Ultrafast excited-state dynamics of 4-hydroxychalcone: role of intramolecular charge transfer and photoacidity†
Received
28th November 2024
, Accepted 16th March 2025
First published on 18th March 2025
Abstract
Diarylketones such as benzophenones, oxybenzones, chalcones and their derivatives exhibit promising applications as UV filters/sunscreen agents due to their effective absorption in the UV region and dissipation through non-radiative pathways. However, elucidation of the underlying photoreactive mechanism is non-trivial due to the ultrafast lifetimes of transient species, involvement of non-adiabatic curve crossings among the potential surfaces, etc. In this context, we investigate the excited-state photoreaction dynamics of 4-hydroxychalcone (4-HC) under various environments through femtosecond-transient absorption (fs-TA), nanosecond-transient absorption (ns-TA), and femtosecond-fluorescence upconversion (fs-FL) measurements. Steady-state fluorescence measurements of 4-HC in the presence of 1-methylimidazole (MI)/tert-butylamine (TBA) exhibit dual band emission. The fs-TA measurements of 4-HC in the presence of MI/TBA exhibit distinct spectral and associated lifetimes as compared to 4-HC alone indicating a significant interaction of the hydroxyl proton with bases and influencing the reaction dynamics. The 4-HC:MI/TBA adduct undergoes excited-state intermolecular proton transfer within a time scale of ∼500 fs and subsequently relaxes back to the ground state through a long-lived triplet state. The experimental observations of excited-state reaction dynamics of 4-HC in the presence of MI/TBA bases have been well corroborated with the computational analysis.
Introduction
Adequate sunlight exposure is vital for the synthesis of vitamin D and has been linked to potential mood-boosting effects.1 However, extreme or unshielded exposure to ultraviolet radiation can have detrimental consequences such as oxidative stress and sun tanning promoting the production of reactive oxygen species, which can lead to adverse skin effects including inflammation, photoaging, and skin cancer.2–4 Ultraviolet (UV) radiation is categorized into three spectral regions: UVA (320–400 nm), UVB (280–320 nm), and UVC (100–280 nm).2,5 While UVA contributes to triggering melanin overproduction, photoaging, and immune suppression, UVB is more effective in causing sunburns.5–8 UVC, though highly intense, is largely absorbed by the ozone layer. Photoprotective agents, specifically sunscreens, are vital in shielding the skin from UV damage and are broadly categorized as inorganic or organic sunscreens. Inorganic filters like ZnO and TiO2 scatter, reflect, and absorb UV rays but are susceptible to photocorrosion.9 Organic sunscreens are composed of aromatic compounds, including benzophenones,10–13 ethylhexylmethoxycinnamate,14,15 and para-aminobenzoic acid derivatives.16–18 They absorb UV light and dissipate energy via diverse non-radiative relaxation pathways, including cis–trans photoisomerization,19–25 conformational relaxation,26 torsions, keto–enol tautomerization, and excited-state proton transfer.27–30 Elucidating these relaxation pathways is crucial for understanding sunscreen efficacy and broadening UV protection.
Chalcones are flavonoid chemicals found in nature. Chalcones are chemicals with several health benefits; they possess cytotoxic, anti-inflammatory, antibacterial, antioxidant, antipyretic, anticancer, antimicrobial, antimalarial, and anti-inflammatory properties.31–42 According to reports, chalcone derivatives absorb light in the UV region, which consists of two key absorption bands. Such electronic transitions are very crucial for the sunscreen agent to avoid direct exposure to the skin. 4-Hydroxychalcone (4-HC) is one such chalcone compound, a flavonoid derivative possessing two aryl rings connected to α and β unsaturated ketone groups in their structure.43–48 2′-Hydroxychalcones have been studied extensively due to their ability to undergo excited-state intramolecular proton transfer (ESIPT); however, a significant gap exists in understanding the excited-state dynamics of 4-hydroxychalcone or its derivatives, particularly with limited utilization of femtosecond time-resolved measurements.49–57 Abdul Karim and co-workers published research exploring the application of 4-hydroxychalcone gels as potential sunscreens and photoprotective agents.44–46 Recently, Gabriela Fehn Fiss and co-workers have also reported the strategic synthesis of ten novel 4-hydroxychalcone derivatives.53 These compounds exhibited molar absorptivity (εmax) greater than 104 mol−1 L cm−1 in the UV region, indicating their potential effectiveness as UV filters. Notably, several of these derivatives demonstrated significant sun protection factors (SPF-UVB) ranging from 18.04 to 21.06. This performance surpasses that of para-aminobenzoic acid (PABA), a commonly used commercial sunscreen agent, which has an SPF-UVB of 14.34. Wai-Ming Kwok and co-workers have employed ultrafast spectroscopic techniques like fs-transient absorption spectroscopy to explore the excited-state dynamics of 4-aminobenzoic acid (PABA), 4-dimethylaminoacetophenone (DMAAP), and 4-dimethylaminobenzaldehyde (DMABA) under various solvent environments.17 The TAS results revealed that, in aqueous environments, PABA undergoes rapid protonation within ∼0.5 ps after excitation, forming a neutral excited-state species that efficiently transition to a long-lived triplet state via intersystem crossing (ISC). In contrast, in CH3CN, ISC from the LE state to the T1 state remains the dominant non-radiative deactivation pathway, occurring without any deprotonation. DMAAP and DMABA exhibited solvent-dependent deactivation pathways, where hydrogen bonding in water suppressed ISC and eliminated the twisted intramolecular charge transfer (TICT) state. This facilitated an efficient internal conversion (IC) mechanism, allowing ∼80% of the excited-state population to return to the ground state, thereby enhancing photostability safely. This study demonstrates at the molecular level that PABA when used as a sunscreen ingredient may pose risks due to its long-lived triplet state, which has the potential to cause nucleic acid damage.17,58–61
Guangjiu Zhao and co-workers have reported non-adiabatic excited-state dynamics mechanism and photoprotective properties of a novel chalcone analog sunscreen molecule (E)-3-(5-bromofuran-2-yl)-1-(2-hydroxyphenyl)prop-2-en-1-one (FPPO-HBr) by combining the steady-state and femtosecond transient absorption spectroscopy and theoretical calculations.27 The efficacy of sunscreen molecules is often limited by a single energy relaxation pathway as well as a lack of effective UVA light protection. Upon UV excitation, FPPO-HBr mainly decays via ESIPT, followed by an ultrafast conformational twist.27
Despite existing research, the UV excitation dynamics of 4-hydroxychalcone and its derivatives, particularly when investigated using femtosecond transient absorption and femtosecond fluorescence upconversion techniques, remains largely unexplored. This gap represents a significant oversight, as understanding the excited-state behavior of these compounds is essential for elucidating their photoprotective as well as energy deactivation mechanisms. Investigating the excited-state properties, such as fluorescence characteristics, fluorescence lifetime, intersystem crossing, relaxation pathways, and excitation energy levels, could provide valuable insights into the mechanisms of their interaction with light and can enhance their application in sunscreen formulations. The objective of this current work is to investigate the excited-state dynamics of 4-hydroxychalcone, a molecule identified as a potential photoprotective agent. Understanding its excitation and relaxation pathways is crucial for assessing its effectiveness in UV protection. Thus, the current work aims to explore various photo-initiated solvent-dependent intramolecular charge transfer (ICT) dynamics and associated proton-transfer dynamics in molecular adducts of the 4-HC photoacid with external bases such as tert-butylamine (TBA) and 1-methylimidazole (MI). We employed femtosecond transient absorption and femtosecond fluorescence up-conversion spectroscopy to elucidate the intricate ultrafast dynamics and subsequently validated the experimental results through computational analysis.
