
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural evolution and electronic properties of boron sulfides (B2S3)n (n = 1–6): insights from DFT calculations†
Jingxin
Hu
 ,
Lin
Zhang
and
Zexing
Cao
*
,
Lin
Zhang
and
Zexing
Cao
*
State Key Laboratory of Physical Chemistry of Solid Surfaces and Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, China 361005. E-mail: zxcao@xmu.edu.cn
First published on 12th March 2025
Abstract
The low-energy isomers of boron sulfides (B2S3)n (n = 1–6) were constructed by using B2S3 as the building block, and their structure, stability, and reactivity toward small molecules have been explored by density functional theory (DFT) calculations. It is found that the low-energy isomers of (B2S3)n clusters are of rich bonding characteristics for their structural constituents, such as [S−B−S], [B2S3], [B3S3], [S], etc. The planar or non-planar B2S2 rings, as the basic structural units, are bridged together by the S atoms to form the most stable structures of (B2S3)n clusters with n ≥ 2. These low-energy (B2S3)n clusters are predicted to be stable both structurally and electronically, and their boron centers show relatively high activity to the binding of small molecules NH3 and CO, until all boron atoms are ligated by NH3 or CO. The present results provide new insights into the structure and properties of (B2S3)n clusters, which are beneficial to the development of boron chemistry and the application of boron-based materials.
1. Introduction
Boron clusters and boron-containing compounds have attracted considerable attention due to their intriguing physical and chemical properties,1–3 and numerous studies of theory and experiments have been conducted to explore the structural and bonding properties, spectroscopic characteristics, and chemical reactivity of boron clusters,4–16 borophenes,17–23 boron oxide clusters,24–36 and so on. In particular, boron sulfide clusters and materials are of great interest because of their diverse structural features and bonding modes.37–43In the 1960s and 1970s, White and Gilles et al. confirmed the existence of boron sulfides, including BS2+,44 B2S2+,45,46 B2S3+,47,48 and so on, making a significant advancement in boosting the development of boron sulfide chemistry. The existing boron sulfide clusters can be categorized into three main types: proportional, sulfur-rich, and boron-rich boron sulfide clusters. In 2015, Li and co-workers presented a set of perfectly planar dicyclic B6S60/−/2− clusters featuring a twin B3S2 five-membered ring as the core. It was found that the closed-shell B6S62− exhibits a delocalized characteristic of 10π-electrons, closely analogous to the bonding pattern of the aromatic naphthalene.49 Subsequently, Zheng and co-workers investigated the structural, electronic, and bonding properties of BS0/− and BS30/− by combining experimental and theoretical methods.50 The results reveal that the ground state of BS3− has a planar structure with C2v symmetry, whereas BS3 presents an S−B−S−S bending structure. Recently, Chen and co-workers explored the structural evolution of BnS20/− (n = 2–13) clusters through the DFT calculations combined with particle swarm optimization algorithm software, and the predicted global minima for these clusters suggest a conformational evolution from a linear-chain structure to planar or quasi-planar ones.51 Obviously, while the above theoretical and experimental studies show that boron sulfide clusters exhibit very rich chemical bonding modes, depending on the B/S atomic ratio and the charged state, our understanding of B2S3-type clusters remains insufficient.
Notably, B2O3-type clusters were proved to play a crucial role in boron chemistry and oxidative dehydrogenation of propane.52–57 In 2013, Li and Cheng theoretically studied the structure evolution and electronic properties of (B2O3)n (n = 1–6) clusters, and found that the global minima structures of these clusters are planar up to n = 3, and cages at n = 4–6.58 Given the similarity in the valence electron configurations of sulfur and oxygen atoms, (B2S3)n and (B2O3)n may exhibit similar structural and electronic features. This also implies that B2S3-type clusters could display analogous chemical properties and stability, making them ideal candidates for organic catalysis and materials science.
Recently, B2S3-type two-dimensional (2D) materials were predicted to be promising catalysts for splitting water59,60 and suppressing the shuttle effect in lithium–sulfur batteries.61 However, our knowledge of B2S3-type clusters in terms of their structural evolution and chemical properties is still very limited. Further investigation into the growth of B2S3-type clusters could lead to the development of more stable and efficient materials for these applications.
Under this background, B2S3 has been chosen as a fundamental structural unit to search for the stable structures of a series of (B2S3)n (n = 1–6) clusters by using the artificial bee colony algorithm.62,63 Subsequent DFT calculations were used to determine the structural and electronic properties of the corresponding low-energy structures of (B2S3)n. Additionally, the interaction of selected clusters with small molecules was discussed to explore their potential applications in small molecule activation and capture. The present work aims to further understand the structures, properties, and potential applications of boron sulfide clusters, and to provide a theoretical basis for the development of 2D boron-based materials.
