
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEstimating protein binding upon treatment with radionuclide ions
Ran
Friedman

Department of Chemistry and Biomedical Sciences, Linnaeus University, SE-391 82 Kalmar, Sweden. E-mail: ran.friedman@lnu.se
First published on 4th June 2025
Abstract
Several types of radioactive isotopes are used for cancer treatment. While most are embedded in chelating agents, 223Ra is given as RaCl2 salt and 90Y in microspherical particles. If ionic radionuclides are free, they have the potential to bind to proteins instead of their endogenous ions, interfere with their activity and be transported by them. In this study, a computational approach was used to estimate the binding affinities of Y3+, Ra2+ and Pb2+ (207Pb is the decay product of 223Ra) to proteins, instead of their native cofactors Ca2+ and Mn2+. Y3+ was found to bind strongly to proteins with the ability to replace Ca2+ and to some degree also Mn2+. Ra2+ does not bind to the studied proteins but Pb2+ can replace Ca2+ in Ca2+ binding proteins. A recently identified coordination compound was found to be highly selective for 223Ra.
1 Introduction
Radionuclides have been used in cancer therapy since the beginning of the 20th century. Initially, radium salts were used for skin cancers.1 Thereafter, instruments were developed to deliver the radiation directly to the tumour,2 whereby the use of solid salts had a benefit over radiation therapy. The common isotope of radium, 226Ra, however, has a long half-life of 1599 years and is not considered safe for therapy. Nevertheless, radioactive salts have emerged as potential therapies and in 1951 the use of 131I was approved by the U.S. Food and Drug Administration (FDA) for thyroid cancer.3 With a half-life of 8 days and decay through β- and γ- rays to stable and inert 131Xe, 131I has been shown to be a safe and effective therapy. Meanwhile, treatment with radium isotopes, was reintroduced and has been used since 2013 for castration-resistant prostate cancer with symptomatic bone metastases. To this end, the less stable isotope 223Ra is given as RaCl2 salt. 223Ra has a half-life of 11.43 days and decays by α- and β-radiation to form stable 207Pb. It is believed that the radioactive ions adsorb preferentially to the bones. Early clinical studies have shown improved overall survival4 (albeit by a median of 3 months), and a good safety profile in a three years follow-up study.5 On the other hand, a recent study6 reported haematological toxicities in ∼15% of the patients and second primary malignancies in ∼1%.Delivery of radionuclides to the tumour can be made directly when the radionuclide is given as salt (as is the case for 131I), through chelating agents that strongly bind a radioactive ion (these can be further attached to peptides or antibodies for specific delivery), or through the use of microspheres. The latter method has so far been used exclusively with the 90Y isotope. This therapy, known as radioembolisation, is used for liver tumours. The radioactive microspheres are injected directly into blood vessels that supply blood to the tumour and get stuck in tiny blood vessels in the tumour. 90Y decays to stable 90Zr through release of β radiation. The isotope has a half-life of 2.67 days. There are two different ways to deliver 90Y to the tumour, namely glass spheres which encapsulate the radionuclides and resin spheres where 90Y is bound to the surface. These treatments are effective and relatively safe. However, a systemic review showed gastrointestinal, hepatic and respiratory toxicities with both types of microspheres.7 It is likely that the decay product is released from the microsphere with the impact from the radiation. In addition, the release of energy can lead to unbinding of 90Y3+ ions from microspheres in the immediate vicinity of the β emitter.
Multivalent metal ions such as Mg2+, Ca2+, Zn2+, Mn2+ and others are important cofactors of proteins and maintain proper protein structure and function. The binding of xenobiotic ions instead of endogenous ones can lead to toxicities, by modifying the protein's structure, hindering catalysis, or reducing the affinity to substrates. Treatment with radionuclides that are multivalent metal ions could lead to the radionuclide binding to proteins, thereby being transported and also affecting protein activity. With respect to the latter risk, not only the metal ions themselves but also their decay products should be considered. 223Ra poses a particular risk since it is given as soluble salt, whereas 90Y could mediate such risks if unbound from the microspheres. In the case of 223Ra, especially with repeated therapy, even the decay product 207Pb might cause a risk. 90Y however decays to 90Zr. The latter form stable Zr4+ ions that are strongly hydrated and carry low if any health risk.
