
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of the Ru concentration on the CO tolerance and the oxidizability of a composition spread PtRu/Pt(111) near-surface alloy†
Valentin
Schwaab
 ab,
Fabian
Düll
a,
Phiona
Bachmann
a,
Felix
Hemauer
ab,
Hans-Peter
Steinrück
a and
Christian
Papp
*b
ab,
Fabian
Düll
a,
Phiona
Bachmann
a,
Felix
Hemauer
ab,
Hans-Peter
Steinrück
a and
Christian
Papp
*b
aLehrstuhl für Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Egerlandstr. 3, 91058 Erlangen, Germany
bAngewandte Physikalische Chemie, Freie Universität Berlin (FU Berlin), Arnimallee 22, 14195 Berlin, Germany. E-mail: christian.papp@fu-berlin.de
First published on 15th April 2025
Abstract
We prepared a Pt-terminated PtRu near-surface alloy with a lateral concentration gradient on Pt(111). The near-surface alloy is exposed to CO at 100 K or O2 at 600 K and subsequently investigated using synchrotron-based high-resolution X-ray photoelectron spectroscopy. By moving the sample laterally under the focused X-ray beam (spot size ∼0.05 mm), we were able to study locally different alloy compositions under identical measurement conditions. With increasing subsurface Ru concentration, we observe a gradual decrease in the CO occupancy of Pt bridge sites, while the amount of on-top adsorbed CO remains constant over the investigated compositional range. Oxidation of the alloy reveals a clear increase in the fraction of oxidized Ru atoms (the RuOx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru ratio) with increasing Ru content.
:Ru ratio) with increasing Ru content.
Introduction
Platinum–ruthenium catalysts are used as state-of-the-art anode materials for the electrooxidation of alcohols in proton exchange membrane fuel cells (PEM-FCs).1–4 Different promoting roles of Ru are responsible for the high performance of this bimetallic combination in the oxidation of primary and secondary alcohols, which exceeds that of pure Pt as the most active single metal catalyst in these reactions.5A key challenge in primary alcohol-based fuel cells (e.g., in the direct methanol fuel cell, where MeOH is oxidized to CO2) is the oxidative removal of the site-blocking reaction intermediate CO.6 Compared to bare Pt catalysts, bimetallic PtRu catalysts show an enhanced activity in MeOH and CO oxidation, which has been explained by two concepts: (1) as more oxophilic element, Ru binds oxy-/hydroxy-species at lower positive potentials than Pt; these species in return facilitate the oxidation of CO molecules at neighboring Pt sites (the so-called bifunctional mechanism);7,8 (2) the binding energy of CO to Pt sites within the alloy is weakened;9,10 this is attributed to electronic ligand- and/or compressive strain effects in the binary system, both of which lead to an energetic downshift of the Pt d-band center, which results in weaker metal–adsorbate interactions.11–16
Secondary alcohols have recently attracted considerable attention as rechargeable electrofuels in the context of liquid organic hydrogen carriers (LOHCs).3,4,17–21 Isopropanol can be selectively oxidized to acetone in a PEM-FC to produce electricity (electrons and protons) without notable CO2 formation.3,4 Since acetone can be hydrogenated again to isopropanol, the process is not only carbon-neutral but also recyclable. When the oxidation of isopropanol is carried out over a PtRu anode, an additional peak at low overpotentials (onset at 0.05 VRHE) is observed together with the peak at 0.75 VRHE (onset >0.30 VRHE), which is also found with pure Pt.18 The peak at low overpotentials has been associated with ultrasmall Pt aggregates that are stabilized by Ru.20 The performance of this fuel cell is, however, still limited to comparably low current maxima; poisoning of the active site by the product acetone and the oxidation of Ru are assumed to be the limiting factors here.18,20,22
Finally, also for the thermal dehydrogenation of alcohol-functionalized LOHCs the bimetallic combination of Pt and Ru appears to be a very effective catalyst. In a recent study, Maurer et al. investigated the performance of carbon-supported PtRu catalysts in the thermal dehydrogenation of dicyclohexylmethanol to benzophenone and observed an up to 65% increased hydrogen release productivity compared to the monometallic Pt/C ref. 23. The authors explained this by the formation of Ru-stabilized Pt nanoclusters and a weakened binding of adsorbates to Pt sites (in the concept of the aforementioned d-band model13,24).
With our study, we aim to contribute to a better fundamental understanding of this bimetallic system. Specifically, we are interested in how the Ru content affects the CO tolerance and the oxidizability of PtRu catalysts on the atomic scale. To this end, we prepared a PtRu near-surface alloy with a concentration gradient on a Pt(111) substrate. The near-surface alloy was exposed to CO at 100 K or O2 at 600 K and subsequently investigated using synchrotron-based high-resolution X-ray photoelectron spectroscopy (HR-XPS). By moving the sample laterally under the focused X-ray beam (spot size ∼ 0.05 mm), we were able to probe locally different alloy compositions under identical measurement conditions.
Experimental
All experiments were performed at the beamline UE56-2 PGM-2 of the BESSY II synchrotron facility, Helmholtz-Zentrum Berlin (Germany). Our transportable UHV apparatus has been described in detail by Denecke et al.25 It is equipped with a hemispherical electron energy analyzer (Omicron EA125 U7 HR), a sputter gun (Ar+), and a three-stage supersonic molecular beam. The latter was used in this work to dose CO and O2. Additionally, an electron beam evaporator for the deposition of Ru and a quartz crystal microbalance (QCM) for the calibration of the deposition rate were installed.Prior to Ru deposition, the Pt(111) single crystal (MaTecK, 99.999%) was cleaned by Ar+ ion sputtering (1 keV, pAr = 5 × 10−6 mbar, 20 min) and subsequent flash annealing to 1200 K. Remaining carbon impurities were removed by exposure to O2 at 900 K. The cleanness of the substrate was confirmed by XPS.