2. Computational details
The low-energy structures of the studied (B2S3)n clusters are predicted by using the artificial bee colony algorithm, implemented in the ABCluster software for the global optimization of chemical clusters.62,63 The initial configurations, ranging from 1000 to 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 depending on the number of atoms in the studied clusters, are auto-generated and pre-optimized by the semiempirical PM6 and GFN2-xTB method with ABCluster.64–67 Then 80 searched structures with relatively low energy for the studied clusters are selected and fully optimized with the more accurate DFT method at the M06-2X-D368,69/6-31G(d,p)70,71 level of theory, implemented in the Gaussian 16 package.72 The final energy ranking of low-energy structures is obtained through a combination of PM6 and GFN2-xTB methods, effectively reducing the risk of missing important structures. All the optimized cluster structures underwent harmonic vibrational analysis at the same level, confirming that all the related species are stable without imaginary frequency. In addition, the composite quantum chemical method G4(MP2)-6X73 is adopted to evaluate the reliability of the M06-2X-D3/6-31G(d,p) level. Mayer bond order,74 adaptive natural density partitioning (AdNDP),75 and orbital composition analyses are performed by the Multiwfn program.76 Geometric structures and molecular orbitals were visualized by using the CYLview,77 IboView,78 and VMD programs.79
000 depending on the number of atoms in the studied clusters, are auto-generated and pre-optimized by the semiempirical PM6 and GFN2-xTB method with ABCluster.64–67 Then 80 searched structures with relatively low energy for the studied clusters are selected and fully optimized with the more accurate DFT method at the M06-2X-D368,69/6-31G(d,p)70,71 level of theory, implemented in the Gaussian 16 package.72 The final energy ranking of low-energy structures is obtained through a combination of PM6 and GFN2-xTB methods, effectively reducing the risk of missing important structures. All the optimized cluster structures underwent harmonic vibrational analysis at the same level, confirming that all the related species are stable without imaginary frequency. In addition, the composite quantum chemical method G4(MP2)-6X73 is adopted to evaluate the reliability of the M06-2X-D3/6-31G(d,p) level. Mayer bond order,74 adaptive natural density partitioning (AdNDP),75 and orbital composition analyses are performed by the Multiwfn program.76 Geometric structures and molecular orbitals were visualized by using the CYLview,77 IboView,78 and VMD programs.79
3. Results and discussion
Low-energy structures
Based on the results searched by using ABCluster, some low-energy structures of (B2S3)n (n = 1–6) clusters have been identified. Prior to investigating the structural and electronic properties of these clusters in detail, a comparative analysis was conducted on the equilibrium structures and relative energy order of selected low-energy clusters evaluated by both the M06-2X-D3/6-31G(d,p) and G4(MP2)-6X methods, and there are no qualitative differences between the results obtained by the two methods, thereby confirming that the M06-2X-D3/6-31G(d,p) approach is capable of accurately evaluating the structures and properties of the studied boron-based clusters (see more details in Fig. S1–S6, ESI†). Herein, the following discussion mainly focuses on clusters with relative Gibbs free energies below 30.0 kcal mol−1 at the M06-2X-D3/6-31G(d,p) level, as well as the isomers similar to the most stable (B2O3)n cluster structures in previous studies.25,58As shown in Fig. 1, the most stable isomer I of the B2S3 cluster exhibits a V-shaped structure with alternating bonding between B and S atoms. In contrast, isomers II (T-shaped) and III (Y-shaped) with a B−B motif are less stable than I by 22.5 and 29.0 kcal mol−1, respectively. For the B4S6 cluster, the most stable isomer I features a B2S2 rhombic core with two linear [S−B−S] arms in a trans-configuration, and its corresponding cis-configuration II is almost isoenergetic with I. The isomer III of B4S6 with two non-planar B2S2 units linked by S atoms is lying at 12.8 kcal mol−1 above I. Notably, both isomers IV and V without the B2S2 moiety, with the boron-centered structure connecting three linear [S−B−S] motifs, are higher in energy than I by 19.4 and 22.3 kcal mol−1, respectively. For relatively large clusters of B6S9, B8S12, B10S15, and B12S18, their most stable isomers generally consist of several planar or non-planar B2S2 rings as the main structural units, connected by S atoms to form a closed structure. Obviously, their higher-energy structures tend to feature six-membered [B3S3] rings or linear [S−B−S] structural units. Overall, the low-energy isomers of (B2S3)n clusters with n = 1 and 2 mainly exhibit a planar configuration, whereas a non-planar or cage configuration is adopted for the large-sized clusters with n = 3–6.
 | ||
| Fig. 1 The optimized low-energy structures and relative free energies (in kcal mol−1) of the studied boron sulfide clusters, where B and S atoms are in pink and yellow, respectively. | ||
Interestingly, it was found that the most stable isomers of B2S3 and B4S6 have the same configuration as that of the corresponding B2O3 and B4O6 clusters.25,58 Conversely, for relatively large clusters such as B6S9, B8S12, B10S15, and B12S18, the isomers with structures similar to the corresponding boron oxide clusters are less stable than the most stable species by more than 20.0 kcal mol−1. For instance, VI in B6S9, V in B8S12, III in B10S15, and II in B12S18 are higher in energy than their respective most stable isomers by 45.8, 63.0, 47.8, and 23.9 kcal mol−1, respectively. Presumably, there may be significant differences in structural and electronic properties between the boron sulfide (B2S3)n and boron oxide (B2O3)n clusters.
