DOI:
10.1039/D4CP04656K
(Communication)
Phys. Chem. Chem. Phys., 2025,
27, 1772-1777
Theoretical and machine learning models for reaction-barrier predictions: acrylate and methacrylate radical reactions†
Received
10th December 2024
, Accepted 26th December 2024
First published on 27th December 2024
Abstract
We propose density functional theory (DFT)- and random forest (RF)-based theoretical and machine learning (ML) models, respectively, for predicting reaction barriers (ΔETS) using acrylate and methacrylate radical reactions as representatives. DFT is used to determine 100 transition state (TS) structures of both radicals, after which the obtained data are used to determine theoretical relationships (explained with Bell–Evans–Polanyi or Brønsted–Evans–Polanyi (BEP) and Marcus-like models) between ΔETS and stabilization energy of the product. Next, we construct several theoretical regression models for predicting ΔETS of the representative reactions based on our theoretical analyses, presenting an RF-based ML model that eases ΔETS predictions by circumventing time-consuming DFT calculations. These theoretical and RF-based ML approaches will accelerate the advancement of material development.
1. Introduction
Radical reactions are employed for diverse applications across various fields, including chemistry, medicine, and materials science, owing to the high reactivities and versatilities of radicals.1–3 Typically, these highly selective reactions proceed under relatively mild conditions. Furthermore, the high reactivities of radicals under mild conditions facilitate the extensive adoption of radical polymerization in the syntheses of various acrylic polymers with numerous applications in products, such as paints, adhesives, medical materials, plastics, and fibers.4–7 Here, we discussed the radical reactions of acrylate (ACR) and/or methacrylate (MA), which are essential to the synthesis of acrylic polymers. Generally, the efficient development of acrylic polymers requires sophisticated and precise controls of the radical reactions, and the detailed mechanisms of these reactions must be understood. Furthermore, effective density functional theory (DFT)-based transition state (TS) searches are required for the investigation of radical reaction mechanisms. The DFT-based TS analyses of simple MA monomers8 and catalyzed radical polymerization products9 have been reported. However, the high computational cost of TS search complicates the calculations of many structures for material developments, as TS-structure determination typically requires numerous trials and errors together with highly expensive DFT calculations. To avoid heavy calculations, simple theoretical models have been employed for reaction analysis. For example, the Bell–Evans–Polanyi or Brønsted–Evans–Polanyi (BEP) model10–12 is widely utilized in catalysis reactions, which is based on an a empirical linear relationship between the activation energy and the reaction enthalpy to predict the catalytic activity. Conversely, the utilization of databases and machine learning (ML) for efficient material development has garnered substantial interest in recent years.13–24 For example, in the experimental syntheses of polymers with desired thermal conductivities, ML was deployed for the screening of several promising materials among numerous candidates,25 representing a promising approach for accelerating and advancing material development.
Therefore, in this study, we considered the radical reactions between ACR and MA. First, we performed DFT calculations to determine the TSs of these reactions, after which we discussed the general trends of the obtained computational results. Next, we confirmed that theoretical regression models for performing easy reaction-barrier predictions can be constructed based on the results, after which we constructed a random forest (RF)-based ML model for predicting their reaction barriers (ΔETS) using simple descriptors based on their chemical structures and circumventing complex DFT calculations.
2. Computational methods
The structures of the reactants, products, and TSs were optimized using the B3LYP functional26,27 with Grimme's empirical dispersion28 and the 6-31+G* basis set (B3LYP+D3/6-31+G*). All the DFT calculations were performed using the Gaussian16 package.29 The ML algorithm (RF)30 with scikit-learn liberally (version 0.22.1)31 was used to construct the ML model for predicting ΔETS. Here, the number of trees in the forest (n_estimators) was set to 30, and tenfold cross-validation was used to evaluate the models.
3. Results and discussion
3.1. Trends of reaction barriers and energy of the products obtained via density functional theory calculations
In this study, we performed DFT calculations to determine ΔETS and the product–reactant energy difference (ΔEprod) for the radical reactions between radical monomers X˙ and Y (Fig. 1(a) and (b)), where X and Y represent ACR and/or MA monomers, respectively, including an acrylic acid/methacrylic acid (Fig. 1(c)). Notably, X˙ was generated by adding a hydrogen radical to the monomer X. Additionally, the calculated ΔETS and ΔEprod values of all the combinations of the 10 monomers, i.e., 100 reactions, are listed in Table S1 ESI.† Here, the energy references were set to the pre-reactant complex of each system. For example, the lowest ΔETS (3.9 kcal mol−1) was obtained from the reaction between an ethylcyclohexyl ACR radical and methacrylic acid. In comparison, the highest ΔETS (10.1 kcal mol−1) was obtained from the reaction between a γ-butyrolactone MA radical and γ-butyrolactone ACR. These radical reactions can be categorized into four types based on whether the chemical structures of reactants X˙ and Y correspond to ACR or MA, respectively.