Experimental methods
Materials
4-Hydroxychalcone (4-HC), 1-methylimidazole (MI), tert-butylamine (TBA), and KOH (potassium hydroxide) were obtained from Sigma-Aldrich and used without further purification. HPLC grade solvents acetonitrile (MeCN), toluene (TOL), ethyl acetate (EtAc) and methanol (MeOH) were purchased from Spectrochem.
Steady-state spectroscopic measurements
Steady-state UV-vis absorption spectra were recorded using a Shimadzu UV-3600 UV-vis-NIR spectrophotometer in a quartz cuvette of 1 cm path length and fluorescence spectra were recorded using a Horiba Jobin Yvon–Fluorolog 3 spectrofluorimeter. The fluorescence quantum yield (Φ) is determined using quinine sulfate with a concentration of 0.05 M H2SO4, which served as the reference molecule having Φ(ref) = 0.546.62 The Φ is subsequently calculated using the following equation:| | 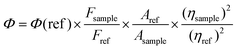 | (1) |
where Φ(ref) is the quantum yield of the reference, and Fsample and Fref are the integrated emission intensities of the sample and the reference, respectively; Asample and Aref correspond to the optical densities of the sample and the reference, respectively; also, ηsample and ηref are the refractive indices of the solvent used for the sample and the reference, respectively.
Femtosecond transient absorption (fs-TA) measurements
The setup consists of a Ti-sapphire based amplifier system from Spectra Physics, which produces fundamental laser light having a central wavelength of 790 nm, a pulse width of 100 fs, a pulse energy of 2.2 mJ and a repetition rate of 1 kHz. The output beam is split into two parts, one beam is used to pump an optical parametric amplifier (OPA) and a small portion of the remaining energy is used to generate a white-light continuum from a CaF2 crystal that is further split into two parts, one of which acts as a probe while the other acts as a reference beam. The OPA is used to generate a pump beam in the ultraviolet region of 320 nm with beam parameters including a pulse-width of 100 fs, a repetition rate of 1 kHz and a pulse energy of 10 μJ and to create an excited-state population. Spatio-temporal overlapping of the pump and the probe beams in a 1.0 mm thick flow-type sample cell is achieved. The probe beams are dispersed in the spectrometer and detected using a dual-array detector. The excited-state absorption signal is obtained by modulating the pump beam with a mechanical chopper at 500 Hz for each time-delay point. The TA measurements were carried out using a pump energy of ∼800 nJ per pulse with a focal spot size of 100 μm. The sample concentration of 0.5 mM of 4-HC was employed for transient absorption.
Nanosecond laser flash photolysis measurements
Nanosecond laser flash photolysis experiments were performed using an Applied Photophysics Model LKS-60 laser kinetic spectrometer. The excitation source is the third harmonic of a Quanta Ray INDI-40-10 series pulsed Nd:YAG laser. The excitation pulse possesses a central wavelength at λex = 335 nm and a pulse duration of ∼8 ns. Measurements were carried out for the case of argon and oxygen-purged conditions for 4-HC (100 μM) in acetonitrile. Exponential fitting of the decay traces at 550 nm was performed with the help of OriginPro software.63
Femtosecond fluorescence upconversion measurements
Fluorescence up-conversion measurements are carried out using a commercial setup, FOG-100 from CDP systems. A second harmonic generation of the fundamental output from MaiTai-HP (centered at 800 nm, with a pulse width of 100 fs and a repetition rate of 80 MHz) at 400 nm is used to excite the sample. The fluorescence from the molecular system and the residual fundamental beam are overlapped in a nonlinear BBO crystal to generate a sum frequency signal, which is essentially an up-converted fluorescence signal. The up-converted signal is effectively allowed to pass through a double monochromator and is ultimately detected using a highly sensitive photomultiplier tube coupled with a photon counter.57,64
Computational analysis
The computational analysis was carried out using the Gaussian16 package.65 The ground state geometries of 4-HC in the gas phase and MeCN were optimized using the CAM-B3LYP/6-311+G(d,p) level of theory. The potential energy surfaces at the ground and the first excited singlet states have also been computed using density functional theory (DFT) and time-dependent DFT (TDDFT) methods, respectively.
Results and discussion
Steady-state measurements
The steady-state absorption and fluorescence spectra of 4-HC in TOL, EtAc, MeCN, and MeOH solvents are shown in Fig. 1a and b, respectively. The absorption spectrum of 4-HC in the spectral range of 300–380 nm appears similar in all these solvents, and the absorption maximum occurs at ∼343 nm. A slight redshift is observed in the case of MeOH, and such a redshift in the absorption maximum could arise due to the hydrogen-bonding effect. The emission spectrum in a non-polar solvent, TOL (ε = 2.38), possesses a structured vibronic band centered at 389 nm. A significant red-shift in the emission spectrum is evident from 389 nm to 463 nm upon increasing the solvent polarity. Such a large Stokes shift (∼4108 cm−1) in the emission spectrum indicates a prominent intramolecular charge transfer (ICT) characteristic in the excited-state. The emission in a non-polar solvent could be attributed to the emission from the locally excited (LE) state. However, in a polar solvent, fluorescence originates predominantly from a highly polar ICT state. An additional intriguing observation pertains to the widths of the emission bands. The broader fluorescence spectra of 4-HC could arise due to twisting about one or more single bonds (conformational relaxation) and relaxation of the excited state along a shallow potential energy surface.66 The excited states of differently twisted conformers, populated during conformational relaxation, collectively contribute to the overall fluorescence spectrum. Despite the pronounced ICT characteristic, fluorescence quantum yield measurements showed surprising results. The fluorescence quantum yield in TOL was 1.4%, which decreased to 0.13% in MeCN, Φ = 0.17% in EtAc. The diminished fluorescence quantum yield in polar solvents strongly implies that conformational relaxation plays a crucial role in the excited-state dynamics.
