
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interactions of lycopene with β-cyclodextrins: Raman spectroscopy and theoretical investigation†
Laurynas
Diska
a,
Alma
Bockuviene
b,
Rūta
Gruskiene
c,
Tatjana
Kavleiskaja
b,
Jolanta
Sereikaite
c,
Goda
Bankovskaite
a and
Mindaugas
Macernis
 *a
*a
aInstitute of Chemical Physics, Faculty of Physics, Vilnius University, Sauletekio ave. 3, Vilnius, LT-10257, Vilnius, Lithuania. E-mail: mindaugas.macernis@ff.vu.lt
bInstitute of Chemistry, Faculty of Chemistry, Vilnius University, Sauletekio ave. 3, Vilnius, LT-10257, Vilnius, Lithuania
cDepartment of Chemistry and Bioengineering, Vilnius Gediminas Technical University, Sauletekio ave. 11, Vilnius, LT-10257, Vilnius, Lithuania
First published on 17th March 2025
Abstract
Carotenoids (Cars) are essential molecules with diverse biological roles, and their solubility can be enhanced by forming complexes with cyclodextrins, which incorporate them into their cavities without chemical bonds. In this study, we investigated the interactions of lycopene with β-cyclodextrins using Raman spectroscopy, molecular dynamics and DFT. We simulated models of simplified structures, including various β-cyclodextrins and trans and cis lycopene configurations, to better understand how structural changes influence the v1 band shift observed in Raman spectroscopy. Our focus was on reducing interactions such as van der Waals interactions, hydrogen bonds, and electrostatic effects to isolate the impact of structural alterations on the methyl group in carotenoids. We measured solid lycopene and its complexes with β-cyclodextrins, which exhibited Raman v1 shifts. According to the modelling data, this can be attributed to the monomer band of lycopene.
Introduction
Carotenoids (Cars) are indispensable molecules that fulfill diverse biological roles that are fundamental to the integrity and efficiency of living systems.1,2 Lycopene (Fig. 1A) (LYC) is predominantly present in mature fruits, where it serves as a key pigment contributing to their characteristic coloration.3 Despite extensive research, many of its properties remain uncertain, inviting deeper investigation into their nature and function.4 The phenomenon of singlet exciton fission represents a potential photoprotective mechanism that has been investigated in LYC.4–8 Also, LYC is related to action against prostate cancer.9 | ||
| Fig. 1 Structures in the study: (A) all-trans-lycopene (all-trans); (B) β-cyclodextrin (R = –H) and 2-hydroxypropyl-β-cyclodextrin (R = –CH2–CH(OH)CH3). | ||
Cyclodextrins (Fig. 1B) (CDs) are highly effective in complexing with carotenoids through a process in which Cars are incorporated into the cavities of the CDs without chemical bond formation, rendering Cars water-soluble.10–13 β-cyclodextrin (β-CD) consists of seven cyclic oligomers of α-1,4,-D-glucose13–15 and represents an effective model system for computational studies of the complexation of 2-hydroxypropyl-β-cyclodextrin with various molecules.16–20
The Raman spectra of Cars (as well as of LYC) are characterised by four main bands, referred to as ν1 through ν4. The ν1 band, appearing around 1500 cm−1, corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond stretching modes. The ν2 band, near 1160 cm−1, is due to the combined effects of C–C stretching and in-plane C–C bending. The ν3 band, situated at about 1000 cm−1, is attributed to the coupling of in-plane rocking vibrations of methyl groups on the conjugated chain with neighbouring C–H in-plane bending.2,4,21
C bond stretching modes. The ν2 band, near 1160 cm−1, is due to the combined effects of C–C stretching and in-plane C–C bending. The ν3 band, situated at about 1000 cm−1, is attributed to the coupling of in-plane rocking vibrations of methyl groups on the conjugated chain with neighbouring C–H in-plane bending.2,4,21
Using well-established extraction protocols, LYC can be obtained from fresh tomatoes for study.22,23 The preparation and characterization of Cars complexed with CDs to achieve water solubility have been reported in ref. 24. Similar methodology has been used together with computational study previously,25–27 and we presented an analysis based on analysing the β-ring dihedral angles and revealing three key all-trans-β-carotene conformers (trans–trans, trans–cis, and cis–cis of β-ring), with ν1 differing by 4 cm−1 among them.26 However, the unique properties of LYC arise from a different origin—its distinct terminal group structures instead of β-rings—resulting in unique complexation characteristics which are different for crystals and solutions as observed from their Raman spectra.2,28,29
Noncovalent interactions, encompassing van der Waals forces, electronic effects, and hydrophobic interactions, play a crucial role in forming CDs and Cars complexes.30 Many authors have observed the self-association of CD complexes,31–33 which has been confirmed through various analytical13 and microscopy techniques.30 Various studies have also explored CD/drug complexes using molecular modelling approaches.34–36 These products are often termed “inclusion complexes,” although the specific form of the CD is frequently not well characterized.37 The LYC Cars are good candidates for inclusion characterisation as they exhibit Raman ν1 band shift from their crystal to their monomer structures and visually change colour from red to yellow.4,29
This work combines Raman spectroscopy, molecular dynamics (MD), and density functional theory (DFT) to investigate complexes of LYC with β-CD, HP-CD, and artificial CD-like structures. We examine the shifts in the Raman ν1 band and identify potential influences contributing to these variations. The study concludes with an evaluation of structural modifications, followed by an analysis and discussion of the influence of complexation on LYC and its broader significance.
