 group instead of the benzene ring. Note that relevant points in the PES are highlighted with a coloured bar: Min 1 in red, Min 2 in blue and TS 1 in green. | ||
Fig. 4(a) shows a straightforward water roaming pathway starting from Min 2 with subsequent H3O+ release. The proton abstraction causes the initial benzene ring to be transformed into a five-membered ring with a cyclopentadiene configuration. The emission of neutral water is also depicted here and the energy required for the barrierless release of H2O lies 1.73 eV above the vertical double ionisation potential (VIP in Fig. 4). Apart from the water emission, the highest energy point in this pathway corresponds to the transition state TS 1, so H3O+ release becomes accessible with an internal energy of 1.05 eV. A more complicated series of hydrogen and water migrations is depicted in Fig. 4(b), but the endpoint is the same five-atom-ring structure as in Fig. 4(a). The highest energy point in this figure is 23.73 eV, so the pathway is accessible with a slightly higher internal energy of 1.96 eV, which is also higher than the H2O emission energy of 1.73 eV. Finally, Fig. 4(c) depicts water roaming pathways starting from Min 2 and the initial water-producing transition states towards the NH2 group, from which either proton may be abstracted by the roaming water to produce H3O+. The highest energy point here is again the first transition state TS 1, so the pathways given in this panel are also accessible with an internal energy of 1.05 eV, which is in rather good agreement with the experimental peak separation between the stable dication and H3O+ signals (see Fig. 3). A comparison of the microcanonical coefficient rates of the pathways in Fig. 4 indicates that proton abstraction from the –NH2 group is the most efficient process (see ESI† for more details). A partial charge analysis was also performed to investigate the flow of charge during the fragmentation pathways. The results can be found in the ESI.†
Among the aminobenzoic acid isomers, oABA (specifically the rotamer 1) is the most stable in terms of total energy in the ground state. It has been suggested that the reason for its greater stability is the intramolecular hydrogen bonding between the functional groups H–N–H⋯O–C–O–H.56 Based on our calculations, the relaxation from the VIP to the dicationic minimum Min 1 further strengthens this bond. Specifically, the N–O and H–O distances in the hydrogen bond are shortened by 0.17 Å and 0.37 Å, respectively, creating a sort of double-ring structure for the dicationic oABA1. To verify this, we have performed a bonding analysis in the Min 1 structure using the quantum theory of atoms in molecules, QTAIM,57,58 as implemented in the AIMAll program.59 The results of this analysis are presented in Fig. 5. Part of the positive charge is located in the amino group, which undergoes planarization. Electrons in one of the lone pair of the carboxylic oxygen are attracted with higher intensity towards the hydrogen atom in the amino group, thus giving rise to a strengthening of the hydrogen bond with respect to the neutral molecule, creating a double-ring structure. A similar effect has been observed in the ionization of γ-aminobutyric acid.6