 |
| | Fig. 1 Normalised steady-state (a) absorption and (b) fluorescence spectra of 4-HC in TOL, EtAc, MeCN and MeOH obtained using 330 nm excitation wavelength. (c) Optimized molecular structure of 4-HC obtained at the CAM-B3LYP/6-311+G(d,p) level of theory. | |
Time-resolved measurements
Femtosecond transient absorption (fs-TA) spectra of 4-HC in TOL and MeCN upon excitation at ∼320 nm are shown in Fig. 2 and 3, respectively. TA exhibits the emergence of an excited-state absorption (ESA) band in the spectral regions of 400–450 nm and 500–650 nm during the initial time-delays until 1.6 ps. Such positive ESA bands could be due to transitions from the S1 to higher-lying Sn states. For further time-delays, ESA evolves into a single band centered at ∼541 nm. The 541 nm peak exhibits an increase in intensity until 7 ps, followed by a subsequent decrease in intensity. Furthermore, the amplitude of the ESA remains significant even for a time-delay of ∼2.0 ns. It clearly indicates that 4-HC is essentially evolving into long lived species, such as a triplet state. The fs-TA data were analysed using singular-value decomposition followed by a global fitting analysis through the Glotaran program package.67 Such analysis reveals that fs-TA data are best fit with three distinct species. Such species are also described through associated spectral profiles and associated rates or time-constants. Each species is characterized by associated spectral profiles that are labelled as species-associated spectra (SAS1, SAS2 and SAS3). Fig. 2b presents the species-associated spectra for 4-HC in TOL. SAS1 possesses two bands centered at ∼420 nm and ∼600 nm, while SAS2 exhibits a single peak centered at ∼550 nm, and SAS3 exhibits a similar peak at ∼550 nm with a reduced amplitude. Fig. 2c presents the kinetic decay profiles associated with SAS1, SAS2, and SAS3. It is clearly depicted that SAS1, SAS2 and SAS3 possess kinetic decays with time-constants of ∼2.4 ps, ∼32 ps, and >2 ns. The global fit was optimized while considering the long time-component to be above 2 ns. SAS1 possessing two bands at ∼420 nm and ∼600 nm with a time-constant of ∼2.4 ps is attributed to the time-scale of triplet state formation from a singlet state. SAS2, predominantly possessing a single peak at ∼550 nm with a time-constant of ∼32 ps, is attributed to the vibrational relaxation in the triplet-state. Furthermore, SAS3 has evolved to be narrower as compared to SAS2 possessing a band centered at ∼550 nm with a time-constant of >2 ns. Therefore, it is safely attributed to the lifetime of the triplet state. Importantly, the narrowing of the excited-state absorption (ESA) band (SAS2 to SAS3) reveals that the excited molecule undergoes vibrational relaxation, which would be significantly affected by the polarity and nature of the solvent environment. Fig. 3a presents the fs-TA data of 4-HC in the MeCN solvent, while SAS and associated kinetic decay profiles are shown in Fig. 3b and c, respectively. The global analysis of fs-TA data of 4-HC in MeCN is also best fit with three distinct species and associated time constants. The fs-TA data in MeOH are depicted in Fig. S1 in the ESI.† The global-analysis fit parameters of fs-TA of 4-HC in all three solvents are tabulated in Table 1. It is interesting to note that the spectral profiles of SAS for 4-HC appear very similar in all the solvents. However, the associated time constants appear to be different.
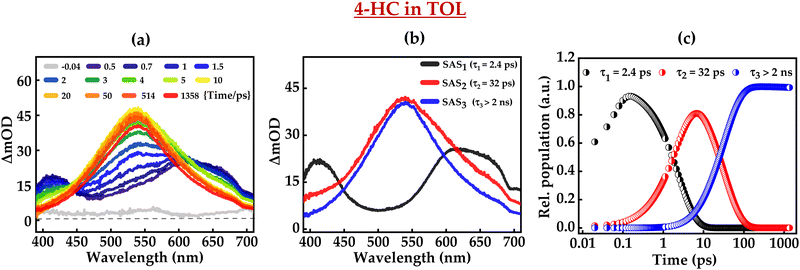 |
| | Fig. 2 (a) fs-TA of 4-HC in TOL, (b) species-associated spectra, and (c) kinetic decay profiles. The concentration of 4-HC is 0.5 mM. | |
 |
| | Fig. 3 (a) fs-TA of 4-HC in MeCN, (b) species-associated spectra, and (c) kinetic decay profiles. The concentration of 4-HC is 0.5 mM. | |
Table 1 Kinetic parameters obtained from the global-fit analysis of fs-TA data and fs fluorescence upconversion of 4-HC in TOL, MeCN and MeOH
| Solvent |
TA |
Up-conversion |
|
τ
1 (ps) |
τ
2 (ps) |
τ
3 (ns) |
τ
1 (ps) |
τ
2 (ps) |
|
Below instrument response function (IRF).
|
| TOL |
2.4 |
32 |
>2.0 |
0.07a |
1.4 |
| MeCN |
2.5 |
10 |
>2.0 |
0.04a |
0.2 |
| MeOH |
2.9 |
16 |
>2.0 |
2 |
13 |
Single-wavelength amplitude kinetics along with best-fitted decay profiles of fs-TA data in all solvents is presented in Fig. S2 (ESI†).
Fs-fluorescence upconversion (fs-FL) measurements probing at 440 nm for 4-HC were conducted in TOL, MeCN, and MeOH, while exciting at a wavelength of 387 nm. The amplitude decay profiles and the corresponding best-fit models are presented in Fig. S3 (ESI†). In all the solvents, the decay profiles were well-described by a bi-exponential function, yielding two ultrafast time constants (τ1 and τ2) of 0.07 ps and 1.4 ps in TOL, 0.04 ps and 0.2 ps in MeCN, and 2.0 ps and 13 ps in the case of MeOH. The ultrafast component (τ1) in TOL and MeCN represents processes too rapid to be resolved with the current instrumental limits. τ2 can be attributed to the lifetime of the emissive S1 state. These experimentally observed short-lived emissive excited state lifetimes, along with poor fluorescence quantum yields, are indicative of the involvement of ultrafast non-radiative decay pathways. These pathways, which are often ultrafast and dissipative in nature, likely compete with fluorescence emission, thereby reducing the overall fluorescence efficiency.
Furthermore, it is very apparent from the fs-TA measurements in all solvents and the global-fit analysis that upon photo-excitation, 4-HC eventually evolves into a long-lived state.
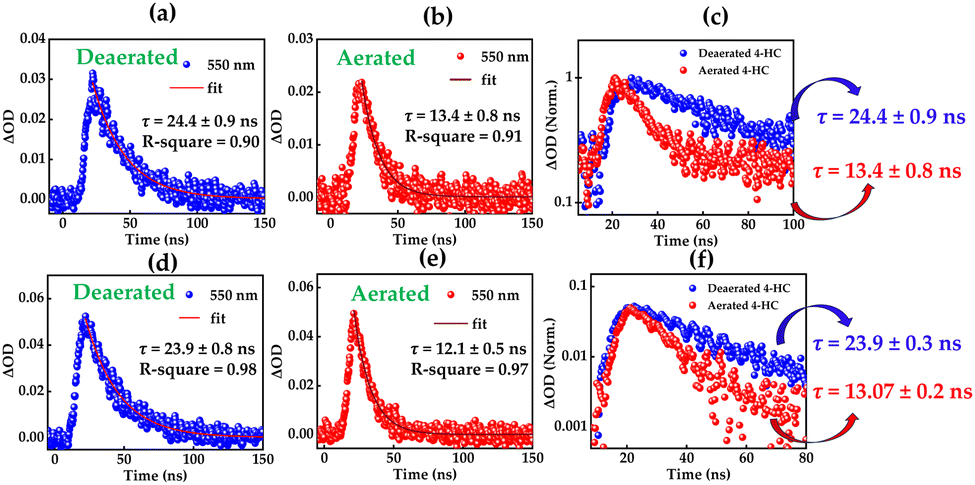
In order to characterize such long-lived species, we carried out an investigation with the nanosecond-transient absorption (ns-TA) measurements of 4-HC in TOL and MeCN. Fig. 4 presents the amplitude of the single-wavelength kinetics at ∼550 nm which was noted to be the band centre for longer time delays in the case of fs-TA measurements. The nanosecond transient absorption spectra for various time delays are shown in Fig. S4 (ESI†). For argon-purged conditions in TOL, single-exponential kinetics offer the best fit, with a time constant τ of around 24 ns. When oxygen is purged, the time constant τ decreases significantly to ∼13 ns. Similarly, τ is significantly reduced from 23 ns to 13 ns in the case of the MeCN solvent on oxygen purging. Such a significant reduction in the excited-state lifetime in the presence of O2 is a clear indication of the existence of an excited triplet state in 4-HC upon photoexcitation.