All XP spectra were recorded in normal emission (0°), with a light incident angle of 50°. The excitation energies were 380 eV for the Ru 3d/C 1s region with a resolution of ∼260 meV and 150 eV for the Pt 4f region with a resolution of ∼100 meV. All binding energies (BEs) were referenced to the Fermi level and a linear background was subtracted from each spectrum. The spectra were then fitted with a convolution of asymmetric Doniach–Šunjić and Gaussian functions.26
Results and discussion
Preparation and characterization of the composition spread near-surface alloy
The composition spread near-surface alloy was prepared by depositing a wedge-shaped Ru gradient onto the clean Pt(111) substrate.27–29 The gradient was achieved by positioning the sample with a lateral offset from the focus of the electron beam evaporator during Ru deposition at 100 K. After flashing the sample to 600 K to desorb CO impurities from the evaporation process, the concentration gradient was determined by measuring Pt 4f and Ru 3d XP spectra at different sample positions, that is, while laterally shifting the sample. More specifically, the sample was moved in the y direction in steps of 0.10 mm under the focused X-ray beam (spot size ∼ 0.05 mm) to probe the lateral extension and local concentration of the Ru deposit. The gradual growth of the Ru 3d signal in the corresponding y scan (y = 0.00 → 1.40 mm) in Fig. S1 in the ESI,† indeed confirms the presence of a lateral Ru gradient on the Pt(111) surface.The local Ru coverages, that is, the thicknesses along the wedge were deduced from the attenuation of the Pt 4f7/2 signal while accounting for a linear damping in the submonolayer regime. Details of this quantification procedure are provided in the ESI.† The maximal wedge thickness was calculated to be 1.19 Å (at y = 1.40 mm), which, considering a monolayer thickness of 2.15 Å based on the interlayer spacing in Ru(0001),30 corresponds to a local Ru coverage of θRu = 0.56 ML. The corresponding Pt 4f7/2 and Ru 3d5/2 spectra at y = 1.40 mm, before and after Ru deposition, are shown at the bottom and in the middle of Fig. 1, respectively. In addition to the main peaks at 70.93 and 280.07 eV (Pt 4f7/2/Ru 3d5/2), the Pt 4f7/2 level of the clean surface displays an additional peak at lower binding energy (70.53 eV), which is attributed to the surface-core-level-shift (SCLS) component of the Pt atoms of the clean surface. After Ru deposition, this Pt 4f7/2 SCLS peak shows significantly lower intensity, since about half of the surface is covered by Ru, which quenches the SCLS. Note that the observed decrease of the Pt 4f7/2 SCLS to ∼59% of its original value is lower than what we expect for a Ru film of 0.56 ML (that is, ∼44%). We attribute this deviation to a fraction of the Ru being present in the form of 3-dimensional (bilayer) islands and to some extent likely also incorporated in the first substrate layer (beneath the islands) due to a vertical exchange of Pt and Ru atoms (see discussion on the growth of Ru on Pt(111) below). From this observation, we conclude that the calculated maximal Ru coverage of 0.56 ML underestimates the actual Ru coverage by at most 0.10 ML.
 | ||
| Fig. 1 Comparison of the observed Ru 3d5/2 and Pt 4f7/2 spectra at different stages of the experiment, recorded at the position y = 1.40 mm. From bottom to top: pristine Pt(111) substrate; after deposition of the Ru wedge and flash-annealing to 600 K; after subsequent flash-annealing to 850 K. | ||
After the preparation of the Ru wedge, the sample was flashed to 850 K to promote intermixing and alloy formation of the two metals in the near-surface region. The comparison of the Pt 4f7/2 and Ru 3d5/2 regions at y = 1.40 mm before (bottom) and after (top) the aforementioned preparation steps shows the complete disappearance of the SCLS in the Pt 4f7/2 region after flashing to 850 K, which we attribute to the formation of a near-surface alloy based on the following consideration: it is known that alloying Pt with Ru causes a shift of the Pt 4f7/2 signals towards higher BEs. This effect has been explained in the context of the d-band model with a downshift of the Pt d-band center due to electronic ligand and compressive strain effects in the bimetallic system.14 Assuming that the intermixing of the metals in our sample occurred predominantly in the near-surface region, as is to be expected after the short annealing period, the binding energy shift should be most pronounced for Pt atoms in the surface layer, which display the SCLS. In other words, after annealing to 850 K, the Pt 4f7/2 peaks of the surface-alloyed atoms have a similar binding energy to the bulk Pt atoms. We can exclude that the surface was simply covered by a Ru film, e.g., due to a change in surface morphology from 3-dimensional Ru islands to a purely 2-dimensional Ru adlayer upon flashing the wedge to 850 K: while such a spreading of the Ru would also cause a diminution of the SCLS Pt 4f7/2 component, it should result in a clear increase in the Ru 3d signal intensity; however, the opposite is observed here; moreover, also the coverage of only 0.56 ± 0.10 ML is not sufficient to form a closed film, as only roughly half of the surface would be covered by Ru.
