
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of fluid and lattice oxygen mediated by interstitial atoms at the TiO2(110)–water interface†
Heonjae
Jeong
 bc,
Ian I.
Suni
a,
Raylin
Chen
a,
Marina
Miletic
a,
Xiao
Su
a and
Edmund G.
Seebauer
*a
bc,
Ian I.
Suni
a,
Raylin
Chen
a,
Marina
Miletic
a,
Xiao
Su
a and
Edmund G.
Seebauer
*a
aDepartment of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: eseebaue@illinois.edu
bDepartment of Mechanical Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
cDepartment of Electronic Engineering, Gachon University, Seongnam, Gyeonggi 13120, South Korea
First published on 10th April 2025
Abstract
O2 interacts with TiO2 surfaces in numerous aqueous reactions for clean hydrogen production, wastewater cleanup, reduction of CO2 and N2, and O2 sensing. In many cases, these reactions involve reversible exchange of O with the solid, whose participation is usually thought to require oxygen vacancies (VO). Based on measurements of oxygen isotopic self-diffusion in rutile TiO2 under water, this work proposes a different perspective centered on O interstitial atoms (Oi). Experiments with varying concentrations of O2 and mole fractions of 18O show that the (110) surface facilitates O exchange with both the H2O liquid and its dissolved O2. First-principles calculations indicate that on-top and “surface Oi” configurations of adsorbed O participate sequentially in the exchange process. Adsorbed OH appears to provide a single pathway for H2O and O2 to contribute oxygen, although fitting the diffusion data to simple models indicates that H2O contributes more. Because rutile TiO2 is a prototypical metal oxide, this picture based on Oi probably generalizes in many cases to other oxides − explaining important aspects of their thermal, electrochemical, and photochemical reactions with dissolved O2.
1. Introduction
O2 interacts with water-submerged TiO2 surfaces in many aqueous reactions for production of clean hydrogen,1–5 cleanup of wastewater,6–11 reduction of N2 to NH3,12–16 reduction of CO2 to hydrocarbons,17,18 production of H2O2,19 and sensing of O2.20 For the oxygen evolution reaction (OER) by photocatalysis21,22 and electrocatalysis,23 significant attention focuses on participation of lattice oxygen from TiO2. This attention extends to other metal oxides24–28 for the OER, the oxygen reduction reaction29–31 (ORR) and advanced oxidation processes (AOP).11 Many authors use the term “lattice oxygen mechanism” (LOM) to describe the chemical pathways for exchange of atoms between O2 and the solid,21–23,25–27,32,33 although “Mars–van Krevelen mechanism” prevails among other authors.31,34 Such mechanisms contrast with pathways involving atoms derived only from the liquid, termed an “adsorbate evolution mechanism” in the OER.10,26,35Regardless of the oxide or driving force (thermal, electrochemical, or photochemical), O vacancies (VO) are usually thought to mediate participation of lattice oxygen.25,27,33,35 However, this physical picture has been questioned recently,29 partly because O interstitial atoms (Oi) appear to facilitate lattice O participation in some cases.29,30 This literature struggles to reconcile the notion of “active oxygen” in the lattice with the assumed need for surface VO to assist the reaction. Furthermore, mediation by vacancies contains implicit inconsistencies. For example, the OER on TiO2 and perovskites often operates under intensely oxidizing conditions26,35 (strongly alkaline pH, high applied potential, and high O2 concentration) that are not hospitable for surface VO, which reflect chemical reduction. Moreover, increasing the VO concentration in O-deficient perovskites increases the barrier for participation of lattice O.26 In other words, the reaction seems to require vacancies, yet increasing their concentration diminishes the rate (e.g., in SrCoO336).
As described elsewhere,29,30 an alternate perspective based on Oi merits consideration in many cases. Under O-rich conditions, Oi mediates O exchange between several different binary oxides and liquid H2O.37,38 Yet no physical picture has been proposed by which O atoms transfer between the lattice, diffusing Oi, the surface, and dissolved O2.
The mediating form of adsorbed O remains especially murky. Adsorbed OH (anion and neutral) and molecular H2O dominate adsorbate populations on submerged metal oxides,39–42 but reaction mechanisms for O2 atop oxides assume participation of adsorbed O,35 whose existence has been inferred from experiments and density functional theory (DFT) calculations.43–48 The literature for catalysis and surface science considers O on rutile TiO2 to sit directly atop a metal cation.49–56 This species protonates readily. In contrast, DFT calculations for three different oxides37,57,58 reveal a “surface interstitial” that converts into bulk Oi by diffusive hopping. The surface interstitial's high bond coordination implies little propensity for protonation.
Interconversion between these forms of adsorbed O provides the foundation for a self-consistent physical picture to describe atom exchange between the lattice and dissolved O2. To support this picture, the present work employs isotopic self-diffusion measurements in submerged rutile TiO2(110) single crystals with varying concentrations of 18O isotopic label. We selected the (110) surface termination because extensive literature already exists for this orientation, and its nonpolar structure should minimize surface reconstruction upon immersion.59 These experiments show that O exchanges between the solid and both dissolved O2 and H2O liquid, mediated within the solid by Oi. At the surface, DFT calculations suggest that adsorbed O exists in both on-top and surface interstitial configurations. Adsorbed OH appears to provide a single pathway for H2O and O2 to contribute oxygen, although fitting the diffusion data to simple models indicates that H2O contributes more. Because rutile TiO2 is a prototypical metal oxide,39,60 this picture based on Oi probably generalizes in many cases to other oxides − explaining important aspects of their thermal, electrochemical, and photochemical reactions with dissolved O2.
2. Methods
2.1 Experiments
Many published experiments seeking to measure participation of lattice O have focused on detecting 18O in the fluid or on the solid surface.24,32,61 In contrast, the present work focuses on measuring 18O self-diffusion within the solid. Such measurements offer a complementary perspective more directly attuned to defect behavior. Our protocols for the 18O self-diffusion experiments have been detailed previously.37 The experiments were performed at varying temperatures involving no photochemical perturbations. Undoped single-crystal rutile TiO2(110) specimens (5 × 5 × 0.5 mm) purchased from MTI Corp. were de-greased by 10 min of ultrasonic agitation in a succession of solvents (acetone, isopropanol, ethanol, and methanol) followed by wet etching (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 30% NH4OH:H2O) at room temperature for 40 min to remove elemental poisons that inhibit Oi injection. Surface analysis by X-ray photoelectron spectroscopy (XPS) showed only Ti, O, and C within the detection limit, both before and after Oi diffusion. Consistent with previous studies, no variations in surface composition with experimental conditions were observed. Atomic force microscopy (AFM) revealed a root mean square (rms) roughness of 0.23 ± 0.03 nm for as-received TiO2 and 0.14 ± 0.08 nm after wet etching.58
:2, 30% NH4OH:H2O) at room temperature for 40 min to remove elemental poisons that inhibit Oi injection. Surface analysis by X-ray photoelectron spectroscopy (XPS) showed only Ti, O, and C within the detection limit, both before and after Oi diffusion. Consistent with previous studies, no variations in surface composition with experimental conditions were observed. Atomic force microscopy (AFM) revealed a root mean square (rms) roughness of 0.23 ± 0.03 nm for as-received TiO2 and 0.14 ± 0.08 nm after wet etching.58
We discovered empirically that, during submersion in water, application of potential bias in a conventional three-electrode cell configuration increases the Oi injection rate and thereby aids the measurement of profile metrics. TiO2 specimens were immersed for 60 min at constant temperature (70 °C) with a potential bias applied using a Biologic SP200 potentiostat. The electrical connection to the TiO2 specimens was a Cu wire attached with double-stick carbon tape covered with Kapton. An Ag/AgCl reference electrode and a Pt counter electrode were employed. The water contained no electrolyte, and the pH was about 7. Before immersion, 10 min of gas bubbling through the aqueous solution with simultaneous gas flow through the headspace established liquid–gas equilibrium.
The solid TiO2 was undoped and assumed to be insulating. In addition, no electrolyte was employed, so no faradaic electrochemistry occurred during the experiments. Potentiostat measurements of the current were very small (10–100 nA) and uncorrelated with any profile metric. The potentiostat itself typically displayed an overload warning characteristic of an open circuit. As shown in Fig. S1 of the ESI,† measurements of pH before and after each self-diffusion experiment revealed only small changes. The starting pH averaged 6.96 with a standard deviation of 0.30, and the pH change during diffusion was most commonly 0.25 or less.
In water submersion without applied bias, injected fluxes of Oi are sufficiently high to yield isotopic fractionation within the first few nanometers of the surface.38 In this region, the concentration of 18O dips below the natural abundance level – a counterintuitive phenomenon, because the ambient solution is enriched in 18O. Fractionation originates from high gradients in Oi concentration combined with the statistics of diffusion by an interstitialcy mechanism, wherein two O atoms share a single lattice site and either atom can hop into an adjacent site in a diffusive event. The existence of fractionation provides added features in the diffusion profiles for quantifying profile behavior.
To ascertain the relative contributions of O2 and H2O to injection and diffusion of Oi, the following combinations of isotopic labels and water–gas ambient were investigated:
• Case I: 10% 18O-labeled H2O (Sigma-Aldrich), natural abundance N2 (19 profiles)
• Case II: 10% 18O-labeled H2O, natural abundance O2 (8 profiles)
• Case III: 10% 18O-labeled H2O, natural abundance air (74 profiles)
• Case IV: natural O-abundance (0.2%) D2O (Sigma-Aldrich), 97% labeled 18O2 (Sigma-Aldrich, 12 profiles)
These four cases contain differing concentrations of 18O in the liquid and gas phases, allowing inference of the source of 18O.
18O concentration profiles were measured ex situ by secondary ion mass spectrometry (SIMS) in time-of-flight mode using a PHI-TRIFT III instrument with a 3 keV Cs ion beam source and a spot size of 0.5 mm. 18O concentrations were calibrated to the known natural abundance of 18O (0.2%) in the as-received TiO2 specimens. Profiles were measured at 2–5 different locations on the surface of each specimen.