Fig. 2a presents key bond lengths, Mayer bond orders, and bond angles for the most stable isomers of (B2S3)n (n = 1–6) clusters. For planar B2S3 and B4S6 featuring linear [S−B−S] arms, the bonding between the terminal S and B atoms is intermediate between a double and a triple bond, with a Mayer bond order of 2.453 and bond lengths around 1.605 Å. For relatively large clusters, their most stable isomers are composed of planar or non-planar B2S2 cyclic structures bonded together by the sulfur atom, such as B4S6, B6S9, B8S12, B10S15, and B12S18, where the B−S single bonds vary from 1.809 to 1.832 Å. Furthermore, the natural adaptive orbital (NAdO) analyses are performed to describe the distribution, character, and intensity of the chemical bonding.80,81 The NAdO analysis reveals that the terminal B−S bond in B4S6 is characterized by three major NAdOs, one σ-type (0.914) and two π-type (0.621 and 0.601) bonding categories, and the occupancy sum of σ and π NAdOs is 2.116, indicating that the terminal B−S bond is between double and triple bonds, as shown by the Mayer bond order (2.453) analysis.
 | ||
| Fig. 2 (a) The optimized global minimum configurations of the studied (B2S3)n clusters, where the key bond lengths (Å), Mayer bond orders, and bond angles (°) are given in black, blue, and red, respectively. (b) The NAdO analysis on the bonding modes of B4S6 is given, where B and S atoms are in pink and yellow, respectively. | ||
Bonding and stability
In order to gain a deeper understanding of the bonding properties of the most stable isomer of the studied clusters, the adaptive natural density partitioning (AdNDP) method is employed to describe the key delocalized orbitals and bonding characteristics, as depicted in Fig. 3 and Fig. S7 (ESI†). For B2S3 in Fig. 3a, there are two lone pairs (LPs) of electrons on the S atoms with the occupied number (ON) of 1.96 |e|, four two-center two-electron (2c–2e) σ-bonds (ON = 1.99–2.00 |e|) from the inner B−S bond, four 2c–2e π-bonds (ON = 1.98–2.00 |e|) from the terminal B−S bond, and one 3c–2e π-bond (ON = 1.96–1.97 |e|). For B4S6 in Fig. 3b, there are four lone pairs of electrons mainly originating from the S atoms, ten 2c–2e σ bonds, four 2c–2e π bonds, two 3c–2e π bonds, one 4c–2e π bond, and one 2c–2e π* bond. It was found that the cyclic B2S2 structure is characterized by σ, π, and π* bonds. Note that the large-sized B6S9, B8S12, B10S15, and B12S18 clusters have similar bonding characteristics, mainly including LPs on the S atoms, 2c–2e σ-bonds for the B−S single bonds, delocalized 3c–2e π-bonds contributed by the B−S−B units, and 4c–2e π-bonds and 2c–2e π*-bonds from the planar or non-planar B2S2 rings. Overall, the AdNDP analysis further deepens our understanding of the structures and bonding properties of the selected clusters. | ||
| Fig. 3 AdNDP delocalized bonding patterns and the corresponding occupied numbers for (a) B2S3, (b) B4S6, (c) B6S9, and (d) B8S12. The color of atoms: B-pink; S-yellow. | ||
Additionally, the bonding strength between (B2S3)n clusters and the B2S3 monolayer52 are compared under the same theoretical level (see more details in Table S3, ESI†). The results show that the bonding energy of the B−S bonds in (B2S3)n clusters is generally stronger than that of the B2S3 monolayer (except B6S9 cluster). The B6S9 cluster has a lower bonding energy than the B2S3 monolayer, indicating its lower stability.
The key frontier molecular orbitals and the corresponding energy levels of the most stable clusters are shown in Fig. 4. It was found that the HOMO of B2S3 originates from the terminal B−S π bonding and the non-bonding 3p orbital of the meso S atoms, whereas the LUMO is characterized by the B−S antibonding (π*). Similarly, the HOMO of B4S6 is derived from the terminal B−S π bonding and the 3p orbitals of the connected S atoms, and its LUMO is mainly contributed by the S atoms of the B2S2 moiety. For the large-sized clusters, such as B6S9, B8S12, B10S15, and B12S18, their HOMOs and LUMOs are primarily composed of the 3p orbitals of S atoms and the 2p orbitals of B atoms, respectively. As shown in Fig. 4b, these boron sulfide clusters generally have relatively large HOMO–LUMO gaps (EGAP = 4.36–5.14 eV), indicating that all the studied (B2S3)n (n = 1–6) clusters may possess highly stable electronic structures. As the number of B2S3 basic units increases, the EGAP value decreases first and then maintains at about 4.4 eV.
 | ||
| Fig. 4 (a) The key molecular orbitals (isosurface = 0.05) and (b) the corresponding energy levels (in eV) of the studied (B2S3)n clusters. | ||
In order to understand the electronic gain and loss characteristics as well as the relative stability of these studied boron sulfide clusters, their adiabatic/vertical electron affinities (EAa and EAv) and adiabatic/vertical ionization potentials (IPa and IPv), average binding energies (Eb), and second-order energy difference (Δ2E) were calculated as follows:
| EAa = E0therm − E−1therm |
| EAv = E0 − E−1 |
| IPa = E1therm − E0therm |
| IPv = E1 − E0 |
| Etherm = Eelec + Ecorr |

| Δ2E([B2S3]n) = E([B2S3]n−1) + E([B2S3]n+1) − 2E([B2S3]n) |
As shown in Table 1, as the cluster size increases, the EAv values gradually increase overall, while the IPa and IPv values correspondingly decrease first and then remain less varied. Additionally, the EAa value of B12S18 is exceptionally low at 1.25 eV, primarily attributed to significant structural reorganization upon the formation of the anion cluster B12S18− (see more details in Fig. S16, ESI†). Notably, the relatively low EAa and EAv values suggest a diminished capability of the molecule to accommodate additional electrons, whereas the relatively large IPa and IPv values indicate that the corresponding clusters are highly stable in their neutral state. These observations collectively underscore the high chemical stability of the studied boron sulfide clusters. Note that the evaluated EAs and IPs are closely correlated with the frontier molecular orbital properties of the studied molecules. There is a sharp drop in IPs from B4S6 to B6S9, which can be ascribed to the transition of the HOMO composition from predominant π orbitals associated with B−S bonds to the 3p orbitals of S atoms.