The growth of Ru on Pt(111) has been the subject of several STM studies by Hoster, Behm, Berkó, and Bergbreiter:31–33 For sub-monolayer Ru coverages, the authors observed a change in growth behavior with increasing sample temperature, from the preferred formation of Ru bilayer islands at room temperature to monolayer islands when Ru was deposited at temperatures >523 K. A clear trend of an increasing Pt content of the islands with increasing deposition temperature was observed and attributed to a vertical exchange of Ru atoms from the islands and Pt atoms from the underlying substrate layer. For deposition temperatures ≥773 K, the resulting surfaces were predominantly smooth, which was ascribed to alloy formation in the near-surface region. Moreover, Ru bulk dissolution was observed >773 K.33
Taking these observations into account, we assume that after the initial annealing step to 600 K, the Ru on our sample was mostly present in the form of 2-dimensional islands, and to some extent also in the form of bilayer islands or incorporated in the first subsurface layer (beneath the islands), as a result of the aforementioned vertical exchange of Ru and Pt atoms above 523 K. However, judging from the well-visible SCLS in the Pt 4f7/2 region (Fig. 1, middle), the degree of intermixing at this stage was likely minimal. This conclusion is further supported by a comparison of the Ru 3d5/2 signals at 100 K and subsequent flashing to 600 K, which suggests that Ru diffusion to below the surface upon flashing to 600 K occurs only to a minor extent (see ESI,† Fig. S3). Notably, the intensity of the Ru 3d5/2 signal even increases by about 3% after flashing to 600 K, which we attribute to the desorption of CO impurities from the evaporation process and spreading of the randomly deposited Ru atoms to a more uniform 2D film; the observed change in line shape in Fig. S3 (ESI†) is attributed to the transition of a very rough Ru film with inequivalent atom environments after deposition at 100 K to a flat, homogeneous film upon flashing to 600 K. The proposed formation of a near-surface alloy after flashing the wedge to 850 K aligns well with the literature.33
After forming the near-surface alloy, we analyzed the sample by measuring XP spectra along the concentration gradient of the Ru deposit. Fig. 2a shows a waterfall plot of the Ru 3d/C 1s spectra acquired from y = 0.00 to 1.40 mm (bottom to top) in steps of 0.10 mm. An exemplary fit of the spectrum at y = 1.30 mm is provided in Fig. 2c. The spin–orbit splitting of the Ru 3d5/2 and Ru 3d3/2 components leads to two distinct signals at 280.05 and 284.23 eV, respectively. The doublet is fitted with two peaks, using a splitting of 4.18 eV, and a constrained area ratio of 6:4, consistent with the multiplicities of both states. The peaks have the same Gaussian width, however, their full-width at half-maximum (FWHM) differs due to a Coster–Kronig decay of the 3d3/2 core hole,34 which due to a shorter lifetime results in a larger FWHM as compared to the 3d5/2 peak. A third peak at ∼284.0 eV was introduced to account for carbide impurities (<0.04 ML). This peak was clearly resolved at the position of pure Pt (y = 0.00 mm), where the Ru 3d components are entirely absent (cf.Fig. 3a and b). All peak and fit parameters are detailed in the ESI.†
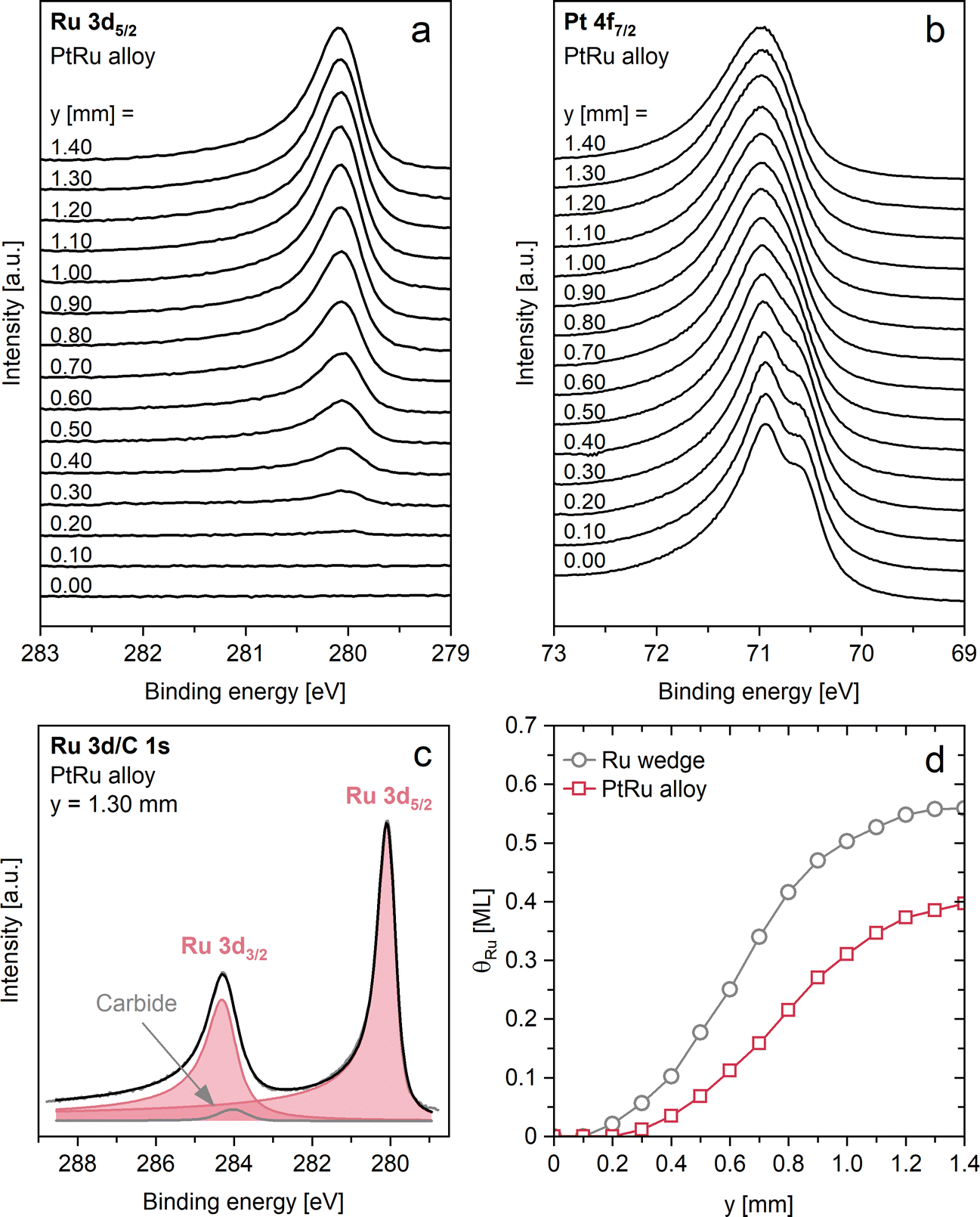 | ||
| Fig. 2 (a) Ru 3d5/2/C 1s y scan of the as-prepared near-surface alloy in 0.10 mm steps. (b) Corresponding Pt 4f7/2y scan of the as-prepared near-surface alloy. (c) Selected Ru 3d/C 1s spectrum at the position y = 1.30 mm to illustrate the applied fit model. (d) Plot of the determined Ru coverage (θRu) as a function of the measurement position: after Ru deposition and flash-annealing to 600 K (Ru wedge, grey); after subsequent flash-annealing to 850 K (PtRu alloy, pink). | ||
 | ||
| Fig. 3 (a) C 1s/Ru 3d3/2y scan of the PtRu near-surface alloy after exposure to CO at 100 K (y = 0.00 mm → 1.40 mm, in 0.1 mm steps). The peak at 284.0 eV in the bottom spectrum (y = 0.00 mm) stems from carbide. (b) Selected XP spectra at y = 0.00 and 1.40 mm to illustrate the applied fit model. (c) Corresponding quantitative analysis; (COPtbridge) and (COPton-top) are normalized to 0.25 ML at y = 0.00 mm, in accordance with the proposed c(4 × 2)–CO structure at this position. | ||
The local Ru coverages after heating to 850 K were determined by referencing the Ru 3d5/2 peak areas at each y position to the Ru 3d5/2 peak of the wedge at y = 1.40 mm (θRu = 0.56 ML). The results are plotted in Fig. 2c. The comparison of the near-surface alloy to the initially prepared wedge yields a consistent decrease in θRu at all examined y positions, e.g., 0.39 ML for the alloy versus 0.56 ML for the non-alloyed wedge, at the position of maximum Ru coverage, y = 1.40 mm. This apparent decrease of θRu is attributed to the diffusion of Ru atoms beneath the surface during annealing to 850 K, in agreement with ref. 33. Note that the absence of the SCLS in the corresponding Pt 4f7/2 scan indicates that the influence of the Ru component on the surface Pt atoms is still significant. Here, it is important to mention that not only Ru atoms in the topmost layer but also from lower layers in the near-surface region can contribute to a shift of the Pt surface contribution through electronic and/or strain effects. This will become evident in the following section.