 |
| | Fig. 1 (a) Energy diagram and (b) scheme of the radical reaction between ACR and/or MA: X˙ + Y → XY˙, where X˙ represents the radical monomer. (c) Target reactant monomers of acrylic acid, ACR, methacrylic acid, and MA. Here, acrylic and methacrylic acids were categorized as ACR and MA, respectively. | |
Table 1 presents the average ΔETS and ΔEprod for each category. In this study, we categorized acryl and methacrylic acids as ACR and MA, respectively. As presented in Table 1, ΔETS and ΔEprod tended to be lower when the reactant radical (X) was ACR rather than MA. For example, in the case of Y = ACR, average ΔETS values of 5.4 and 7.2 kcal mol−1 were obtained for X = ACR and MA, respectively. Conversely, average ΔEprod values of −16.6 and −11.6 kcal mol−1 were obtained for X = ACR and MA, respectively. Additionally, we observed a decreasing trend in ΔEprod of the Y species. These trends were roughly consistent with those reported in the literature1,32 and could be explained by the stabilities of the radical species: as ACR˙ and MA˙ are secondary and tertiary radicals, respectively, the latter would be more stable than the former owing to hyperconjugation.
Table 1 Average ΔETS and ΔEprod (kcal mol−1) for each category. Their standard deviations are listed in parentheses
| Category (X˙ + Y) |
Average ΔETS |
Average ΔEprod |
| ACR˙ + ACR |
5.4 (0.6) |
−16.6 (1.2) |
| ACR˙ + MA |
4.5 (0.3) |
−18.4 (1.6) |
| MA˙ + ACR |
7.2 (1.3) |
−11.6 (1.5) |
| MA˙ + MA |
7.3 (1.0) |
−12.6 (1.3) |
To gain an insight into the trend of the computational results, we plotted the datasets with ΔETS and ΔEprod on the vertical and horizontal axes (Fig. 2). In the figure, the colors and markers correspond to the reaction categories. Namely, the blue and red colors represent ACR˙ and MA˙ for the reactant radical (X˙), respectively. Conversely, the circle (●) and cross (×) markers represent ACR and MA for the monomer (Y), respectively. Some trends can be easily confirmed from Fig. 2 (see also Table 1). For example, the computational results were largely clustered into two regions, i.e., X = ACR and MA, which were represented by blue and red colors, respectively; ΔETS and ΔEprod were influenced by the stability of the reactant radical (X˙). Moreover, the stability of the reactant in the product (Y) caused a slight difference between ● and × within the blue and red regions. In these reactions, ACR˙, a secondary radical, was more unstable than MA˙, a tertiary radical. Thus, in the reactant, X˙ = ACR˙ was relatively more reactive, i.e., its ΔETS value became lower than that of X˙ = MA˙.
 |
| | Fig. 2 Scatter plot of the ΔETS with respect to the DFT-obtained ΔEprod. The blue and red colors represent ACR˙ and MA˙ for the reactant radical (X˙), respectively. Conversely, ● and × represent ACR and MA for the reactant monomer (Y), respectively. | |
Furthermore, ΔEprod, which is the relative energy between the product and reactant, tended to become unstable when X˙ = ACR˙; thus a larger ΔEprod would be obtained compared with the case of X˙ = MA˙, following the polymerization reaction. Similarly, the decreasing tendency of Y would also be due to the stability of the product (XY˙). After the polymerization reaction, the radical moves from X to Y, and hence, the secondary radical (Y = ACR) yields more unstable products than Y = MA. However, radical reactions are affected by the stability of the radical species and other factors, such as steric hindrance and electronic effects, especially with the increasing polymer-chain length. For example, we could not ignore the steric hindrance effects from the methyl groups on the radical reactions of MA˙ + MA. In practical cases, other factors, such as the solvents and electronic effects from the side chains, must be considered. Although the precise assessments of all the aforementioned factors were challenging, our assessments of ΔETS and ΔEprod offered a valuable overview of radical reactions.