| Cluster | EAa (eV) | EAv (eV) | IPa (eV) | IPv (eV) | E b (eV) | Δ2E |
|---|---|---|---|---|---|---|
| a Adiabatic electron affinities EAa, vertical electron affinities EAv, adiabatic ionization potentials IPa, vertical ionization potentials IPv, average binding energies Eb, and second-order energy difference Δ2E. | ||||||
| B2S3 | 1.79 | 0.69 | 9.60 | 9.73 | 0.1860 | — |
| B4S6 | 1.63 | 1.17 | 9.13 | 9.23 | 0.1922 | 0.2720 |
| B6S9 | 1.77 | 1.18 | 8.08 | 8.51 | 0.1936 | −0.9400 |
| B8S12 | 1.87 | 1.30 | 8.26 | 8.42 | 0.1961 | 0.2335 |
| B10S15 | 2.00 | 1.77 | 8.50 | 8.45 | 0.1972 | 0.2345 |
| B12S18 | 1.25 | 2.03 | 8.51 | 8.42 | 0.1976 | — |
The slightly increasing Eb suggests that the stability of (B2S3)n (n = 1–6) clusters gradually enhances with the increase in cluster size. Furthermore, the second-order differences (Δ2E) reveal a different perspective on stability. Although most isomers have positive Δ2E values, B6S9 stands out with a significantly negative Δ2E value of −0.9400, suggesting a higher reactivity structurally, compared to the other clusters. Overall, these studied clusters show relatively high stability both structurally and electronically.
Infrared spectroscopy
To facilitate future identification of B2S3-type clusters, the predicted infrared spectra of the selected clusters are collected in Fig. 5. Note that the infrared spectrum of B2S3 shows two notable absorption bands, where the stronger peak at 1299 cm−1 and the weaker one at 1337 cm−1 arise from the asymmetric and symmetric stretching vibrations of both [S−B−S] units, respectively. The infrared spectrum of B4S6 displays three key vibrational absorption bands, where the adsorptions at 841 and 1320 cm−1 are attributed to the symmetric and asymmetric stretching vibrations of the [S−B−S] unit, respectively, whereas the peak at 893 cm−1 arises from the symmetric deformation of the B2S2 ring along the direction of B atoms. Additionally, the infrared spectra of B6S9, B8S12, B10S15, and B12S18 share a common pattern due to their primary cyclic B2S2 structural units, and their main vibrational peaks all located at 842 cm−1, which are attributed to the breathing vibrations of the B2S2 unit. Overall, the infrared spectral characteristics are closely tied to the core structural units of each cluster, which provide a theoretical basis to identify and distinguish the structure of B2S3-type clusters. | ||
| Fig. 5 Predicted infrared spectra of the studied (B2S3)n clusters at the B3LYP/def2-TZVP level of theory. | ||
Interactions of (B2S3)n with small molecules
Due to the electron-deficient nature of boron, boron clusters and their derivatives often exhibit chemical reactivity toward small molecules.82–86 Herein, it was found that the selected boron sulfide clusters can interact with small molecules, particularly NH3 and CO, and the main binding modes were elucidated, based on DFT calculations. The binding energies (ΔE) were determined as follows:| ΔE = EB2S3Xm − (EB2S3 + m × EX) |
As shown in Fig. 6, main interaction modes between (B2S3)n (n = 1–6) clusters and NH3 are explored. Note that the complexation between NH3 and the B atom of (B2S3)n is actually a Lewis acid–base interaction. As shown in Fig. 6b, the complex of NH3 and B2S3 exhibits two configurations, arising from the nucleophilic attack of NH3 on the B site from both inside and outside the V-shaped structure, while the overall structure remains planar. Energetically, [B2S3][NH3]-I is a more stable configuration. According to the relative energies of various binding modes between B4S6 and NH3 in Fig. 6c, it was found that NH3 more readily binds to the cyclic B2S2 moiety rather than the linear [S−B−S] unit, with a difference in ΔE exceeding 10.0 kcal mol−1. The binding interaction of NH3 with the global minimum structure of clusters B6S9, B8S12, B10S15, and B12S18, forms a similar stable configuration, with the B atom in the B2S2 moiety as the main active site. Notably, the N−H bond (1.015 Å) and the H−N−H bond angle (106°) of NH3 increase to some extent, upon binding to the selected clusters, as shown in Fig. 6. Overall, the studied clusters can stably adsorb NH3 molecules through the Lewis acidity of their B atoms, which may be used for capture and separation of small molecules.