Adsorption of CO
To study the adsorption of CO on the near-surface alloy, the sample was first exposed to CO at 100 K, and the adsorption process was monitored in situ by XPS in the C 1s region at the position of maximum Ru concentration, y = 1.40 mm. The objective of this step was to saturate the sample under conditions where CO forms a well-defined c(4 × 2) superstructure on Pt(111).35,36 Ru-free regions on the substrate can then be used as a reference of known coverage and adsorption site occupancy to analyze CO adsorption on the surface alloy. CO was dosed via the molecular beam (1.7 L of indirect exposure at a background pressure of 6 × 10−9 mbar, followed by 9 L of direct exposure at an effective local pressure of 2 × 10−7 mbar).25 The dosing process was stopped when the CO coverage reached saturation, as determined by XPS. Spectra from this experiment are provided in Fig. S4 of the ESI.† In the following, we focus on the y scan depicted in Fig. 3a, which was obtained immediately after CO exposure.The bottom spectrum in Fig. 3a (y = 0.00 mm) represents a Ru-free region on the Pt(111) substrate, where CO adsorption led to the evolution of two characteristic peaks at 286.10 and 286.77 eV. The peaks correspond to CO molecules adsorbed on Pt bridge and on-top sites, respectively.35,36 The line shape and the relative intensities of both peaks align well with that of a previous study, showing the c(4 × 2)-CO superstructure with a coverage of 0.5 ML CO, which forms under the applied exposure conditions. In this superstructure, the CO molecules are equally distributed on Pt bridge and on-top sites (0.25 ML each);37 the difference in peak area (that is, the COPtbridge signal is by ∼17% larger than that of COPton-top) is well known and has been attributed to photoelectron diffraction effects;35,36 this has been taken into account in the quantitative analysis (see below).
In the region of the near-surface alloy (y = 0.20 ↔ 1.40 mm), CO adsorption on Ru sites must be considered. The saturation coverage of CO on Ru(0001) under UHV conditions is 0.66 ML, typically achieved at ∼10 L of exposure, with CO molecules exclusively adsorbed on Ru on-top sites.38 According to ref. 39, CORuon-top produces a C 1s signal at ∼285.8 eV, and bridge bound species form only at pressures >10−6 mbar, yielding a peak between that of CORuon-top and the Ru 3d3/2 component at 284.23 eV.38 For PtRu/Ru(0001) near-surface alloys, the C 1s signal was reported at the same energy of ∼285.8 eV.39 Notably, in our experiment, these contributions are not observed. The absence of CORu peaks in the y scan indicates a comparably low Ru concentration in the topmost layer of the sample. This assumption aligns well with the proposed bulk diffusion of Ru after annealing to 850 K and is further supported by the observations of ref. 39, which will be addressed later in the discussion. Note that a small CORuon-top contribution at ∼285.8 eV might still be present but is not resolved beneath the more intense COPtbridge signal at 286.10 eV.
At first sight, the absence of a Ru-bound CO species may seem to contradict our hypothesis of an influence of neighboring Ru atoms on surface Pt atoms as reason for the absence of the Pt 4f7/2 SCLS peak at positions on the surface where a notable Ru 3d intensity is observed. However, as indicated above, an explanation could be Ru neighbors largely residing in the near-surface region, that is, beneath the surface Pt atoms. This explanation agrees perfectly with the observations by Berkó et al., who found that a predominantly Pt-terminated, smooth surface alloy is formed when Ru is deposited onto Pt(111) at temperatures ≥773 K.33 Interestingly, assuming only minor bulk diffusion after flash-annealing to 600 K, the observed decrease in the apparent Ru coverage after flash-annealing to 850 K (from 0.56 to 0.39 ML, that is, by only ∼30%) suggests that most of the deposited Ru has moved to the 2nd layer. (For this scenario, one would expect an attenuation of the Ru 3d signal by ∼40%; the deviation is again explained by some of the Ru in the non-annealed wedge being present in the form of bilayer islands and/or incorporated the first subsurface layer.) An enrichment of Ru atoms in the 2nd layer could also be rationalized in the context of the STM results: there, bulk dissolution was exclusively observed at temperatures ≥773 K; however, already at deposition temperatures of 573 K, the Ru content of the 2-dimensional islands was found to be significantly lowered. The authors proposed that this could be attributed to a vertical exchange of Ru adlayer atoms with Pt atoms from the underlying substrate layer, which is more facile than Ru bulk dissolution due to the less stringent geometric confinement of the surface atoms compared to atoms in the bulk.33 In this sense, the short annealing period above 773 K in our experiment could explain an accumulation of Ru atoms in the 2nd layer due to limited bulk dissolution of Ru but facile Pt ↔ Ru surface exchange.