Little is known about the biological activity of Ra2+ and Y3+ and whether these ions can bind to proteins. The scarcity and radioactivity of radium, and the difficulty to separate Y3+ from other rare-earth metals make it difficult to experimentally study their effects in biology in general and binding to proteins in particular. Here, a computational approach is used to estimate the risks with binding of the radioactive ions to proteins in the body. Since Ra2+ binds to bone instead of Ca2+, and as lanthanides were shown to bind strongly to Ca2+ binding proteins,8 the bindings to such proteins were studied first. To this end, calmodulin (CaM) was used as a prototype of Ca2+ binding proteins, as it exhibits the common EF-hand domain that binds Ca2+ in many proteins. EF domains are remarkably similar between different proteins and the binding to the EF domain in calmodulin can thus inform on the interference of ions with most Ca2+-binding proteins in the human body. Protein kinase A (PKA) is also considered, as it exhibits a different Ca2+ binding site and can accommodate larger ions as well. Finally, Mn2+ can form coordination complexes with varied (often high) coordination numbers where it binds to hard ligands and hence DNA polymerase ι that preferentially uses Mn2+ as a cofactor was also considered in this study. The binding of Ra2+'s decay product Pb2+ to those proteins was examined as well. For the Ca2+ binding proteins, the affinity to Sr2+ was considered a reference for the calculations, since it is known that Sr2+ is chemically similar to Ca2+ and might replace it.
2 Theory and methods
2.1 Theory
| ΔΔGbind = ΔfG°(prot·M) − ΔfG°(prot·L) − [G°(M) − G°(L)] | (1) |
In reality, it is not possible to consider the full environment (protein, other cofactors, substrate(s), water, additional ions, etc.) for the complex. In calculations of ion binding affinities it is therefore assumed that the coordinating groups have the most significant influence on the interaction energy and selectivity for ions.8–10 Thus, eqn (1) is approximated as:
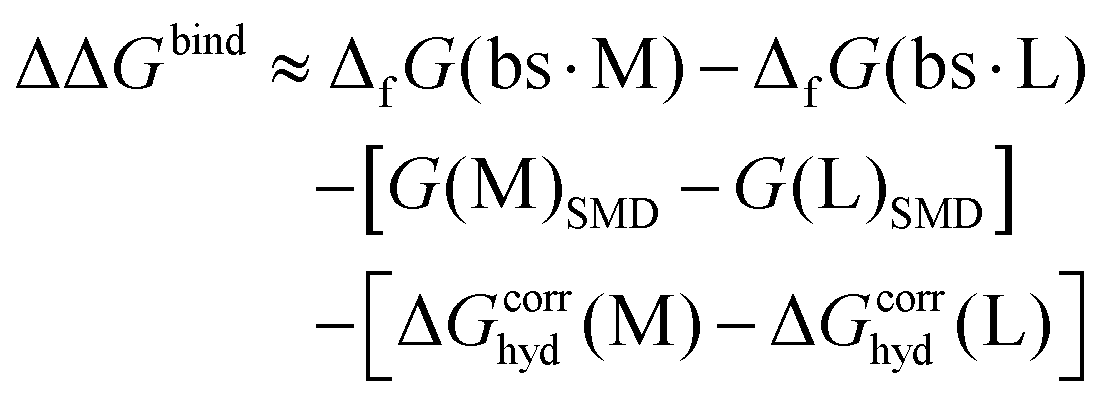 | (2) |
Here, ΔfG°(prot·□) was replaced by ΔfG(bs·□) where bs stands for ‘binding site’, surrounded by implicit solvent (here, using the SMD model11). Gibbs energies of the ions in solvent were replaced by their values in the same solvent model, G(□)SMD. The approximate nature of the implicit solvent model necessitates careful calibration of the radii of ions to reproduce the correct hydration energy,12 while the ionic radius has a limited effect in the complex because the ion is totally surrounded by larger moieties. Since the ionic radii are hard-coded in some quantum chemistry software packages, instead of modifying the radius it is possible to correct for the difference between the real value of the hydration energy (estimated from experiments) and the value calculated with the implicit solvent model which is referred to here as ΔGcorrhyd(□). Explicitly, the values were calculated as:
| ΔGcorrhyd = ΔGexphyd − ΔGSMDhyd | (3) |
ΔGexphyd is the experimental value for the hydration energy of an ion and ΔGSMDhyd is the calculated value.
To mimic the environment inside a protein metal binding site, which is partially exposed to the solvent and includes many polar residues, ethanol (ε = 24.5) was used as the solvent of the complex.
The free energies of formation of the binding site here include the internal (Gibbs) energy calculated with a quantum mechanical (QM) method of choice (here, density functional theory, DFT) in implicit solvent. Although a single point energy calculation without considering thermochemical corrections is formally the Gibbs energy in solvent, corrections for the enthalpy in finite temperature (here, 300 K), vibrational, rotational and translational entropies of the complex are included in the values for ΔfG(bs·□).
| ΔGtotal = ΔGele + ΔGex + ΔGrep + ΔGpol + ΔGcorr + ΔGdisp + ΔGdesolv | (4) |
It is important to note that thermochemistry (corrections for the enthalpy and entropy of the solutes in finite temperature) is not included in the EDA. In addition, calculating the EDA necessitates the use of the same geometry for the complex and its components, i.e. the protein binding site is assumed to be pre-formed. This is however not important when comparing between multiple ions binding to the same protein, since the free protein (or binding site) always has the same structure.
2.2 Computational methods
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. All optimisations were carried out in implicit solvent model (SMD11), with ethanol (ε = 24.5) as a solvent to model the protein environment.