 |
| | Fig. 4 Amplitude-decay kinetics of the 550 nm probed in TOL: (a) deaerated and (b) aerated environments and in MeCN: (d) deaerated and (e) aerated environments. Combined amplitude-decay kinetic plots in (c) TOL and (f) MeCN. Excitation is at 355 nm. | |
Computational analysis
To corroborate our studies further, TDDFT calculations were performed on the optimized ground-state (S0) and optimized excited-state (S1) to discern the excited-state dynamics of 4-HC. The vertical excitation energies for the ground-state geometry, 3.752 eV and 3.878 eV, possess oscillator strengths of 0.02 and 0.95, respectively. The simulated UV absorption spectrum shows that the molecule is predominantly excited to the S2 state upon photoexcitation. Both calculated and experimental steady-state UV-vis absorption spectra show that 4-HC exhibits excellent absorption in the UV region. The change in the electron density upon excitation is computed and depicted in terms of frontier molecular orbitals as shown in Fig. 5b. Additionally, the energy of triplet state T4 was observed to be 3.7335 eV, which is in close proximity to the S1 energy. In addition, the natural transition orbital (NTO) analysis (Fig. S5 and S6, ESI†) confirms the S1–T4 transition in accordance with El-Sayed's rules,68 through the ISC process. The lower fluorescence quantum yield further substantiates the involvement of such an ISC process. Analysing the redistribution of electron density upon excitation is also crucial in assessing the change in the photo-acidity nature of 4-HC. Typically, photoacids show a reduction in electron density around the X–H bond (X can be any electronegative element such as O and N) in the excited-state, facilitating the easy removal of proton to promote excited-state proton transfer (ESPT). A reduction in the electron density is observed around the O–H bond indicating the increase in acidity of 4-HC upon photo-excitation. The transfer of electron density from the hydroxyl group to the other phenyl ring causes enhanced acidity in the excited state. Therefore, a nearby proton acceptor can abstract a proton from 4-HC in the excited-state more efficiently than the proton abstraction in the ground state. Fig. 5b also reveals that the electron density in the S0 state is concentrated on the hydroxyl (OH) group, whereas in the S1 state, it is localized on the carbonyl group and phenyl ring. The TDDFT-optimized geometry of the S1 state in 4-HC closely resembles that of the S0 state, differing only in the bond lengths and angles, thereby ruling out the possibility of substantial conformational alterations in the excited-state. To investigate further, the potential energy surface (PES) was constructed with respect to the dihedral angle coordinate for various angles around C8–C9, which is depicted in Fig. 5c. In a PES scan, the potential energy surface of a system is scanned against one or multiple degrees of freedom while relaxing all other degrees of freedom. In a relaxed potential energy surface scan, the geometry is optimized at each step while the scanned variable is kept constant. It was observed that the molecule attains maximum stability with a planar geometry at a dihedral angle (θ) of 180° in both the S0 and S1 states. Conversely, a 90° dihedral orientation exhibits the least stability in the S0 state, while in the excited-state, it significantly stabilizes the molecule.
 |
| | Fig. 5 (a) S0, S1, S2, T3, and T4 energies (in eV) of 4-HC in MeCN and (b) frontier molecular orbitals (MOs) of 4-HC. The isovalue is set as 0.2. (c) Computed potential energy surfaces in the S0 and S1 states of 4-HC in MeCN with respect to the twisting dihedral angle coordinate, θ = C11–C9–C8–C7 obtained at the CAM-B3LYP/6-311+G(d,p) level of theory. | |
Recently, Hongyu Zhang and Andong Xia reported the excited-state dynamics of 4-(dimethyl/diethylamino)methoxychalcone (DEAMC) and its derivative, 4-(dimethyl/diethylamino)hydroxychalcone (DEAHC), in various solvents.54 Notably, DEAMC exhibits a bathochromic shift in both its absorption and emission spectra, attributed to intramolecular charge transfer (ICT) transitions. The fs-TA and nanosecond transient absorption (ns-TA) measurements revealed that DEAMC undergoes ISC in non-polar solvents with a time constant of approximately ∼5.2 ps, followed by relaxation from the triplet state (T1) to the ground state occurring over a timescale greater than 1 ns. In polar solvents, relaxation to the ground state via a solvent-stabilized ICT state is reported. In contrast to DEAMC, DEAHC exhibits intramolecular hydrogen bonding (IMHB), facilitating efficient excited-state intramolecular proton transfer (ESIPT) reactions. This is evidenced by a keto-stimulated emission observed at 650 nm, with a lifetime of approximately 0.8 ps in MeCN. 4-HC, a structural analogue to DEAMC, may exhibit similar excited-state dynamics; however, it does not exhibit the potential ESIPT process. The extent of intermolecular excited-state proton transfer and its impact on the excited-state dynamics of 4-HC in the presence of external bases such as methylimidazole (MI) and tert-butylamine (TBA) will be analyzed in the following section (Scheme 1).
 |
| | Scheme 1 Illustration of the photo-initiated intermolecular proton transfer between 4-HC and MI. | |
Excited-state intermolecular proton transfer: steady-state measurements
Steady-state absorption and fluorescence spectra of 4-HC in the presence of an external base, methylimidazole (MI), were recorded in the MeCN solvent and are shown in Fig. 6a and b. The absorption spectrum of 4-HC exhibits a band around 335 nm, which undergoes a mild red-shift to 345 nm upon addition of the base MI, whose pKa for conjugate acid is 7.4. The 345 nm band is attributed to hydrogen bonding interactions between the hydroxyl group of 4-HC and the base and this hydrogen bonded adduct is represented as 4-HC:MI. The emission spectrum on the other hand exhibits dual bands centered at ∼430 nm and ∼550 nm with increasing concentrations of MI. The band centered at 550 nm is tentatively attributed to the deprotonated form of 4-HC. The fluorescence spectrum of 4-HC in the absence of MI is also shown as an inset in Fig. 6b. Similar experiments were conducted in the presence of a comparatively stronger base, tert-butylamine (TBA), with a pKa of 10.6 for the conjugate acid, and the results are shown in Fig. 6c and d. Steady-state absorption studies showed a slight red-shift with an increase in the concentration of TBA due to absorption of the hydrogen-bonded adduct, 4-HC:TBA. Additionally, the fluorescence spectrum exhibits an emergence of a new band at 550 nm. Such a band also exhibits a drastic increase in its amplitude upon increasing the concentration of TBA, while the intensity of the band centered at ∼450 nm remains unchanged. Excitation spectra for two emission wavelengths showed no significant change with the addition of base, indicating the absence of two different ground state species (Fig. S7, ESI†). In order to confirm that the emergence of the 550 nm band is due to a deprotonated form, control steady-state experiments were performed, in MeOH and the MeOH:KOH solvent mixture as shown in Fig. S8(a and b) (ESI†). It is observed that the absorption spectrum exhibits a red-shift to 424 nm from 347 nm in the presence of KOH. Furthermore, the emission spectrum is significantly affected and shows a new peak at 550 nm upon addition of KOH. pH-dependent control experiments were also carried out, which confirmed the presence of the 550 band in the buffer solution with pH 13 as shown in Fig. S8(c and d) (ESI†). These observations clearly confirm that the ∼550 nm band is predominantly due to a deprotonated form of 4-HC.