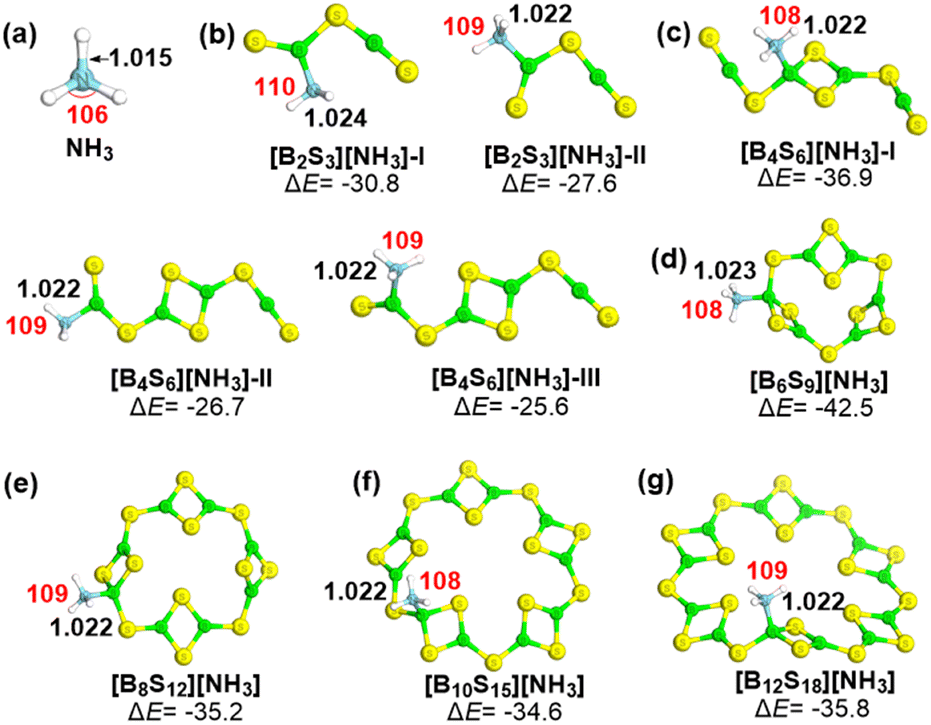 | ||
| Fig. 6 The optimized structures and the relative energies (ΔE, in kcal mol−1) of the NH3 molecule (a) and studied complexes [B2S3]n[NH3] (b–g), taking the energy sum of individual components as the reference. Key N−H bond lengths (Å) and H−N−H angles (°) are given in black and red, respectively. The color of atoms: S-yellow; B-green; N-blue; H-white. | ||
Subsequently, the interactions of (B2S3)n (n = 1–6) clusters with carbon monoxide (CO) are further explored, as depicted in Fig. 7. We note that the bond length of the CO adsorbed at the B atom of the [S−B−S] unit is almost unchanged (1.132∼1.134 Å), compared to the free molecule, but the CO molecule bound to the B atom of the cyclic B2S2 moiety has a shortened C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond with the bond length of about 1.27 Å, suggesting that the formation of a B−CO dative bond is beneficial to the dispersion of excess electrons on the carbon atom and thus further enhances the CO bonding interaction. The predicted binding energies (ΔE) range from −13.3 to −4.7 kcal mol−1, where the B6S9 cluster has the strongest interaction toward CO among these clusters considered here.
O bond with the bond length of about 1.27 Å, suggesting that the formation of a B−CO dative bond is beneficial to the dispersion of excess electrons on the carbon atom and thus further enhances the CO bonding interaction. The predicted binding energies (ΔE) range from −13.3 to −4.7 kcal mol−1, where the B6S9 cluster has the strongest interaction toward CO among these clusters considered here.
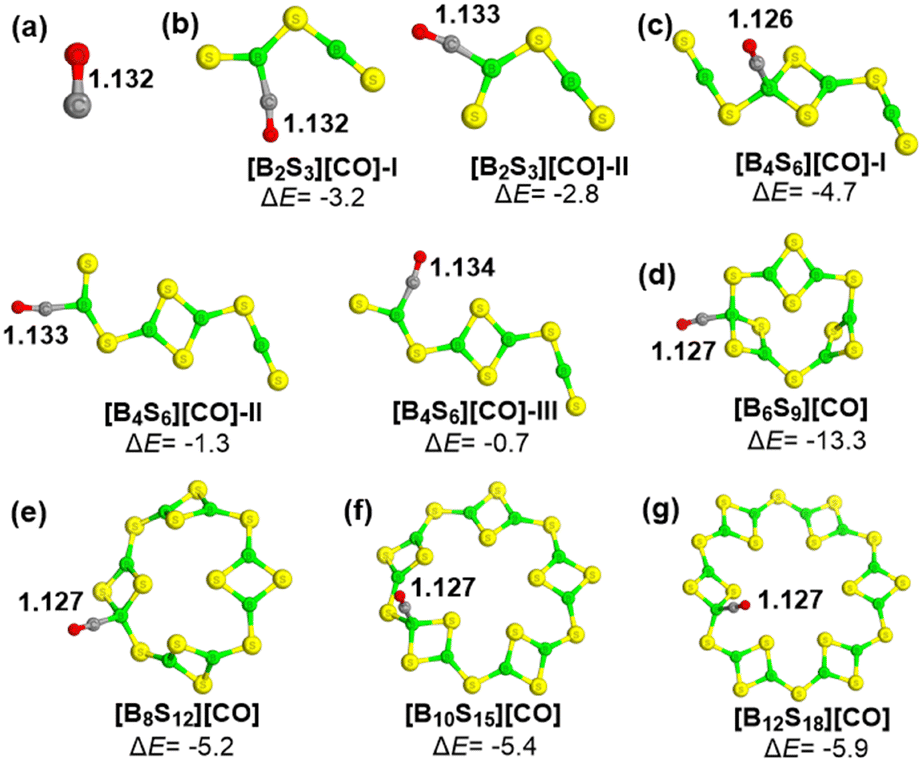 | ||
| Fig. 7 The optimized structures of the CO molecule (a) and studied complexes [B2S3]n[CO] (b–g), and their corresponding CO binding energies (ΔE, in kcal mol−1), where key C−O bond lengths (Å) are given in black. The color of atoms: S-yellow; B-green; C-gray; O-red. | ||
As Fig. 6 and 7 show, B6S9 has the strongest binding interaction with both NH3 and CO molecules among the boron sulfide clusters here. In addition, we investigated the interactions of (B2S3)n clusters with multiple NH3 or CO molecules, and the optimized structures and corresponding bind energies of these complexes [B2S3]n[NH3]m and [B2S3]n[CO]m, are presented in Fig. S8 and S9 (ESI†). Overall, all B atoms of these boron sulfide clusters can bind these small molecules until the boron sites are saturated.