2.2 DFT calculations
DFT calculations were performed using the Vienna Ab Initio Simulation Package (VASP).62,63 The bulk rutile TiO2 structure was obtained from the Materials Project.64 The structure was optimized using the Perdew–Burke–Ernzerhof65 (PBE) generalized gradient approximation (GGA) exchange–correlation functional, and the projector augmented wave66 (PAW) method was employed. Details of the optimized structure were previously published.67 For the slab modeling, the (110) plane in its 2 × 1 in vacuo reconstruction was employed. Some computational evidence suggests the surface reconstructs to 1 × 1 periodicity upon submersion,68 but most first principles simulations of this surface upon submersion continue to employ the 2 × 1 structure.46,56 A plane wave cutoff energy of 520 eV was used, together with Monkhorst–Pack k-point sampling having a 4 × 4 × 1 mesh size. All atoms were relaxed until the forces on each atom were smaller than 0.01 eV Å−1. The slab thickness was set to 15.74 Å and included five trilayers of (O–Ti2O2–O) to allow for a net charge-neutral stoichiometry.57 A vacuum thickness of 15.0 Å was employed to isolate each slab from its images originating from periodic boundary conditions.To model specific adsorption cases, all potential adsorption sites were examined. The (110) plane includes Ti5f, O3f and O2f sites, although only the first two lie in-plane and are of direct concern here. Adsorbed O atoms adopt an “on-top” geometry at the Ti5f site. At the O3f site, adsorbed O atoms can adopt a “dumbbell” geometry (neutral charge) containing an O–O bond or a “split” geometry (−2 charge) that contains bonds only to surrounding metal cations. For the split, we employed a previous protocol57 that places an O vacancy on the backside of the slab to facilitate adsorbate charging while maintaining the simulation's Fermi energy (EF) at the top of the valence band. The adsorption energy was computed as described previously,57 with the oxygen chemical potential computed under standard conditions (25 °C and 1 atm) for both O-rich (μO = −4.68 eV) and Ti-rich (μO = −10.11 eV) environments.
3. Results
Fig. 1 illustrates typical 18O SIMS profiles following diffusion. The general features match those observed previously in the absence of applied potential bias,37,38 with the most dramatic being the isotopic fractionation represented by the near-surface “valley” wherein the 18O concentration drops well below natural abundance (Cnat = 1.29 × 1020 cm−3) as described at length elsewhere.38 This phenomenon is transient and represents a manifestation of uphill diffusion69 in the solid state. | ||
| Fig. 1 (a) Example 18O profiles at several locations on a single specimen of case II (H2O with 10 at% 18O) with Vappl = −0.8 V vs. Ag/AgCl, 70 °C, t = 60 min, illustrating “valley” regions (having 18O concentrations below natural abundance). The variability is characteristic as discussed in the text. (b) Example 18O profiles comparing case II (“H2O labeled,” 10 at% 18O) and case IV (“O2 labeled,” 97 at% 18O) at 70 °C, t = 60 min. Although Vappl varies, this condition does not affect the profiles; the variation has the same origin as in (a). Typical O2-labeled profiles extend much deeper than H2O-labeled profiles and exhibit deeper and wider valleys near the surface. (c) Example profile showing definitions of the metrics W and Cmin, F18, λ1 and λ2. Profile was measured in air (case I) at Vappl = −0.4 V, 70 °C, t = 60 min. For all profiles here, the water contained no electrolyte, and was air-equilibrated prior to diffusion by bubbling for 10 min. | ||
All features are stable during long-term storage. Fig. 1a shows examples of profiles for labeled water with natural abundance O2 (case II). Details of the profile shapes vary with position on a given specimen. Such variations have been discussed at length elsewhere58,70 and arise in large part from differences in the level of surface contamination, especially adventitious carbon, which can poison the surface sites that facilitate injection. Fig. 1b shows typical profiles comparing cases II and IV, wherein the gas ambient is pure O2 and either the water or the gas, respectively, is isotopically labeled. Differences between the two cases are evident despite the variability; profiles for case IV extend much deeper than case II and exhibit deeper and wider valleys near the surface. This latter behavior seems quite counterintuitive, as greater 18O enrichment in the gas and no enrichment in the liquid enhances the degree of fractionation.
Fig. 1c illustrates five metrics that were used to quantify the profiles:
• F18: net 18Oi injection flux.
• Cmin: near-surface minimum of the 18O concentration in the “valley.”
• W: width of the region for which the 18O concentration is below its natural abundance.
• λ1 and λ2: characteristic lengths describing bi-exponential decay, with λ1 ≪ λ2.
W and Cmin characterize the extent of isotopic fractionation, while λ1 and λ2 describe the penetration of 18O into the solid. The ESI† details the fitting procedures employed to obtain these metrics.
As shown in Fig. S2 and Table S2 of the ESI,† the metrics change regardless of the applied potential's value. The mechanism by which the applied potential bias operates remains unknown. One possibility is that the electrode system unintentionally removes a contaminant that would normally poison injection sites. Alternatively, a form of electrostatic electrochemistry71,72 operates, wherein redox reactions occur on statically charged insulators. The static charge in published reports is usually created by mechanical rubbing,73,74 and whether charge transfer involves ions or electrons is still debated. A third possibility is that an electric field enhances injection.
Knowledge of the mechanism is not important for the focus of this work, which seeks to demonstrate the importance of bulk and surface O interstitials in mediating the exchange of O atoms between TiO2 and the liquid. Bulk and surface interstitials are already known to serve this purpose for TiO2 in water without applied bias, both thermally37 and with UV stimulation.70 As the sections below will show, applying potential bias does not change this picture. Applying bias simply accentuates the profile metrics compared to water without bias, thereby making measurement easier. For example, applying potential bias increases the net injection fluxes by over an order of magnitude, which steepens the gradient for fractionation and greatly extends the penetration of interstitials up to roughly 10 μm into the solid. Accordingly, the 18O-depleted region's width increases from an average of 2.8 nm to as high as 90 nm. Isotopic depletion increases from an average factor of 1.4 below natural abundance to as high as 3.
3.1 Statistical tests for isotope and O2 concentration effects
Table 1 shows the mean and standard deviation for each of the five metrics (F18, W, Cmin, λ1, λ2) for the four ambient conditions studied. The results in Table 1 appear as bar graphs in Fig. 2. The variability among profiles induces large standard deviations for each metric. Hence, statistical tests were employed to determine whether changes in the experimental conditions between cases lead to a significant change in the mean. Such tests yield a probability (“p-value”) that quantifies the likelihood of the null hypothesis being true. The null hypothesis is that a difference in the mean between two cases arises from random chance.| Case I | Case II | Case III | Case IV | |
|---|---|---|---|---|
![[F with combining macron]](https://www.rsc.org/images/entities/i_char_0046_0304.gif) 18 (cm−2 s−1)
18 (cm−2 s−1) |
1.6(1.0) × 1011 | 1.2(0.6) × 1011 | 3.0(2.9) × 1011 | 5.1(4.5) × 1011 |
![[W with combining macron]](https://www.rsc.org/images/entities/i_char_0057_0304.gif) (nm) (nm) |
8.3(5.3) | 6.1(3.0) | 13.6(14.4) | 27.5(16.1) |
![[C with combining macron]](https://www.rsc.org/images/entities/i_char_0043_0304.gif) min (cm−3)
min (cm−3) |
7.5(1.8) × 1019 | 8.3(1.4) × 1019 | 8.3(2.0) × 1019 | 4.2(1.0) × 1019 |
|
C
nat–min (cm−3) |
5.4(1.8) × 1019 | 4.6(1.4) × 1019 | 4.6(2.0) × 1019 | 8.6(1.0) × 1019 |
![[small lambda, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cc.gif) 1 (nm)
1 (nm) |
21(14) | 17(10) | 45(45) | 80(79) |
|
2 (nm) |
390(270) | 240(140) | 420(350) | 1160(850) |
 | ||
| Fig. 2 Bar graphs showing means and standard deviations for profile metrics in each of the four cases: (a) F18 (b) λ1 (c) W (d) Cmin (e) λ2. For all metrics, conditions are 70 °C, t = 60 min. | ||
Histograms of the metric distributions for the largest data set (case III) exhibit considerable deviations from normality even after logarithmic transformation (Fig. S3 and S4 in the ESI†). The classical “parametric” Student's t-test for comparing means exhibits considerable robustness to deviations from a normal distribution.75,76 Nevertheless, we also employed a common “non-parametric” Mann–Whitney U-test that does not presuppose normality. There is no reason to believe cases I–IV have differently shaped distributions.
In such situations, a non-parametric test compares medians77 rather than means. However, this difference in data aggregation affects the statistical power of t and Mann–Whitney tests (i.e., the likelihood of detecting a statistically significant effect). The values for most of the metrics in Table 1 vary over 1.5 to 2 orders of magnitude (e.g., 0.6 to 94 nm for W). The means typically exceed the medians by a factor of 1.3 to 1.6. Thus, closely spaced small values are weighted more heavily than widely spaced large ones in a statistical test that compares medians instead of means. This property decreases the vulnerability of Mann–Whitney tests to type I errors that reject the null hypothesis when it is true but increases their vulnerability to type II errors that fail to reject the null hypothesis when it is false.
