
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topochemical reduction of FeCo-oxide to FeCo-alloy nanosystems into a SiO2 matrix†
Jean Pierre
Miranda Murillo
a,
Alexander
Omelyanchik
 ab,
Gianni
Barucca
c,
Gaspare
Varvaro
b,
Ayda Ghary
Haghighat
bd,
Sara
Laureti
b,
Aldo
Capobianchi
b,
Antonio
Comite
a,
Diego
Colombara
a,
Nikolaos
Ntallis
e,
Kalliopi N.
Trohidou
e,
Fabio
Canepa
a,
Pierfrancesco
Maltoni
*ab and
Davide
Peddis
*ab
ab,
Gianni
Barucca
c,
Gaspare
Varvaro
b,
Ayda Ghary
Haghighat
bd,
Sara
Laureti
b,
Aldo
Capobianchi
b,
Antonio
Comite
a,
Diego
Colombara
a,
Nikolaos
Ntallis
e,
Kalliopi N.
Trohidou
e,
Fabio
Canepa
a,
Pierfrancesco
Maltoni
*ab and
Davide
Peddis
*ab
aDepartment of Chemistry and Industrial Chemistry & INSTM RU, University of Genoa, 16146 Genoa, Italy. E-mail: pierfrancesco.maltoni@unige.it; davide.peddis@unige.it
bInstitute of Structure of Matter, National Research Council, nM2-Lab, Via Salaria km 29.300, Monterotondo Scalo 00015, Rome, Italy
cDepartment of Science and Engineering of Matter, Environment and Urban Planning, University Politecnica delle Marche, Via Brecce Bianche 12, 60131 Ancona, Italy
dDepartment of Science, University of Roma Tre, Via della Vasca Navale 84, 00146, Rome, Italy
eInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, Agia Paraskevi, Attiki 153 10, Greece
First published on 10th March 2025
Abstract
This study focuses on the synthesis of metallic magnetic nanosystems embedded in mesoporous silica (SiO2), and the impact of matrix porosity, controlled by temperature treatment, on the efficiency of H2 reduction process. The reduction of FeCo oxides to the corresponding alloy nanosystems was first optimized, identifying FeCo with 50 at% Fe as the optimal composition due to its high saturation magnetization (∼242 A m2 kg−1) and oxidation onset temperature (∼440 °C). Then, the FeCo-oxide nanocomposites were synthesized into SiO2via sol–gel self-combustion under thermal treatments, to properly tune the surface area of the silica matrix. By controlling the annealing temperature, the specific surface area (SA) of the matrix decreases from ∼512(1) m2 g−1 to ∼345(1) m2 g−1 when annealed to 900 °C in air. Following topochemical reduction in H2, the structural properties of the obtained FeCo–SiO2 nanocomposites have been analyzed using X-ray powder diffraction and magnetic properties were evaluated to establish a correlation between matrix SA and reduction capability. The decrease of SA leads to incomplete reduction at higher temperatures, with the formation of FeYOX/CoXOY intermediates. This work underscores the critical role of matrix porosity in achieving a delicate balance to ensure both the efficient conversion of nanostructured oxide to their metallic state and the preservation of their magnetic and structural integrity.
Introduction
The synthesis of nanostructured FeCo alloys has attracted considerable interest due to their soft magnetic properties (i.e., low coercivity Hc, high magnetic moment and large magnetic susceptibility) compared to their oxide counterparts.1,2 Technological applications demand a large magnetic energy density and the ability to operate at both large magnetic fields and high frequencies, thus requiring a large magnetic moment and a high Curie temperature.3–6 This applies to several sectors such as medical,7 power conversion,8,9 media recording,10 and catalysis,11,12 owing to the number of techniques that can allow the preparation of nanosystems with various morphologies.13–15 FeCo-alloys can be integrated into multiple matrices or substrates to create composite materials with enhanced functionalities.16–18The FeCo-alloy is a ferromagnetic system with a saturation magnetization, Ms, which may reach up to ∼245 A m2 kg−1 at 300 K, the highest value among binary alloys. This is more than twice that of common bulk oxides ferrimagnetic iron oxides (e.g., maghemite, γ-Fe2O3, Ms = 72 A m2 kg−1 and magnetite, Fe3O4, Ms = 83 A m2 kg−1) or cobalt ferrite (CoFe2O4, Ms = 80 A m2 kg−1).19–24 Furthermore, it has a high Curie temperature Tc up to ∼1200 K. However, these metallic systems at the nanoscale face challenges such as chemical instability and oxidation.25 Typically, these issues have been addressed by encapsulating the particles within protective matrices, with amorphous silica (SiO2) being a commonly employed material.26–31 Recently, metal-embedded silica nanostructures have attracted considerable attention as biocompatible and stable templates, particularly for embedding noble metals.32 While the reduction of oxide nanosystems within these matrices, often carried out by hydrogen (H2) treatment during sintering, is well-established, the role of matrix textural properties (i.e. surface area) in this process has been overlooked in the literature. The permeation of H2 driving the reduction of oxide nanoparticles depends on the thickness and porosity of the silica matrix, but also on the reaction conditions (annealing temperature and H2 flow).33–35 The reported reduction temperature is usually very high, in the 600–900 °C range,36–40 which is not only energy consuming, but may also generate by-products such as fayalite (Fe2SiO4),26 that deteriorate the magnetic properties.