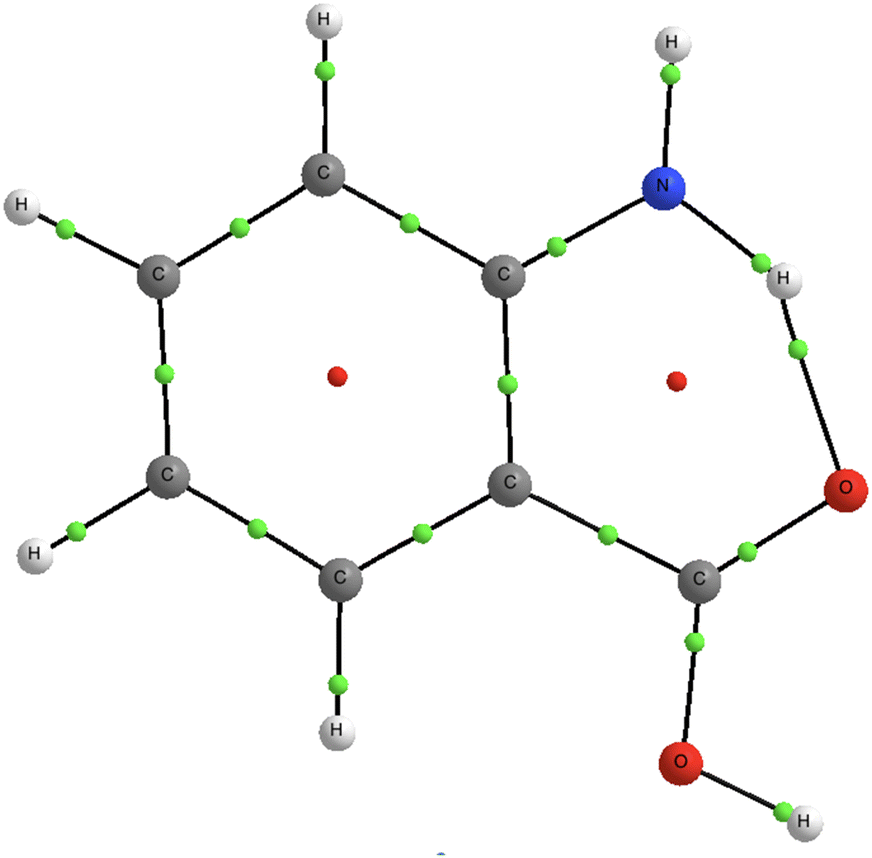 | ||
| Fig. 5 QTAIM analysis of Min 1. Black lines are bond paths; green points are bond critical points and small red points are ring critical points. | ||
The key differences in fragmentation pathways between the ABA isomers can be seen in the TOF spectra in Fig. 2. The mABA and pABA spectra are missing the 59.5 m/z peak (i.e. [M–H2O]2+), but instead have notable peaks at 54.5 m/z, which can either be [M–CNH2]2+ or [M–CO]2+, both of which imply a breakup of one of the functional groups. For pABA specifically the 68.5 m/z peak (i.e. [M]2+) is very weak, indicating that the dication is very unstable. We propose that the double ring structure that is formed in dicationic oABA1 stabilises the functional groups, suppressing the competing fragmentation channels that are observed in the meta- and para-isomers, allowing for the migration and roaming processes that are necessary for H3O+ and H2O production.
The fragmentation processes were also simulated using molecular dynamics simulations. A small sample of trajectories were computed with internal energies of 2 or 5 eV and propagation times of 500–1000 fs. However, with these energies and propagation times, the simulations produced no fragmentation. Therefore the bulk of simulations were run using internal energies of 10, 20 and 30 eV. These higher internal energies were needed in the simulations to produce fragmenting trajectories within short propagation times, but this is problematic because in the Auger electron–photoion coincidences, we observe the H3O+ channel already at 1.2 eV. A propagation time of 250 fs was chosen for the computational feasibility of a large number of trajectories. For the 10, 20 and 30 eV simulations ∼90%, ∼60% and ∼20% of trajectories remained unfragmented, respectively. The full results of the molecular dynamics simulations can be found in the ESI.† We can thus conclude that 250 fs may be enough time to describe dynamics at higher energies but is insufficient closer to the potential energy surface. The Auger electron–photoion coincidence measurements also show that this is exactly the energy range where H3O+ fragments are produced. In the experimental setup, the flight time of ionized particles is on the order of microseconds, so a molecule will have orders of magnitude more time to reach the available fragmentation pathways. Thus, the low energy dynamics are far better described by the MP2 PES exploration rather than the molecular dynamics simulations. Some H2O and a handful of H3O+ producing trajectories were seen in the molecular dynamics, however without any notable isomer discrepancy. One such trajectory is depicted in Fig. 6, which closely resembles the pathway in Fig. 4(a), thus confirming the proposed mechanism.