Table 2 shows the p-values for each metric obtained from t-tests and Mann–Whitney U-tests for all pairwise permutations of ambient conditions. The comparison of cases II–IV has particular importance because no change occurs in the chemical composition of either gas (pure O2) or liquid. Only the isotopic composition changes. Both tests yield p < 0.02 for all profile metrics. In other words, the likelihood is very high that changing only the 18O mole fractions in the gas and liquid (at constant partial pressure of O2) suffices to influence all aspects of the 18O profiles. This result shows that O2 gas supplies some of the injected oxygen. Furthermore, the effects extend at least as deep as the largest individual value of λ2, i.e., >1 μm.
| Ambient pair | F 18 (cm−2 s−1) | C min (cm−3) | W (nm) | λ 1 (nm) | λ 2 (nm) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| p t-test | p MW | p t-test | p MW | p t-test | p MW | p t-test | p MW | p t-test | p MW | |
| Cases I–II | 0.19 | 0.44 | 0.24 | 0.33 | 0.18 | 0.37 | 0.47 | 0.94 | 0.075 | 0.19 |
| Cases I–III | 8 × 10−4 | 0.15 | 0.12 | 0.45 | 0.018 | 0.20 | 1 × 10−4 | 0.038 | 0.73 | 0.83 |
| Cases I–IV | 0.022 | 7 × 10−4 | 9 × 10−7 | 8 × 10−5 | 1 × 10−3 | 2 × 10−4 | 0.027 | 6 × 10−4 | 0.010 | 6 × 10−4 |
| Cases II–III | 4 × 10−5 | 0.087 | 0.85 | 0.65 | 4 × 10−4 | 0.092 | 4 × 10−5 | 0.13 | 0.011 | 0.18 |
| Cases II–IV | 0.012 | 8 × 10−4 | 1 × 10−5 | 2 × 10−4 | 4 × 10−4 | 4 × 10−4 | 0.019 | 0.0014 | 0.0056 | 5 × 10−4 |
| Cases III–IV | 0.14 | 0.041 | 1 × 10−9 | 4 × 10−4 | 0.011 | 1 × 10−3 | 0.17 | 0.078 | 0.012 | 7 × 10−5 |
Cases I, II and III entail no change in isotopic composition of either gas or liquid; only the chemical concentration of O2 varies in the gas phase. The t-test p-values are rather large for I–II but are quite small for many metrics involving I–III and II–III. The corresponding Mann–Whitney p-values are noticeably larger for these latter two comparisons. However, the p-values for λ1 in I–III are quite small at 0.038 and are also rather small for F18 and W in II–III at 0.087 and 0.092, respectively. In combination, the two tests therefore agree that O2 concentration affects some metrics of the profiles.
3.2 DFT simulations
Fig. S5 (ESI†) shows surface geometries of both pristine and bridging hydroxylated TiO2(110) surfaces. For the bridging hydroxylated surface, two hydrogen atoms occupy bridging O2f sites to represent the fragments of liquid water dissociation. Fig. 3 displays the metastable surface geometries of O atoms adsorbed on pristine TiO2(110), which include dumbbell, split and on-top structures. The dumbbell and split configurations both associate two O atoms with a surface O3f site originally occupied by a single O atom. The two atoms represent distorted versions of corresponding Oi configurations in the bulk reported previously, and thus act like “surface interstitials.”57 The structures remain minimally changed upon hydroxylation of the surface, as shown in Fig. 4. Table 3 summarizes the calculated adsorption energies and Bader charges in the limit of maximally O-rich conditions investigated here. Adsorption energies appear for values of EF at the valence band maximum (VBM, EF = 0 eV) and conduction band minimum (CBM, EF = 3.1 eV). For completeness, Table S3 (ESI†) shows parallel results for maximally Ti-rich conditions. | ||
| Fig. 3 Geometry of metastable adsorbed O configurations on pristine TiO2(110) terraces, including (a) and (b) dumbbell (Odum), (c) and (d) split (Osp) and (e) and (f) on-top (Otop). Panels (a), (c) and (e) show top views and (b), (d) and (f) show side views. Shading colors respectively represent blue for Ti and red for O. | ||
 | ||
| Fig. 4 Geometry of metastable adsorbed O configurations with co-adsorbed bridging hydroxyls on TiO2(110) terraces, including (a) and (b) dumbbell (Odum), (c) and (d) split (Osp) and (e) and (f) on-top (Otop). Panels (a), (c) and (e) show top views and (b), (d) and (f) show side views. Shading colors respectively represent blue for Ti, red for O, and white for H. | ||
| TiO2(110) surface conditions | Adsorption configuration | Adsorption energy (eV) | Bader charge (e−) | Charge state | ||
|---|---|---|---|---|---|---|
| E F = 0 eV | E F = 3.1 eV | Adsorbed O atom | Neighboring O atom | |||
| Pristine | Dumbbell | 0.80 | 0.80 | −0.43 | −0.69 | 0 |
| Split | 0.95 | −5.11 | −1.11 | −0.94 | −2 | |
| On-top | 2.63 | −0.40 | −0.61 | −1.09 | −1 | |
| Bridging hydroxylated | Dumbbell | 0.48 | 0.48 | −0.54 | −0.71 | 0 |
| Split | −0.30 | −6.36 | −1.12 | −1.03 | −2 | |
| On-top | −2.21 | −5.23 | −0.87 | −1.13 | −1 | |
Bader charge analysis of the dumbbell surface interstitial structure on the pristine surface shows −0.43e− on the topmost atom and −0.69e− on its nearest neighbor just below, for a sum of −1.12e−. Previous analysis57 has demonstrated this species to be neutral. By contrast, the split structure is known to be charged −2. The topmost atom has a Bader charge of −1.11e−, and −0.94 resides on the underlying nearest-neighbor O just below for a total of −2.05e−. For the on-top geometry, the adsorbed O has a Bader charge of −0.61e−. The symmetry of this structure does not associate this O with any particular in-plane atom, but each of the four equivalent O3f nearest- neighbor atoms in the surface plane have a Bader charge of −1.09e−. Thus, a sum comparable to those of the dumbbell and split configurations is −1.70e−, which lies intermediate between the values for the dumbbell and the split. The calculations therefore suggest an on-top charge state of −1, consistent with previously published DFT results for isolated O.49 For the hydroxylated surface, the Bader charges all become slightly more electron-rich, but the ordering does not change. Thus, we infer no change in charge state induced by hydroxylation.
For nearly all values of EF, the adsorption energies on the pristine surface in Table 3 show that the split geometry is most stable (most negative). The dumbbell becomes most stable by a small margin when EF is near the VBM. On the hydroxylated surface, the on-top geometry is most stable for EF near the VBM, while the split is most stable for EF near the CBM. In contrast with the surface interstitial structures, for the on-top configuration the adsorbed O atom pulls the underlying Ti atom significantly above the surface plane. Fig. 4e and f show that hydroxylation stabilizes the resulting bond stretching by pulling the adsorbed O laterally toward a nearby H atom. Much less stabilization is possible for the surface interstitial structures.
4. Discussion
4.1 Contributions of injected O from H2O and O2
The 18O profiles provide strong evidence that both H2O and dissolved O2 contribute O that diffuses into submerged TiO2. Do O2 and H2O contribute independently or employ the same pathway? Adsorbate populations on submerged TiO2 are dominated by OH (either anion or neutral) and molecular H2O.39–42 Adsorbed OH is not very acidic,78 yet experiments with deuterated water have shown37 that oxygen injects as Oi, not OH. Similar experiments performed here with applied bias yield the same results. This apparent paradox can be reconciled by the capacity for hydrogen bonding79 and acid–base chemistry80 afforded by the liquid phase. These solvation effects stabilize intermediates such as adsorbed O,35 as evidenced by electron spin resonance81 and photoluminescence.45 Submersion can reduce the activation energy for O2 dissociation82 and increase the acidity of OH83 by reducing the barrier for deprotonation.84 The temperature dependence of Oi injection in submersion is much less than that of elementary-step injection of adsorbed O, which suggests the rate-limiting step is kinetically upstream.37 Thus, there is ample reason to believe that deprotonation of OH to O limits the overall rate of Oi creation, meaning that adsorbed OH serves as a single kinetic conduit for movement of O between the solid and fluid.Note that solvation effects limit the mechanistic insights afforded by the large literature for in vacuo adsorption of H2O and O2 on TiO2. As with submerged TiO2, those studies show that H2O can dissociate on TiO2 and other common metal oxides. On TiO2, surface VO aids dissociation by providing adjacent cation and anion sites that bond readily to H2O and the products OH and H.40 However, OH appears as the only oxygenated product39,61,85 because no solvation is possible to stabilize adsorbed O. Without doping or photoexcitation, molecular O2 chemisorption in vacuo requires electron donation from VO or other surface defects.40 Dissociation of O2 to O is favored by conditions that increase the electronegativity of the surface,40,86 including the presence of electron donors such as surface VO.40,50 However, gaseous O2 does not adsorb and dissociate fast enough on TiO2(110) to inject Oi unless temperatures exceed about 600 °C.87 In submersion, solvation effects enable dissociation and injection of Oi even at 70 °C.
The aftereffects of ion sputtering for surface cleaning also limit the direct translation of in vacuo mechanistic findings to TiO2 submersion. Sputtering preferentially removes O from the solid, which chemically reduces some of the Ti and forms VO.40 Reduction of the surface layer is irreversible in vacuo below 400 K40,88 unless the ion fluence is low.40,89,90 With low fluence, submersion quickly removes surface VO with dissolved O240,91,92 or H2O itself.93–95 However, submersion reverses VO formation in the near subsurface (2–3 nm) slowly and only partially.96 This failure to reverse subsurface reduction reflects the suppression of Ti interstitials, which are central to in vacuo reversal of chemical reduction above 400 K.88 Ti interstitials play no role at submersion temperatures because there is no generation mechanism, and their high mobility ensures their sequestration in bulk traps or at the surface.97
Two mathematical models were formulated and compared to test the hypothesis that OH serves as a single kinetic conduit for O. Model derivations appear in the ESI.† One model is agnostic about the injection mechanism (thereby not aligning with the hypothesis) and assumes that H2O and O2 contribute Oi and isotopic labels in linear proportion to their respective total concentrations in the fluid. The net injection flux F18 of 18O (time-averaged) is then given by:
| F18 = Y18,WxFW + Y18,O2XO2(1 − x)FW, | (1) |
The second model aligns with the hypothesis by presupposing a Langmuir-style dual pathway mechanism wherein both O2 and H2O inject through the same adsorbed OH intermediate whose surface coverage does not vary. This differs slightly from “competitive adsorption,” wherein O2 and H2O compete for the same surface sites but have different coverages. For simplicity, first order kinetics are assumed for dissociative adsorption and desorption of H2O and O2 to form adsorbed O. First order kinetics are also assumed for conversion of OH to injected O. The rate constant kinj for injection is a composite quantity that implicitly contains the rate constants for deprotonation of OH and injection of O, and the concentration of adsorbed O. The resulting expression for F18 is:
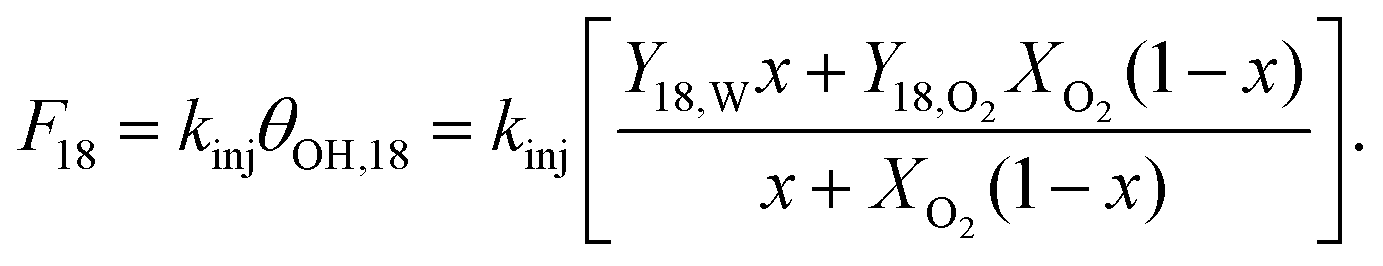 | (2) |
In the asymptotic limit of x = 1, eqn (1) from the linear proportion model represents a limiting case of eqn (2) from the dual pathway model with FW = kinj. The dual pathway model has the same number of fitting parameters (two) as the linear proportion model but allows for possible “saturation” of adsorbed OH with gas-derived O as the O2 concentration rises. Thus, fitting eqn (1) and (2) to the means values from experiments offers a direct comparison with the same number of fitting parameters of two different models: one that allows for saturation and one that does not.