O. All optimisations were carried out in implicit solvent model (SMD11), with ethanol (ε = 24.5) as a solvent to model the protein environment.
2.2.1.1 Calmodulin. The PDB structure 1CLL14 was used to build the model. Binding site 1 was modelled with the side chains of residues Asp20, Asp22, Asp24 and Glu31, the backbone carbonyl of Thr26, and one water molecule (36 atoms). Binding site 2 was modelled with the side chains of residues Asp56, Asp58, Asn60 and Glu67, the backbone carbonyl of Thr62, and one water molecule (38 atoms).
2.2.1.2 Protein kinase A. The structure of PKA with two Ba2+ ions (PDB code 4IAZ15) was used as a starting structure for optimisations. Preliminary calculations showed that starting the optimisations with a structure where PKA binds to Sr2+ did not modify the results. The two cations in the binding site were modelled with the backbone of Asn171 (modelled as acetamide), the side chain of Asp184, ADP modelled as diphosphoricacid, monomethylester with −3 charge, the side chain of pSer621 modelled as methylphosphate with −3 charge and six water molecules. Residue Lys72 and four additional water molecules were included for solvation of the negative charges. There were 81 atoms in the structure.
2.2.1.3 DNA polymerase ι. The non-truncated structure of DNA polymerase ι (residues 1–455) with a short DNA sequence, a DNA template and two Mn2+ with PDB code 5KT716 was used. The side chains of residues Asp34, Asp126 and Glu127, the backbone carbonyl of Leu35 (all residues in the PDB structure numbering), a dCTP analogue truncated at the phosphates with a methyl, and a hydroxyl oxygen from the DNA primer were included, for a total of 50 atoms.
| Ion | ΔGexphyd |
|---|---|
| Ca2+ | −363.5 |
| Mn2+ | −437.4 |
| Sr2+ | −329.8 |
| Y3+ | −824.6 |
| Ba2+ | −298.8 |
| Pb2+ | −340.6 |
| Ra2+ | −289.5 |
Galland and co-workers34 pointed out the importance of spin orbit coupling (SOC) for calculations of binding energies of chelators to Ra2+. Indeed, preliminary calculations with a recently described radium binding complex39 have shown that the binding energy of Ra2+ to this complex was lowered (i.e. became more favourable) by −10.9 kcal mol−1 when SOC was considered. For the same complex with Pb2+, the second heaviest ion studied here, SOC was negligible (−0.2 kcal mol−1). Thus, SOC was included when calculating differences in the binding of Ra2+ to other ions in all complexes. A SOC correction, ΔESOC was added to ΔfG(bs·M) in eqn (2) when M was Ra2+. This correction was calculated as:
| ΔESOC = [E(bs·M)SODFT − E(M)SODFT] − [E(bs·M)DFT − E(M)DFT] | (5) |
To this end, the energies of the complexes, E(bs·M), and the ion E(M) were calculated with NWCHEM40,41 twice, once using DFT without SOC and once using spin–orbit DFT (SODFT). The same basis set and DFT functional were used as for the calculation of geometries and binding energies.
3 Results
Given that Ra2+ binds to hydroxyapatite in bones and that lanthanides, which are similar to Y3+ in many aspects, can bind to Ca2+-binding proteins,8 the potential bindings of Ra2+, its decay product Pb2+, and Y3+ were estimated for calmodulin (CaM), a prototype calcium-binding protein with the typical calcium binding sites. Thereafter, the binding of the ions was studied to protein kinase A (PKA), an enzyme that has two metal-ion binding sites in its catalytic pocket where the metals are coordinated to amino acids, water and a phosphate. PKA is known to be able to bind Mg2+, Ca2+, Sr2+ and Ba2+, while maintaining activity.15 Finally, the ability of Ra2+ and Y3+ to bind to an Mn-dependent enzyme, DNA polymerase ι, was also examined.3.1 Calmodulin
CaM binds Ca2+ in four binding sites, each comprising of an EF-hand which is a typical Ca2+-binding motif in proteins (Fig. 1). The sites are overall similar, with seven oxygen ligands, but differ in the number of carboxylate oxygens that coordinate to the metal ion: five in sites 1 and 4, four in sites 2 and 3. The different coordinations can lead to different binding energies and preferences for coordination of ions besides Ca2+. For this reason, sites 1 (prototype of a binding site with five coordinating carboxylate atoms) and 2 (prototype of a binding site with four coordinating carboxylate atoms) were studied here. | ||
| Fig. 1 The structure of calmodulin (PDB code: 1CLL). The location of the four Ca2+ ions is indicated as green spheres. | ||
The Gibbs energy differences with respect to Ca2+, for binding of the ions to the two binding sites of CaM, are shown in Table 2. Sr2+, an alkali earth metal and an analogue of Ca2+ is known to be able to bind CaM when present in high enough concentrations47 and was included in this study as a control. Indeed, the calculations show that binding of the Sr2+ ions to calmodulin is disfavoured by 2.4 to 2.9 kcal mol−1 in comparison to Ca2+, suggesting that the concentration of Sr2+ should be about 100 times larger than that of Ca2+ in order to bind the protein. In contrast, both Pb2+ and Y3+ strongly interact with CaM, with the first preferring site 2 and the second site 1. Finally, with an energy difference of over 6 kcal mol−1 disfavouring its binding, it cannot be expected that Ra2+ will replace Ca2+ in CaM or other EF-hand proteins.