 | ||
| Fig. 6 Snapshots from a simulated 20 eV internal energy trajectory depicting H3O+ formation in oABA. The trajectory is similar to the pathway in Fig. 4(a). | ||
Conclusions
In summary, we present a combined experimental and computational study of the fragmentation of ortho-aminobenzoic acid following C 1s photoionization and Auger decay. We present energy-resolved Auger electron–photoion coincidence (AEPICO) measurements which reveal that H3O+ ions are mostly (and their two-body dissociation counterparts, [M–H3O]+ ions, almost exclusively) detected in coincidence with high kinetic energy Auger electrons with a very narrow energy range around 268 eV. Thus, the fragmentation of ortho-aminobenzoic acid yielding H3O+ ions predominantly occurs within a specific range of internal energies. Modeling the dicationic excited states allows us to identify the electronic states contributing to the stable dication and H3O+ signals in the AEPICO measurements. Our potential energy surface exploration using quantum chemistry methods reveals multiple possible pathways for H3O+ production with activation energies that match the experimental H3O+ signal onset. We also propose an explanation for the isomer dependency in the H3O+ production based on the increased stability due to a double ring structure in the dicationic oABA ground state, which suppresses the competing fragmentation mechanisms seen in the meta- and para-isomers.Author contributions
O. V.: conceptualisation, formal analysis, funding acquisition, investigation, visualisation, writing – original draft. A. R. A.: conceptualisation, investigation, writing – review & editing. M. B., L. P., A. K., N. W.: investigation, writing – review & editing. E. K.: resources, software, investigation, writing – review & editing. M. H., S. U.: funding acquisition, writing – review & editing. M. A.: resources, supervision, writing – review & editing. O. B.: conceptualisation, writing – review & editing. M. P.: conceptualisation, formal analysis, funding acquisition, investigation, visualisation, writing – original draft. S. D.-T.: conceptualisation, formal analysis, funding acquisition, investigation, resources, visualisation, writing – original draft.Data availability
Data for this article are available at the following URL/DOI: https://doi.org/10.23729/42b5718f-10e0-41cb-89c2-0725a95068e9.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the CSC – IT Center for Science, Finland, for computational resources. The research leading to these results has been supported by the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie I4Future (Grant agreement No. 713606). OV acknowledges the COST Action CA18222 (Attosecond Chemistry). This project was also granted travel funding from CALIPSOPlus from the EU Framework Programme for Research and Innovation Horizon 2020 (Grant agreement No. 730872). ARA acknowledges the Väisälä Fund and the Finnish Academy of Science & Letters. We acknowledge the Research Council of Finland for financial support (including The University of Oulu and Research Council of Finland Profi5 – project 326291 and INTRICat project 341288). We thank Drs K. Chernenko, E. Pelimanni and N. Boudjemia for assistance during the experiments. We acknowledge MAX IV Laboratory for time on Beamline FinEstBeAMS under Proposals 20190884 and 20230302. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research Council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. We acknowledge the generous allocation of computer time at the Centro de Computación Científica at the Universidad Autónoma de Madrid (CCC-UAM). This work was partially supported by the MICINN – Spanish Ministry of Science and Innovation – Project PID2022-138470NB-I00 funded by MCIN/AEI/10.13039/501100011033, and the ‘María de Maeztu’ (CEX2023-001316-M) Program for Centers of Excellence in R&D.Notes and references
- A. P. Demchenko, BBA Adv., 2023, 3, 100085 Search PubMed.
- M. B. Soley, P. E. Videla, E. T. J. Nibbering and V. S. Batista, J. Phys. Chem. Lett., 2022, 13, 8254–8263 Search PubMed.
- K.-C. Tang, C.-L. Chen, H.-H. Chuang, J.-L. Chen, Y.-J. Chen, Y.-C. Lin, J.-Y. Shen, W.-P. Hu and P.-T. Chou, J. Phys. Chem. Lett., 2011, 2, 3063–3068 Search PubMed.
- A. R. Abid, O. Veteläinen, N. Boudjemia, E. Pelimanni, A. Kivimäki, M. Alatalo, M. Huttula, O. Björneholm and M. Patanen, J. Phys. Chem. A, 2023, 127, 1395–1401 Search PubMed.