Linear regression using the linear proportion model yields x = 0.78, FW = 2.5 × 1012 cm−2 s−1, and a total residual variance of 1.9 × 1022 cm−4 s−2. Nonlinear regression using the dual pathway model yields x = 0.76, kinj = 2.2 × 1012 cm−2 s−1, and a total residual variance of 1.5 × 1022 cm−4 s−2. The factor of 1022 in the variances can be removed by choosing different units or scaling. The point of comparison centers on the multiplying factors 1.5 vs. 1.9. The difference is modest, but the lower residual fitting error for the dual pathway model is consistent with the hypothesis that adsorbed OH provides the single conduit by which O exchanges between fluid and solid.
The two models yield nearly identical values for x. This factor almost certainly varies with temperature, pH and other experimental variables. Although H2O provides most of the injected O in the present experiments, O2 also contributes significantly. Notably, the concentration of dissolved O2 at 70 °C is only 0.83 × 10−3 mole l−1, while the concentration of H2O is 6.5 × 104 times higher at 54.2 mole l−1. Therefore, the reaction rate constant to form injectable O via adsorbed OH must be considerably lower for H2O than for O2, which is consistent with chemical intuition that H2O is much less reactive than O2.
4.2 Role of bulk Oi in lattice O participation
As discussed elsewhere,37 the diffusive penetration of 18O to hundreds of nanometers at 70 °C is very strong evidence that the diffusing species is Oi. At this temperature, VO does not diffuse quickly enough to penetrate that far. The dominance of Oi implies that VO has been eliminated by Oi annihilation, both at the surface and within the bulk.37The prevalence of Oi to depths of hundreds of nanometers indicates that lattice O from these depths exchanges with O from both H2O and dissolved O2. This can be seen as follows. In TiO2,67,87,98 Oi comprises two atoms symmetrically arranged around a single O lattice site. The atoms assume either a neutral “dumbbell” geometry with an O–O bond, or a charged “split” geometry with bonds only to surrounding metal cations. Diffusion of Oi occurs by an interstitialcy mechanism, in which each constituent atom has a 50% chance of hopping – leaving its partner behind in the lattice. Although the net flow of Oi is toward the deep bulk, numerous random hopping and lattice exchange events enable some O atoms from the deep bulk to reach the surface for transfer to the fluid.
Several factors drive Oi creation at the surface. Some driving forces are energetic, impelled by defect reactions. An example is annihilation of VO by Oi, which oxidizes the solid. Other examples include addition of Oi to extended defects99 and combination with adventitious hydrogen to form Oi–Hi− (ref. 37) or other small clusters.57 Another driving force is entropic. At 70 °C, the solid initially contains virtually no Oi because this species has sufficient mobility to disappear at trap sites or the surface. The fluid maintains a continuous supply of adsorbed O, which converts into Oi with a modest barrier near 0.8 eV29 and spreads entropically into the solid.
The rate of Oi generation observed here may be compared to the rate of a typical reaction involving O2 on TiO2 − the OER in photostimulated water splitting, which several groups have investigated on electrically conductive rutile TiO2(110).45,100–103 Photocurrents range from 0.54 to 2.2 mA-cm−2 under UV irradiation in the range from 0.2 to 7 mW-cm−2. These photocurrents correspond to O2 molecular fluxes in the range 1–3 × 1015 cm−2 s−1. Here, FW or kinj set the rough magnitude the Oi injection fluxes − roughly three orders of magnitude lower. None of the photocurrent studies estimated their current efficiency, so competing reactions could have contributed to the measured current. However, Oi injection seems to constitute only a minor side reaction.
4.3 Role of a “surface interstitial” in lattice O participation
Adsorbed O on TiO2(110) is usually depicted as sitting directly atop an underlying Ti5c atom based on in vacuo scanning probe experiments49–55 and first principles simulations49,50,52–55 including in submersion.46,56 The charge state can be toggled between −1 and −2 with a scanning probe tip,53–55 and this species has been represented in DFT simulations as both −156 and −2.46 The present DFT calculations make plausible the hypothesis that both on-top and surface interstitial (neutral dumbbell and charged split) configurations are metastable. The dominant form depends upon the value of EF, the presence of liquid water, the energy barriers for interconversion, and other factors.The on-top configuration is readily protonated. Protonation of the surface interstitial was not investigated here, but its high bond coordination implies little propensity for this reaction. Under some conditions the on-top and surface interstitial forms could coexist in comparable concentrations. Thus, there is good reason to believe that O moves from the fluid into the solid by a pathway in which H2O or O2 form OH. O atoms produced by O2 dissociation convert rapidly to OH before injecting; in other words, equilibrium between OH and O greatly favors OH. However, that equilibrium involves the on-top form of O. Some of that form converts into the surface interstitial, which then injects as Oi. Diffusion of Oi into the solid ensues, followed by exchange into the lattice. Movement of O from the lattice into the fluid occurs through the reverse of these steps as depicted in Fig. 5.
 | ||
| Fig. 5 Schematic comparison of O atom exchange between rutile TiO2(110) and H2O or dissolved O2 in the fluid – mediated by (a) O vacancies in the conventional picture and (b) O interstitials in the proposed picture. Diagrams do not attempt to represent exact geometric configurations or certain details of reaction stoichiometry, e.g., the fate of H or exchange of O between mobile species and the lattice. Balls depict atoms of Ti (purple) H (pink) and O originating primarily from the fluid (black) or solid lattice (grey). In reality, atoms originating from the fluid and solid are represented to some degree in all reaction and diffusion pathways. | ||
This mechanistic picture can only be considered plausible, not proven. The present calculations simulate neither free water molecules nor the possibilities for hydrogen bonding and protonation. Water molecules in the liquid probably screen hydrogen bonding interactions across rows, which seem especially effective at stabilizing the on-top structure. Moreover, liquid water changes relative stability in other ways, such as affecting the value of EF at the surface via pH or dissolved ions that adsorb.
The foregoing discussion highlights a conceptual disconnect between computational simulations of adsorbed O focused on catalysis, photocatalysis and electrochemistry vs. those focused on defect reaction and diffusion in the solid. The former simulations typically emphasize mechanisms involving VO, with little explicit consideration of EF in the solid. The latter simulations emphasize Oi but treat liquid solvation and pH simplistically if at all. Both approaches yield useful insights, but further progress must account for contributions to the surface thermodynamics and kinetics from both the liquid and solid.