| Ion | ΔΔGbind | ΔΔGbind |
|---|---|---|
| Site 1 | Site 2 | |
| Sr2+ | 2.4 | 2.9 |
| Pb2+ | −6.7 | −9.9 |
| Y3+ | −28.5 | −20.6 |
| Ra2+ | 6.0 | 9.6 |
To better understand the reasons for the preferred binding of Y3+ and Pb2+ to CaM, and the inability of Ra2+ to replace Ca2+, the interactions of the ions with the binding site were subject to energy decomposition analysis (EDA) calculations using the PCM-EDA approach.13 The EDA results (Table 3) reveal that pure electrostatic interactions are weaker for Pb2+ when compared with Ca2+, but polarisation for Pb2+ is stronger and there is a lower cost of desolvation for the binding of this ion. Y3+ binds better due to increased electrostatics and polarisation and in spite of the higher cost of desolvating the ion. Sr2+ behaves as Ca2+ in general. Finally, Ra2+ behaves like a hard ion, with a less pronounced contribution from polarisation relative to electrostatics and otherwise weaker interactions than all other ions (except for dispersion and correlation that do not constitute important contributions to ion binding). Of note, the total interaction energies as calculated by EDA are different than those used to obtain the values presented in Table 2. This is due to the fact that the binding energies calculated in EDA do not include rotational and vibrational entropy, and corrections to the enthalpy. Basis set differences account for a smaller share of the difference.
| Ca | Sr | Pb | Y | Ra | |
|---|---|---|---|---|---|
| Site 1 | |||||
| ΔGele | −929.8 | −893.7 | −774.5 | −1412.3 | −825.1 |
| ΔGexrep | 89.7 | 90.6 | 102.4 | 177.0 | 72.8 |
| ΔGpol | −120.5 | −101.8 | −233.3 | −340 | −77.9 |
| ΔGcorr | −0.5 | −6.8 | −28.3 | −24.4 | −12.9 |
| ΔGdisp | −3.5 | −4.2 | −5.8 | −3.6 | −5.5 |
| ΔGdesolv | 736.2 | 690.8 | 683.4 | 1229.0 | 679.2 |
| ΔEsoc | −6.4 | ||||
| ΔGtotal | −228.4 | −225.2 | −256.1 | −374.3 | −175.8 |
| Site 2 | |||||
| ΔGele | −786.2 | −754.9 | −686.1 | −1175.5 | −705.4 |
| ΔGexrep | 91.1 | 91.2 | 99.0 | 176.6 | 67.1 |
| ΔGpol | −123.2 | −104.2 | −205.5 | −352.8 | −68.5 |
| ΔGcorr | 0.8 | −4.9 | −31.6 | −23 | −9.6 |
| ΔGdisp | −3.5 | −4.2 | −5.3 | −3.8 | −5.5 |
| ΔGdesolv | 609.4 | 563.3 | 577.0 | 1014.3 | 570.5 |
| ΔEsoc | −5.8 | ||||
| ΔGtotal | −211.5 | −213.7 | −252.6 | −364.2 | −157.2 |
3.2 Protein kinase A