This study aims to fill this gap by systematically investigating the influence of matrix porosity, controlled through the thermal treatment, on the efficacy of topochemical H2 reduction of nanostructured oxide with spinel structure to metal alloys. The study focuses on nanocomposites of FeCo-oxides in mesoporous silica synthesized via sol–gel self-combustion, with thermal treatment selectively altering the surface area of the silica matrix. By controlling matrix porosity, we demonstrated that the reduction process is driven by the textural features of the matrix controlling the diffusion of H2 and the conversion of oxide particles to their metallic form. Through this work, we highlight the critical role of matrix porosity in facilitating solid-state chemical transformation due to H2 access. Overall, our findings underscore the importance of balancing matrix porosity to ensure both efficient conversion of oxide nano-systems to metallic states and preservation of their magnetic and structural integrity.
Experimental
FeCo-oxides and alloys
The synthesis of FeCo-oxides and FeCo-alloys followed procedures outlined in previous studies,41 with detailed information provided in the ESI.† In summary, Fe(NO3)3·9H2O (Sigma Aldrich, 98%) and Co(NO3)2·6H2O (Sigma Aldrich, 98%) were dissolved in deionized water in the desired ratio, and C6H8O7 (citric acid, Sigma Aldrich, 99.5%) was introduced as a chelating agent.42 The ratio of metallic ions was carefully selected to adjust the Fe3+ at% in the final oxide, ensuring that Fe3+ at% + Co2+ at% equaled 100 (with Fe3+ at% varying as 100, 67, 50, 33, or 0). Following adjustment of the pH to 7 using NH4OH (ammonia solution 30% (aq.), Sigma Aldrich), the solution was dried to form a thick gel, which was subsequently heated to induce crystallization, yielding FeCo-oxides powders.These powders underwent grinding and annealing in a hydrogen (H2) flow environment (83% H2 in N2, flow rate of 65 mL min−1) for 5 hours at 500 °C to obtain metal particles.
FeCo-oxides in SiO2
Nanocomposites of FeCo-oxides in silica with a silica content of 70 wt% were synthesized by following a procedure similar to that outlined by Cannas et al.43 Initially, 5 mmol of Fe(NO3)3·9H2O and 5 mmol of Co(NO3)2·6H2O were dissolved in 23 mL of deionized water along with 10 mmol of citric acid in a Teflon beaker under magnetic stirring. The molar concentration ratios were carefully adjusted, with both Fe3+/Co2+ and (Co2+ + Fe3+)/citric acid set to 1, resulting in a total molar concentration in water (Co2+ + Fe3+ + citric acid) of 1. The pH of the solution was then adjusted to 2 by adding NH4OH (30% water solution) dropwise. Subsequently, tetraethyl orthosilicate (TEOS, Sigma Aldrich, 98%) was dissolved in approximately 45 mL of absolute ethanol, and this solution was added to the nitrate solution in water. The pH was once again adjusted to 2, and the mixture was stirred for 40 minutes at room temperature. The resulting solution was then allowed to dry in a preheated oven set at 40 °C for 24 hours. The dried gel obtained was transferred to a pyrex beaker and heated on a hot plate set to 350 °C. After a short time, the gel underwent self-combustion, yielding a black powder. This powder was subsequently ground using an agate mortar, and annealed in air at different temperatures, reported in Table 1. The oxide samples were denoted as NCFO_X00 (N = nanocomposite, C = cobalt, F = iron, O = oxygen), with the subscript X00 indicating the annealing temperature in air (X00 °C with X = 3, 5, 7, 9).| Sample name | Precursor | Temperature (°C) | Time (h) | Atmosphere |
|---|---|---|---|---|
| NCFO | Post synthesis | |||
| NCFO_300 | NCFO | 300 | 1 | Air |
| NCFO_500 | NCFO | 500 | 1 | Air |
| NCFO_700 | NCFO | 700 | 1 | Air |
| NCFO_900 | NCFO | 900 | 1 | Air |
| NCF_300 | NCFO_300 | 500 | 5 | H2/N2 |
| NCF_500 | NCFO_500 | 500 | 5 | H2/N2 |
| NCF_700 | NCFO_700 | 500 | 5 | H2/N2 |
| NCF_900 | NCFO_900 | 500 | 5 | H2/N2 |
FeCo-alloys in SiO2
NCFO_X00 oxide samples underwent reduction under identical annealing conditions, specifically with a flow rate of 65 mL min−1 of 83% H2 – 17% N2 for 5 hours at 500 °C (Table 1). The corresponding reduced samples were labelled as NCF_X00. To prevent oxidation, the samples were stored inside an Ar-filled glove box.Methods
X-ray powder diffraction (XRPD) was conducted with a MiniFlex (Rigaku) X-ray diffractometer equipped with a Cu Kα anode (λ = 1.54184 Å) in the 2θ geometry using a step size of 0.01° and 1 second per step. Phase identification was performed by comparison with literature patterns using the Pearson Crystal Database. The average size of the crystallites was extracted from Scherrer equation.44 The lattice constant of cubic phases was calculated using equation . It should be underlined that all the XRD patterns reported in this paper contain a strong background due to the presence of amorphous silica. In this scenario, both crystallite size and lattice constant should be considered as a rough estimation. Transmission electron microscopy (TEM) images were acquired using a Philips CM200 microscope operating at 200 kV and equipped with a LaB6 filament. The differential thermal analysis and thermogravimetry (DTA/TG) were performed by using a LabsysEvo 1600 Setaram between 25 and 1000 °C. The surface area (SA) of the samples was calculated using an ASAP 2020 Plus MP, Micromeritics, USA, with the Brunauer–Emmett–Teller (BET) method at 300 K. Inductively coupled plasma – atomic emission spectroscopy (ICP-AES) measurements were performed using an iCAP 6300 DUP ICP-AES spectrometer (ThermoScientific) to check the composition of each sample.