4.4 Surface sites active for Oi production
What kinds of surface sites other than VO promote dissociation of H2O and O2 in submersion? Clues come from fully amorphous metal oxides, which enhance activity for the OER through a wide variety of surface site geometries and their structural flexibility. Such sites are unfortunately difficult to characterize,104 which is scientifically unsatisfying.61 However, the design of “high-entropy” oxides embraces such complexity by adding five or more dopant metals to tune the behavior of active sites.23 With rms surface roughness of 0.14 ± 0.08 nm, the etched TiO2 employed here also supports ill-defined sites having a wide range of effectiveness for injecting Oi.58What accounts for the susceptibility of TiO2 to poisoning of Oi creation? The most reactive sites on catalytic surfaces are often most vulnerable to deactivation by poisoning.105 An example afflicts TiO2 photocatalysts used for the ORR to degrade aqueous pollutants,8,106,107 wherein the poisons derive from dissolved organic matter. Deprotonation of OH and subsequent injection of Oi entails a form of heterogeneous catalysis, and a similar correlation between injection activity and poisoning susceptibility appears to hold for injection of Oi from oxide surfaces.58
4.5 Implications for other oxides and reaction conditions
Because rutile TiO2 is a prototypical metal oxide, this picture based on Oi probably generalizes in many cases to other oxides − explaining important aspects of their thermal, electrochemical, and photochemical reactions with dissolved O2. Examples include oxides fabricated to be hyperstoichiometric, including spinel ferrites,108–111 perovskites,112 and Ruddlesden–Popper oxides.29,30,113–115 Hyperstoichiometric oxides permanently contain mobile Oi, and often exhibit enhanced catalysis and photocatalysis rates.29,30In stoichiometric oxides, Oi adopts dumbbell or split configurations like those within TiO2 in wurtzite ZnO,116–121 corundum Al2O3,122–124 fluorite CeO2,125 corundum Cr2O3,126 hematite Fe2O3,127,128 perovskite SrTiO3,129–131 fluorite ThO2,125,132 fluorite UO2,133,134 tetragonal135 and cubic ZrO2,136,137 and Ruddlesden–Popper oxides.138 Following the behavior of TiO2, the formation energy for Oi in these oxides is typically lower than for VO in O-rich environments,87,117,130,139,140 as is the Oi hopping barrier.37,141 To existing barrier compilations, low barriers for Oi hopping (0.13–0.47 eV) may be added for corundum Cr2O3,126 hematite Fe2O3,128 perovskite SrTiO3,129 fluorite ThO2,125 and fluorite UO2.142
Counterintuitively, Oi may mediate aqueous reactions of O even in reduced oxides. The high diffusional mobility of Oi would make its dominance temporary and difficult to detect. Thus, Oi may dominate near the surface of vacancy-rich perovskites during reaction and then be replaced by VO from the bulk after the reaction ends. Such behavior would accord with the malleability and rapid reversibility of perovskite surface composition and structure observed in other contexts.32
5. Conclusions
Isotopic self-diffusion measurements in submerged rutile TiO2 single crystals with varying concentrations of 18O isotopic label show that O exchange occurs between rutile TiO2(110) and dissolved O2 as well as the H2O liquid. Diffusion in the TiO2 is mediated by oxygen interstitials (Oi). First-principles calculations strongly suggest that adsorbed O in on-top and “surface interstitial” configurations mediate the surface reaction rather than VO. Adsorbed OH appears to provide a single pathway for H2O and O2 to contribute injectable oxygen, although fitting the diffusion data to simple models indicates that H2O contributes more. The most active sites on the surface for OH deprotonation and Oi injection are probably ill-defined and vulnerable to deactivation by poisoning. Compared to the rate of a typical reaction between O2 and TiO2 such as photostimulated OER, the rate of Oi injection is small. Because rutile TiO2 is a prototypical metal oxide, this physical picture helps to explain many aspects of thermal, electrochemical, and photochemical reactions between dissolved O2 and a variety of oxides.Data availability
The data that support the findings of this study are included within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. National Science Foundation under grant DMR 17-09327 and DMR 23-22121, and the Air Force Office of Scientific Research under grant FA9550-21-0078. SIMS and XPS measurements were performed in the Materials Research Laboratory Central Research Facilities, University of Illinois at Urbana-Champaign. We gratefully acknowledge the use of high-performance computing (HPC) resources, such as Nurion and Neuron, provided by the R&D Innovation-Support Program at the Korea Institute of Science and Technology Information (KISTI). We are grateful to Dr. Stephen D. Bond for helpful advice on curve-fitting the 18O profiles.References
- M. Rafique, S. Hajra, M. Irshad, M. Usman, M. Imran, M. A. Assiri and W. M. Ashraf, Hydrogen Production Using TiO2-Based Photocatalysts: A Comprehensive Review, ACS Omega, 2023, 8, 25640–25648 CrossRef CAS PubMed.
- Y. Nosaka, Water Photo-Oxidation over TiO2—History and Reaction Mechanism, Catalysts, 2022, 12, 1557 CrossRef CAS.
- H. Eidsvåg, S. Bentouba, P. Vajeeston, S. Yohi and D. Velauthapillai, TiO2 as a Photocatalyst for Water Splitting—An Experimental and Theoretical Review, Molecules, 2021, 26, 1687 CrossRef PubMed.
- A. Miyoshi, S. Nishioka and K. Maeda, Water Splitting on Rutile TiO2-Based Photocatalysts, Chem. – Eur. J., 2018, 24, 18204–18219 CrossRef CAS PubMed.
- S. Chen and Y. H. Hu, Color TiO2 Materials as Emerging Catalysts for Visible-NIR Light Photocatalysis, A Review, Catal. Rev., 2024, 66, 1951–1991 CrossRef CAS.
- M. A. Al-Nuaim, A. A. Alwasiti and Z. Y. Shnain, The photocatalytic process in the treatment of polluted water, Chem. Pap., 2023, 77, 677–701 CrossRef CAS PubMed.
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang, P. Huo and X. Li, A review on heterogeneous photocatalysis for environmental remediation: From semiconductors to modification strategies, Chin. J. Catal., 2022, 43, 178–214 CrossRef CAS.
- L. Ren, W. Huo, G. Li, W. Choi and T. An, Photocatalytic mechanisms and photocatalyst deactivation during the degradation of 5-fluorouracil in water, Catal. Today, 2023, 410, 45–55 CrossRef CAS.
- G. Ren, H. Han, Y. Wang, S. Liu, J. Zhao, X. Meng and Z. Li, Recent Advances of Photocatalytic Application in Water Treatment: A Review, Nanomaterials, 2021, 11, 1804 CrossRef CAS PubMed.
- H. Sheng, H. Ji, W. Ma, C. Chen and J. Zhao, Direct Four-Electron Reduction of O2 to H2O on TiO2 Surfaces by Pendant Proton Relay, Angew. Chem., Int. Ed., 2013, 52, 9686–9690 CrossRef CAS PubMed.
- H. Du, H. Luo, M. Jiang, X. Yan, F. Jiang and H. Chen, A review of activating lattice oxygen of metal oxides for catalytic reactions: Reaction mechanisms, modulation strategies of activity and their practical applications, Appl. Catal., A, 2023, 664, 119348 CrossRef CAS.
- J. Chen, W. Zhang, H. Li, W. Li and D. Zhao, Recent advances in TiO2-based catalysts for N2 reduction reaction, SusMat, 2021, 1, 174–193 CrossRef CAS.
- X.-Y. Xie, P. Xiao, W.-H. Fang, G. Cui and W. Thiel, Probing Photocatalytic Nitrogen Reduction to Ammonia with Water on the Rutile TiO2(110) Surface by First-Principles Calculations, ACS Catal., 2019, 9, 9178–9187 CrossRef CAS.
- E. Tayyebi, Á. B. Höskuldsson, A. Wark, N. Atrak, B. M. Comer, A. J. Medford and E. Skúlason, Perspectives on the Competition between the Electrochemical Water and N2 Oxidation on a TiO2(110) Electrode, J. Phys. Chem. Lett., 2022, 13, 6123–6129 CrossRef CAS PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, Photocatalytic Conversion of Nitrogen to Ammonia with Water on Surface Oxygen Vacancies of Titanium Dioxide, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- P.-W. Huang and M. C. Hatzell, Prospects and good experimental practices for photocatalytic ammonia synthesis, Nat. Commun., 2022, 13, 7908 CrossRef CAS PubMed.
- R. B. Domínguez-Espíndola, D. M. Arias, C. Rodríguez-González and P. J. Sebastian, A critical review on advances in TiO2-based photocatalytic systems for CO2 reduction, Appl. Therm. Eng., 2022, 216, 119009 CrossRef.
- J. Chen, T. Li, X. An and D. Fu, Photocatalytic Reduction of CO2 by TiO2 Modified Materials: Recent Advances and Outlook, Energy Fuels, 2024, 38, 7614–7636 CrossRef CAS.
- Z. Chen, H. Chen, K. Wang, J. Chen, M. Li, Y. Wang, P. Tsiakaras and S. Song, Enhanced TiO2 Photocatalytic 2 e – Oxygen Reduction Reaction via Interfacial Microenvironment Regulation and Mechanism Analysis, ACS Catal., 2023, 13, 6497–6508 CrossRef CAS.
- B. Bastug Azer, A. Gulsaran, J. R. Pennings, R. Saritas, S. Kocer, J. L. Bennett, Y. Devdas Abhang, M. A. Pope, E. Abdel-Rahman and M. Yavuz, A Review: TiO2 based photoelectrocatalytic chemical oxygen demand sensors and their usage in industrial applications, J. Electroanal. Chem., 2022, 918, 116466 CrossRef CAS.
- Y.-F. Li and A. Selloni, Pathway of Photocatalytic Oxygen Evolution on Aqueous TiO2 Anatase and Insights into the Different Activities of Anatase and Rutile, ACS Catal., 2016, 6, 4769–4774 CrossRef CAS.
- C. Zhao, H. Tian, Z. Zou, H. Xu and S.-Y. Tong, Understanding oxygen evolution mechanisms by tracking charge flow at the atomic level, iScience, 2023, 26, 107037 CrossRef CAS PubMed.
- S. C. Pathan, J. S. Shaikh, M. Rittiruam, T. Saelee, V. Márquez, P. Khajondetchairit, S. S. Mali, J. V. Patil, C. K. Hong, P. Praserthdam and S. Praserthdam, High entropy metal oxide/TiO2 nanocomposite for electrocatalytic overall water splitting, J. Alloys Compd., 2024, 176811 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Water electrolysis on La1−xSrxCoO3−δ perovskite electrocatalysts, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- J. S. Yoo, Y. Liu, X. Rong and A. M. Kolpak, Electronic Origin and Kinetic Feasibility of the Lattice Oxygen Participation During the Oxygen Evolution Reaction on Perovskites, J. Phys. Chem. Lett., 2018, 9, 1473–1479 CrossRef CAS PubMed.
- A. M. Frandsen, K. M. Macounová, J. Rossmeisl and P. Krtil, Lattice oxygen evolution in rutile Ru1−xNixO2 electrocatalysts, Electrochim. Acta, 2024, 497, 144567 CrossRef CAS.
- X. Tan, M. Zhang, D. Chen, W. Li, W. Gou, Y. Qu and Y. Ma, Electrochemical Etching Switches Electrocatalytic Oxygen Evolution Pathway of IrOx/Y2O3 from Adsorbate Evolution Mechanism to Lattice-Oxygen-Mediated Mechanism, Small, 2023, 19, 2303249 CrossRef CAS PubMed.
- B. Yin, Y. Li, N. Sun, X. Ji, Y. Huan, D. Dong, X. Hu and T. Wei, Activating ORR and OER in Ruddlesden–Popper based catalysts by enhancing interstitial oxygen and lattice oxygen redox reactions, Electrochim. Acta, 2021, 370, 137747 CrossRef CAS.
- Y. Huan, S. Chen, R. Zeng, T. Wei, D. Dong, X. Hu and Y. Huang, Intrinsic Effects of Ruddlesden–Popper-Based Bifunctional Catalysts for High-Temperature Oxygen Reduction and Evolution, Adv. Energy Mater., 2019, 9, 1901573 CrossRef.