PKA was crystallised with Mg2+, Ca2+, Sr2+ and Ba2+. The structures and metal binding sites are overall highly similar, with small differences due to the different size of the ions (Fig. 2). The coordinating moieties for the two ions included the amide oxygen of Asn171, one carboxylate oxygen of Asp184, phosphate groups from ADP, the substrate phosphate oxygens, and six water oxygens. The binding energies for the ions, relative to Ca2+ are shown in Table 4 (values are for a pair of ions in each case). The results reveal that Sr2+ binds somewhat worse than Ca2+, with Ba2+ binding even less, Pb2+ binds better and will likely impair the protein, and Y3+ even better than Pb2+. Surprisingly, Ra2+ binds only slightly weaker than Ca2+ and better than Sr2+ and Ba2+. This is due to SOC effects, that amount to −17.5 kcal mol−1. However, the high physiological concentration of Ca2+, about 1 mM in the blood, makes it unlikely that 223Ra will bind PKA in appreciable amounts.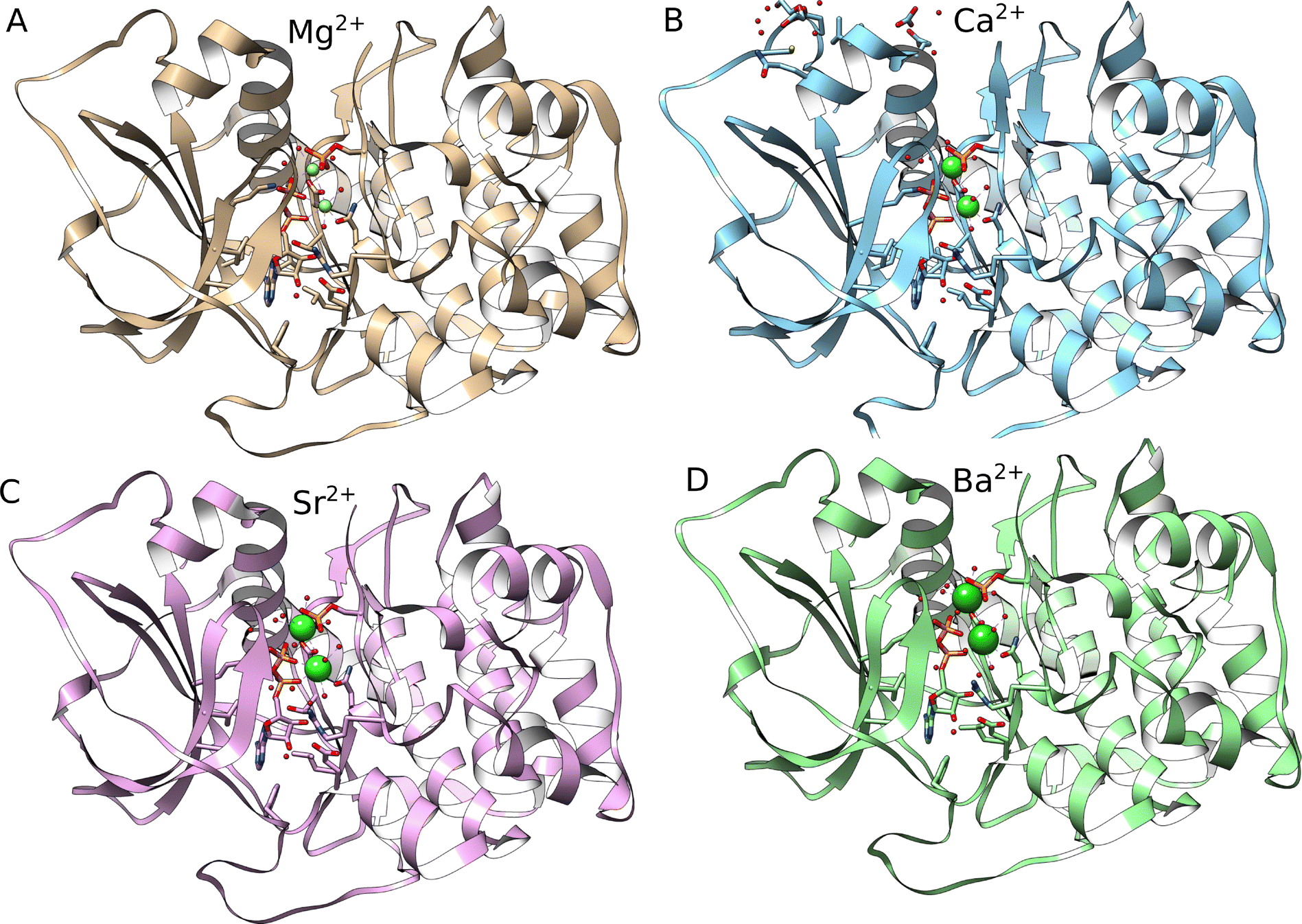 | ||
| Fig. 2 The crystal structures of PKA with (A) Mg2+, (B) Ca2+, (C) Sr2+ and (D) Ba2+ (PDB codes: 4IAF, 4IAI, 4IAL and 4IAZ, respectively). The locations of the ions are indicated as green spheres. | ||
| Ion | ΔΔGbind |
|---|---|
| Sr2+ | 3.4 |
| Ba2+ | 10.8 |
| Pb2+ | −7.3 |
| Y3+ | −43.0 |
| Ra2+ | 1.1 |
As it is clear from the experiment that PKA can bind Ba2+, the values in Table 4 indicate that it can bind all ions. Thus, it is interesting to examine the structures of the optimised complexes. Overall, the ion binding sites are quite similar (Fig. 3). There are some differences when compared to the X-ray structures, that are due to the optimisation. It is likely that in solution the binding site of the ions can adopt multiple conformations that cannot all be adequately sampled here, as also shown in the two structures of the same protein with Sr2+. In the optimised complexes, the ion–ligand distances increase within the alkali earth series, as expected (Table 5). The coordination number (CN) is smaller for Ca2+ in comparison with the larger alkali-earth ions. Interestingly, while Sr2+ and Ba2+ bind to the second site in PKA with a higher CN, Ra2+ on the contrary binds to the first site with higher CN; this reveals some differences in the binding. Pb2+ binds with higher CN to the first site, in similarity with Ra2+. Y3+ binds to PKA as Ca2+ but with smaller ion–ligand distances.