. It should be underlined that all the XRD patterns reported in this paper contain a strong background due to the presence of amorphous silica. In this scenario, both crystallite size and lattice constant should be considered as a rough estimation. Transmission electron microscopy (TEM) images were acquired using a Philips CM200 microscope operating at 200 kV and equipped with a LaB6 filament. The differential thermal analysis and thermogravimetry (DTA/TG) were performed by using a LabsysEvo 1600 Setaram between 25 and 1000 °C. The surface area (SA) of the samples was calculated using an ASAP 2020 Plus MP, Micromeritics, USA, with the Brunauer–Emmett–Teller (BET) method at 300 K. Inductively coupled plasma – atomic emission spectroscopy (ICP-AES) measurements were performed using an iCAP 6300 DUP ICP-AES spectrometer (ThermoScientific) to check the composition of each sample.
Field-dependent magnetization loops (M vs. H) were recorded at 300 K using a Model 10 Microsense Vibrating Sample Magnetometer (VSM) equipped with an electromagnet producing a maximum magnetic field of 2 T.
Results and discussion
FeCo-oxides and alloys
Five different metallic samples (with Fe at% varying as 100, 67, 50, 33, and 0) were prepared by reduction in H2 of the corresponding FeCo-oxides powders, previously obtained by sol–gel self-combustion method. Fig. 1 shows the patterns for the starting oxides and the corresponding reduced metal particles, revealing the complete reduction (i.e., absence of oxide phases) achieved under the explored annealing conditions (see Table 1). | ||
| Fig. 1 XRPD patterns of the nanostructured FeCo-oxides before (left) and after (right) the reduction process in H2. | ||
The synthetized oxides are predominantly composed of cubic spinel ferrite phase (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group). Upon reduction, the samples underwent a significant structural transformation. The XRPD patterns of the reduced samples reveal characteristic reflections corresponding to a body-centered cubic (bcc) structure, which is typical for FeCo alloys with Pmm space group.45 This bcc structure was observed across the composition range, with two notable exceptions: the pure iron (Imm) and the pure cobalt sample. In the case of pure Co, the XRPD pattern was indexed to a face-centered cubic (fcc) structure belonging to the Fdm space group. The lattice constant a decreases gradually from 2.870(1) Å for pure Fe to 2.845(1) Å for FeCo with 33 at% of Fe as expected for FeCo alloys with more than 20% cobalt content.46 For pure Co, the lattice constant (3.547(1) Å) is close to the values reported earlier for fcc Co(0) nanoparticles.45 The crystal size 〈D〉 of all metal nanoparticles is around 30 nm. As expected, all metallic particles exhibit a higher saturation magnetization (Ms), as extracted from the magnetic hysteresis loops reported in ESI† (Fig. S1 and S2), compared to the starting oxide particles (Fig. 2), with a maximum value for the FeCo-alloys with Fe at% of 50 and 67. The observed values, also reported in Table 2, closely align with the theoretical Ms for those FeCo compositions,19,47 also predicted by DFT calculations,48,49 which highlights the maximum of the saturation magnetization in the region of 35–50 Co at% for systems above 20 nm in size (see ESI,† for more details, Fig. S3) closer to bulk values,50 confirming the effective reduction within the used conditions.