- S. Ding, Y. Zhang, F. Lou, M. Li, Q. Huang, K. Yang, B. Xia, C. Tang, J. Duan, M. Antonietti and S. Chen, Oxygen-vacancy-type Mars–van Krevelen mechanism drives ultrafast dioxygen electroreduction to hydrogen peroxide, Mater. Today Energy, 2023, 38, 101430 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, A review on fundamentals for designing oxygen evolution electrocatalysts, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- Y. Yang, K. Xu and X. Ma, Catalytic Mechanism of Oxygen Vacancy Defects in Metal Oxides, Prog. Chem., 2023, 35, 543–559 CAS.
- J. Ferreira de Araújo, F. Dionigi, T. Merzdorf, H.-S. Oh and P. Strasser, Evidence of Mars-Van-Krevelen Mechanism in the Electrochemical Oxygen Evolution on Ni-Based Catalysts, Angew. Chem., Int. Ed., 2021, 60, 14981–14988 CrossRef PubMed.
- T. E. Jones, D. Teschner and S. Piccinin, Toward Realistic Models of the Electrocatalytic Oxygen Evolution Reaction, Chem. Rev., 2024, 124, 9136–9223 CrossRef CAS PubMed.
- H. A. Tahini, X. Tan, U. Schwingenschlögl and S. C. Smith, Formation and Migration of Oxygen Vacancies in SrCoO3 and Their Effect on Oxygen Evolution Reactions, ACS Catal., 2016, 6, 5565–5570 CrossRef CAS.
- H. Jeong, E. Ertekin and E. G. Seebauer, Surface-Based Post-synthesis Manipulation of Point Defects in Metal Oxides Using Liquid Water, ACS Appl. Mater. Interfaces, 2022, 14, 34059–34068 CrossRef CAS PubMed.
- H. Jeong and E. G. Seebauer, Strong Isotopic Fractionation of Oxygen in TiO2 Obtained by Surface-Enhanced Solid-State Diffusion, J. Phys. Chem. Lett., 2022, 13, 9841–9847 CrossRef CAS PubMed.
- U. Diebold, Perspective: A controversial benchmark system for water-oxide interfaces: H2O/TiO2(110), J. Chem. Phys., 2017, 147, 40901 CrossRef PubMed.
- O. Berger, Understanding the fundamentals of TiO2 surfacesPart II. Reactivity and surface chemistry of TiO2 single crystals, Surf. Eng., 2022, 38, 846–906 CrossRef CAS.
- Z. Zeng, F. Wodaczek, K. Liu, F. Stein, J. Hutter, J. Chen and B. Cheng, Mechanistic insight on water dissociation on pristine low-index TiO2 surfaces from machine learning molecular dynamics simulations, Nat. Commun., 2023, 14, 6131 CrossRef CAS PubMed.
- C. Di Valentin, A. Tilocca, A. Selloni, T. J. Beck, A. Klust, M. Batzill, Y. Losovyj and U. Diebold, Adsorption of Water on Reconstructed Rutile TiO2(011)-(2 × 1): Ti
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O Double Bonds and Surface Reactivity, J. Am. Chem. Soc., 2005, 127, 9895–9903 CrossRef CAS PubMed.
O Double Bonds and Surface Reactivity, J. Am. Chem. Soc., 2005, 127, 9895–9903 CrossRef CAS PubMed. - N. Siemer, D. Muñoz-Santiburcio and D. Marx, Solvation-Enhanced Oxygen Activation at Gold/Titania Nanocatalysts, ACS Catal., 2020, 10, 8530–8534 CrossRef CAS.
- F. Liu, N. Feng, Q. Wang, J. Xu, G. Qi, C. Wang and F. Deng, Transfer Channel of Photoinduced Holes on a TiO2 Surface As Revealed by Solid-State Nuclear Magnetic Resonance and Electron Spin Resonance Spectroscopy, J. Am. Chem. Soc., 2017, 139, 10020–10028 CrossRef CAS PubMed.
- A. Imanishi, T. Okamura, N. Ohashi, R. Nakamura and Y. Nakato, Mechanism of Water Photooxidation Reaction at Atomically Flat TiO2 (Rutile) (110) and (100) Surfaces: Dependence on Solution pH, J. Am. Chem. Soc., 2007, 129, 11569–11578 CrossRef CAS PubMed.
- Y.-B. Zhuang and J. Cheng, Deciphering the Anomalous Acidic Tendency of Terminal Water at Rutile(110)–Water Interfaces, J. Phys. Chem. C, 2023, 127, 10532–10540 CrossRef CAS.
- P. Gono, F. Ambrosio and A. Pasquarello, Effect of the Solvent on the Oxygen Evolution Reaction at the TiO2–Water Interface, J. Phys. Chem. C, 2019, 123, 18467–18474 CrossRef CAS.
- L. J. Falling, J. J. Velasco-Vélez, R. V. Mom, A. Knop-Gericke, R. Schlögl, D. Teschner and T. E. Jones, The ladder towards understanding the oxygen evolution reaction, Curr. Opin. Electrochem., 2021, 30, 100842 CrossRef CAS.
- I. Sokolović, M. Reticcioli, M. Čalkovský, M. Wagner, M. Schmid, C. Franchini, U. Diebold and M. Setvín, Resolving the adsorption of molecular O2 on the rutile TiO2(110) surface by noncontact atomic force microscopy, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14827–14837 CrossRef PubMed.
- E. Lira, J. Ø. Hansen, P. Huo, R. Bechstein, P. Galliker, E. Lægsgaard, B. Hammer, S. Wendt and F. Besenbacher, Dissociative and molecular oxygen chemisorption channels on reduced rutile TiO2(110): An STM and TPD study, Surf. Sci., 2010, 604, 1945–1960 CrossRef CAS.
- H. F. Wen, H. Sang, Y. Sugawara and Y. J. Li, Imaging oxygen molecular adsorption and dissociation on the Ti site of rutile TiO2(110) surface with real configuration at 78 K by atomic force microscopy, Phys. Chem. Chem. Phys., 2020, 22, 19795–19801 RSC.
- I. Lyubinetsky, N. A. Deskins, Y. Du, E. K. Vestergaard, D. J. Kim and M. Dupuis, Adsorption states and mobility of trimethylacetic acid molecules on reduced TiO2(110) surface, Phys. Chem. Chem. Phys., 2010, 12, 5986–5992 RSC.
- Q. Zhang, H. F. Wen, Y. Adachi, M. Miyazaki, Y. Sugawara, R. Xu, Z. H. Cheng and Y. J. Li, Electrical Engineering of the Oxygen Adatom and Vacancy on Rutile TiO2(110) by Atomic Force Microscopy at 78 K, J. Phys. Chem. C, 2019, 123, 28852–28858 CrossRef CAS.
- Q. Zhang, Y. J. Li, H. F. Wen, Y. Adachi, M. Miyazaki, Y. Sugawara, R. Xu, Z. H. Cheng, J. Brndiar, L. Kantorovich and I. Štich, Measurement and Manipulation of the Charge State of an Adsorbed Oxygen Adatom on the Rutile TiO2(110)-1 × 1 Surface by nc-AFM and KPFM, J. Am. Chem. Soc., 2018, 140, 15668–15674 CrossRef CAS PubMed.
- Y. Adachi, J. Brndiar, H. F. Wen, Q. Zhang, M. Miyazaki, S. Thakur, Y. Sugawara, H. Sang, Y. Li, I. Štich and L. Kantorovich, Electron dynamics of tip-tunable oxygen species on TiO2 surface, Commun. Mater., 2021, 2, 71 CrossRef CAS.
- D. Wang, T. Sheng, J. Chen, H.-F. Wang and P. Hu, Identifying the key obstacle in photocatalytic oxygen evolution on rutile TiO2, Nat. Catal., 2018, 1, 291–299 CrossRef CAS.
- H. Jeong, E. G. Seebauer and E. Ertekin, Fermi level dependence of gas–solid oxygen defect exchange mechanism on TiO2(110) by first-principles calculations, J. Chem. Phys., 2020, 153, 124710 CrossRef CAS PubMed.
- H. Jeong and E. G. Seebauer, Effects of adventitious impurity adsorption on oxygen interstitial injection rates from submerged TiO 2 (110) and ZnO(0001) surfaces, J. Vac. Sci. Technol., A, 2023, 41, 033203 CrossRef CAS.
- R. L. Kurtz, R. Stock-Bauer, T. E. Msdey, E. Román and J. L. De Segovia, Synchrotron radiation studies of H2O adsorption on TiO2(110), Surf. Sci., 1989, 218, 178–200 CrossRef CAS.
- R. Wang, B. Wang, A. S. Abdullahi and H. Fan, Understanding the prototype catalyst TiO2 surface with the help of density functional theory calculation, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2024, 14, e1686 CrossRef CAS.
- Z. Jakub, M. Meier, F. Kraushofer, J. Balajka, J. Pavelec, M. Schmid, C. Franchini, U. Diebold and G. S. Parkinson, Rapid oxygen exchange between hematite and water vapor, Nat. Commun., 2021, 12, 6488 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. Jeong, E. G. Seebauer and E. Ertekin, First-principles description of oxygen self-diffusion in rutile TiO2: Assessment of uncertainties due to enthalpy and entropy contributions, Phys. Chem. Chem. Phys., 2018, 20, 17448–17457 RSC.
- U. Aschauer and A. Selloni, Structure of the Rutile TiO2(011) Surface in an Aqueous Environment, Phys. Rev. Lett., 2011, 106, 166102 CrossRef CAS PubMed.
- R. Krishna, Uphill diffusion in multicomponent mixtures, Chem. Soc. Rev., 2015, 44, 2812–2836 RSC.
- H. Jeong and E. G. Seebauer, Effects of Ultraviolet Illumination on Oxygen Interstitial Injection from TiO2 under Liquid Water, J. Phys. Chem. C, 2022, 126, 20800–20806 CrossRef CAS.
- J. Zhang, S. Ferrie, S. Zhang, Y. B. Vogel, C. R. Peiris, N. Darwish and S. Ciampi, Single-Electrode Electrochemistry: Chemically Engineering Surface Adhesion and Hardness To Maximize Redox Work Extracted from Tribocharged Silicon, ACS Appl. Nano Mater., 2019, 2, 7230–7236 CrossRef CAS.