For a quantitative analysis of the CO coverage within the near-surface alloy, it was assumed that CORu contributions are negligible. In other words, the CO features in all C 1s spectra of the y scan were fitted only with the two COPt peaks, using the optimized parameters obtained from the spectrum at the position of Ru-free, pristine Pt(111), y = 0.00 mm. Exemplary fits are illustrated in Fig. 3b; all peak parameters are detailed in the ESI.† In Fig. 3c, the obtained coverages are plotted against the measurement position. The coverages for COPtbridge (orange) and COPton-top (blue) were normalized individually such that both are 0.25 ML at y = 0.00 mm, in accordance with the proposed c(4 × 2) structure at this position.37 This individual normalization is necessary because the experimentally observed peak heights differ due to photoelectron diffraction effects, as mentioned above.35,36
The analysis reveals a significant decrease in the total CO coverage (black) with increasing Ru concentration, from 0.5 ML at y = 0.00 mm to 0.35 ML at y = 1.40 mm. This observation is solely attributed to a ∼60% decrease in the intensity of the peak at 286.10 eV, that is, (neglecting potential contributions from CORu) to an equivalent decrease in the COPtbridge population. In contrast, the COPton-top peak at ∼286.77 eV remains nearly unaffected. The slight binding energy shift from 286.77 to 286.69 eV (y = 0.00 → 1.40 mm) is of similar magnitude as the coverage-dependent shift of both C 1s peaks of CO on Pt(111);36 in addition, the change in the electronic environment due to neighboring Ru atoms could also contribute. It is also important to note that for the COPton-top peak no overlap with the CORu contribution is conceivable, as the former is shifted by ∼900 meV to higher BEs compared to the literature value for CORuon-top (>286.69 eV vs. ∼285.8 eV39). Along the concentration gradient, the intensity of the COPton-top peak is constant, that is, the density in Pt adsorption sites does not change to a detectable extent. Thus, we conclude that the Ru concentration in the topmost layer of the sample is negligible.
The observed decrease in the CO coverage of Pt bridge sites with increasing Ru concentration in the near-surface alloy aligns well with previous findings by Starr and Bluhm,39 who investigated the adsorption of CO on PtRu/Ru(0001) near-surface alloys of three distinct compositions (θPt = 0.36, 0.73, 0.94 ML): they found a COPtbridge species only on the alloy with the highest Pt concentration (0.94 ML), that is, lowest Ru content. Notably, they also observed a decrease in the CO adsorption on Pt on-top sites in a comparison of the 0.94 ML Pt alloy with those of 0.73 and 0.36 ML Pt.39 This latter aspect is in contrast to our own findings of a constant COPton-top coverage for the inverse system of PtRu/Pt(111).
Because our near-surface alloy is essentially purely Pt terminated, that is, the Ru concentration in the topmost layer of the sample is negligible, the observed decrease in the COPtbridge population cannot be explained by an ensemble effect. Instead, the trend appears to be the result of vertical ligand- and/or compressive strain effects, similar to what has been previously reported for pseudomorphic Pt films on Ru(0001), where a clear decrease in the binding strength of adsorbates on thin Pt films was observed as compared to adsorption on bare Pt(111).10,11,15,16 Interestingly, with a continuous gradient in the subsurface Ru concentration, our experiment offers a gradual picture on the influence of these effects on the adsorption of CO.
As a final step, CO was removed again from the sample by annealing to 450 K in a temperature-programmed XPS experiment. Details can be found in the ESI† (see Fig. S6). Briefly, comparison with an equivalent TPXPS experiment of CO on bare Pt(111) revealed a decreased desorption temperature of CO for the PtRu near-surface alloy (investigated at y = 1.40 mm). The finding can be interpreted as a weaker adsorption energy for this latter system, which is in excellent agreement with the results of earlier reports on the adsorption of CO on PtRu/Ru(0001) surface alloys40 and Pt monolayer film covered Ru(0001) surfaces.11,12
Exposure to O2 at 600 K
To study the effect of the Ru concentration on the oxidizability of the surface alloy, the sample was exposed to O2 at 600 K (9.3 L of indirect exposure at 3 × 10−8 mbar).Fig. 4a depicts the Ru 3d/C 1s spectra collected during the y scan of the oxygen-treated surface alloy. Two main observations can be made from this scan: (i) compared to the pristine surface alloy, the Ru 3d features of the oxygen-treated surface alloy are broadened towards higher binding energy. This broadening is attributed to the presence of an additional doublet with contributions at 280.62 and 284.79 eV, which is ascribed to the spin–orbit split 3d5/2 and 3d3/2 components of oxidized Ru (RuOx).20,34 (ii) The intensity ratio of this oxide contribution and the unreacted metal contribution (at 280.05 and 284.23 eV, respectively), RuOx:Ru, decreases towards lower Ru concentrations (from top to bottom in Fig. 3a). This finding is analyzed in more detail below. It should also be noted that the Ru 3d contributions in this scan diminish at y ≤ 0.40 mm, whereas in the scan of the pristine surface alloy, they are visible down to y = 0.20 mm. We attribute this to be the result of a hysteresis effect, since the sample position was slightly adjusted in x and z direction for the duration of the O2 exposure in order to obtain a better signal intensity in the concurrent in situ O 1s measurement, and then re-adjusted to the original values before the scan. This shift does, however, not affect our following interpretation.