 | ||
| Fig. 3 Optimised complexes of the ion binding site of PKA with Ca2+, Sr2+, Ba2+, Y3+, Pb2+ and Ra2+. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
3.3 DNA polymerase ι
DNA polymerase ι achieves high activity with Mn2+ as cofactor. The structure of the protein reveals that it binds two Mn2+ ions through one backbone oxygen, five carboxylate oxygens, three phosphate groups, and one oxygen from the primer that is added to the substrate, yielding a 5 + 6 coordinated binding site (Fig. 4). The results of the calculation of the binding energies show that the protein binds somewhat better to Y3+ than Mn2+ and much less to Pb2+ and Ra2+. In practice it cannot be expected that the protein will bind Ra2+ at all (Table 6). | ||
| Fig. 4 The ion binding site of DNA polymerase ι. Mn2+ ions are shown as violet spheres. | ||
| Ion | ΔΔGbind |
|---|---|
| Pb2+ | 17.6 |
| Y3+ | −16.0 |
| Ra2+ | 53.0 |
4 Discussion
4.1 Radium does not significantly replace other ions in proteins
The results show that Ra2+ binds to proteins less than Ca2+ and Mn2+. It was further shown to be a hard ion, displaying low polarisability while at the same time its electrostatic interactions are weaker than those of Ca2+. Interestingly, analysing binding to water as a ligand, Matsuda and Mori have shown a similar trend with contributions from electrostatics and polarisation steadily declining along the alkaline-earth series.48 Toxicities of 223Ra therapy can thus be attributed to direct effects of its decay outside of the tumour tissue. It is not likely that it is transported by proteins or affects protein activity.4.2 Pb2+ binds to proteins instead of Ca2+
Unlike 223Ra, its decay product 207Pb can bind to proteins as a doubly charged ion and shows affinity to typical (CaM) Ca2+-bind proteins and to PKA that is higher than the affinities of these proteins to Ca2+. Its binding energy was estimated to be up to ∼10 kcal mol−1 more favourable than that of Ca2+. Pb2+ is a soft ion, and contribution from polarisation has been high (almost twice that of Ca2+) even with hard ligands such as oxygen. On the other hand, Pb2+ is also adsorbed to the bone tissue and its release is slow. It remains to be seen if repeated exposure to 223Ra treatment poses a risk of Pb2+ poisoning. However, this study points out that accidental overdose of 223Ra Cl2 can mitigate not only too much radiation (acute toxicity) but also lead exposure (chronic toxicity).4.3 Yttrium binds strongly to proteins
Despite its strong hydration Y3+ has shown significant binding to proteins, with the ability to replace Ca2+ and potentially also Mn2+. The short lifetime of 90Y is beneficial in this respect, since it is mostly trapped in the liver. Toxicities associated with 90Y therapy were indicated to be somewhat higher with resin compared to glass microspheres,6 which might have to do with the binding of the ions at the surface of such spheres from which they can presumably more easily escape and thereafter bind to proteins. Considering the accuracy of the calculations, it should be noted that Y3+ is the only ion that differs in charge from the others. Estimations of the hydration free energies in this work were taken from the seminal work of Marcus.35 Such estimations depend on the value that is used for calculating the hydration energy of a proton (ΔGhyd(H+) = −252.4 kcal mol−1). As Marcus noted, other estimations were more negative (by up to 11.2 kcal mol−1). Comparing between ions, the choice of this value will only affect ΔΔGbind values for Y3+, reducing its Gibbs hydration energy by the same amount. Had the lowest estimation of ΔGhyd(H+) been used, the results with respect to binding of Y3+ would have been shifted up. Qualitatively, this would have meant that Y3+ could still have replaced Ca2+ and Mn2+ in proteins.4.4 Targeted delivery of radium
While 223Ra is used for bone metastases it is difficult to see how it can be used to target other organs than the bone. For targeted delivery, there is a need to design chelators that will bind to 223Ra. It has been difficult to develop coordination complexes with high affinity to 223Ra that must not be replaced by physiological ions such as Ca2+. The radioactivity and scarcity of Ra on the one hand, and its lower ability to bind many ligands on the other have hindered the development of Ra-chelators, with the first crystallographic structure of a radium complex described only recently.39 The neutral crown ether cage that binds the ion in such host–guest complexes (Fig. 5) was shown to be able to bind 223Ra and Ba2+ with preferences that depend on the co-anions.39 For such complexes that are injected or ingested in the body, it is important to verify if other ions might displace 223Ra. Examination of the affinities of the ions Ba2+, Pb2+, Y3+ and Ca2+ to this crown ether complex leads to the conclusion that this particular host–guest complex binds Ra2+ better than those other ions (Table 7). Nevertheless, it is important to analyse the risks to release of decay products in the body. The high energy release with the first alpha-particle upon decay from 223Ra to 219Rn will likely break down the coordination compound and release gaseous, radioactive 219Rn with a half-life of ∼4 s. The radon atom will diffuse in the nearby tissue before decaying further. Thus, the risks associated with radon diffusion also need to be taken into account when considering targeted delivery of radium. | ||
| Fig. 5 A crown ether complex bound to Ra2+. Optimised structure, with the coordinates from ref. 39. | ||
| Ion | ΔΔGbind |
|---|---|
| Ca2+ | 18.5 |
| Pb2+ | 7.6 |
| Ba2+ | 6.9 |
| Y3+ | 28.1 |
Data availability
All calculations were performed with open source or academic licence software as described in the main text. Optimised structures of metal complexes are available at https://dx.doi.org/10.6084/m9.figshare.27613200.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computations (except EDA) were enabled by resources provided by LUNARC, The Centre for Scientific and Technical Computing at Lund University, and the National Academic Infrastructure for Supercomputing in Sweden (NAISS), partially funded by the Swedish Research Council through grant agreement no. 2022-06725. EDA calculations were run on the Xiamen Atomistic Computing Suite (XACS).Notes and references
- J. H. Sequeira, BMJ, 1915, 1, 365–366 CrossRef CAS PubMed.