m space group). Upon reduction, the samples underwent a significant structural transformation. The XRPD patterns of the reduced samples reveal characteristic reflections corresponding to a body-centered cubic (bcc) structure, which is typical for FeCo alloys with Pmm space group.45 This bcc structure was observed across the composition range, with two notable exceptions: the pure iron (Imm) and the pure cobalt sample. In the case of pure Co, the XRPD pattern was indexed to a face-centered cubic (fcc) structure belonging to the Fdm space group. The lattice constant a decreases gradually from 2.870(1) Å for pure Fe to 2.845(1) Å for FeCo with 33 at% of Fe as expected for FeCo alloys with more than 20% cobalt content.46 For pure Co, the lattice constant (3.547(1) Å) is close to the values reported earlier for fcc Co(0) nanoparticles.45 The crystal size 〈D〉 of all metal nanoparticles is around 30 nm. As expected, all metallic particles exhibit a higher saturation magnetization (Ms), as extracted from the magnetic hysteresis loops reported in ESI† (Fig. S1 and S2), compared to the starting oxide particles (Fig. 2), with a maximum value for the FeCo-alloys with Fe at% of 50 and 67. The observed values, also reported in Table 2, closely align with the theoretical Ms for those FeCo compositions,19,47 also predicted by DFT calculations,48,49 which highlights the maximum of the saturation magnetization in the region of 35–50 Co at% for systems above 20 nm in size (see ESI,† for more details, Fig. S3) closer to bulk values,50 confirming the effective reduction within the used conditions.
 | ||
| Fig. 2 Saturation magnetization, MS, and coercivity, μ0Hc, of different samples of nanostructured FeCo-oxides before (black dots) and after (blue dots) the reduction process in H2 atmosphere. | ||
| Fe (at%) | Fe (at%) [ICP-AES] | M s (A m2 kg−1) | μ 0 H c (mT) | 〈D〉 (nm) | a (Å) |
|---|---|---|---|---|---|
| 100 | 219(4) | 6(1) | 31(6) | 2.870(1) | |
| 67 | 62.8(6) | 242(6) | 10(1) | 29(4) | 2.861(1) |
| 50 | 45.4(5) | 242(5) | 14(1) | 29(3) | 2.854(1) |
| 33 | 29.7(3) | 222(1) | 9(1) | 33(6) | 2.845(1) |
| 0 | 166(4) | 10(1) | 30(4) | 3.547(1) |
Then, to assess the stability of the obtained alloys against oxidation, TG experiments were conducted (see Fig. S4 in ESI,† for TG analysis). Particularly, the onset temperature of oxidation for FeCo with 50 at% of Fe was found at ∼440 °C, relatively higher than that for other compositions, suggesting its suitability for applications requiring higher thermal stability and oxidation resistance. Considering also the notable Ms value, this alloy was chosen as a model sample for investigating the structural and magnetic properties when it is embedded in a silica matrix.
FeCo-oxides and alloys in SiO2
At first, a sample of FeCo-oxide in SiO2 30/70 wt%, denoted as NCFO, was prepared with a synthetic approach integrating traditional sol–gel techniques, involving the hydrolysis and condensation of alkoxide precursors, with nitrate–citrate sol–gel self-combustion.43 This procedure has successfully yielded nanoparticles consisting of CoFe2O4/Co3O4 spinel phases (due to the Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe 1:1 ratio51), dispersed within an amorphous silica matrix. The XRPD patterns for the NCFO samples are reported in Fig. 3. Only the (311) reflection of the Fdm space group of the main spinel CoFe2O4 phase appears in the pattern of the NCFO nanocomposite, whereas the other expected reflections are hidden by the amorphous silica matrix (70 wt%), whose hump is visible between 20°–30° (Fig. 3). The predominance of this spinel phase becomes more conspicuous following annealing up to 900 °C, attributed to growth and possibly potential reorientation of the initial particles. However, the average crystallite size remains relatively small (<7 nm) due to the constraining effect of the silica matrix, which prevents significant particle growth.52 The confirmed high porosity of the matrix for NCFO, as indicated by BET analysis in Table 3, diminishes post-annealing in air, from 512 m2 g−1 for NFO to 345 m2 g−1 for NCFO_900 (∼30%), implying that the lower SA will potentially affect the subsequent reduction process making it less effective.33–35
:Fe 1:1 ratio51), dispersed within an amorphous silica matrix. The XRPD patterns for the NCFO samples are reported in Fig. 3. Only the (311) reflection of the Fdm space group of the main spinel CoFe2O4 phase appears in the pattern of the NCFO nanocomposite, whereas the other expected reflections are hidden by the amorphous silica matrix (70 wt%), whose hump is visible between 20°–30° (Fig. 3). The predominance of this spinel phase becomes more conspicuous following annealing up to 900 °C, attributed to growth and possibly potential reorientation of the initial particles. However, the average crystallite size remains relatively small (<7 nm) due to the constraining effect of the silica matrix, which prevents significant particle growth.52 The confirmed high porosity of the matrix for NCFO, as indicated by BET analysis in Table 3, diminishes post-annealing in air, from 512 m2 g−1 for NFO to 345 m2 g−1 for NCFO_900 (∼30%), implying that the lower SA will potentially affect the subsequent reduction process making it less effective.33–35
 | ||
| Fig. 3 (Left) XRPD patterns of the samples obtained by the sol–gel self-combustion synthesis and after thermal treatment at 300 °C, 500 °C, 700 °C, and 900 °C for 1 h in air. (Right) corresponding XRPD patterns after treatment in H2 at 500 °C for 5 h. | ||
| Sample name | Fe (at%) | SA (m2 g−1) | Av. pore size (nm) |
|---|---|---|---|
| NCFO | 55(3) | 512(8) | 5(1) |
| NCFO_500 | 55(3) | 413 (7) | 5(1) |
| NCFO_900 | 55(3) | 345(5) | 4(1) |
The composites annealed at several temperatures were then treated in a reducing H2 atmosphere at 500 °C. XRPD patterns of the reduced NCF samples reveal distinct reflections at 2θ = 44.7, 65.2, and 82.4°, corresponding to the Pmm spacegroup of the FeCo alloy with 50 Fe at%, validating the successful reduction to the metallic alloy phase (up to NCF_500). Nonetheless, additional reflections were observed in the XRPD patterns of samples NCF_700 and NCF_900 at 2θ ∼36.3, ∼42.0, ∼61.7, and ∼78.1°, suggesting the presence of intermediate phases such as Fe or Co monoxides (FeO and CoO, respectively) or a spinel (Fe1−xCox)3O4 phase, possibly arising from incomplete reduction attributed to increased particle size, and decreased SA. Additionally, the estimated crystallite size of the alloy nanocomposites remains quite constant across the series independently of the thermal treatment in H2 (see Table 4). This lack of dependence is presumably due to the silica matrix and its confinement effect, which however makes the size estimation not trivial due to the amorphous contribution in the pattern. TEM images (see Fig. 4) offer valuable insights into the morphology of both pure FeCo-alloy particles and their corresponding nanocomposites.