Overall, the present results suggest that B2S3-type clusters exhibit potential applications in organic catalysis and materials engineering, specifically reflected in the following aspects: (a) the strong Lewis acidity of electron-deficient boron centers may facilitate the catalytic activation of small molecules, analogous to established B–N catalytic systems;87,88 (b) the structural stability and delocalized electron characteristics observed in these clusters provide design principles for developing novel 2D boron–sulfide materials with graphene-like architectures; (c) the bonding energy of the B−S bonds in (B2S3)n clusters is generally stronger than that in the reported B2S3 monolayer,52 suggesting that the predicted clusters may offer broader application prospects compared to the B2S3 monolayer materials due to their enhanced stability.
4. Conclusions
Herein, DFT calculations combined with the artificial bee colony algorithm were used to investigate the structural evolution, bonding and reactivity of (B2S3)n clusters. The computational results indicate that the low-energy isomers of these boron sulfides have different structural features as the number of B2S3 basic units increases. In particular, the global minimum structures of B2S3 and B4S6 present a planar configuration, while for the larger-sized clusters of (B2S3)n with n = 3–6, their global minimum configurations are mainly non-planar or cage-like structures. Overall, the linear [S−B−S] and cyclic B2S2 structures are the primary constituent units of these clusters. These structural features differ from those observed in (B2O3)n-type clusters to some extent. The most stable structures of relatively large (B2S3)n clusters are generally assembled by the cyclic B2S2 motifs through S atoms as the linker, and the multiple bonding interactions, including 2c-2e σ and π bonds, 3c-2e and 4c-2e π bonds, lone pairs, etc., are responsible for their relatively high stability both structurally and electronically. These boron sulfides show high activity toward the capture of small molecules NH3 and CO. The computational findings suggest that these (B2S3)n clusters are promising for the development of boron-based functional materials.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22373078 and 21933009).References
- R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 Search PubMed.
- V. Avdeeva, E. Malinina and N. Kuznetsov, Coordin. Chem. Rev., 2022, 469, 214636 CAS.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, Chem. Rev., 2024, 124, 2441–2511 CAS.
- S. Pan, J. Barroso, S. Jalife, T. Heine, K. R. Asmis and G. Merino, Acc. Chem. Res., 2019, 52, 2732–2744 CrossRef CAS PubMed.
- N. G. Szwacki, A. Sadrzadeh and B. I. Yakobson, Phys. Rev. Lett., 2007, 98, 166804 CrossRef.
- A. Ray, I. Howard and K. Kanal, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 14247–14255 CrossRef CAS PubMed.
- A. P. Sergeeva, Z. A. Piazza, C. Romanescu, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 18065–18073 CrossRef CAS PubMed.
- S. Li, Z. Zhang, Z. Long, G. Sun and S. Qin, Sci. Rep., 2016, 6, 25020 CrossRef CAS PubMed.
- A. P. Sergeeva, D. Y. Zubarev, H. J. Zhai, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 7244–7246 CrossRef CAS PubMed.
- J. I. Aihara, H. Kanno and T. Ishida, J. Am. Chem. Soc., 2005, 127, 13324–13330 CrossRef CAS PubMed.
- A. P. Sergeeva, I. A. Popov, Z. A. Piazza, W. L. Li, C. Romanescu, L. S. Wang and A. I. Boldyrev, Acc. Chem. Res., 2014, 47, 1349–1358 CrossRef CAS PubMed.
- I. Boustani, Chem. Phys. Lett., 1995, 240, 135–140 CrossRef CAS.
- I. Boustani, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 16426–16438 CrossRef CAS.
- T. Jian, X. Chen, S. D. Li, A. I. Boldyrev, J. Li and L. S. Wang, Chem. Soc. Rev., 2019, 48, 3550–3591 RSC.