Results and discussion
Experimental data
For the preparation of the lycopene/2-hydroxypropyl-β-cyclodextrin complex (LYC/HP-CD), LYC was obtained from fresh tomatoes as previously described.23 Based on 1H and 1H–1H-COSY NMR spectra, LYC was identified as an (all-trans)-isomer (Fig. 2A). The chemical shifts were in good accordance with previously published spectral data of (all-trans) lycopene.38,39 | ||
| Fig. 2 1H NMR (A) and 1H–1H-COSY NMR (B) spectra of the (all-trans)-lycopene in CDCl3. | ||
The 1H NMR spectrum of the symmetric LYC indicates an overlapping multiplet for the protons 11/11′ and 15/15′ at 6.60–6.67 ppm. The overlapped doublets at 6.25–6.3 ppm were attributed to the 8/8′ and 14/14′ protons. The signal of the 7/7′ protons appears as a doublet of doublets (dd) at 6.45–6.55 ppm. The multiplet at 5.11 ppm and three doublets at 5.95, 6.18 and 6.35 ppm correspond to the protons 2/2′, 6/6′, 10/10′ and 12/12′, respectively. The coupling constants J6/7 (11.0 Hz), J7/8 (15.0 Hz), J10/11 (10.9 Hz) and J11/12 (14.9 Hz) are in the region typical for trans conjugated protons in Cars. The 1H–1H-COSY NMR spectrum shows the correlated COSY cross-peaks between protons 7/8 and 11/12, while the cross-peaks of 6/7 and 10/11 overlap due to their close NMR shifts (Fig. 2B).
The purity of the LYC extracted from tomatoes was analysed by HPLC chromatography using the same column and mobile phase as in the previously developed method for determining LYC content in tomatoes and resolving all-trans LYC and its cis isomers.40 The obtained LYC used in the experiments had a purity of 94.8% with the retention time for all-trans isomers (Fig. 3A). After extraction from the LYC/HP-CD complex, the LYC purity reached 92.8% with the same retention time (Fig. 3B).
 | ||
| Fig. 3 The HPLC chromatograms of the initial lycopene extracted from tomatoes (A) and lycopene from the lycopene/2-hydroxypropyl-β-cyclodextrin complex (B). | ||
The Raman spectra of LYC, the LYC/HP-CD complex and HP-CD are presented in Fig. 4. The intense vibrational bands at 1510 and 1157 cm−1 were attributed to the stretching of CC and C–C in the polyene chain of LYC, respectively. The band at 1006 cm−1 occurs due to methyl in-plane rocking modes. In the Raman spectrum of the LYC/HP-CD complex, the band at 1510 cm−1 shifts to a higher frequency of 1516 cm−1 due to the interaction of LYC with HP-CD. Thus, the Raman spectra of LYC and the LYC/HP-CD complex crystal structure results can be interpreted by assuming that there are no longer lycopene–lycopene interactions.