- A. Dronsfield and P. Ellis, Educ. Chem., 2011, 48, 56 CAS.
- P. Borges de Souza and C. J. McCabe, Endocr.-Relat. Cancer, 2021, 28, T121–T124 Search PubMed.
- C. Parker, S. Nilsson, D. Heinrich, S. Helle, J. OSullivan, S. FossÅ, A. Chodacki, P. Wiechno, J. Logue, M. Seke, A. Widmark, D. Johannessen, P. Hoskin, D. Bottomley, N. James, A. Solberg, I. Syndikus, J. Kliment, S. Wedel, S. Boehmer, M. DallOglio, L. Franzén, R. Coleman, N. Vogelzang, C. OBryan-Tear, K. Staudacher, J. Garcia-Vargas, M. Shan, Ø. Bruland and O. Sartor, N. Engl. J. Med., 2013, 369, 213–223 CrossRef CAS PubMed.
- C. C. Parker, R. E. Coleman, O. Sartor, N. J. Vogelzang, D. Bottomley, D. Heinrich, S. I. Helle, J. M. OSullivan, S. D. FossÅ, A. Chodacki, P. Wiechno, J. Logue, M. Seke, A. Widmark, D. C. Johannessen, P. Hoskin, N. D. James, A. Solberg, I. Syndikus, J. Kliment, S. Wedel, S. Boehmer, M. DallOglio, L. Franzén, Ø. S. Bruland, O. Petrenciuc, K. Staudacher, R. Li and S. Nilsson, Eur. Urol., 2018, 73, 427–435 CrossRef.
- C. S. Higano, D. J. George, N. D. Shore, O. Sartor, K. Miller, P. S. Conti, C. N. Sternberg, F. Saad, J. P. Sade, J. Bellmunt, M. R. Smith, K. Chandrawansa, P. Sandström, F. Verholen and B. Tombal, eClinicalMedicine, 2023, 60, 101993 CrossRef.
- J. R. Kallini, A. Gabr, K. Thorlund, C. Balijepalli, D. Ayres, S. Kanters, S. Ebrahim, E. Mills, R. J. Lewandowski and R. Salem, Cardiovasc. Intervent. Rad., 2017, 40, 1033–1043 CrossRef PubMed.
- R. Friedman, J. Phys. Chem. B, 2021, 125, 2251–2257 CrossRef CAS.
- R. Friedman, Dalton Trans., 2014, 43, 2878–2887 RSC.
- L. Moretto, M. Ušaj, O. Matusovsky, D. E. Rassier, R. Friedman and A. Månsson, Nat. Commun., 2022, 13, 4575 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- R. Friedman, ChemPhysChem, 2022, 24, e202200516 CrossRef PubMed.
- P. Su, H. Liu and W. Wu, J. Chem. Phys., 2012, 137, 034111 CrossRef PubMed.
- R. Chattopadhyaya, W. E. Meador, A. R. Means and F. A. Quiocho, J. Mol. Biol., 1992, 228, 1177–1192 CrossRef CAS PubMed.
- O. Gerlits, M. J. Waltman, S. Taylor, P. Langan and A. Kovalevsky, Biochemistry, 2013, 52, 3721–3727 CrossRef CAS PubMed.
- J.-Y. Choi, A. Patra, M. Yeom, Y.-S. Lee, Q. Zhang, M. Egli and F. P. Guengerich, J. Biol. Chem., 2016, 291, 21063–21073 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2e, 73–78 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2025, 15, e70019 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- J. Koput and K. A. Peterson, J. Phys. Chem. A, 2002, 106, 9595–9599 CrossRef CAS.
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed.
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2006, 125, 074110 CrossRef PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef CAS.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- Y. Osanai, E. Soejima, T. Noro, H. Mori, M. S. Mon, M. Klobukowski and E. Miyoshi, Chem. Phys. Lett., 2008, 463, 230–234 CrossRef CAS.