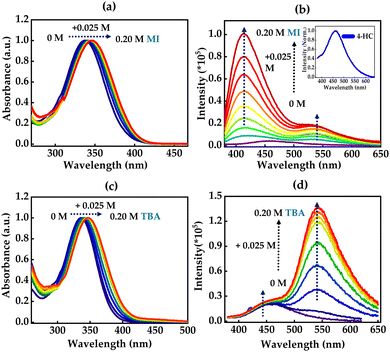 |
| | Fig. 6 (a) Steady-state absorption and (b) fluorescence spectra of 4-HC in MeCN (70 μM) for different concentrations of the MI base. The inset shows the emission from 4-HC in MeCN. (c) Steady-state absorption and (d) fluorescence spectra of 4-HC in MeCN for different concentrations of the TBA base. Excitation is at 330 nm for the emission measurements. | |
Time-resolved measurements
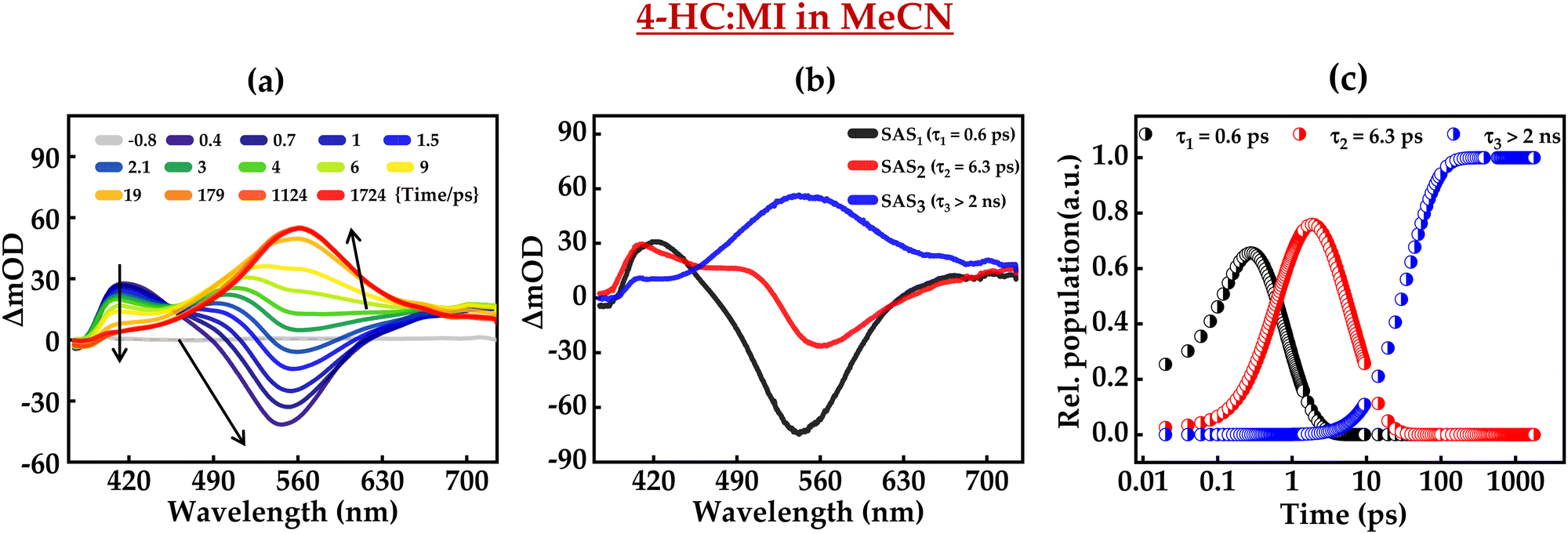
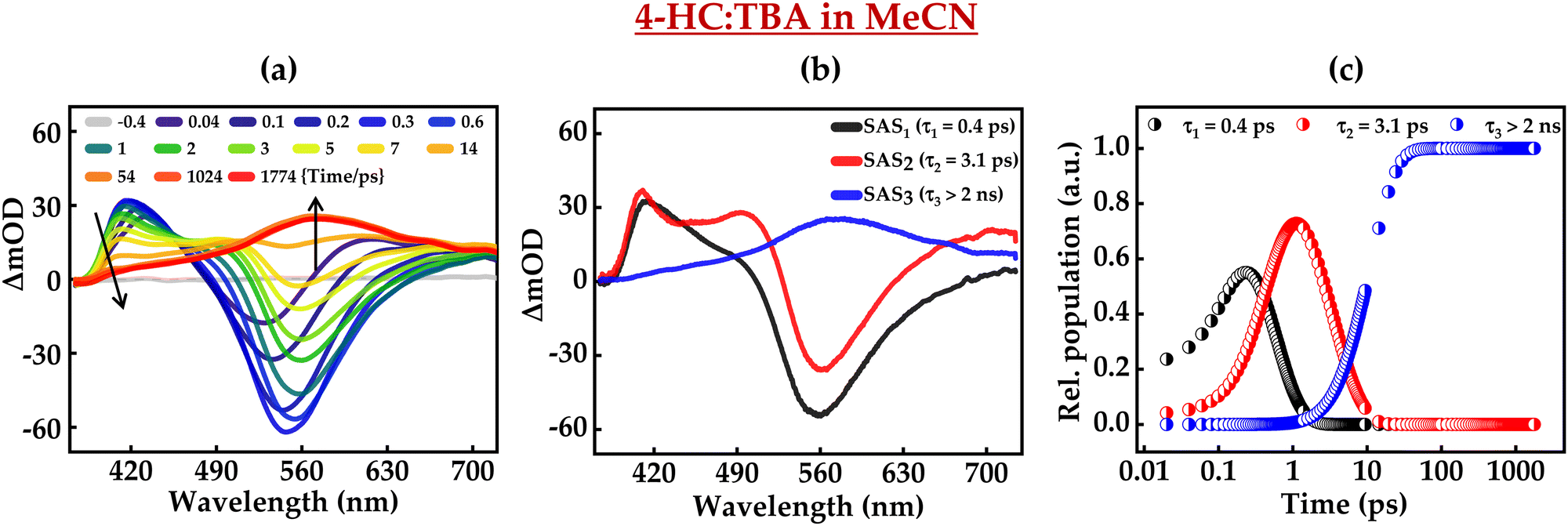
Furthermore, the excited-state intermolecular proton transfer dynamics were probed by fs-TA measurements. Fig. 7 and 8 present the fs-TA spectra of 4-HC in the MeCN solution in the presence of MI and TBA, respectively. These plots depict distinct TA spectral profiles as compared to the TA profile of 4-HC in the absence of a base. In the case of MI, for initial time-delays, the TA spectrum primarily possesses one positive band centered around ∼420 nm and a negative band around ∼550 nm. The positive band is labelled as ESA, while the negative band is labelled as the stimulated emission (SE) band as the spectral profile of this band matches with the steady-state emission spectrum of 4-HC in the presence of MI. The amplitude of the 550 nm band diminishes significantly within a time-delay of ∼2 ps. For subsequent time-delays, the TA spectrum converges to a single band with a broad spectrum centered at ∼560 nm. This band exists with significant amplitude even at a time-delay of ∼2 ns. In the case of Fig. 8a, the spectral features are noticed to be very similar to those of Fig. 7a. However, the amplitude of the SE band exhibits a decline at a time scale of ∼5 ps unlike that of ∼2 ps in the case of Fig. 7a. A global-fitting analysis of the fs-TA spectra reveals that the data are best fit with three distinct species having associated three-time constants. The kinetic fit parameters for both the cases are summarized in Table 2. Fig. 7b and c present the species-associated spectra and kinetic decay profiles, respectively for 4-HC in the presence of MI. Similarly, Fig. 8b and c present the SAS and kinetic decay profiles, respectively for 4-HC in the presence of TBA. In the case of 4-HC in MI, SAS1 with a time-constant of ∼0.6 ps predominantly possessing a negative TA band (SE) is attributed to the formation of the deprotonated state of 4-HC. SAS2 is attributed to the deprotonated species evolving into a triplet-state with a time constant of ∼6.3 ps. SAS3 possessing a single broad ESA spectrum is attributed to the triplet state which has a lifetime of >2 ns. In the case of 4-HC in the presence of TBA, SAS1, SAS2 and SAS3 are attributed similarly to the case of 4-HC in the presence of MI. However, SAS1 and SAS2 occur at time constants slightly faster as compared to 4-HC in MI. Single-wavelength amplitude kinetics along with best-fitted decay profiles of fs-TA data in MeCN with the addition of MI and TBA is depicted in Fig. S9 (ESI†).