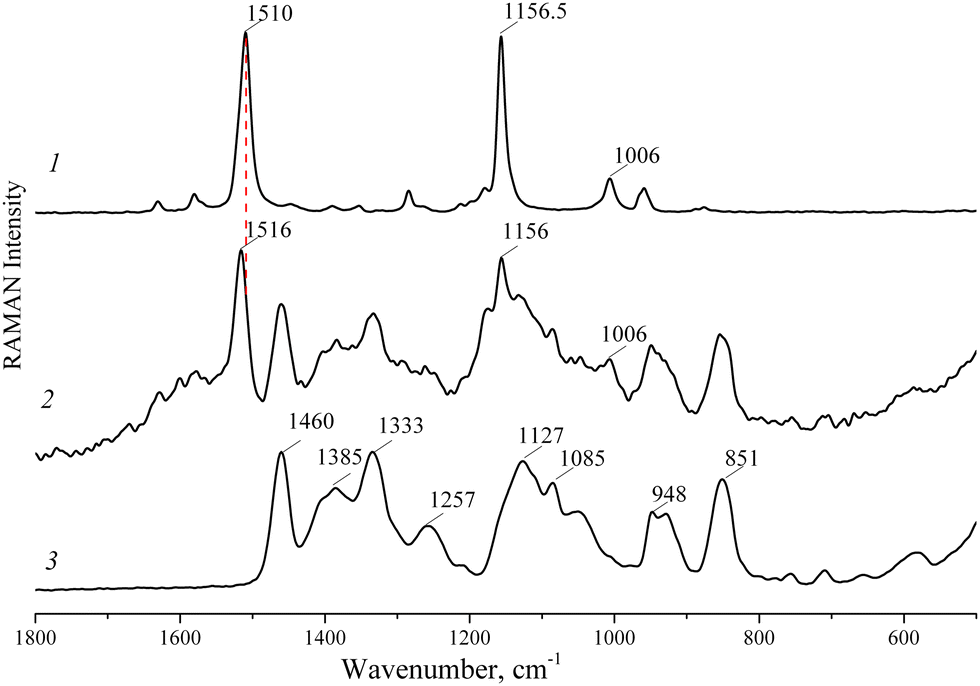 | ||
| Fig. 4 Raman spectra of pure (all-trans)-lycopene (1), the lycopene/2-hydroxypropyl-β-cyclodextrin complex (2) and 2-hydroxypropyl-β-cyclodextrin (3). | ||
Computational data
 | ||
| Fig. 5 Analysis using RMSD and MOC (mass of center) methods for the lycopene inside artificial CD simulations: MOC clearly shows several different possible complexes; in both cases it is clear that after 1 ns the structures are separate (inset). | ||
The solvent was modelled with water molecules around the structures, as water molecules create strong hydrogen bonding, which can lead to different energetic surfaces.21,41 We placed various amounts of water molecules (1000, 2500, 4000, 4500, 5000 or 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules) around Lyc and the artificial CD complex. The criteria were water molecules filling the simulation box and, at the same time, water molecules surrounding the complex. We found for a small number or molecules (1000, 2500, or 4000 water molecules), the water molecules formed water clusters. MD simulations of LYC in an artificial CD complex with 10,000 water molecules were successfully performed up to 0.5 ns, with no evidence of water clustering throughout the trajectory. After detailed analysis, we found that the major differences were in the artificial CD structure itself, and these new artificial structures.
000 molecules) around Lyc and the artificial CD complex. The criteria were water molecules filling the simulation box and, at the same time, water molecules surrounding the complex. We found for a small number or molecules (1000, 2500, or 4000 water molecules), the water molecules formed water clusters. MD simulations of LYC in an artificial CD complex with 10,000 water molecules were successfully performed up to 0.5 ns, with no evidence of water clustering throughout the trajectory. After detailed analysis, we found that the major differences were in the artificial CD structure itself, and these new artificial structures.
Next, computations were performed by choosing different positions along the LYC polyene chain (Fig. 5). We found that the largest possible ν1 was 1554.58 cm−1, and the smallest was 1550.96 cm−1. The monomer computations of LYC were used as a reference, for which the computed Raman v1 band was 1553.66 cm−1 (Table 1). Thus, we successfully determined the shift to lower frequency by modelling LYC packing. The value 1550.96 cm−1 was achieved when the artificial CD directly blocked the vibration availability of both central methyl groups in the polyene chain of LYC. This would explain the shifts in the Raman ν1 bands of crystal and crystalloid LYC relative to the monomer values as being from structural changes, which can be true in large-scale J-aggregate type structures.4,42–44 Moreover, by artificially surrounding LYC with several LYC molecules, we observed lower shifts of 3 cm−1 and larger shifts of 8 cm−1 (using the PBE045 functional and QM/MM with UFF46); a more detailed analysis will be reported separately.