- N. Kling, S. Díaz-Tendero, R. Obaid, M. Disla, H. Xiong, M. Sundberg, S. Khosravi, M. Davino, P. Drach, A. M. Carroll, T. Osipov, F. Martín and N. Berrah, Nat. Commun., 2019, 10, 2813 Search PubMed.
- M. Capron, S. Díaz-Tendero, S. Maclot, A. Domaracka, E. Lattouf, A. Ławicki, R. Maisonny, J.-Y. Chesnel, A. Méry, J.-C. Poully, J. Rangama, L. Adoui, F. Martn, M. Alcam, P. Rousseau and B. A. Huber, Chem. – Eur. J., 2012, 18, 9321–9332 Search PubMed.
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, Bioenerg., 2001, 1504, 196–219 Search PubMed.
- T. J. Wasowicz and B. Pranszke, J. Phys. Chem. A, 2016, 120, 964–971 Search PubMed.
- K. Aplin and R. McPheat, J. Atmos. Sol.-Terr. Phys., 2005, 67, 775–783 Search PubMed.
- D. Catone, M. C. Castrovilli, F. Nicolanti, M. Satta and A. Cartoni, Phys. Chem. Chem. Phys., 2023, 25, 25619–25628 Search PubMed.
- F. Arnold, D. Krankowsky and K. Marien, Nature, 1977, 267, 30–32 Search PubMed.
- R. Martinez, A. N. Agnihotri, P. Boduch, A. Domaracka, D. Fulvio, G. Muniz, M. E. Palumbo, H. Rothard and G. Strazzulla, J. Phys. Chem. A, 2019, 123, 8001–8008 CrossRef CAS PubMed.
- T. E. Cravens, R. L. McNutt Jr., J. H. Waite Jr., I. P. Robertson, J. G. Luhmann, W. Kasprzak and W.-H. Ip, Geophys. Res. Lett., 2009, 36, L08106 Search PubMed.
- N. Indriolo, D. A. Neufeld, M. Gerin, P. Schilke, A. O. Benz, B. Winkel, K. M. Menten, E. T. Chambers, J. H. Black, S. Bruderer, E. Falgarone, B. Godard, J. R. Goicoechea, H. Gupta, D. C. Lis, V. Ossenkopf, C. M. Persson, P. Sonnentrucker, F. F. S. van der Tak, E. F. van Dishoeck, M. G. Wolfire and F. Wyrowski, Astrophys. J., 2015, 800, 40 CrossRef.
- J. Tang and T. Oka, J. Mol. Spectrosc., 1999, 196, 120–130 Search PubMed.
- F. F. S. van der Tak, S. Aalto and R. Meijerink, Astron. Astrophys., 2008, 477, L5–L8 Search PubMed.
- D. P. Glavin, J. P. Dworkin, C. M. O. Alexander, J. C. Aponte, A. A. Baczynski, J. J. Barnes, H. A. Bechtel, E. L. Berger, A. S. Burton, P. Caselli, A. H. Chung, S. J. Clemett, G. D. Cody, G. Dominguez, J. E. Elsila, K. K. Farnsworth, D. I. Foustoukos, K. H. Freeman, Y. Furukawa, Z. Gainsforth, H. V. Graham, T. Grassi, B. M. Giuliano, V. E. Hamilton, P. Haenecour, P. R. Heck, A. E. Hofmann, C. H. House, Y. Huang, H. H. Kaplan, L. P. Keller, B. Kim, T. Koga, M. Liss, H. L. McLain, M. A. Marcus, M. Matney, T. J. McCoy, O. M. McIntosh, A. Mojarro, H. Naraoka, A. N. Nguyen, M. Nuevo, J. A. Nuth, Y. Oba, E. T. Parker, T. S. Peretyazhko, S. A. Sandford, E. Santos, P. Schmitt-Kopplin, F. Seguin, D. N. Simkus, A. Shahid, Y. Takano, K. L. Thomas-Keprta, H. Tripathi, G. Weiss, Y. Zheng, N. G. Lunning, K. Righter, H. C. Connolly and D. S. Lauretta, Nat. Astron., 2025, 9, 199–210 Search PubMed.