| Annealing X00 °C | NCFO | NCF | ||
|---|---|---|---|---|
| 〈D〉 (nm) | a (nm) | 〈D〉 (nm) | a (Å) | |
| Post synthesis | n.d. | n.d. | 23(5) | 2.865(11) |
| 300 | n.d. | n.d. | 15(4) | 2.841(13) |
| 500 | 5(3) | 8.38(6) | 19(3) | 2.840(11) |
| 700 | 4(3) | 8.50(6) | 20(2) | 2.875(6) |
| 900 | 7(3) | 8.41(7) | 23(6) | 2.867(7) |
 | ||
| Fig. 4 TEM images of the CoFe-alloy with 50 at% (left, (a)) and the CoFe-alloy/SiO2 (NCF) composite (right, (b) obtained by the NCFO sample reduction. | ||
FeCo-alloy grains have irregular shapes with sizes ranging from 50 to 400 nm (Fig. 4a) and are interconnected giving rise to highly porous structures. The inherently high porosity observed in the FeCo-alloy validates the selection of this material as the ideal starting system for this topochemical reduction process. When the silica matrix is added to form the nanocomposites, the FeCo-alloy grains are trapped inside the porous amorphous matrix. Fig. 4b shows a typical cluster of FeCo-alloy grains (dark arrows) inside a finely porous amorphous matrix (for sample NCF). In principle, the abundant active sites provided by the finely dispersed nanophase within this structurally open matrix should enhance diffusion kinetics and catalytic activity.53,54 The same trend was confirmed for the set of samples in Table 1. In particular, when NCFO is annealed at higher temperatures, ferrite particles crystallise while remaining confined in the matrix, Fig. 5a. Selected area electron diffraction (SAED) measurements were performed (inset of Fig. 5a). All the visible diffraction rings can be attributed to the cobalt ferrite phase, suggesting that the Co3O4 phase, if present, is poorly crystallised. TEM dark-field images were performed to evidence the crystallites in the amorphous matrix. Fig. 5b was obtained with the diffraction spots encircled in the SAED image of Fig. 5a's inset and the crystallites corresponding to those diffraction spots appear brighter in the image. The subsequent reduction process yields the expected FeCo phase (NCF_500 in Fig. 5c), without changing the morphology of the composite. If we consider a higher annealing temperature in air, as shown for NCFO_700 in Fig. 5d, we did not observe relevant changes, in agreement with the results from XRPD analysis.
 | ||
| Fig. 5 (a) Bright-field TEM image showing the crystalline phase (arrows) inside the silica amorphous matrix. The inset is a typical selected area electron diffraction (SAED) pattern taken on a large sample area. (b) Corresponding dark-field image obtained with the diffraction spots circled in (a) insetM (c) bright-field TEM image showing the reduced crystalline phase (arrows) inside the silica amorphous matrix. (d) Bright-field TEM image of the sample NCFO_700: the crystallites inside the silica amorphous matrix are indicated by arrows. | ||
Further details about the reduction process can be obtained from field-dependence magnetization loops at 300 K (see Fig. 6). The composite NCFO_900 shows an increase of both Ms and μ0Hc from ∼5 to 12 A m2 kg−1 and from ∼8 to 21 mT, respectively, when annealed in air at 900 °C for 1 h. This behavior can be ascribed to the particle's crystallization and consequent growth inside the matrix compared to NCFO.55 After the reduction process (Fig. 6 right), the Ms of the NCF sample increases from ∼5 to 60 A m2 kg−1, specifically by ∼12 times compared to NCFO, consistently with the transformation in FeCo-alloy. The measured coercivity (∼40 mT) is a bit higher to what is expected for the FeCo alloy, suggesting an incomplete reduction of the sample or the existence of a thin passive layer. This was also observed in previous studies, where they show possible passivation of the alloy inside the matrix.36 On the other hand, the pattern of NCF_900 reveals a mixture of phases, making the comparison complex: this is presumably due to the presence of FeYOX/CoXOY and Co ferrite as the FeCo alloy is probably obtained in low yield or rich in iron,45 because of the much lower SA of NCF_900.