| Structure/complex | ν 1, cm−1 | ν 2, cm−1 | Additional (ν2-b), cm−1 |
|---|---|---|---|
| LYC (all-trans-lycopene) | 1553.66 | 1191.16 | |
| 5-cis-lycopene | 1553.53 | 1191.17 | |
| 7-cis-lycopene | 1552.91 | 1191.44 | |
| 9-cis-lycopene | 1557.74 | 1190.81 | 1201.83 |
| 11-cis-lycopene | 1551.32 | 1190.07 | 1202.15 |
| 13-cis-lycopene | 1560.13 | 1577.07 | 1182.88, 1169.94, 1162.72 |
| 15-cis-lycopene | 1556.99, 1565.95, (1607.04 weak) | 1607.04 | 1202.80, 1270.49 |
| All-trans + β-CD (central) | 1552.81 | 1192.36 | |
| LYC + β-CD (pos1) | 1552.83 | 1188.42 | |
| LYC + β-CD (pos2) | 1552.62 | 1191.15 |
 | ||
| Fig. 6 Major cis isomers of LYC (A) and their relative energies (B) and (C). | ||
• Type A includes the 9-cis and 13-cis-isomers. These cis-isomers were slightly less stable (within 0.05 eV) than the all-trans-form.
• Type A′ includes the 5-cis isomer. Relative to the all-trans-form, this was the only cis-isomer that was slightly unstable (within 0.003 eV).
• Type B includes the 15-cis-isomer, which was about 0.1 eV less stable than the all-trans form.
• Type C includes the 7-cis and 11-cis-isomers. These cis-isomers were about 0.23 eV higher on the energy scale than the all-trans form.
Their stability followed the order all-trans < 5-cis < 9-cis < 13-cis < 15-cis < 7-cis < 11-cis. The same schema (Fig. 6B and C) of stability was achieved when we omitted the ZPE. According to B3LYP47–50 with the basis set cc-pVDZ,51 6-311++G**,52–59 6-311++G(2d,p);52–59 and PBE1PBE45,60 or CAM-B3LYP61 with cc-pVDZ51 and 6-31G**,52–59 levels of calculation, all-trans < 5-cis in all cases.
The results of Raman studies for the all-trans-lycopene (all-trans), 5-cis-lycopene (5-cis), 7-cis-lycopene (7-cis), 9-cis-lycopene (9-cis), 11-cis-lycopene (11-cis), 13-cis-lycopene (13-cis), 15-cis-lycopene (15-cis) structures are presented in Table 1. One group (9-cis-lycopene, 13-cis-lycopene and 15-cis-lycopene) has values up to 6.47 cm−1 larger relative to all-trans-lycopene. The second group (5-cis-lycopene, 7-cis-lycopene and 11-cis-lycopene) has values up to 2.34 cm−1 lower relative to all-trans-lycopene.
We positioned the open and closed forms of β-CD at three locations along the polyene chain of LYC, placing the primary structures near the methyl groups. The positions were labelled from the centre to the ending group (central), position 1 (pos1), and position 2 (pos2). According to the modelling data described in the previous section, these configurations were expected to have the greatest impact on the Raman ν1 band.
As expected, the β-CD open form with LYC was energetically >1 eV higher than the closed form. In contrast to the monomer, the complexes did not have imaginary frequencies. Among the positions of the open form complexes, pos1 had the lowest energy, while the energy of pos2 was higher by 0.12 eV and that of the central position by 0.04 eV. The Raman ν1 bands were 1551.33 cm−1, 1550.96 cm−1, and 1552.62 cm−1 for central, pos1 and pos2 complexes, respectively. These findings indicate that the open form influences the methyl groups of LYC. Although the ring has a diameter of about 9 Å, its oxygen atoms can interact with LYC.
The β-CD closed form (or β-CD) with LYC was energetically favourable, with the lowest energy observed at pos2 (Fig. 7). Position pos1 was higher in energy by 0.05 eV, and the central position was higher by 0.12 eV. The complexes also did not have imaginary frequencies. The Raman ν1 bands were 1552.81 cm−1, 1552.83 cm−1, 1552.62 cm−1 for the central, pos1 and pos2 complexes, respectively (Table 1). The differences from the monomer (all-trans-polyene) was up to 1.04 cm−1. The Raman ν2 band was also in the 1 cm−1 range, except for the pos2 complex, which differed by 2.74 cm−1.
 | ||
| Fig. 7 β-Cyclodextrin structures and complexes: (A) from side; (B) from top; (C), at the terminal group of lycopene; (D) at the central position of lycopene. | ||
The computational data was analysed not including the scaling factor, as experimental data for LYC in the monomer is quite complex,42 as the solid-state can have H-aggregate and J-aggregate type crystal systems. Thus, LYC monomer DFT computations represent a scenario without interactions, which is not representative of solid-state LYC. Our data indicate that the fixation of methyl groups by the surrounding environment can influence the Raman ν1 band shifts; this phenomenon is likely applicable to solid-state LYC.