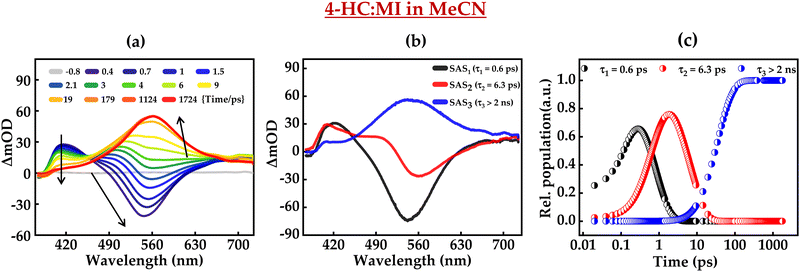 |
| | Fig. 7 (a) fs-TA of 4-HC in MeCN in the presence of MI, (b) species-associated spectra, and (c) kinetic decay profiles. The concentrations of 4-HC and MI are 0.5 mM and 0.31 M, respectively. | |
 |
| | Fig. 8 (a) fs-TA of 4-HC in MeCN in the presence of TBA, (b) species-associated spectra, and (c) kinetic decay profiles. The concentrations of 4-HC and TBA are 0.5 mM and 0.23 M, respectively. | |
Table 2 Kinetic parameters derived from fs-TA and fs-FL measurements of 4-HC in MeCN following base additions
|
|
4-HC in MeCN |
4-HC + 0.31 M MI |
4-HC + 0.23 M TBA |
| TA |
Up-conversion |
TA |
Up-conversion |
TA |
Up-conversion |
| 440 (λProbe) |
450 (λProbe) |
490 (λProbe) |
530 (λProbe) |
590 (λProbe) |
490 (λProbe) |
530 (λProbe) |
590 (λProbe) |
|
τ
1/ps |
2.5 |
0.1 |
0.2 |
0.6 |
— |
0.3 |
0.1 |
0.4 |
0.1 |
0.3 |
0.2 |
|
τ
2/ps |
10 |
— |
— |
6.3 |
0.5 |
6.1 |
5.8 |
3.1 |
2.3 |
5.4 |
5.6 |
|
τ
3/ps |
>2 ns |
— |
— |
>2 ns |
— |
— |
— |
>2 ns |
— |
— |
— |
Femtosecond fluorescence upconversion measurements were also conducted to investigate the evolution of emissive states upon photoexcitation. The fluorescence decay kinetics of 4-HC were examined both in the presence and in the absence of a base, as illustrated in Fig. 9. Fig. 9a presents the decay-kinetics for emissions at 440 nm and 450 nm for 4-HC in MeCN. The decay kinetics at various emission wavelengths for 4-HC in the presence of MI and TBA are shown in Fig. 9b and c, respectively. In the case of 4-HC alone, the decay profiles were best fitted with single exponentials, yielding a time constant of less than 1.0 ps. Conversely, in the case of the 4-HC:MI adduct, decay profiles were best fitted with two exponentials. The corresponding time constants of emission decay profiles for 4-HC in the presence and in the absence of MI are summarized in Table 2. We had chosen three wavelengths spanning the broad emission spectrum for the analysis. The amplitude of emission at 490 nm exhibits similar decay kinetics for 4-HC both in the absence and in the presence of MI. However, the amplitude of emission at 530 nm and 590 nm exhibits distinct kinetic profiles for 4-HC in MI. It is a clear indication that the 4-HC:MI adduct evolves through distinct excited-states upon photoexcitation. It also suggests the formation of a new emissive species characterized by an extended fluorescence lifetime. In the case of 4-HC:MI or 4-HC:TBA, the time constant τ1 of 0.3 ps is attributed to the relaxation of the proton-transferred species. While the time constant τ2 of 5–6 ps is attributed to the lifetime of the emissive proton-transferred state.
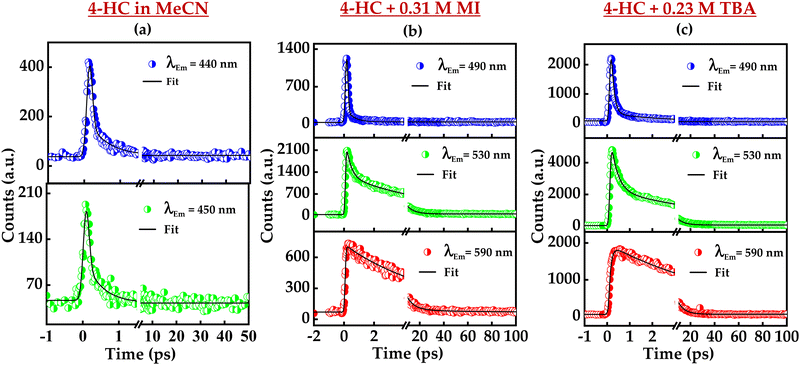 |
| | Fig. 9 Fluorescence upconversion decay profiles along with fits of 4-HC in (a) MeCN, (b) MeCN + 0.31 M MI, and (c) MeCN + 0.23 M TBA, while exciting at 400 nm. | |
Computational analysis
Proton-transfer dynamics is further corroborated with computational analysis. A potential energy surface (PES) was constructed for S0 and S1 states along the r(O12–H34) coordinate while changing the distance from 0.8 to 1.8 Å, which is depicted in Fig. 10. A PES scan provides insights into the feasibility and the energy barrier associated with a proton transfer reaction. In a relaxed PES scan, the geometry is optimized at each step while the scanned variable is kept constant. The energy barrier for a proton transfer process in the ground electronic state is found to be 0.24 eV, whereas the barrier is reduced to 0.072 eV in the excited S1 state. A reduction in the observed energy barrier facilitates the proton transfer in the excited-state better as compared to the ground state. Based on the above PES scan, we propose that upon excitation of the 4-HC:MI or 4-HC:TBA adduct, a nearly barrierless proton transfer event occurs between 4-HC and MI/TBA. Molecular electrostatic potential surface (MEPS) maps can provide a quantitative analysis of the electron density redistribution occurring during the process of photoexcitation. MEPS maps provide a three-dimensional illustration of the charge distribution of a molecule. In an MEPS map, the charge distribution of a molecule is illustrated as a gradient of colours. Blue coloured regions indicate the regions of higher positive charge and red colour regions indicate higher negative charge. The MEPS maps for 4-HC were derived on the van der Waals surface, as depicted in Fig. S10 (ESI†). It is very apparent from Fig. S10 (ESI†) that there is an undeniable negative potential around O10 and O12. However, the extent of negative charge is more prominent on carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) oxygen, indicating that such a site behaves with an enhanced photobasicity.