 | ||
| Fig. 6 Magnetization (M) vs. applied field (H) at 300 K for NCFO/NCFO_900 (left), and NCF/NCFO (right). | ||
Our investigations demonstrate that annealing in air before reduction critically influences the matrix porosity, directly impacting hydrogen diffusion and, consequently, the efficiency of oxide-to-alloy conversion (see the scheme of Fig. 7). This, in turn, determines the final magnetic properties of the embedded nanoparticles, with higher annealing temperatures leading to incomplete reduction and lower saturation magnetization.
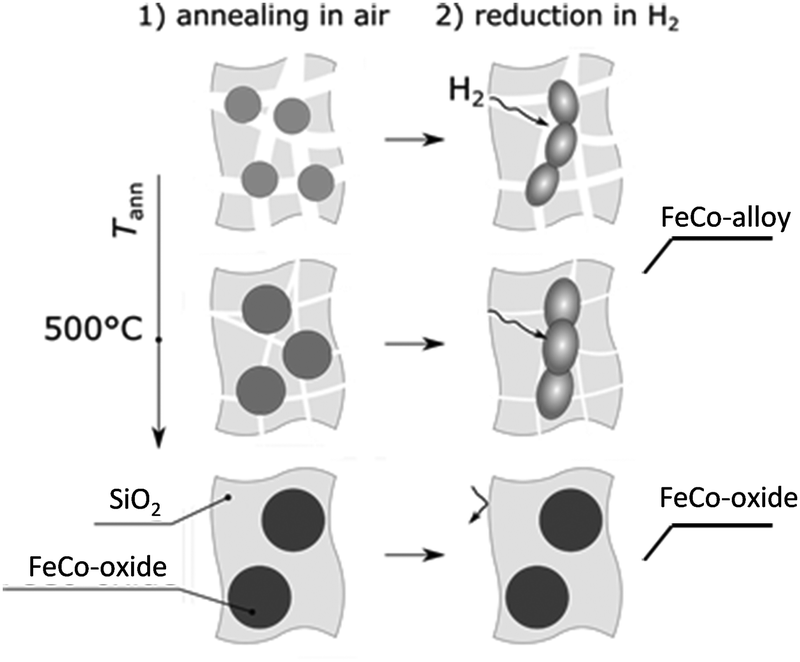 | ||
| Fig. 7 Schematic representation of the annealing process. | ||
To corroborate and expand the applicability and effectiveness of the method, we applied the reducing treatment to a composite, referred to as NFO, having the same composition in silica and iron oxides (i.e., a mixture of maghemite/hematite), as shown in Fig. 8. The results suggest a complete reduction of the oxide phases (right panel) to Iron Imm (left panel), wherein the reflections corresponding to the oxides disappear as previously observed for the oxide phases without silica matrix. The emergence of the metallic phase is accompanied by the enhancement of Ms visible in the hysteresis loop in the bottom panel of Fig. 8, and thus confirming the reproducibility of the reducing treatment on composites of different spinel oxides phases embedded in a porous silica matrix.
 | ||
| Fig. 8 XRPD patterns of the iron-based samples obtained by the sol–gel self-combustion synthesis (top, left) and after thermal treatment in H2 (top, right). Field dependence of magnetization at 300 K for NFO and NF_500 samples (bottom). | ||
Conclusions
This study highlights the role of matrix porosity and thermal treatment in governing the topochemical reduction of oxides to metal alloys. By tuning the surface area through annealing, we controlled the diffusion of H2, which directly impacts the extent of reduction and the resulting magnetic properties. When the specific surface area of the matrix decreases due to high-temperature annealing (from ∼512 m2 g−1 post-synthesis, to ∼413 m2 g−1 at 500 °C and finally to ∼345 m2 g−1 at 900 °C), the reduction becomes incomplete, as the limited porosity restricts hydrogen penetration, preventing the full conversion of the oxide phase. This effect is particularly evident in samples annealed above 900 °C, where metallic alloy formation is hindered, and residual spinel oxide phases persist, leading to lower Ms (∼80%) in response to the ∼30% decrease in SA.Author contributions
J. M., A. O., P. M., F. C. and D. P. designed the experiments; P. M., A. O. and D. P. coordinated the data analysis and discussion. J. M. and A. G. synthesized the nanoparticles and characterized them by XRPD and magnetometry techniques, whose results were discussed with G. V., S. L. and D. C. G. B. performed structural characterization by TEM. A. C. performed BET analysis. D. P. acquired the funding, designed and supervised the whole activity. All authors contributed to the results, discussion, and revision of the article.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was partially developed in the framework of the PRIN 2022-PNRR project HyperMag (prot. P2022RRRT4) and supported by European Union – NextGenerationEU. D. P. acknowledges project code PE0000021, Concession Decree No. 1561 of 11.10.2022 adopted by the “Ministero dell’Università e della Ricerca (MUR)”, according to attachment E of Decree No. 1561/2022, Project title “Network 4 Energy Sustainable Transition-NEST” in the frame of a study on surface wettability properties, and REMAP, which has received funding from the European Commission PathFinder Open programme under grant agreement No. 101046909. Funded by the European Union. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or European Innovation Council and SME Executive Agency (EISMEA). Neither the European Union nor the granting authority can be held responsible for them.References
- J. M. Silveyra, E. Ferrara, D. L. Huber and T. C. Monson, Science, 2018, 362(6413) DOI:10.1126/science.aao0195.