Despite the fact that cis-type LYC exhibits Raman v1 shifts to larger values, the experimental data suggest that the cis-type LYC can be neglected. An additional argument is the absence of additional Raman bands and the influence of experimental preparations.
Based on the modelling data of the LYC/β-CD complexes (Table 1), it can be concluded that β-CD encapsulates LYC either at the central position of the Cars chain or at the terminal group. This structural arrangement ensures that the Raman v1 band remains close to that of the monomer. Such a situation would manifest in a Raman experiment as a shift toward the Raman values of the LYC monomer, as observed in the LYC/HP-CD experiment, where a shift of 6 cm−1 was recorded.
Methods
Computational methods
The initial equilibrated clusters were generated using the classical molecular dynamics (MD) approach in the software AMBER 22.62 MD force fields were not optimised structures. Hence, the force field was reoptimised for the molecular models described below. DFT was used to optimise the reference molecules and dimers at the B3LYP47–50/cc-pVDZ51 level. Partial atomic charges were determined with the RESP procedure as required in AMBER's antechamber program. Structures were generated by using the generalized amber force field (GAFF), and the TIP3P Force Field was used for water molecules. An MD simulation box (size 100 × 100 × 100 Å3), filled with the chosen number of molecules (LYC, CD and/or water), was used for MD simulations. Water molecules were generated using the Packmol package.63 MD simulations were performed at a temperature of 333 K with a Langevin thermostat for up to 0.6 simulations (which varied for different simulations). The SHAKE algorithm was used, in which all bonds involving hydrogen are constrained. The nonbonded cutoff was 10 Å. The interaction between complexes primarily was modelled by MD, which has van der Waals interactions modelled by Lennard-Jones potential.62The final geometries and Raman spectra calculations were performed using the density functional theory (DFT) within the Gaussian 16 package.64 All structures were optimised and calculated using CAM-B3LYP61 with the basis set cc-pVDZ;51 frequency and Raman computations were performed at the same level. As the main task was to understand the Raman ν1 shifts, it was decided not to include the basis set superposition error (BSSE) results.64 As the major task was to describe ν1 shifts and obtain data on what can cause them, the computational data were not scaled. According to previous studies, various scaling factors can be used to fit experimental values (see ref. 21, 29 and 65). All structures that had imaginary frequencies were neglected. Due to the fact that the experimental data was for crystal structures, solvent models such as PCM or SMD were not included.
Regarding the complex system, Grimme's dispersion corrections66 should at least be included, since dispersion effects can be important for complex molecules. However, they must be carefully studied, as Raman and excitonic effects are sensitive to the chosen DFT methodology.29,67,68
Experimental methods and sample preparation
HPLC chromatography was performed using a C30 YMC carotenoid column (250 × 4.6 mm I.D., 5 μm particle size, YMC Co., Ltd., Kyoto, Japan). A mobile phase of 1-butanol–acetonitrile–methylene chloride (30/70/10, v/v/v), isocratic elution at 2 mL min−1 and detection at 476 nm were used. To extract LYC from the LYC/HP-CD complex, its aqueous solution was mixed with acetone in a ratio of 1/0.1 (v/v). The LYC was extracted with diethyl ether/petroleum ether (1/1, v/v). After gentle mixing, the organic fraction was separated. The extraction procedure was repeated until the aqueous phase became colourless. The collected organic fractions were combined. The solvent was then removed using a rotary evaporator, and the resulting solid residue was redissolved in the mobile phase of HPLC. The same solvent mixture was used to dissolve the initial LYC extracted from tomatoes. The solutions obtained were analysed using HPLC.
Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker 400 Ascend™ 400 MHz (UltraShield™ Plus 9.4 T magnet) instrument. Samples were dissolved in CDCl3 (1H NMR δ = 7.26 ppm). 1H spectra were recorded with an acquisition time of 4.09 s and with 64 scans. 1H–1H COSY spectra were recorded using the cosygpppqf pulse program with an acquisition time of 0.27 s, 8 scans and a relaxation delay of 2 s.