 | ||
| Fig. 4 (a) C 1s/Ru 3d y scan of the PtRu near-surface alloy after exposure to O2 at 600 K (y = 0.00 mm → 1.40 mm, in 0.10 mm steps). (b) Selected C 1s/Ru 3d spectra at y = 1.40, 1.10, and 0.80 mm to illustrate the applied fit model. (c) Corresponding quantitative plot of the peak areas against the measurement position. (d) Plot of the relative oxide amount, RuOx:Ru, as a function of the combined normalized Ru intensity (Ru 3d + RuOx). | ||
The spectra were fitted in order to analyze the decreasing relative oxide amount with decreasing Ru concentration in the surface alloy in more detail. Two doublets were introduced to account for the 3d5/2/3d3/2 components of Ru (280.05/284.23 eV) and RuOx (280.62/284.79 eV). Each doublet was fitted with a fixed splitting of 4.18 eV and a constrained area ratio of 3d5/2:3d3/2 = 6:4, according to the multiplicities of both states; an uncertainty of ≤5% was allowed in this area ratio to achieve better fits. Exemplary spectra illustrating the fit model are shown in Fig. 3b. The normalized peak areas are depicted in Fig. 3c as a function of the measurement position.
Fig. 3d depicts the RuOx:Ru ratio plotted against the combined intensity of both Ru species. Please note that due to the low signal intensities at small y values and the associated inaccuracies in the quantitative analysis, only datapoints of y ≥ 0.70 mm were included. For y = 1.40 → 0.80 mm, the analysis shows a linear decrease in the relative oxide amount with a decreasing Ru concentration in the surface alloy. Given the negligible Ru concentration in the topmost layer of the alloy, it is reasonable to assume that the observed formation of RuOx involved the diffusion of Ru atoms to the surface. At first sight, this assumption might seem to contradict our previous observations of a clear preference for Ru bulk dissolution/Pt surface segregation at elevated temperatures. However, theoretical studies suggest that exposure of the bimetallic system to oxygen can indeed effect an inversion in the thermodynamically favored surface termination;41,42 that is, from Pt (under vacuum) to Ru (in the presence of oxygen). Due to the strong bond between Ru and O, the oxide formation energy overcompensates for the higher surface free energy of Ru compared to Pt, which is the driving force for Pt surface segregation in vacuum. Experimentally, the surface segregation of Ru with RuOx (RuO2) formation under exposure to air at TS > 520 K has already been observed for carbon supported PtRu alloy crystallites; interestingly, the oxygen-treated crystallites were found to show an enhanced activity in the electrochemical methanol oxidation.43,44
The herein observed decrease in the RuOx:Ru ratio with decreasing local Ru concentration could be interpreted as a direct correlation between the Ru content and the oxidizability of the near-surface alloy.
Conclusion
We studied the influence of the local Ru concentration on the CO tolerance and the oxidizability of a PtRu/Pt(111) near-surface alloy with a concentration gradient using synchrotron-based HR-XPS. The alloy was prepared by physical vapor deposition of a wedge-shaped Ru gradient on Pt(111) and subsequent flash-annealing to 850 K to promote the intermixing of both metals. We found this procedure to result in a predominantly Pt-terminated surface alloy, with a continuous wedge-like Ru gradient in the near-surface region. Significant changes in the electronic structure of the surface Pt atoms upon alloy formation, most likely due to vertical electronic ligand and/or strain effects arising from subsurface Ru, are evidenced by the disappearance of the surface core level shift in the Pt 4f region.Exposure of the as-prepared surface alloy to CO revealed a gradual decrease in the COPtbridge population with increasing local Ru concentration. The COPton-top population was found to be constant over the investigated compositional range. No CORu was observed, which we attribute to a negligible amount of Ru adsorption sites, that is, a negligible amount of Ru in the surface layer.
Exposure to O2 at 600 K resulted in RuOx formation, most likely through Ru surface segregation, driven by the strong binding between Ru and O. We found a linear increase in the relative amount of oxidized Ru atoms, that is, the RuOx:Ru ratio, with increasing local Ru concentration in the sample. This trend might be interpreted as a direct correlation between the Ru content and the oxidizability of the near-surface alloy.
Our study contributes to a better fundamental understanding of the properties of bimetallic PtRu catalysts. With a negligible Ru concentration in the topmost layer but a continuous Ru gradient in the near-surface region, the herein prepared and studied sample is a particularly interesting model system for Pt-terminated PtRu catalysts, e.g., core–shell type structures.
Author contributions
Valentin Schwaab: conceptualization (equal); formal analysis (lead); investigation (lead); visualization (lead); writing – original draft (lead); writing – review & editing (equal). Fabian Düll: conceptualization (equal); investigation (supporting). Phiona Bachmann: investigation (supporting). Felix Hemauer: investigation (supporting). Hans-Peter Steinrück: funding acquisition (supporting), project administration (supporting), supervision (supporting); writing – original draft (supporting); writing – review & editing (supporting). Christian Papp: conceptualization (equal); project administration (equal); supervision (lead); writing – original draft (supporting); writing – review & editing (equal).Data availability
The data that support the findings of this study are openly available in Zenodo at https://doi.org/10.5281/zenodo.14713107.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Bavarian Ministry of Economic Affairs, Regional Development and Energy, and by the Deutsche Forschungsgemeinschaft (Project No. 419654270). We want to thank Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime and the BESSY II staff for their support.References
- P. Joghee, J. N. Malik, S. Pylypenko and R. O’Hayre, A review on direct methanol fuel cells–In the perspective of energy and sustainability, MRS Energy Sustainability, 2015, 2, E3 CrossRef.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Anode catalysts for direct methanol fuel cells in acidic media: do we have any alternative for Pt or Pt–Ru?, Chem. Rev., 2014, 114(24), 12397–12429 CrossRef CAS PubMed.
- G. Sievi, D. Geburtig, T. Skeledzic, A. Bösmann, P. Preuster, O. Brummel, F. Waidhas, M. A. Montero, P. Khanipour, I. Katsounaros, J. Libuda, K. J. J. Mayrhofer and P. Wasserscheid, Towards an efficient liquid organic hydrogen carrier fuel cell concept, Energy Environ. Sci., 2019, 12(7), 2305–2314 RSC.