- J. M. D. Coey, Magnetism and Magnetic Materials, Cambridge University Press, 2001 Search PubMed.
- C. Desvaux, C. Amiens, P. Fejes, P. Renaud, M. Respaud, P. Lecante, E. Snoeck and B. Chaudret, Nat. Mater., 2005, 4, 2–5 Search PubMed.
- D. L. Huber, Small, 2005, 1, 482–501 CrossRef CAS PubMed.
- J. Liu, D. Su, J. Wang and K. Wu, J. Nanopart. Res., 2020, 22, 1–16 Search PubMed.
- G. Reiss and A. Hütten, Nat. Mater., 2005, 4, 725–726 CrossRef CAS PubMed.
- Ö. Çelik and T. Fırat, J. Magn. Magn. Mater., 2018, 456, 11–16 CrossRef.
- H. Lewis and L. F. Jimenez-Villacorta, Metall. Mater. Trans. A, 2013, 44A, 1–20 Search PubMed.
- C. J. Guzman-Minguez, S. Ruiz-Gomes, M. L. Vicente-Arche, C. Granados-Miralles, C. Fernandez-Fonzalez, F. Mompean, M. Garcia-Hernandez, S. Erohkin, D. Berkov, D. Mishra, C. Julian Fernandez, F. Fernandez, J. L. Perez and A. Quesada, ACS Appl. Nano Mater., 2020, 3, 9842–9851 CrossRef.
- T. Burkert, O. Nordstrom, L. Eriksson and O. Heinonen, Phys. Rev. Lett., 2004, 1, 1–4 Search PubMed.
- Z. Liang, T. Qi, H. Lui, L. Wang and Q. Li, Sci. Total Environ, 2022, 844, 1–11 Search PubMed.
- Y. Liu, X. Wu, X. Guo, K. Lee, Q. Sun, X. Li, C. Zhang, Z. Wang, J. Hu, Y. Zhu, M. K. H. Leung and Z. Zhu, Mater. Today Energy, 2021, 19, 1–8 Search PubMed.
- Y. Chen and A. El-Ghazaly, Small, 2023, 19, 2205079 CrossRef CAS PubMed.
- G. Varvaro, D. Peddis, G. Barucca, P. Mengucci, V. Rodionova, K. Chichay, A. M. Testa, E. Agostinelli and S. Laureti, ACS Appl. Mater. Interfaces, 2015, 7, 22341–22347 CrossRef CAS PubMed.
- B. Muzzi, M. Albino, C. Innocenti, M. Petrecca, B. Cortigiani, C. de, J. Fernández, G. Bertoni, R. Fernandez-Pacheco, A. Ibarra, C. Marquina, M. R. Ibarra and C. Sangregorio, Nanoscale, 2020, 12, 14076–14086 RSC.
- H. K. Daima, P. N. Navya, S. Ranjan, N. Dasgupta and E. Lichtfouse, Nanosci. Med., 2020, 1, 39 Search PubMed.
- J. Bai, Y. Xu, J. Thomas and J. Wang, Nanotechnology, 2007, 18, 1–5 Search PubMed.
- G. Song, M. Kenney, Y. S. Chen, X. Zheng, Y. Deng, Z. Chen, S. X. Wang, S. S. Gambhir, H. Dai and J. Rao, Nat. Biomed. Eng., 2020, 4, 325–334 CrossRef CAS PubMed.
- J. M. D. Coey, Magnetism and Magnetic Materials, Cambridge University Press, 2001 Search PubMed.
- E. Fish, Proc. IEEE, 1990, 78, 947–972 CrossRef.
- T. Sourmail, Prog. Mater. Sci., 2005, 50, 816–880 CrossRef CAS.
- R. S. Sundar and S. C. Deevi, Int. Mater. Rev., 2005, 50, 157–192 CrossRef CAS.
- J. M. MacLaren, T. C. Schulthess, W. H. Butler, R. Sutton and M. McHenry, J. Appl. Phys., 1999, 85, 4833–4835 Search PubMed.
- B. D. Cullity and C. D. Graham, Introduction to Magnetic Materials, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008 Search PubMed.
- L. Leyssens, B. Vinck, C. Van Der Straeten, F. Wuyts and L. Maes, Toxicology, 2017, 17, 1–54 Search PubMed.