3.2. Relationships between the reaction-barrier and product–reactant energy difference
In the BEP model, the potential energy surfaces of the reactant and product were assumed to be two intersecting linear functions, and the ΔETS (ΔyTS) was obtained using coefficients of a and b as follows, We confirmed that ΔETS (ΔyTS) represents a linear relationship with respect to the relative energy of the product (Δyprod). In computational results in Fig. 2, we observed a BEP linear relationship between ΔETS and ΔEprod (see also Fig. S3(a) in the ESI†).
Conversely, we noticed that the relationship between ΔETS and ΔEprod seems to be described as a downwardly convex quadratic curve from the computational results in Fig. 2 (see also Fig. S3(b) in the ESI†). The chemical meaning behind this relationship may be explained using the Marcus-like model.33,34 In the model, the potential energy surfaces of the reactant and product were assumed to be parabolic functions with the same coefficient (c), described as yreac = cx2 and yprod = c (x − xprod)2 + Δyprod, respectively (Fig. 3). Here, x and y represent the reaction coordinate and potential energy, respectively. The y surface of the product deviates by Δyprod from the most stable energy of the reactant at the reaction coordinate (xprod). From these assumptions, the ΔETS (ΔyTS) was obtained from the intersection of these parabolas, as follows,
| |  | (2) |
Here,
λ ≡
cx2prod. Furthermore, Δ
yTS represents a quadratic function with respect to the relative energy of the product (Δ
yprod). The Marcus-like model could explain the quadratic relationship between Δ
ETS and Δ
Eprod (
Fig. 2). Thus, the relationship between Δ
ETS (Δ
yTS) and Δ
Eprod (Δ
yprod) is obtained by assuming the simple analytic function for the potential energy surface of the reactant and product in the Marcus-like model. This theoretical treatment is similar to that in the BEP model. However, linear and quadratic functions are assumed to describe the potential energy surfaces in the BEP and Marcus-like models, respectively.
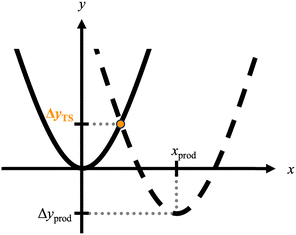 |
| | Fig. 3 Model potential energy surfaces of the reactant and product represented by solid and dashed lines, respectively. These energy surfaces are approximately described as parabolic functions. The reaction barrier in this case is described as  , where λ ≡ cx2prod. Thus, ΔyTS exhibits a quadratic relationship with Δyprod. , where λ ≡ cx2prod. Thus, ΔyTS exhibits a quadratic relationship with Δyprod. | |
3.3. Prediction of reaction-barriers: theoretical models based on the product–reactant energy difference
The BEP and Marcus-like models may be useful for constructing a simple regression model to significantly ease the predictions of ΔETS for those radical reactions. Here, we proposed the following two theoretical regression models using ΔEprod (and its squared value) based on BEP and Marcus-like models:| | | ΔETS = 0.42ΔEprod + 12.3 | (3) |
| | | ΔETS = 0.0341ΔE2prod + 1.44ΔEprod + 19.5 | (4) |
Furthermore, we compared ΔETS predicted by these theoretical models and DFT-based calculations (Fig. 4(a) and (b)). We obtained determination coefficients (R2) of 0.83 and 0.88 from the BEP and Marcus-like models, respectively. To evaluate the predictive performance of these models, we also calculated the Akaike's Information Criterion (AIC).35 We obtained AICs of 648.9 and 618.0 for the BEP and Marcus-like regression models. In the AIC analysis, a model with a lower criterion value is considered to be superior. Thus, the regression model of eqn (4) based on the Marcus-like theory may show slightly better predictive performance for the reaction barrier. In fact, the BEP-based regression model tends to slightly underestimate ΔETS in the high value region, as seen in Fig. 4(a).