- C. Liu and A. J. Bard, Electrons on dielectrics and contact electrification, Chem. Phys. Lett., 2009, 480, 145–156 CrossRef CAS.
- C. Liu and A. J. Bard, Chemical Redox Reactions Induced by Cryptoelectrons on a PMMA Surface, J. Am. Chem. Soc., 2009, 131, 6397–6401 CrossRef CAS PubMed.
- J. Zhang, F. J. M. Rogers, N. Darwish, V. R. Gonçales, Y. B. Vogel, F. Wang, J. J. Gooding, M. Chandramalika, R. Peiris, G. Jia, J.-P. Veder, M. L. Coote and S. Ciampi, Electrochemistry on Tribocharged Polymers Is Governed by the Stability of Surface Charges Rather than Charging Magnitude, J. Am. Chem. Soc., 2019, 141, 5863–5870 CrossRef CAS PubMed.
- M. J. Blanca, R. Alarcón, J. Arnau, R. Bono and R. Bendayan, Datos no normales: ¿es el ANOVA una opción válida?, Psicothema, 2017, 29, 552–557 CrossRef PubMed.
- M. M. Ali and S. C. Sharma, Robustness to nonnormality of regression F-tests, J. Econom., 1996, 71, 175–205 CrossRef.
- M. P. Perme and D. Manevski, Confidence intervals for the Mann–Whitney test, Stat. Methods Med. Res., 2018, 28, 3755–3768 CrossRef PubMed.
- R. L. Kurtz, R. Stock-Bauer, T. E. Msdey, E. Román and J. L. De Segovia, Synchrotron radiation studies of H2O adsorption on TiO2(110), Surf. Sci., 1989, 218, 178–200 CrossRef CAS.
- H. Sheng, H. Zhang, W. Song, H. Ji, W. Ma, C. Chen and J. Zhao, Activation of Water in Titanium Dioxide Photocatalysis by Formation of Surface Hydrogen Bonds: An In Situ IR Spectroscopy Study, Angew. Chem., Int. Ed., 2015, 54, 5905–5909 CrossRef CAS PubMed.
- D. Rosa, G. Manetta and L. Di Palma, Experimental assessment of the pH effect and ions on the photocatalytic activity of iron-doped titanium dioxide supported on polystyrene pellets: Batch and continuous tests, Chem. Eng. Sci., 2024, 291, 119918 CrossRef CAS.
- F. Liu, N. Feng, Q. Wang, J. Xu, G. Qi, C. Wang and F. Deng, Transfer Channel of Photoinduced Holes on a TiO2 Surface As Revealed by Solid-State Nuclear Magnetic Resonance and Electron Spin Resonance Spectroscopy, J. Am. Chem. Soc., 2017, 139, 10020–10028 CrossRef CAS PubMed.
- N. Siemer, D. Muñoz-Santiburcio and D. Marx, Solvation-Enhanced Oxygen Activation at Gold/Titania Nanocatalysts, ACS Catal., 2020, 10, 8530–8534 CrossRef CAS.
- Y.-B. Zhuang and J. Cheng, Deciphering the Anomalous Acidic Tendency of Terminal Water at Rutile(110)–Water Interfaces, J. Phys. Chem. C, 2023, 127, 10532–10540 CrossRef CAS.
- P. Gono, F. Ambrosio and A. Pasquarello, Effect of the Solvent on the Oxygen Evolution Reaction at the TiO2–Water Interface, J. Phys. Chem. C, 2019, 123, 18467–18474 CrossRef CAS.
- Y. Wang, M. Muhler and C. Wöll, Spectroscopic evidence for the partial dissociation of H2O on ZnO(1
![[0 with combining macron]](https://www.rsc.org/images/entities/char_0030_0304.gif) 0), Phys. Chem. Chem. Phys., 2006, 8, 1521–1524 RSC.
0), Phys. Chem. Chem. Phys., 2006, 8, 1521–1524 RSC. - H. F. Wen, M. Miyazaki, Q. Zhang, Y. Adachi, Y. J. Li and Y. Sugawara, Direct observation of atomic step edges on the rutile TiO2(110)-(1 × 1) surface using atomic force microscopy, Phys. Chem. Chem. Phys., 2018, 20, 28331–28337 RSC.
- H. Jeong, E. Ertekin and E. G. Seebauer, Kinetic Control of Oxygen Interstitial Interaction with TiO2(110) via the Surface Fermi Energy, Langmuir, 2020, 36, 12632–12648 CrossRef CAS PubMed.
- M. A. Henderson, A surface perspective on self-diffusion in rutile TiO2, Surf. Sci., 1999, 419, 174–187 CrossRef CAS.
- F. Li, B. Wang, X. Chen, Y. Lai, T. Wang, H. Fan, X. Yang and Q. Guo, Photocatalytic Oxidative Dehydrogenation of Propane for Selective Propene Production with TiO2, JACS Au, 2022, 2, 2607–2616 CrossRef CAS PubMed.
- Y. Lai, R. Wang, Y. Zeng, F. Li, X. Chen, T. Wang, H. Fan and Q. Guo, Low-Temperature Oxidation of Methane on Rutile TiO2(110): Identifying the Role of Surface Oxygen Species, JACS Au, 2024, 4, 1396–1404 CrossRef CAS PubMed.
- M. Al-Hashem, S. Akbar and P. Morris, Role of Oxygen Vacancies in Nanostructured Metal-Oxide Gas Sensors: A Review, Sens. Actuators, B, 2019, 301, 126845 CrossRef CAS.
- N. G. Petrik, Z. Zhang, Y. Du, Z. Dohnálek, I. Lyubinetsky and G. A. Kimmel, Chemical Reactivity of Reduced TiO2(110): The Dominant Role of Surface Defects in Oxygen Chemisorption, J. Phys. Chem. C, 2009, 113, 12407–12411 CrossRef CAS.
- O. Bikondoa, C. L. Pang, R. Ithnin, C. A. Muryn, H. Onishi and G. Thornton, Direct visualization of defect-mediated dissociation of water on TiO2(110), Nat. Mater., 2006, 5, 189–192 CrossRef CAS.
- T. Minato, Y. Sainoo, Y. Kim, H. S. Kato, K. Aika, M. Kawai, J. Zhao, H. Petek, T. Huang, W. He, B. Wang, Z. Wang, Y. Zhao, J. Yang and J. G. Hou, The electronic structure of oxygen atom vacancy and hydroxyl impurity defects on titanium dioxide (110) surface, J. Chem. Phys., 2009, 130, 124502 CrossRef PubMed.
- H. Shi, Y.-C. Liu, Z.-J. Zhao, M. Miao, T. Wu and Q. Wang, Reactivity of the Defective Rutile TiO2(110) Surfaces with Two Bridging-Oxygen Vacancies: Water Molecule as a Probe, J. Phys. Chem. C, 2014, 118, 20257–20263 CrossRef CAS.
- Y. Zhang, Z. Xu, G. Li, X. Huang, W. Hao and Y. Bi, Direct Observation of Oxygen Vacancy Self-Healing on TiO2 Photocatalysts for Solar Water Splitting, Angew. Chem., Int. Ed., 2019, 58, 14229–14233 CrossRef CAS PubMed.
- H. Jeong and E. G. Seebauer, Influence of interstitial cluster families on post-synthesis defect manipulation and purification of oxides using submerged surfaces, J. Chem. Phys., 2024, 161, 121103 CrossRef CAS PubMed.
- H.-Y. Lee, S. J. Clark and J. Robertson, Calculation of point defects in rutile TiO2 by the screened-exchange hybrid functional, Phys. Rev. B:Condens. Matter Mater. Phys., 2012, 86, 75209 CrossRef.
- K. L. Gilliard and E. G. Seebauer, Manipulation of native point defect behavior in rutile TiO2 via surfaces and extended defects, J. Phys.: Condens. Matter, 2017, 29, 445002 CrossRef PubMed.
- R. Nakamura, N. Ohashi, A. Imanishi, T. Osawa, Y. Matsumoto, H. Koinuma and Y. Nakato, Crystal-Face Dependences of Surface Band Edges and Hole Reactivity, Revealed by Preparation of Essentially Atomically Smooth and Stable (110) and (100) n-TiO2 (Rutile) Surfaces, J. Phys. Chem. B, 2005, 109, 1648–1651 CrossRef CAS PubMed.
- R. Nakamura, T. Okamura, N. Ohashi, A. Imanishi and Y. Nakato, Molecular Mechanisms of Photoinduced Oxygen Evolution, PL Emission, and Surface Roughening at Atomically Smooth (110) and (100) n-TiO2 (Rutile) Surfaces in Aqueous Acidic Solutions, J. Am. Chem. Soc., 2005, 127, 12975–12983 CrossRef CAS PubMed.
- Y. Nakabayashi and Y. Nosaka, OH Radical Formation at Distinct Faces of Rutile TiO2 Crystal in the Procedure of Photoelectrochemical Water Oxidation, J. Phys. Chem. C, 2013, 117, 23832–23839 CrossRef CAS.
- C. Haisch, C. Günnemann, S. Melchers, M. Fleisch, J. Schneider, A. V. Emeline and D. W. Bahnemann, Irreversible surface changes upon n-type doping – A photoelectrochemical study on rutile single crystals, Electrochim. Acta, 2018, 280, 278–289 CrossRef CAS.
- Y. Zhou and H. J. Fan, Progress and Challenge of Amorphous Catalysts for Electrochemical Water Splitting, ACS Mater. Lett., 2021, 3, 136–147 CrossRef CAS.
- M. Králik, Adsorption, chemisorption, and catalysis, Chem. Pap., 2014, 68, 1625–1638 Search PubMed.
- S. Carbonaro, M. N. Sugihara and T. J. Strathmann, Continuous-flow photocatalytic treatment of pharmaceutical micropollutants: Activity, inhibition, and deactivation of TiO2 photocatalysts in wastewater effluent, Appl. Catal., B, 2013, 129, 1–12 CrossRef CAS.