- H. Kato, K. Yamashita and K. Morokuma, Chem. Phys. Lett., 1992, 190, 361–366 CAS.
- W. L. Li, Y. F. Zhao, H. S. Hu, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2014, 53, 5540–5545 CrossRef CAS PubMed.
- W. L. Li, X. Chen, T. Jian, T. T. Chen, J. Li and L. S. Wang, Nat. Rev. Chem., 2017, 1, 0071 CAS.
- T. Jian, W. L. Li, X. Chen, T. T. Chen, G. V. Lopez, J. Li and L. S. Wang, Chem. Sci., 2016, 7, 7020–7027 CAS.
- J. Wang, T. Yu, Y. Gao and Z. Wang, Sci. China. Mater., 2017, 60, 1264–1268 CAS.
- L. S. Wang, Int. Rev. Phys. Chem., 2016, 35, 69–142 Search PubMed.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu and Y. B. Wu, Nat. Chem., 2014, 6, 727–731 CAS.
- Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu and H. J. Zhai, ACS Nano., 2015, 9, 754–760 CAS.
- Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai and S. D. Li, J. Chem. Phys, 2016, 144, 064307 Search PubMed.
- H. J. Zhai, S. D. Li and L. S. Wang, J. Am. Chem. Soc., 2007, 129, 9254–9255 CrossRef CAS.
- B. I. Loukhovitski, A. V. Pelevkin and A. S. Sharipov, Phys. Chem. Chem. Phys., 2022, 24, 13130–13148 RSC.
- S. D. Li, H. J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 2573–2579 CrossRef CAS PubMed.
- D. Z. Li, L. Y. Feng, L. J. Zhang, L. Pei, W. J. Tian, P. F. Li and H. J. Zhai, J. Phys. Chem. A, 2018, 122, 2297–2306 CAS.
- T. B. Tai, M. T. Nguyen and D. A. Dixon, J. Phys. Chem. A, 2010, 114, 2893–2912 Search PubMed.
- S. J. Gao, P. F. Han, J. C. Guo and H. J. Zhai, J. Mol. Struct., 2024, 1295, 136769 CAS.
- T. B. Tai and M. T. Nguyen, Chem. Phys. Lett., 2009, 483, 35–42 Search PubMed.
- W. J. Tian, X. R. You, D. Z. Li, T. Ou, Q. Chen, H. J. Zhai and S. D. Li, J. Chem. Phys, 2015, 143, 064303 Search PubMed.
- W. J. Tian, Z. Y. Guo and M. Yan, J. Chem. Phys, 2024, 161, 084301 Search PubMed.
- S. J. Gao, J. C. Guo and H. J. Zhai, Front. Chem., 2022, 10, 868782 CAS.
- M. L. Drummond, V. Meunier and B. G. Sumpter, J. Phys. Chem. A, 2007, 111, 6539–6551 CAS.
- M. T. Nguyen, M. H. Matus, V. T. Ngan, D. J. Grant and D. A. Dixon, J. Phys. Chem. A, 2009, 113, 4895–4909 CAS.
- H. J. Zhai, Q. Chen, H. Bai, S. D. Li and L. S. Wang, Acc. Chem. Res., 2014, 47, 2435–2445 CAS.
- B. M. Gimarc and N. Trinajstic, Inorg. Chem., 1982, 21, 21–25 CrossRef CAS.
- W. Z. Yao, J. C. Guo, H. G. Lu and S. D. Li, Int. J. Quantum Chem., 2010, 110, 2689–2696 CrossRef CAS.
- X. Y. Tang, Z. H. Cui, C. B. Shao and Y. H. Ding, Int. J. Quantum Chem., 2012, 112, 1299–1306 CrossRef CAS.
- D. Z. Li, R. Li, L. J. Zhang, T. Ou and H. J. Zhai, Phys. Chem. Chem. Phys., 2016, 18, 21412–21420 RSC.
- D. Z. Li, S. G. Zhang and C. C. Dong, Chem. – Eur. J., 2016, 2016, 1103–1107 CAS.
- L. J. Zhang, L. Y. Feng, H. Bian, L. Pei, D. Z. Li and H. J. Zhai, Int. J. Quantum Chem., 2020, 120, e26229 CrossRef CAS.
- S. P. Kuntar, A. Ghosh and T. K. Ghanty, J. Phys. Chem. A, 2022, 126, 7888–7900 CrossRef CAS PubMed.
- A. Sommer, P. N. Walsh and D. White, J. Chem. Phys, 1960, 33, 296 CrossRef CAS.
- F. T. Greene and P. W. Gilles, J. Am. Chem. Soc., 1962, 84, 3598–3599 CrossRef CAS.
- H. Y. Chen and P. W. Gilles, J. Am. Chem. Soc., 1970, 92, 2309–2312 CrossRef CAS.
- F. T. Greene and P. W. Gilles, J. Am. Chem. Soc., 1964, 86, 3964–3969 CAS.
- H. Y. Chen and P. W. Gilles, J. Phys. Chem., 1972, 76, 2035–2038 CAS.
- D. Z. Li, H. Bai, T. Ou, Q. Chen, H. J. Zhai and S. D. Li, J. Chem. Phys, 2015, 142, 014302 Search PubMed.