 |
| | Fig. 4 Comparisons of ΔETS values predicted via DFT-based calculations and the theoretical models (a) BEP model in eqn (3) and (b) Marcus-like model in eqn (4). (c) Comparisons of ΔETSs predicted via DFT-based calculations and the RF model using four descriptors; the dummy parameter (DP(m)) and molecular weight of each reactant. Here, the blue and red colors represent ACR˙ and MA˙ for the reactant radical (X˙), respectively. Conversely, ● and × represent ACR and MA for the reactant monomer (Y), respectively. (d) Feature-importance values in the RF-based model. | |
3.4. Prediction of reaction-barriers: a random forest-based machine learning model based on simple descriptors from the reactant monomers
The theoretical models established in eqn (3) and (4) can ease the estimation of ΔETS; however, it still requires DFT calculations to determine ΔEprod. Therefore, we constructed an ML model for predicting ΔETS while circumventing DFT calculations. To employ the ML approach, we first established some reaction-related descriptors (feature vectors). To do this, we examined several physicochemical properties of the reactant monomers, which were estimated from several group contribution methods implemented in OpenBabel (version 3.3.1)36 and RDkit (vers. 2021.03.5)37 libraries. Here, for simplicity, we used the properties of the “X monomer,” not X˙ itself. Additionally, we considered a dummy parameter (DP(m)) to represent ACR or MA, using m to specify X or Y. For example, DP(X) = 0 represented X = ACR, and DP(Y) = 1 was used to describe Y = MA. We employed the tenfold cross-validation technique to evaluate the performance of ML models. In the cross-validation technique, data are divided into several groups in the first step. Then, a group is used for model evaluation and the others for model training. This operation is carried out while replacing the groups for the model evaluation. As a first trial, we obtained a model with R2 = 0.84 to predict ΔETS using RF with 50 descriptors, using the cross-validation technique. For each reactant species, 24 descriptors from the group contribution method and one DP(m) were used (S3 in the ESI†). Moreover, after multiple trials using the RF-based feature-importance guideline, we finally obtained a simpler model (Fig. 4(c)) with R2 = 0.80 using 4 descriptors, where only the molecular weight and DP(m) for each reactant species were utilized. As shown in Fig. 4(c), we compared the RF- and DFT-based ΔETS predictions.
Here, we examined another approach to predict ΔETS. We first predict ΔEprod using a RF-based ML model. Then, ΔETS is estimated using the BEP and Marcus-like regression models (eqn (3) and (4)), respectively. From this procedure, we can predict ΔETS values with R2 of 0.75 and 0.76 for the BEP and Marcus-like models, respectively (see also S6 in the ESI†). Thus, we confirmed the validity of the BEP and Marcus-like models. Conversely, data related to the ΔEprod property are easier to collect compared with data related to ΔETS. Therefore, the collaborative approach between theoretical and ML models may be useful to construct a more convenient means for estimating the reaction energy.
We demonstrated the feature-importance values of the descriptors obtained using RF (Fig. 4(d)). Even without the DFT calculations, the results revealed that the ML model achieved an accuracy that was comparable with that of the DFT-based theoretical model (eqn (2)). Among the feature-importance values (Fig. 4(d)), DP(X) was the most significant. It might be related to the reactant stability, as it determined whether the reactant radical was a secondary or tertiary one. Similarly, DP(Y) was related to the product stability but less significant. This trend correlates with the results in Table 1 and Fig. 2. In this study, we employed the same basic chemical framework for the reactant molecules, and only side chains are different, as shown in Fig. 1(b). When the molecular weight descriptor together with DP(m) to distinguish ACR or MA is given, we can obtain some information about side chains related to the bulkiness (steric effect) of the monomer. For example, acrylic acid and ethylcyclohexyl methacrylate molecules have molecular weights of 72.1 and 196.3, respectively. Here, ethylcyclohexyl methacrylate has a bulkier side chain. ML models may use such information to predict the reaction barrier. Thus, DP(m) and molecular weight descriptors may comprise some chemical features, such as the stability and bulkiness of the radical.
4. Conclusion
In this study, we analyzed 100 radical reactions determined by combining ten types of ACRs and/or MAs whose TSs were assessed using DFT. To analyze calculation data, we employed the BEP model. In addition, the computational results revealed the quadratic relationship between ΔETS and ΔEprod, and this relationship was explained with a Marcus-like model. Based on these theoretical analyses, we constructed theoretical regression models; BEP and Marcus-like models yield R2 = 0.83 and 0.88 to predict ΔETS. However, these models still require time-consuming DFT-based calculations to obtain ΔEprod. Therefore, we constructed an ML model with R2 = 0.80 without DFT calculations using simple monomer descriptors, namely the dummy parameter (DP(m)) and molecular weight. We believe that our theoretical and ML approaches for radical reaction predictions will benefit future material developments.
Author contributions
Makito Takagi: data curation, investigation, methodology, software, visualization, writing – original draft, and writing – review & editing. Tomomi Shimazaki: methodology, project administration, resources, software, supervision, and writing – review & editing. Osamu Kobayashi: methodology and writing – review & editing. Takayoshi Ishimoto: project administration, supervision, and writing – review & editing. Masanori Tachikawa: funding acquisition, project administration, resources, supervision, and writing – review & editing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study is partially supported by a Grant-in-Aid for Scientific Research (KAKENHI) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Numbers 21H00026 and 22K05038. This work was also partially supported by MEXT through the “Program for Promoting Researches on the Supercomputer Fugaku” (JPMXP1020230318). This study utilized the computational resources of ITO provided by the Research Institute for Information Technology, Kyushu University, through the HPCI System Research Project (Project ID: hp220061) and Supercomputer Center, the Institute for Solid State Physics, the University of Tokyo.