- C. Potiszil, T. Ota, M. Yamanaka, C. Sakaguchi, K. Kobayashi, R. Tanaka, T. Kunihiro, H. Kitagawa, M. Abe, A. Miyazaki, A. Nakato, S. Nakazawa, M. Nishimura, T. Okada, T. Saiki, S. Tanaka, F. Terui, Y. Tsuda, T. Usui, S.-I. Watanabe, T. Yada, K. Yogata, M. Yoshikawa and E. Nakamura, Nat. Commun., 2023, 14, 1482 CrossRef CAS PubMed.
- T. Koga and H. Naraoka, Sci. Rep., 2017, 7, 636 CrossRef PubMed.
- A. Pernet, J. Pilmé, F. Pauzat, Y. Ellinger, F. Sirotti, M. Silly, P. Parent and C. Laffon, Astron. Astrophys., 2013, 552, A100 Search PubMed.
- J. Wang, A. A. Nikolayev, J. H. Marks, A. M. Turner, S. Chandra, N. F. Kleimeier, L. A. Young, A. M. Mebel and R. I. Kaiser, J. Am. Chem. Soc., 2024, 146, 28437–28447 CAS.
- J. H. Marks, J. Wang, B.-J. Sun, M. McAnally, A. M. Turner, A. H.-H. Chang and R. I. Kaiser, ACS Cent. Sci., 2023, 9, 2241–2250 Search PubMed.
- T. Shirota, N. Mano, M. Tsuge and K. Hoshina, Rapid Commun. Mass Spectrom., 2010, 24, 679–686 CrossRef CAS PubMed.
- K. Hoshina and M. Tsuge, Chem. Phys. Lett., 2010, 489, 154–158 CrossRef.
- Y. Tamenori, K. Okada, K. Tabayashi, A. Hiraya, T. Gejo and K. Honma, Chem. Phys. Lett., 2006, 433, 43–47 CrossRef CAS.
- C. Ma, J. Zhou, E. Wang, T. Yang, Z. Xu, S. Jia, A. Dorn and X. Ren, Laser Part. Beams, 2021, 2021, e23 Search PubMed.
- P. Suwannakham, S. Chaiwongwattana and K. Sagarik, RSC Adv., 2018, 8, 36731–36744 RSC.
- P. Intharathep, A. Tongraar and K. Sagarik, J. Comput. Chem., 2006, 27, 1723–1732 Search PubMed.
- T. M. Di Palma and A. Bende, J. Mass Spectrom., 2014, 49, 700–708 CrossRef CAS PubMed.
- Y. Tamenori, K. Okada, K. Tabayashi, A. Hiraya, T. Gejo and K. Honma, J. Chem. Phys., 2011, 134, 204302 Search PubMed.
- S. Maclot, D. G. Piekarski, A. Domaracka, A. Méry, V. Vizcaino, L. Adoui, F. Martín, M. Alcamí, B. A. Huber, P. Rousseau and S. Díaz-Tendero, J. Phys. Chem. Lett., 2013, 4, 3903–3909 Search PubMed.
- D. G. Piekarski, R. Delaunay, S. Maclot, L. Adoui, F. Martín, M. Alcamí, B. A. Huber, P. Rousseau, A. Domaracka and S. Díaz-Tendero, Phys. Chem. Chem. Phys., 2015, 17, 16767–16778 RSC.
- S. Maclot, R. Delaunay, D. G. Piekarski, A. Domaracka, B. A. Huber, L. Adoui, F. Martín, M. Alcamí, L. Avaldi, P. Bolognesi, S. Díaz-Tendero and P. Rousseau, Phys. Rev. Lett., 2016, 117, 073201 CrossRef CAS PubMed.