Raman spectra of samples were recorded using a MultiRAM (Bruker Optik GmbH, Ettlingen, Germany) spectrometer equipped with a 1000-mW Nd:YAG laser (1064 nm) as the excitation source. All the samples were analysed in the form of powder, and the spectra were collected with a laser power of 50 mW and an exposure time of 15 s (about 100 scans).
Conclusions
We studied LYC/β-CD complexes by modelling various details and comparing them with the experimental data for LYC/HP-CD. Experimental data show a clear shift of the Raman v1 bands when comparing solids and complexes with β-CD structures. We successfully simulated various shifts using the modelling methodology and identified methyl groups as the key structural components. This work enhanced our understanding of potential LYC/β-CD complexes. Notably, it revealed that β-CD provides a means to investigate LYC as a monomer. However, the solid-state behaviour of LYC remains an open question, requiring further investigation to identify internal interactions, which we anticipate addressing in future studies. This work demonstrates that LYC/β-CD complexes offer new insights into structural characteristics.Author contributions
All authors agreed to the final version of the manuscript. Goda Bankovskaite: methodology, data curation, formal analysis. Laurynas Diska: methodology, data curation, formal analysis, and writing – editing. Alma Bočkuvienė: investigation. Rūta Gruškienė: investigation, visualisation, validation. Tatjana Kavleiskaja: investigation, visualisation. Jolanta Sereikaitė: methodology, writing – original draft. Mindaugas Macernis: supervision, conceptualization, project administration, methodology, data curation, and writing – original draft.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Research Council of Lithuania (Grant No. S-MIP-23-48). Computations were performed on resources of the supercomputer ‘‘VU HPC’’ Saulėtekis in Vilnius University at Faculty of Physics.References
- A. Vershinin, BioFactors, 1999, 10, 99–104, DOI:10.1002/biof.5520100203.
- T. Polívka and V. Sundström, Chem. Rev., 2004, 104, 2021–2071, DOI:10.1021/cr020674n.
- L. U. Hansen and M.-C. M. Chiu, J. Agri, Food Chem., 2005, 53, 6678–6682, DOI:10.1021/jf058013p.
- M. J. Llansola-Portoles, K. Redeckas, S. Streckaité, C. Ilioaia, A. A. Pascal, A. Telfer, M. Vengris, L. Valkunas and B. Robert, Phys. Chem. Chem. Phys., 2018, 20, 8640–8646, 10.1039/C7CP08460A.
- W. Barford, J. Phys. Chem. Lett., 2023, 14, 9842–9847, DOI:10.1021/acs.jpclett.3c02435.
- A. J. Musser, M. Maiuri, D. Brida, G. Cerullo, R. H. Friend and J. Clark, J. Appl. Chem. Sci., 2015, 137, 5130–5139, DOI:10.1021/jacs.5b01130.
- W. Barford, Phys. Rev. B, 2022, 106, 035201, DOI:10.1103/PhysRevB.106.035201.
- D. Manawadu, D. J. Valentine, M. Marcus and W. Barford, J. Phys. Chem. Lett., 2022, 13, 1344–1349, DOI:10.1021/acs.jpclett.1c03812.
- A. Kaiser, C. Haskins, M. M. Siddiqui, A. Hussain and C. D'Adamo, Curr. Opin. Oncol., 2019, 31, 222–229, DOI:10.1097/CCO.0000000000000519.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025, DOI:10.1021/js950534b.
- G. P. Blanch, M. L. R. Del Castillo, M. Del Mar Caja, M. Perez-Mendez and S. Sanchez-Cortes, Food Chem., 2007, 105, 1335–1341, DOI:10.1016/j.foodchem.2007.04.060.
- V. T. D'Souza and K. B. Lipkowitz, Chem. Rev., 1998, 98, 1741–1742, DOI:10.1021/cr980027p.
- T. Loftsson and M. E. Brewster, J. Pharm. Sci., 1996, 85, 1017–1025, DOI:10.1021/js950534b.
- B. Ivanova and M. Spiteller, Int. J. Biol. Macromol., 2014, 64, 383–391, DOI:10.1016/j.ijbiomac.2013.12.026.
- E. Sabadini, T. Cosgrove and C. E. Fdo, Carbohydr. Res., 2006, 341, 270–274, DOI:10.1016/j.carres.2005.11.004.