- L. J. Zhao, X. L. Xu, H. G. Xu, G. Feng and W. J. Zheng, New J. Chem., 2018, 42, 16021–16026 CAS.
- S. X. Li, Y. J. Yang and D. L. Chen, ACS Omega, 2023, 8, 30757–30767 CAS.
- W. D. Lu, D. Wang, Z. Zhao, W. Song, W. C. Li and A. H. Lu, ACS Catal., 2019, 9, 8263–8270 CAS.
- R. W. Dorn, L. O. Mark, I. Hung, M. C. Cendejas, Y. Xu, P. L. Gor’kov, W. Mao, F. Ibrahim, Z. Gan and I. Hermans, J. Am. Chem. Soc., 2022, 144, 18766–18771 CAS.
- P. Han, R. Yan, Y. Wei, L. Li, J. Luo, Y. Pan, B. Wang, J. Lin, S. Wan and H. Xiong, J. Am. Chem. Soc., 2023, 145, 10564–10575 CAS.
- J. Tian, G. Collinge, S. F. Yuk, J. Lin, V. A. Glezakou, M. S. Lee, Y. Wang and R. Rousseau, ACS Catal., 2023, 13, 8219–8236 CAS.
- J. Tian, J. Tan, Z. Zhang, P. Han, M. Yin, S. Wan, J. Lin, S. Wang and Y. Wang, Nat. Commun., 2020, 11, 5693 CAS.
- H. Yan, S. Alayoglu, W. Wu, Y. Zhang, E. Weitz, P. C. Stair and J. M. Notestein, ACS Catal., 2021, 11, 9370–9376 CAS.
- L. Li and L. Cheng, J. Chem. Phys, 2013, 138, 094312 CrossRef PubMed.
- J. Guan, X. Zhang, Q. Li, K. Deng, P. Jena and E. Kan, Nanoscale, 2021, 13, 3627–3632 RSC.
- X. Li, B. Cui, W. Zhao, Y. Xu, D. Zou and C. Yang, Nanotechnology, 2021, 32, 225401 CrossRef CAS.
- L. Li, X. Chen, Y. Huang, P. Zhang, D. Zhou, G. Zhang and B. Xiao, Appl. Surf. Sci., 2022, 602, 154295 CrossRef CAS.
- J. Zhang and M. Dolg, Phys. Chem. Chem. Phys., 2015, 17, 24173–24181 RSC.
- J. Zhang and M. Dolg, Phys. Chem. Chem. Phys., 2016, 18, 3003–3010 RSC.
- J. P. Doye and D. J. Wales, Phys. Rev. Lett., 1998, 80, 1357 CrossRef CAS.
- S. Kirkpatrick, C. D. Gelatt Jr and M. P. Vecchi, Science, 1983, 220, 671–680 CrossRef CAS.
- J. J. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys, 2010, 132, 154104 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta., 1973, 28, 213–222 Search PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys, 1972, 56, 2257–2261 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision A.03, Wallingford CT, 2016, vol. 2016 Search PubMed.
- B. Chan, J. Deng and L. Radom, J. Chem. Theory Comput., 2011, 7, 112–120 CrossRef CAS PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 Search PubMed.
- C. Legault, CYLview20, 2020, https://www.cylview.org Search PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 Search PubMed.
- E. Francisco, A. Martín Pendás, M. García-Revilla and R. Álvarez Boto, Comput. Theor. Chem, 2013, 1003, 71–78 Search PubMed.
- M. Menéndez, R. Álvarez Boto, E. Francisco and A. Martín Pendás, J. Comput. Chem., 2015, 36, 833–843 CrossRef PubMed.
- E. V. Bukovsky, A. M. Pluntze and S. H. Strauss, J. Fluorine. Chem., 2017, 203, 90–98 CrossRef CAS.
- J. C. Axtell, L. M. Saleh, E. A. Qian, A. I. Wixtrom and A. M. Spokoyny, Inorg. Chem., 2018, 57, 2333–2350 CrossRef CAS PubMed.
- K. R. Asmis, B. B. Beele, C. Jenne, S. Kawa, H. Knorke, M. C. Nierstenhöfer, X. B. Wang, J. Warneke, Z. Warneke and Q. Yuan, Chem. – Eur. J., 2020, 26, 14594–14601 CrossRef CAS PubMed.
- M. E. Kilic and P. J. Jena, Phys. Chem. Lett., 2023, 14, 8697–8701 CAS.
- M. E. Kilic and P. J. Jena, Phys. Chem. A, 2024, 128, 1993–2002 CAS.
- B. Miao, Z. Qiu, Z. Zhen, Y. Yang, Z. Yang, T. Xiao, J. Lv, S. Huang, Y. Wang and X. Ma, Phys. Chem. Chem. Phys., 2024, 26, 10494–10505 CAS.
- M. M. Shmakov, S. A. Prikhod’ko, V. N. Panchenko, M. N. Timofeeva, V. V. Bardin, V. N. Parmon and N. Y. Adonin, Catal. Rev., 2024, 1–47 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Functional testing; low-energy structures of charged clusters; AdNDP analysis; optimized structures of studied molecular compounds; key interaction analysis between [B2S3] with NH3 and CO molecules; absolute free energies and electronic energies; and Cartesian coordinates. See DOI: https://doi.org/10.1039/d4cp04699d |
| This journal is © the Owner Societies 2025 |