- M.-A. Hervé du Penhoat, A. Souchaud, A. Rajpal, R. Vuilleumier, M.-P. Gaigeot, I. Tavernelli, K. Fujii, A. Yokoya, S. Díaz-Tendero and M.-F. Politis, Phys. Chem. Chem. Phys., 2024, 26, 15693–15704 RSC.
- P. Rousseau, D. G. Piekarski, M. Capron, A. Domaracka, L. Adoui, F. Martn, M. Alcam, S. Díaz-Tendero and B. A. Huber, Nat. Commun., 2020, 11, 3818 CrossRef CAS PubMed.
- E. Kukk, L. Pihlava, K. Kooser, C. Stråhlman, S. Maclot and A. Kivimäki, Phys. Chem. Chem. Phys., 2023, 25, 5795–5807 RSC.
- K. Kooser, A. Kivimäki, P. Turunen, R. Pärna, L. Reisberg, M. Kirm, M. Valden, M. Huttula and E. Kukk, J. Synchrotron Radiat., 2020, 27, 1080–1091 CrossRef CAS PubMed.
- R. Pärna, R. Sankari, E. Kukk, E. Nõmmiste, M. Valden, M. Lastusaari, K. Kooser, K. Kokko, M. Hirsimäki, S. Urpelainen, P. Turunen, A. Kivimäki, V. Pankratov, L. Reisberg, F. Hennies, H. Tarawneh, R. Nyholm and M. Huttula, Nucl. Instrum. Methods Phys. Res., Sect. A, 2017, 859, 83–89 CrossRef.
- K. Chernenko, A. Kivimäki, R. Pärna, W. Wang, R. Sankari, M. Leandersson, H. Tarawneh, V. Pankratov, M. Kook, E. Kukk, L. Reisberg, S. Urpelainen, T. Käämbre, F. Siewert, G. Gwalt, A. Sokolov, S. Lemke, S. Alimov, J. Knedel, O. Kutz, T. Seliger, M. Valden, M. Hirsimäki, M. Kirm and M. Huttula, J. Synchrotron Radiat., 2021, 28, 1620–1630 Search PubMed.
- C. Nicolas and C. Miron, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 267–272 Search PubMed.
- A. Hiltunen, S. Aksela, G. Víkor, S. Ricz, Á. Kövér and B. Sulik, Nucl. Instrum. Methods Phys. Res., Sect. B, 1999, 154, 267–271 CrossRef CAS.
- G. Prümper and K. Ueda, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 574, 350–362 CrossRef.
- E. Kukk, G. Snell, J. D. Bozek, W.-T. Cheng and N. Berrah, Phys. Rev. A:At., Mol., Opt. Phys., 2001, 63, 062702 Search PubMed.
- E. Kukk, K. Ueda, U. Hergenhahn, X.-J. Liu, G. Prümper, H. Yoshida, Y. Tamenori, C. Makochekanwa, T. Tanaka, M. Kitajima and H. Tanaka, Phys. Rev. Lett., 2005, 95, 133001 CrossRef CAS PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 Search PubMed.
- J. Dunning and H. Thom, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- H. Koch and P. Jørgensen, J. Chem. Phys., 1990, 93, 3333–3344 Search PubMed.
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS.
- H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, A. D. Daniels, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 114, 9758–9763 CrossRef CAS.
- S. S. Iyengar, H. B. Schlegel, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 115, 10291–10302 Search PubMed.
- H. B. Schlegel, S. S. Iyengar, X. Li, J. M. Millam, G. A. Voth, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2002, 117, 8694–8704 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.02, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- B. C. M. Maciel and P. Chaudhuri, Int. J. Quantum Chem., 2011, 111, 1709–1718 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019 (https://aim.tkgristmill.com).
Footnote |
| † Electronic supplementary information (ESI) available: Extended experimental details, molecular dynamics simulations, Auger profiles for selected fragments, partial charge analysis and microcanonical rate coefficients. See DOI: https://doi.org/10.1039/d5cp00415b |
| This journal is © the Owner Societies 2025 |