References
- T. Otsu, J. Synth. Org. Chem. Jpn., 1970, 28, 1183–1196 CrossRef.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef.
- T. Pirman, M. Ocepek and B. Likozar, Ind. Eng. Chem. Res., 2021, 60, 9347–9367 CrossRef.
- U. Ali, K. J. B. Abd Karim and N. A. Buang, Polym. Rev., 2015, 55, 678–705 CrossRef.
- S. C. Ligon-Auer, M. Schwentenwein, C. Gorsche, J. Stampfl and R. Liska, Polym. Chem., 2016, 7, 257–286 RSC.
- N. Ballard and J. M. Asua, Prog. Polym. Sci., 2018, 79, 40–60 CrossRef.
- M. S. Zafar, Polymers, 2020, 12, 2299 CrossRef PubMed.
- I. Degirmenci, V. Aviyente, V. Van Speybroeck and M. Waroquier, Macromolecules, 2009, 42, 3033–3041 CrossRef.
- A. Debuigne, C. Michaux, C. Jérôme, R. Jérôme, R. Poli and C. Detrembleur, Chem. – Eur. J., 2008, 14, 7623–7637 CrossRef.
- R. P. Bell, Proc. R. Soc. London, Ser. A, 1936, 154, 414–429 Search PubMed.
- M. Evans and M. Polanyi, Trans. Faraday Society, 1936, 32, 1333–1360 RSC.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- W. A. Warr, Mol. Inform., 2014, 33, 469–476 CrossRef CAS.
- K. Jorner, A. Tomberg, C. Bauer, C. Sköld and P. O. Norrby, Nat. Rev. Chem., 2021, 5, 240–255 CrossRef CAS PubMed.
- J. A. Keith, V. Vassilev-Galindo, B. Q. Cheng, S. Chmiela, M. Gastegger, K. R. Mueller and A. Tkatchenko, Chem. Rev., 2021, 121, 9816–9872 CrossRef CAS PubMed.
- R. X. Wang, X. L. Fang, Y. P. Lu and S. M. Wang, J. Med. Chem., 2004, 47, 2977–2980 CrossRef CAS.
- J. J. Irwin, T. Sterling, M. M. Mysinger, E. S. Bolstad and R. G. Coleman, J. Chem. Inf. Model., 2012, 52, 1757–1768 CrossRef CAS PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Y. Han, J. E. He, S. Q. He, B. A. Shoemaker, J. Y. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2016, 44, D1202–D1213 CrossRef.
- M. Nakata and T. Shimazaki, J. Chem. Inf. Model., 2017, 57, 1300–1308 CrossRef.
- R. Jose and S. Ramakrishna, Appl. Mater. Today, 2018, 10, 127–132 CrossRef.
- C. Draxl and M. Scheffler, J. Phys.: Mater., 2019, 2, 036001 Search PubMed.
- M. Nakata, T. Shimazaki, M. Hashimoto and T. Maeda, J. Chem. Inf. Model., 2020, 60, 5891–5899 CrossRef PubMed.
- T. R. Lane, D. H. Foil, E. Minerali, F. Urbina, K. M. Zorn and S. Ekins, Mol. Pharm., 2021, 18, 403–415 CrossRef.
- S. Wu, Y. Kondo, M. A. Kakimoto, B. Yang, H. Yamada, I. Kuwajima, G. Lambard, K. Hongo, Y. B. Xu, J. Shiomi, C. Schick, J. Morikawa and R. Yoshida, NPJ Comput. Mater., 2019, 5, 66 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- K. S. Anseth, C. M. Wang and C. N. Bowman, Polymer, 1994, 35, 3243–3250 CrossRef.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef.
- D. G. Brooks, Technometrics, 1989, 31(2), 270–271 CrossRef.
- N. M. O’Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminform., 2011, 3, 33 CrossRef.
- RDKit: Open-Source Cheminformatics Software. (https://www.rdkit.org.).
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Tomomi
Shimazaki
*a,
Tomomi
Shimazaki




 , where λ ≡ cx2prod. Thus, ΔyTS exhibits a quadratic relationship with Δyprod.
, where λ ≡ cx2prod. Thus, ΔyTS exhibits a quadratic relationship with Δyprod.