- M. Brodt, K. Müller, J. Kerres, I. Katsounaros, K. Mayrhofer, P. Preuster, P. Wasserscheid and S. Thiele, The 2-Propanol Fuel Cell: A Review from the Perspective of a Hydrogen Energy Economy, Energy Technol., 2021, 9(9), 2100164 CrossRef CAS.
- I. Mangoufis-Giasin, O. Piqué, P. Khanipour, K. J. J. Mayrhofer, F. Calle-Vallejo and I. Katsounaros, Different promoting roles of ruthenium for the oxidation of primary and secondary alcohols on PtRu electrocatalysts, J. Catal., 2021, 400, 166–172 CrossRef CAS.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Recent advances in catalysts for direct methanol fuel cells, Energy Environ. Sci., 2011, 4(8), 2736–2753 RSC.
- M. Watanabe and S. Motoo, Electrocatalysis by ad-atoms: Part III. Enhancement of the oxidation of carbon monoxide on platinum by ruthenium ad-atoms, J. Electroanal. Chem. Interfacial Electrochem., 1975, 60(3), 275–283 CrossRef CAS.
- H. A. Gasteiger, N. Marković, P. N. Ross and E. J. Cairns, Methanol electrooxidation on well-characterized platinum–ruthenium bulk alloys, J. Phys. Chem., 1993, 97(46), 12020–12029 CrossRef CAS.
- J. McBreen and S. Mukerjee, In Situ X-Ray Absorption Studies of a Pt–Ru Electrocatalyst, J. Electrochem. Soc., 1995, 142(10), 3399–3404 CrossRef CAS.
- F. Buatier de Mongeot, M. Scherer, B. Gleich, E. Kopatzki and R. J. Behm, CO adsorption and oxidation on bimetallic Pt/Ru(0001) surfaces – a combined STM and TPD/TPR study, Surf. Sci., 1998, 411(3), 249–262 CrossRef CAS.
- A. Schlapka, M. Lischka, A. Gross, U. Käsberger and P. Jakob, Surface strain versus substrate interaction in heteroepitaxial metal layers: Pt on Ru(0001), Phys. Rev. Lett., 2003, 91(1), 016101 CrossRef CAS PubMed.
- P. Jakob and A. Schlapka, CO adsorption on epitaxially grown Pt layers on Ru(0001), Surf. Sci., 2007, 601(17), 3556–3568 CrossRef CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Role of strain and ligand effects in the modification of the electronic and chemical properties of bimetallic surfaces, Phys. Rev. Lett., 2004, 93(15), 156801 CrossRef CAS PubMed.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, Electronic structures of Pt–Co and Pt–Ru alloys for CO-tolerant anode catalysts in polymer electrolyte fuel cells studied by EC-XPS, J. Phys. Chem. B, 2006, 110(46), 23489–23496 CrossRef CAS PubMed.
- H. Hartmann, T. Diemant, J. Bansmann and R. J. Behm, Interaction of CO and deuterium with bimetallic, monolayer Pt-island/film covered Ru(0001) surfaces, Phys. Chem. Chem. Phys., 2012, 14(31), 10919–10934 RSC.
- J. Klein, F. Argast, A. K. Engstfeld, S. Brimaud and R. J. Behm, Electro-oxidation of methanol on Ru-core Pt-shell type model electrodes, Electrochim. Acta, 2019, 311, 244–254 CrossRef CAS.
- J. Cho, B. Kim, S. Venkateshalu, D. Y. Chung, K. Lee and S. I. Choi, Electrochemically Activatable Liquid Organic Hydrogen Carriers and Their Applications, J. Am. Chem. Soc., 2023, 145(31), 16951–16965 CrossRef CAS PubMed.
- P. Khanipour, F. D. Speck, I. Mangoufis-Giasin, K. J. J. Mayrhofer, S. Cherevko and I. Katsounaros, Electrochemical Oxidation of Isopropanol on Platinum–Ruthenium Nanoparticles Studied with Real-Time Product and Dissolution Analytics, ACS Appl. Mater. Interfaces, 2020, 12(30), 33670–33678 CrossRef CAS PubMed.
- F. Waidhas, S. Haschke, P. Khanipour, L. Fromm, A. Görling, J. Bachmann, I. Katsounaros, K. J. J. Mayrhofer, O. Brummel and J. Libuda, Secondary Alcohols as Rechargeable Electrofuels: Electrooxidation of Isopropyl Alcohol at Pt Electrodes, ACS Catal., 2020, 10(12), 6831–6842 CrossRef CAS.
- A. Simanenko, P. K. Samal, R. Hübsch, J. Škvára, J. Yang, M. Kastenmeier, F. Winkler, T. Skála, N. Tsud, S. Mehl, J. Mysliveček, O. Brummel, Y. Lykhach and J. Libuda, Origin of the Low Overpotential for Isopropanol Oxidation on Pt–Ru Electrocatalysts, ACS Energy Lett., 2024, 9(10), 4875–4882 CrossRef CAS.
- L. Fusek, V. Briega-Martos, M. Minichová, L. Fromm, E. Franz, J. Yang, A. Görling, K. J. J. Mayrhofer, P. Wasserscheid, S. Cherevko, O. Brummel and J. Libuda, Toward High-Energy-Density Fuels for Direct Liquid Organic Hydrogen Carrier Fuel Cells: Electrooxidation of 1-Cyclohexylethanol, J. Phys. Chem. Lett., 2024, 2529–2536 CrossRef CAS PubMed.
- M. Minichová, C. Van Pham, B. Xiao, A. Savan, A. Hutzler, A. Körner, I. Khalakhan, M. G. Rodríguez, I. Mangoufis-Giasin, V. Briega-Martos, A. Kormányos, I. Katsounaros, K. J. J. Mayrhofer, A. Ludwig, S. Thiele and S. Cherevko, Isopropanol electro-oxidation on Pt–Ru–Ir: a journey from model thin-film libraries towards real electrocatalysts, Electrochim. Acta, 2023, 444, 142032 CrossRef.