- J. G. Hill and K. A. Peterson, J. Chem. Phys., 2017, 147, 244106 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. Ahlstrand, D. Spångberg, K. Hermansson and R. Friedman, Int. J. Quantum Chem., 2013, 113, 2554–2562 CrossRef CAS.
- H. Mohaman, S. Happel, G. Montavon and N. Galland, New J. Chem., 2023, 47, 12914–12925 RSC.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- R. M. Noyes, J. Am. Chem. Soc., 1962, 84, 513–522 CrossRef CAS.
- I. Persson, M. Sandström and H. Yokoyama, Z. Naturforsch., A: Phys. Sci., 1995, 50, 21–37 CrossRef CAS.
- R. R. Pappalardo, D. Z. Caralampio, J. M. Martínez and E. Sánchez Marcos, Inorg. Chem., 2021, 60, 13578–13587 CrossRef CAS PubMed.
- F. D. White, N. A. Thiele, M. E. Simms and S. K. Cary, Nat. Chem., 2023, 16, 168–172 CrossRef PubMed.
- M. Valiev, E. Bylaska, N. Govind, K. Kowalski, T. Straatsma, H. V. Dam, D. Wang, J. Nieplocha, E. Apra, T. Windus and W. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- E. Aprà, E. J. Bylaska, W. A. de Jong, N. Govind, K. Kowalski, T. P. Straatsma, M. Valiev, H. J. J. van Dam, Y. Alexeev, J. Anchell, V. Anisimov, F. W. Aquino, R. Atta-Fynn, J. Autschbach, N. P. Bauman, J. C. Becca, D. E. Bernholdt, K. Bhaskaran-Nair, S. Bogatko, P. Borowski, J. Boschen, J. Brabec, A. Bruner, E. Cauët, Y. Chen, G. N. Chuev, C. J. Cramer, J. Daily, M. J. O. Deegan, T. H. Dunning, M. Dupuis, K. G. Dyall, G. I. Fann, S. A. Fischer, A. Fonari, H. Früchtl, L. Gagliardi, J. Garza, N. Gawande, S. Ghosh, K. Glaesemann, A. W. Götz, J. Hammond, V. Helms, E. D. Hermes, K. Hirao, S. Hirata, M. Jacquelin, L. Jensen, B. G. Johnson, H. Jónsson, R. A. Kendall, M. Klemm, R. Kobayashi, V. Konkov, S. Krishnamoorthy, M. Krishnan, Z. Lin, R. D. Lins, R. J. Littlefield, A. J. Logsdail, K. Lopata, W. Ma, A. V. Marenich, J. Martin del Campo, D. Mejia-Rodriguez, J. E. Moore, J. M. Mullin, T. Nakajima, D. R. Nascimento, J. A. Nichols, P. J. Nichols, J. Nieplocha, A. Otero-de-la Roza, B. Palmer, A. Panyala, T. Pirojsirikul, B. Peng, R. Peverati, J. Pittner, L. Pollack, R. M. Richard, P. Sadayappan, G. C. Schatz, W. A. Shelton, D. W. Silverstein, D. M. A. Smith, T. A. Soares, D. Song, M. Swart, H. L. Taylor, G. S. Thomas, V. Tipparaju, D. G. Truhlar, K. Tsemekhman, T. Van Voorhis, Á. Vázquez-Mayagoitia, P. Verma, O. Villa, A. Vishnu, K. D. Vogiatzis, D. Wang, J. H. Weare, M. J. Williamson, T. L. Windus, K. Woliński, A. T. Wong, Q. Wu, C. Yang, Q. Yu, M. Zacharias, Z. Zhang, Y. Zhao and R. J. Harrison, J. Chem. Phys., 2020, 152, 184102 CrossRef PubMed.
- Z. Tang, Y. Song, S. Zhang, W. Wang, Y. Xu, D. Wu, W. Wu and P. Su, J. Comput. Chem., 2021, 42, 2341–2351 CrossRef CAS PubMed.
- Y. Sakai, E. Miyoshi, M. Klobukowski and S. Huzinaga, J. Comput. Chem., 1987, 8, 226–255 CrossRef CAS.
- Y. Sakai, E. Miyoshi, M. Klobukowski and S. Huzinaga, J. Comput. Chem., 1987, 8, 256–264 CrossRef CAS.
- Y. Sakai, E. Miyoshi, M. Klobukowski and S. Huzinaga, J. Chem. Phys., 1997, 106, 8084–8092 CrossRef CAS.
- H. Anjima, S. Tsukamoto, H. Mori, M. Mine, M. Klobukowski and E. Miyoshi, J. Comput. Chem., 2007, 28, 2424–2430 CrossRef CAS PubMed.
- P. Kursula, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 70, 24–30 CrossRef.
- A. Matsuda and H. Mori, J. Comput. Chem., Jpn., 2014, 13, 105–113 CrossRef.
| This journal is © the Owner Societies 2025 |