- N. Ozdemir, C. C. Pola, B. N. Teixeira, L. E. Hill, A. Bayrak and C. L. Gomes, Lwt-Food Sci. Tech., 2018, 91, 439–445, DOI:10.1016/j.lwt.2018.01.046.
- D. H. Li, H. J. Wu, W. Huang, L. Guo and H. T. Dou, Eur. J. Lipid Sci. Tech., 2018, 120, DOI:10.1002/ejlt.201700521.
- M. Ahmad, B. Ashraf, A. Gani and A. Gani, Int, J. Bio. Macromolecules, 2018, 109, 435–442, DOI:10.1016/j.ijbiomac.2017.11.122.
- H. C. Babaoglu, A. Bayrak, N. Ozdemir and N. Ozgun, J. Food Proc. Preserv., 2017, 41, DOI:10.1111/jfpp.13202.
- J. M. López-Nicolás, P. Rodríguez-Bonilla and F. García-Carmona, Crit. Rev. Food Sci. Nutr., 2014, 54, 251–276, DOI:10.1080/10408398.2011.582544.
- S. Streckaite, M. Macernis, F. Li, E. K. Trskova, R. Litvin, C. H. Yang, A. A. Pascal, L. Valkunas, B. Robert and M. J. Llansola-Portoles, J. Phys. Chem. A, 2020, 124, 2792–2801, DOI:10.1021/acs.jpca.9b11536.
- R. Gruskiene, A. Bockuviene and J. Sereikaite, Molecules, 2021, 26, DOI:10.3390/molecules26154601.
- A. Bockuviene, R. Zalneravicius and J. Sereikaite, Food Biosci., 2021, 40, 100854, DOI:10.1016/j.fbio.2020.100854.
- J. Deli, A. Agócs, R. Iványi, K. Németh, J. Visy, J. Szemán, L. Szente and M. Simonyi, Carotenoid Sci., 2008, 12, 201 Search PubMed.
- R. Karpicz, N. Ostapenko, Y. Ostapenko, Y. Polupan, I. Lazarev, N. Galunov, M. Macernis, D. Abramavicius and L. Valkunas, Phys. Chem. Chem. Phus., 2021, 23, 3447–3454, 10.1039/d0cp05436d.
- M. Macernis, A. Bockuviene, R. Gruskiene, T. Krivorotova and J. Sereikaite, J. Mol. Struct., 2021, 1226, DOI:10.1016/j.molstruc.2020.129362.
- V. E. de Oliveira, E. W. Almeida, H. V. Castro, H. G. Edwards, H. F. Dos Santos and L. F. de Oliveira, J. Phys. Chem. A, 2011, 115, 8511–8519, DOI:10.1021/jp2028142.
- M. J. Llansola-Portoles, K. Redeckas, S. Streckaitė, C. Ilioaia, A. A. Pascal, A. Telfer, M. Vengris, L. Valkunas and B. Robert, Phys. Chem. Chem. Phys., 2018, 20, 8640–8646, 10.1039/c7cp08460a.
- M. Macernis, J. Sulskus, S. Malickaja, B. Robert and L. Valkunas, J. Phys. Chem. A, 2014, 118, 1817–1825, DOI:10.1021/jp406449c.
- Y. He, P. Fu, X. Shen and H. Gao, Micron, 2008, 39, 495–516, DOI:10.1016/j.micron.2007.06.017.
- M. Messner, S. V. Kurkov, P. Jansook and T. Loftsson, Int. J. Pharm., 2010, 387, 199–208, DOI:10.1016/j.ijpharm.2009.11.035.
- T. Loftsson, P. Saokham and A. R. Sa Couto, Int. J. Pharm., 2019, 560, 228–234, DOI:10.1016/j.ijpharm.2019.02.004.
- G. González-Gaitano, P. Rodríguez, J. R. Isasi, M. Fuentes, G. Tardajos and M. Sánchez, J. Incl. Phenom. Macrocyclic Chem, 2002, 44, 101–105, DOI:10.1023/A:1023065823358.
- L. Szente, J. Szejtli and G. L. Kis, J. Pharm. Sci., 1998, 87, 778–781, DOI:10.1021/js9704341.
- T. Loftsson, M. Masson and M. E. Brewster, J. Pharm. Sci., 2004, 93, 1091–1099, DOI:10.1002/jps.20047.
- Q. Zhao, N. Miriyala, Y. Su, W. Chen, X. Gao, L. Shao, R. Yan, H. Li, X. Yao, D. Cao, Y. Wang and D. Ouyang, Mol. Pharm., 2018, 15, 1664–1673, DOI:10.1021/acs.molpharmaceut.8b00056.