- L. A. Maurer, C. v Pham, B. Fritsch, H. S. Jeppesen, M. Etter, A. Hutzler, R. B. Neder, M. Wolf, F. Auer, S. Thiele and P. Wasserscheid, Improving Hydrogen Release From Oxygen-Functionalized LOHC Molecules by Ru Addition to Pt/C Catalysts, ChemCatChem, 2024, 16(17), e202400375 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, CO chemisorption at metal surfaces and overlayers, Phys. Rev. Lett., 1996, 76(12), 2141–2144 CrossRef CAS PubMed.
- R. Denecke, M. Kinne, C. M. Whelan and H. P. Steinrück, In Situ Core-Level Photoelectron Spectroscopy of Adsorbates on Surfaces Involving a Molecular Beam—General Setup and First Experiments, Surf. Rev. Lett., 2002, 9(2), 797–801 CrossRef CAS.
- S. Doniach and M. Šunjić, Many-electron singularity in X-ray photoemission and X-ray line spectra from metals, J. Phys. C: Solid State Phys., 1970, 3(2), 285–291 CrossRef CAS.
- S.-H. Yang, B. Balke, C. Papp, S. Döring, U. Berges, L. Plucinski, C. Westphal, C. M. Schneider, S. S. P. Parkin and C. S. Fadley, Determination of layer-resolved composition, magnetization, and electronic structure of an Fe/MgO tunnel junction by standing-wave core and valence photoemission, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84(18), 184410 CrossRef.
- S. Döring, F. Schönbohm, U. Berges, R. Schreiber, D. E. Bürgler, C. M. Schneider, M. Gorgoi, F. Schäfers, C. Papp, B. Balke, C. S. Fadley and C. Westphal, Hard x-ray photoemission using standing-wave excitation applied to the MgO/Fe interface, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(16), 165444 CrossRef.
- F. Düll, M. Meusel, F. Späth, S. Schötz, U. Bauer, P. Bachmann, J. Steinhauer, H. P. Steinrück, A. Bayer and C. Papp, Growth and stability of Pt nanoclusters from 1 to 50 atoms on h-BN/Rh(111), Phys. Chem. Chem. Phys., 2019, 21(38), 21287–21295 RSC.
- Y. He, A. P. Seitsonen and H. Over, Irregular stacking sequence in the initial growth of ultrathin Rh films on Ru(0001), Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(7), 075432 CrossRef.
- H. Hoster, T. Iwasita, H. Baumgärtner and W. Vielstich, Pt–Ru model catalysts for anodic methanol oxidation: Influence of structure and composition on the reactivity, Phys. Chem. Chem. Phys., 2001, 3(3), 337–346 RSC.
- A. Bergbreiter, A. Berkó, P. M. Erne, H. E. Hoster and R. J. Behm, On the origin of Ru bilayer island growth on Pt(111), Vacuum, 2009, 84(1), 13–18 CrossRef CAS.
- A. Berkó, A. Bergbreiter, H. E. Hoster and R. J. Behm, From bilayer to monolayer growth: Temperature effects in the growth of Ru on Pt(111), Surf. Sci., 2009, 603(16), 2556–2563 CrossRef.
- D. J. Morgan, Resolving ruthenium: XPS studies of common ruthenium materials, Surf. Interface Anal., 2015, 47(11), 1072–1079 CrossRef CAS.
- M. Kinne, T. Fuhrmann, C. M. Whelan, J. F. Zhu, J. Pantförder, M. Probst, G. Held, R. Denecke and H. P. Steinrück, Kinetic parameters of CO adsorbed on Pt(111) studied by in situ high resolution X-ray photoelectron spectroscopy, J. Chem. Phys., 2002, 117(23), 10852–10859 CrossRef CAS.
- M. Kinne, Kinetische Untersuchungen von Oberflächenreaktionen mittels hochaufgelöster Röntgen-Photoelektronenspektroskopie – Oxidation von CO auf Pt(111) und zugehörige Elementarschritte, PhD, Friedrich-Alexander Universität Erlangen-Nürnberg, 2004 Search PubMed.
- F. Bondino, G. Comelli, F. Esch, A. Locatelli, A. Baraldi, S. Lizzit, G. Paolucci and R. Rosei, Structural determination of molecules adsorbed in different sites by means of chemical shift photoelectron diffraction: c(4 × 2)-CO on Pt(111), Surf. Sci., 2000, 459(1–2), L467–L474 CrossRef CAS.
- D. E. Starr and H. Bluhm, CO adsorption and dissociation on Ru(0001) at elevated pressures, Surf. Sci., 2013, 608, 241–248 CrossRef CAS.
- D. E. Starr, H. Bluhm and C. O. Adsorption, on PtRu/Ru(0001) Near Surface Alloys from Ultrahigh Vacuum to Millitorr Pressures, J. Phys. Chem. C, 2014, 118(50), 29209–29217 CrossRef CAS.
- H. Rauscher, T. Hager, T. Diemant, H. Hoster, F. Buatier de Mongeot and R. J. Behm, Interaction of CO with atomically well-defined Pt Ru/Ru(0001) surface alloys, Surf. Sci., 2007, 601(19), 4608–4619 CrossRef CAS.
- B. C. Han, A. Van der Ven, G. Ceder and B.-J. Hwang, Surface segregation and ordering of alloy surfaces in the presence of adsorbates, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(20), 205409 CrossRef.
- J. A. Herron and M. Mavrikakis, On the composition of bimetallic near-surface alloys in the presence of oxygen and carbon monoxide, Catal. Commun., 2014, 52, 65–71 CrossRef CAS.
- S. Y. Huang, S. M. Chang, C. L. Lin, C. H. Chen and C. T. Yeh, Promotion of the Electrochemical Activity of a Bimetallic Platinum-Ruthenium Catalyst by Oxidation-Induced Segregation, J. Phys. Chem. B, 2006, 110(46), 23300–23305 CrossRef CAS PubMed.
- Y.-C. Wei, C.-W. Liu, W.-J. Chang and K.-W. Wang, Promotion of Pt–Ru/C catalysts driven by heat treated induced surface segregation for methanol oxidation reaction, J. Alloys Compd., 2011, 509(2), 535–541 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00295h |
| This journal is © the Owner Societies 2025 |