- M. Lu, M. Liu, L. Wang, S. Xu, J. Zhao and H. Li, J. Alloys Compd., 2017, 690, 27–30 CrossRef CAS.
- S. Wu, A. Sun, Z. Lu, C. Cheng and X. Gao, J. Magn. Magn. Mater., 2015, 381, 451–456 CrossRef CAS.
- F. Casula, M. A. Corrias, A. Falqui, V. Serin, D. Gatteschi, C. Sangregorio, C. Fernandez and G. Battaglin, Chem. Mater., 2003, 15, 2201–2207 CrossRef.
- A. K. Giri, C. De Julian and J. M. Gonzalez, J. Appl. Phys., 1994, 76, 6573–6575 CrossRef CAS.
- X. Lu, G. Liang and Y. Zhang, Mater. Lett., 2007, 61, 4928–4931 CrossRef CAS.
- P. Tartaj and C. J. Serna, J. Am. Chem. Soc., 2003, 125, 15754–15755 CrossRef CAS PubMed.
- H.-S. Cho, M. S. Noh, Y.-H. Kim, J. Namgung, K. Yoo, M.-S. Shin, C.-H. Yang, Y. J. Kim, S.-J. Yu, H. Chang, W. Y. Rho and B.-H. Jun, Nanomaterials, 2024, 14, 268 CrossRef CAS PubMed.
- P. Sahu and M. Ali, Mol. Syst. Des. Eng., 2022, 7, 1501–1515 RSC.
- C. Mohr, M. Dubiel and H. Hofmeister, J. Phys.: Condens. Matter, 2001, 13, 525–536 CrossRef CAS.
- C. Briese, L. S. Selle, C. Patzig, Y. Hu, J. Deubener and T. Höche, J. Non Cryst. Solids, 2020, 549, 1–9 CrossRef.
- G. Ennas, A. Falqui, S. Marras and C. Sangregorio, Chem. Mater., 2004, 16, 5659–5663 CrossRef CAS.
- A. Casu, M. F. Casula, A. Corrias, A. Falqui, D. Loche, S. Marras and C. Sangregorio, Phys. Chem. Chem. Phys., 2008, 10, 1043–1052 RSC.
- G. Concas, F. Congiu, G. Ennas, G. Piccaluga and G. Spano, J. Non Cryst. Solids, 2003, 330, 234–241 CrossRef CAS.
- C. Cannas, A. Musinu, D. Peddis and G. Piccaluga, Chem. Mater., 2006, 18, 3835–3842 CrossRef CAS.
- G. Ennas, G. Marongiu, S. Marras and G. Piccaluga, J. Nanopart. Res., 2004, 6, 99–105 CrossRef CAS.
- A. Omelyanchik, G. Varvaro, P. Maltoni, V. Rodionova, J.-P. M. Murillo, F. Locardi, M. Ferretti, C. Sangregorio, F. Canepa, P. Chernavsky, N. Perov and D. Peddis, Appl. Sci., 2022, 12, 1899 CrossRef CAS.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- C. Cannas, A. Musinu, D. Peddis and G. Piccaluga, J. Nanopart. Res., 2004, 6, 223–232 CrossRef CAS.
- S. E. Sandler, B. Fellows and O. T. Mefford, Anal. Chem., 2019, 91, 14159–14169 CrossRef CAS PubMed.
- C. Granados-Miralles, A. Quesada, M. Saura-Múzquiz, H. L. Andersen, J. F. Fernández and M. Christensen, Mater. Chem. Front., 2020, 4, 1222–1230 RSC.
- H. Stuart and N. Ridley, J. Phys. D: Appl. Phys., 1969, 2, 302 Search PubMed.
- D. Bardos, J. Appl. Phys., 1969, 40, 53–55 CrossRef.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrică, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W. -Z. Yu and T. Frauenheim, J. Chem. Phys., 2020, 152, 124101 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- N. Ntallis, D. Peddis and K. Trohidou, DFT calculations of high moment Fe1−xCox alloy and Fe/Co, Co/Fe core shell magnetic nanoparticles, 2025, RSC, in submission.
- T. Block, N. Knoblauch and M. Schmücker, Thermochim. Acta, 2014, 577, 25–32 CrossRef CAS.
- A. Omelyanchik, M. Salvador, F. D’Orazio, V. Mameli, C. Cannas, D. Fiorani, A. Musinu, M. Rivas, V. Rodionova, G. Varvaro and D. Peddis, Nanomaterials, 2020, 10, 1288 CrossRef CAS PubMed.
- J. R. Porter, L. C. De Jonghe and D. Jonghe, Metall. Trans. B, 1981, 12, 299–309 CrossRef.
- A. Corrias, F. M. Casula, G. Ennas, S. Marras, G. Navarra and G. Mountjoy, J. Phys. Chem. B, 2003, 107, 3030–3039 CrossRef CAS.
- T. Dippong, O. Cadar and E. A. Levei, Materials, 2022, 15, 2996 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00305a |
| This journal is © the Owner Societies 2025 |