- S. Clercq, F. Temelli and E. Badens, Mol. Pharm., 2021, 18, 1720–1729, DOI:10.1021/acs.molpharmaceut.0c01227.
- S. Tiziani, S. J. Schwartz and Y. Vodovotz, J. Agric. Food Chem., 2006, 54, 6094–6100, DOI:10.1021/jf061154m.
- M. Takehara, M. Nishimura, T. Kuwa, Y. Inoue, C. Kitamura, T. Kumagai and M. Honda, J. Agric. Food Chem., 2014, 62, 264–269, DOI:10.1021/jf404497k.
- M. T. Lee and B. H. Chen, Chromatographia, 2001, 54, 613–617, DOI:10.1007/BF02492187.
- M. Macernis, L. Mourokh, L. Vinciunas and L. Valkunas, Lith. J. Phys., 2022, 62, 179–193 Search PubMed.
- M. Ishigaki, P. Meksiarun, Y. Kitahama, L. Zhang, H. Hashimoto, T. Genkawa and Y. Ozaki, J. Phys. Chem. B, 2017, 121, 8046–8057, DOI:10.1021/acs.jpcb.7b04814.
- L. Wang, Z. Du, R. Li and D. Wu, Dyes Pigm., 2005, 65, 15–19, DOI:10.1016/j.dyepig.2004.05.012.
- K. Ray and T. N. Misra, J. Photochem. Photobio. A: Chem., 1997, 107, 201–205, DOI:10.1016/S1010-6030(97)00068-3.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170, DOI:10.1063/1.478522.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. I. I. I. Goddard and W. M. Skiff, JACS, 1992, 114, 10024–10035, DOI:10.1021/ja00051a040.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- S. H. Vosko, L. Wilk and M. Nusair, Canad. J. Phys, 1980, 58, 1200–1211, DOI:10.1139/p80-159.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627, DOI:10.1021/j100096a001.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023, DOI:10.1063/1.456153.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648, DOI:10.1063/1.438980.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654, DOI:10.1063/1.438955.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728, DOI:10.1063/1.1674902.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261, DOI:10.1063/1.1677527.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222, DOI:10.1007/BF00533485.
- P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209–214, DOI:10.1080/00268977400100171.
- M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163–168, DOI:10.1016/0009-2614(80)80628-2.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665, DOI:10.1063/1.444267.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036, DOI:10.1063/1.478401.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57, DOI:10.1016/j.cplett.2004.06.011.
- D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros, V. W. D. Cruzeiro, N. Forouzesh, T. J. Giese, A. W. Götz, H. Gohlke, S. Izadi, K. Kasavajhala, M. C. Kaymak, E. King, T. Kurtzman, T.-S. Lee, P. Li, J. Liu, T. Luchko, R. Luo, M. Manathunga, M. R. Machado, H. M. Nguyen, K. A. O’Hearn, A. V. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, A. Risheh, S. Schott-Verdugo, A. Shajan, J. Swails, J. Wang, H. Wei, X. Wu, Y. Wu, S. Zhang, S. Zhao, Q. Zhu, T. E. Cheatham III, D. R. Roe, A. Roitberg, C. Simmerling, D. M. York, M. C. Nagan and K. M. Merz Jr., J. Chem. Inf. Modeling, 2023, 63, 6183–6191, DOI:10.1021/acs.jcim.3c01153.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164, DOI:10.1002/jcc.21224.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Gaussian Inc., Wallingford, CT, 2019 Search PubMed.
- M. Macernis, S. Streckaite, R. Litvin, A. A. Pascal, M. J. Llansola-Portoles, B. Robert and L. Valkunas, J. Phys. Chem. A, 2022, 126, 813–824, DOI:10.1021/acs.jpca.1c09393.
- H. Schroder, A. Creon and T. Schwabe, J. Chem. Theory Comput., 2015, 11, 3163–3170, DOI:10.1021/acs.jctc.5b00400.
- M. Macernis, Open Access J. Chem., 2018, 2, 01–10 CrossRef.
- M. Macernis, Lith. J. Phys., 2018, 58, 358–378 Search PubMed.
- E. Celitan, R. Gruskiene and J. Sereikaite, Molecules, 2021, 26, DOI:10.3390/molecules26247562.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00034c |
| This journal is © the Owner Societies 2025 |