- X. Li, Y. Chen, Y. Tao, L. Shen, Z. Xu, Z. Bian and H. Li, Challenges of photocatalysis and their coping strategies, Chem Catal., 2022, 2, 1315–1345 CrossRef CAS.
- G. Singh, M. Kaur, V. Kumar Garg and A. C. Oliveira, Oxygen hyper stoichiometric trimetallic titanium doped magnesium ferrite: Structural and photocatalytic studies, Ceram. Int., 2022, 48, 24476–24484 CrossRef CAS.
- J. K. Grewal, M. Kaur, R. K. Sharma, A. C. Oliveira, V. K. Garg and V. K. Sharma, Structural and Photocatalytic Studies on Oxygen Hyperstoichiometric Titanium-Substituted Strontium Ferrite Nanoparticles, Magnetochemistry, 2022, 8, 120 CrossRef CAS.
- S. J. Salih and W. M. Mahmood, Review on magnetic spinel ferrite (MFe2O4) nanoparticles: From synthesis to application, Heliyon, 2023, 9, e16601 CrossRef CAS PubMed.
- O. A. Bulavchenko, O. S. Venediktova, T. N. Afonasenko, P. G. Tsyrul’Nikov, A. A. Saraev, V. V. Kaichev and S. V. Tsybulya, Nonstoichiometric oxygen in Mn-Ga-O spinels: Reduction features of the oxides and their catalytic activity, RSC Adv., 2018, 8, 11598–11607 RSC.
- Q. Yin, J. Kniep and Y. S. Lin, Oxygen sorption and desorption properties of Sr–Co–Fe oxide, Chem. Eng. Sci., 2008, 63, 2211–2218 CrossRef CAS.
- W. Li, B. Guan, X. Zhang, J. Yan, Y. Zhou and X. Liu, New mechanistic insight into the oxygen reduction reaction on Ruddlesden–Popper cathodes for intermediate-temperature solid oxide fuel cells, Phys. Chem. Chem. Phys., 2016, 18, 8502–8511 RSC.
- A. J. Majewski, A. Khodimchuk, D. Zakharov, N. Porotnikova, M. Ananyev, I. D. Johnson, J. A. Darr, P. R. Slater and R. Steinberger-Wilckens, Oxygen surface exchange properties and electrochemical activity of lanthanum nickelates, J. Solid State Chem., 2022, 312, 123228 CrossRef CAS.
- S. Lian, L. He, C. Li, J. Ren, L. Bi, M. Chen and Z. Lin, Uncovering the Enhancement Mechanism of the Oxygen Reduction Reaction on Perovskite/Ruddlesden–Popper Oxide Heterostructures (Nd,Sr)CoO3/(Nd,Sr)2CoO4 and (Nd,Sr)CoO3/(Nd,Sr)3Co2O7, J. Phys. Chem. Lett., 2023, 14, 2869–2877 CrossRef CAS PubMed.
- P. Erhart, K. Albe and A. Klein, First-principles study of intrinsic point defects in ZnO: Role of band structure, volume relaxation, and finite-size effects, Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 73, 205203 CrossRef.
- A. Janotti and C. G. Van de Walle, Native point defects in ZnO, Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 76, 165202 CrossRef.
- A. Goyal, P. Gorai, H. Peng, S. Lany and V. Stevanović, A computational framework for automation of point defect calculations, Comput. Mater. Sci., 2017, 130, 1–9 CrossRef.
- A. A. Sokol, S. A. French, S. T. Bromley, C. R. A. Catlow, H. J. J. van Dam and P. Sherwood, Point defects in ZnO, Faraday Discuss., 2007, 134, 267–282 RSC.
- R. Vidya, P. Ravindran, H. Fjellvåg, B. G. Svensson, E. Monakhov, M. Ganchenkova and R. M. Nieminen, Energetics of intrinsic defects and their complexes in ZnO investigated by density functional calculations, Phys. Rev. B:Condens. Matter Mater. Phys., 2011, 83, 45206 CrossRef.
- J. L. Lyons, J. B. Varley, D. Steiauf, A. Janotti and C. G. Van de Walle, First-principles characterization of native-defect-related optical transitions in ZnO, J. Appl. Phys., 2017, 122, 035704 CrossRef.
- R. A. Evarestov, A. Platonenko, D. Gryaznov, Y. F. Zhukovskii and E. A. Kotomin, First-principles calculations of oxygen interstitials in corundum: A site symmetry approach, Phys. Chem. Chem. Phys., 2017, 19, 25245–25251 RSC.
- A. Platonenko, D. Gryaznov, Y. F. Zhukovskii and E. A. Kotomin, Ab initio simulations on charged interstitial oxygen migration in corundum, Nucl. Instrum. Methods Phys. Res., Sect. B, 2018, 435, 74–78 CrossRef CAS.
- A. Kononov, C. W. Lee, E. P. Shapera and A. Schleife, Identifying native point defect configurations in α-alumina, J. Phys.: Condens. Matter, 2023, 35, 334002 CrossRef CAS PubMed.
- H. Y. Xiao, Y. Zhang and W. J. Weber, Stability and migration of charged oxygen interstitials in ThO2 and CeO2, Acta Mater., 2013, 61, 7639–7645 CrossRef CAS.
- B. Medasani, M. L. Sushko, K. M. Rosso, D. K. Schreiber and S. M. Bruemmer, First-Principles Investigation of Native Interstitial Diffusion in Cr2O3, J. Phys. Chem. C, 2018, 122, 12984–12993 CrossRef CAS.
- K. H. Yano, A. A. Kohnert, A. Banerjee, D. J. Edwards, E. F. Holby, T. C. Kaspar, H. Kim, T. G. Lach, S. D. Taylor, Y. Wang, B. P. Uberuaga and D. K. Schreiber, Radiation-Enhanced Anion Transport in Hematite, Chem. Mater., 2021, 33, 2307–2318 CrossRef CAS.
- A. Banerjee, E. F. Holby, A. A. Kohnert, S. Srivastava, M. Asta and B. P. Uberuaga, Thermokinetics of point defects in α-Fe2O3, Electron. Struct., 2023, 5, 024007 CrossRef CAS.
- B. S. Thomas, N. A. Marks and B. D. Begg, Defects and threshold displacement energies in SrTiO3 perovskite using atomistic computer simulations, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 254, 211–218 CrossRef CAS.
- B. Liu, V. R. Cooper, H. Xu, H. Xiao, Y. Zhang and W. J. Weber, Composition dependent intrinsic defect structures in SrTiO3, Phys. Chem. Chem. Phys., 2014, 16, 15590–15596 RSC.
- S. A. Chambers, Y. Du, Z. Zhu, J. Wang, M. J. Wahila, L. F. J. Piper, A. Prakash, J. Yue, B. Jalan, S. R. Spurgeon, D. M. Kepaptsoglou, Q. M. Ramasse and P. V. Sushko, Interconversion of intrinsic defects in SrTiO3(001), Phys. Rev. B, 2018, 97, 245204 CrossRef CAS.
- S. T. Murphy, M. W. D. Cooper and R. W. Grimes, Point defects and non-stoichiometry in thoria, Solid State Ionics, 2014, 267, 80–87 CrossRef CAS.
- R. I. Palomares, M. T. McDonnell, L. Yang, T. Yao, J. E. S. Szymanowski, J. Neuefeind, G. E. Sigmon, J. Lian, M. G. Tucker, B. D. Wirth and M. Lang, Oxygen point defect accumulation in single-phase UO2+x, Phys. Rev. Mater., 2019, 3, 53611 CrossRef CAS.
- L. Yang and B. D. Wirth, Clustering of excess oxygen in uranium dioxide: A first-principles study, J. Nucl. Mater., 2021, 554, 153087 CrossRef CAS.
- X. M. Bai, Y. Zhang and M. R. Tonks, Strain effects on oxygen transport in tetragonal zirconium dioxide, Phys. Chem. Chem. Phys., 2013, 15, 19438–19449 RSC.
- C. Jiang, X.-Y. Liu and K. E. Sickafus, First-principles prediction of the thermodynamic stability of xenon in monoclinic, tetragonal, and yttrium-stabilized cubic ZrO2, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 52103 CrossRef.
- A. Samanta, A theoretical study of the stability of anionic defects in cubic ZrO2 at extreme conditions, J. Mater. Sci., 2016, 51, 4845–4855 CrossRef CAS.
- P. Ding, W. Li, H. Zhao, C. Wu, L. Zhao, B. Dong and S. Wang, Review on Ruddlesden–Popper perovskites as cathode for solid oxide fuel cells, J. Phys.: Mater., 2021, 4, 022002 CAS.
- H.-Y. Lee and J. Robertson, Doping and compensation in Nb-doped anatase and rutile TiO2, J. Appl. Phys., 2013, 113, 213706 CrossRef.
- A. Kyrtsos, M. Matsubara and E. Bellotti, Migration mechanisms and diffusion barriers of vacancies in Ga2O3, Phys. Rev. B, 2017, 95, 245202 CrossRef.
- J. Meng, M. S. Sheikh, R. Jacobs, J. Liu, W. O. Nachlas, X. Li and D. Morgan, Computational discovery of fast interstitial oxygen conductors, Nat. Mater., 2024, 23, 1252–1258 CrossRef CAS PubMed.
- R. K. Behera, T. Watanabe, D. A. Andersson, B. P. Uberuaga and C. S. Deo, Diffusion of oxygen interstitials in UO2+x using kinetic Monte Carlo simulations: Role of O/M ratio and sensitivity analysis, J. Nucl. Mater., 2016, 472, 89–98 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Method for determination of profile metrics; statistical analysis example calculation; adsorption models (linear and dual pathway); table of surface coverages of 18O and 16O; additional supplementary figures and tables including change in pH vs. ln(F18), profile metrics vs. applied potential Vappl, F-tests for null hypothesis on slopes of profile metrics vs. Vappl, histograms of the five profile metrics at 70 °C with and without applied bias, atomic geometries of TiO2(110) with and without adsorbed bridging hydroxyls, adsorption energy and Bader charge of adsorbed O on TiO2(110) under Ti-rich conditions. See DOI: https://doi.org/10.1039/d5cp00319a |
| This journal is © the Owner Societies 2025 |