O) oxygen, indicating that such a site behaves with an enhanced photobasicity.
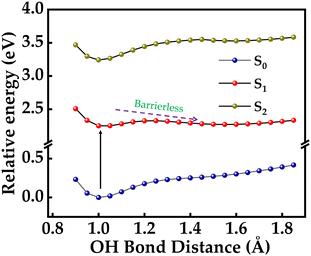 |
| | Fig. 10 PES is constructed for the 4-HC molecule in S0 and S1 states along the O–H coordinate. The energy of the ground state is set as zero. Computation is performed using the CAM-B3LYP/6-311+G(d,p) level of theory. | |
The evolution of the excited 4-HC and 4HC:MI/TBA adduct through various pathways is summarized in Scheme 2 based on the analysis of the results from experimental steady-state measurements, fs-TA, fs-fluorescence upconversion and the computation analysis. Upon photoexcitation of 4-HC in TOL, it exhibits emission from a locally excited (LE) state, followed by rapid intersystem crossing (ISC) with a time constant of ∼2.4 ps. In the case of 4-HC in MeCN, a red-shifted emission occurs at ∼463 nm due to the intramolecular charge-transfer (ICT) state. Subsequently, it undergoes rapid ISC within ∼2.5 ps. In the case of the 4-HC:MI/TBA adduct in MeCN, photoexcitation initially populates the LE state; however, subsequent relaxation occurs through different pathways as compared to 4-HC alone. Excited 4-HC:MI/TBA exhibits dual band emission centered at ∼440 nm and ∼550 nm. As compared with 4-HC in MeCN, the emission band at ∼440 nm is associated with the ICT state. 4-HC:MI undergoes ICT rapidly and evolves into an excited-state proton transfer state with a time constant of ∼0.5 ps, resulting in dual band emission at ∼440 nm and ∼550 nm, respectively. Furthermore, 4-HC:MI/TBA evolves into a triplet state through ISC with a time constant of ∼6 ps, which is slightly at a higher value as compared to that of 4-HC in MeCN. Such spectral evolution and associated kinetics suggest that 4-HC:MI/TBA experiences a slight energy barrier between the ICT and ESPT states, which is well corroborated with computational data analysis.
 |
| | Scheme 2 Schematic representation of the excited-state dynamics of 4-HC and 4-HC:MI/TBA adducts upon photoexcitation. | |
Conclusions
Our investigation explores the excited-state dynamics of 4-HC utilizing steady-state, fs- and ns-time-resolved absorption, fs-fluorescence upconversion, and computational analyses. UV absorption measurements confirm the molecule's excellent photostability and strong UVA absorption, fulfilling the essential criteria for an effective sunscreen. The observed 1.3% fluorescence quantum yield signifies energy dissipation through a non-adiabatic dynamic mechanism, primarily via conformational twisting. The formation of long-lived species in fs-TA indicates triplet-state formation, corroborated by nanosecond laser flash photolysis measurements and computational details. In the presence of the bases MI/TBA, 4-HC exhibits ultrafast excited-state proton transfer within 500 fs, evidenced by the emergence of the SE band in fs-transient absorption. While 4-HC demonstrates desirable UV absorption and minimal fluorescence, the persistence of triplet states raises concerns about potential nucleic acid damage and associated phototoxic effects. Such a photophysical process raises concerns about possible interactions with biological molecules. Current work provides molecular-level evidence suggesting the potential phototoxic effects of 4-HC, highlighting its risks if used as a sunscreen agent. Further studies are needed to assess its potential for oxidative stress or free radical formation. This study not only enhances our understanding of the photo-deactivation mechanism of the model sunscreen molecule but also provides fresh insights for exploring distinct mechanisms in future research. Future work incorporating advanced spectroscopic techniques is under consideration to provide deeper insights into the structural dynamics governing its excited-state behavior.
Author contributions
Preetika Verma: conceptualization (equal); data curation (lead); formal analysis (lead); methodology (lead); software (lead); writing – original draft (lead); and writing – review & editing (supporting). Reshma Mathew: methodology (supporting) and data curation (supporting); Nishant Dhiman: data curation (supporting); Prajoy Kumar Mitra: methodology (supporting); Yapamanu Adithya Lakshmanna: conceptualization (equal); data curation (supporting); formal analysis (lead); funding acquisition (lead); project administration (lead); supervision (lead); and writing – review & editing (lead).
Data availability
Additional experimental/computational data and procedures are available in the ESI.†
Conflicts of interest
The authors have no conflicts to disclose.
Acknowledgements
YAL acknowledges the financial support of this work through the CRG grant (CRG/2020/000321) of SERB. YAL is grateful to Prof. S. Umapathy for granting access to the laser facility at IISc Bangalore and for his constant support. RM, PV, and PKM acknowledge IISER TVM for the fellowships.
References
- K. Biniek, K. Levi and R. H. Dauskardt, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17111–17116 CAS
 .
.
- J. Dahle and E. Kvam, Cancer Res., 2003, 63, 1464–1469 CAS .
- Y. Jiang, M. Rabbi, M. Kim, C. Ke, W. Lee, R. L. Clark, P. A. Mieczkowski and P. E. Marszalek, Biophys. J., 2009, 96, 1151–1158 CAS .
- G. J. Fisher, S. Kang, J. Varani, Z. Bata-Csorgo, Y. Wan, S. Datta and J. J. Voorhees, Arch. Dermatol., 2002, 138, 1462–1470 CAS .
- R. M. Martinez, F. A. Pinho-Ribeiro, V. S. Steffen, C. V. Caviglione, V. Fattori, A. J. Bussmann, C. Bottura, M. J. Fonseca, J. A. Vignoli and M. M. Baracat, Photochem. Photobiol. Sci., 2017, 16, 1162–1173 CrossRef CAS PubMed .
- V. R. Gadgil, A. Darak, S. J. Patil, A. Chopada, R. A. Kulkarni, S. M. Patil, N. A. Gupta, T. N. Mehta and S. V. Joshi, J. Indian Chem. Soc., 2023, 100, 100858 CrossRef CAS .
- T. Maier and H. C. Korting, Skin Pharmacol. Physiol., 2005, 18, 253–262 CAS .
- L. Thomas and H. W. Lim, J. Drugs Dermatol., 2003, 2, 174–177 Search PubMed .
- E. Engel, A. Spannberger, R. Vasold, B. König, M. Landthaler and W. Bäumler, J. Dtsch. Dermatol. Ges., 2007, 5, 583–589 Search PubMed .
- T. G. Smijs and S. Pavel, Nanotechnol. Sci. Appl., 2011, 95–112 CAS .
- L. A. Baker, M. D. Horbury, S. E. Greenough, P. M. Coulter, T. N. Karsili, G. M. Roberts, A. J. Orr-Ewing, M. N. Ashfold and V. G. Stavros, J. Phys. Chem. Lett., 2015, 6, 1363–1368 CAS .
- E. L. Holt and V. G. Stavros, Int. Rev. Phys. Chem., 2019, 38, 243–285 Search PubMed .
- L. A. Baker, B. Marchetti, T. N. Karsili, V. G. Stavros and M. N. Ashfold, Chem. Soc. Rev., 2017, 46, 3770–3791 RSC .
- S. Ramos, V. Homem, A. Alves and L. Santos, Sci. Total Environ., 2015, 526, 278–311 CrossRef CAS PubMed .
- K. Sugiyama, T. Tsuchiya, A. Kikuchi and M. Yagi, Photochem. Photobiol. Sci., 2015, 14, 1651–1659 CAS .
- N. D. Rodrigues, M. Staniforth and V. G. Stavros, Proc. R. Soc. A, 2016, 472, 20160677 CrossRef PubMed .
- C. T.-L. Chan, C. Ma, R. C.-T. Chan, H.-M. Ou, H.-X. Xie, A. K.-W. Wong, M.-L. Wang and W.-M. Kwok, Phys. Chem. Chem. Phys., 2020, 22, 8006–8020 Search PubMed .
- N. d N. Rodrigues, J. M. Woolley, K. M. Krokidi, M. A. Tesa-Serrate, M. A. Turner, N. D. Hine and V. G. Stavros, Phys. Chem. Chem. Phys., 2021, 23, 23242–23255 Search PubMed .
- N. d N. Rodrigues, N. C. Cole-Filipiak, M. A. Turner, K. Krokidi, G. L. Thornton, G. W. Richings, N. D. Hine and V. G. Stavros, Chem. Phys., 2018, 515, 596–602 CAS .
- J. Luo, Y. Liu, S. Yang, A. L. Flourat, F. Allais and K. Han, J. Phys. Chem. Lett., 2017, 8, 1025–1030 CrossRef CAS PubMed .
- Y. Shi, X. Zhao, C. Wang, Y. Wang, S. Zhang, P. Li, X. Feng, B. Jin, M. Yuan and S. Cui, Chem. – Asian J., 2020, 15, 1478–1483 CrossRef CAS PubMed .
- X. Zhao, F. Ji, Y. Liang, P. Li, Y. Jia, X. Feng, Y. Sun, Y. Shi, L. Zhu and G. Zhao, J. Lumin., 2020, 223, 117228 CrossRef CAS .
- F. Ji, X. Zhao, Y. Guo, Z. Wu, M. Wang, Y. Shi and G. Zhao, J. Lumin., 2021, 238, 118215 CrossRef CAS .
- A. L. Whittock, T. T. Abiola and V. G. Stavros, J. Phys. Chem. A, 2022, 126, 2299–2308 CrossRef CAS PubMed .
- T. T. Abiola, N. d N. Rodrigues, C. Ho, D. J. Coxon, M. D. Horbury, J. M. Toldo, M. T. Do Casal, B. Rioux, C. Peyrot and M. M. Mention, J. Phys. Chem. Lett., 2020, 12, 337–344 CrossRef PubMed .
- A. M. Cowden, R. Losantos, A. L. Whittock, B. Peñín, D. Sampedro and V. G. Stavros, Photochem. Photobiol., 2023, 100(2), 298–313 CrossRef PubMed .
- Z. Wu, M. Wang, Y. Guo, F. Ji, C. Wang, S. Wang, J. Zhang, Y. Wang, S. Zhang and B. Jin, J. Phys. Chem. B, 2021, 125, 9572–9578 CrossRef CAS PubMed .
- M. Wang, Z. Wu, F. Ji, C. Wang and G. Zhao, J. Lumin., 2022, 246, 118816 CrossRef CAS .
- Z. Deng, S. Sun, M. Zhou, G. Huang, J. Pang, L. Dang and M.-D. Li, J. Phys. Chem. Lett., 2019, 10, 6499–6503 CrossRef CAS PubMed .
- C. Xu, L. Yu, C. Zhu, J. Yu and Z. Cao, Sci. Rep., 2016, 6, 26768 CrossRef CAS PubMed .
- S. Shukla, A. K. Sood, K. Goyal, A. Singh, V. Sharma, N. Guliya, S. Gulati and S. Kumar, Adv. Anticancer Agents Med. Chem., 2021, 21, 1650–1670 CrossRef CAS PubMed .
- L. Sarbu, L. Bahrin, C. Babii, M. Stefan and M. Birsa, J. Appl. Microbiol., 2019, 127, 1282–1290 CrossRef CAS PubMed .
- N. Raut, P. Dhore, S. Saoji and D. Kokare, Stud. Nat. Prod. Chem., 2016, 48, 287–322 CAS .
- C. Díaz-Tielas, E. Graña, M. Reigosa and A. Sánchez-Moreiras, Planta Daninha, 2016, 34, 607–616 CrossRef .
- M. A. Shalaby, S. A. Rizk and A. M. Fahim, Org. Biomol. Chem., 2023, 21, 5317–5346 RSC .
- A.-M. Katsori and D. Hadjipavlou-Litina, Expert Opin. Ther. Pat., 2011, 21, 1575–1596 CrossRef CAS PubMed .
- C. Karthikeyan, N. S. H. Narayana Moorthy, S. Ramasamy, U. Vanam, E. Manivannan, D. Karunagaran and P. Trivedi, Recent Pat. Anticancer Drug Discov., 2015, 10, 97–115 CAS .
- M. J. Matos, S. Vazquez-Rodriguez, E. Uriarte and L. Santana, Expert Opin. Ther. Pat., 2015, 25, 351–366 CAS .
- W. Dan and J. Dai, Eur. J. Med. Chem., 2020, 187, 111980 CAS .
- M. Xu, P. Wu, F. Shen, J. Ji and K. Rakesh, Bioorg. Chem., 2019, 91, 103133 CAS .
- Y. Ouyang, J. Li, X. Chen, X. Fu, S. Sun and Q. Wu, Biomolecules, 2021, 11, 894 CAS .
- S.-J. Won, C.-T. Liu, L.-T. Tsao, J.-R. Weng, H.-H. Ko, J.-P. Wang and C.-N. Lin, Eur. J. Med. Chem., 2005, 40, 103–112 CAS .
- C. Ning, C. Xiao, R. Lu and P. Zhou, J. Lumin., 2020, 217, 116825 CAS .
- A. K. Zulkarnain, G. T. Kharismawati and F. A. Larasati, Indones. J. Pharm., 2023, 34, 112–119 CAS .
- J. Jumina, R. W. Styaningrum, D. Siswanta, S. Triono, Y. Priastomo, H. Harizal, E. N. Sholikhah and A. K. Zulkarnain, Chem. J. Mold., 2019, 14, 90–96 CAS .
- A. K. Zulkarnain and J. Jumina, J. Food Pharm. Sci., 2023, 11, 803–811 CrossRef .
- S. R. Lemes, L. A. Júnior, D. da Silva Manoel, M. A. M. de Sousa, R. D. Fonseca, R. S. Lima, C. Noda-Perez, P. R. de Melo Reis, C. G. Cardoso and E. de Paula Silveira-Lacerda, Spectrochim. Acta, Part A, 2018, 204, 685–695 CrossRef CAS PubMed .
- Y.-J. Wang, D.-G. Zhou, F.-C. He, J.-X. Chen, Y.-Z. Chen, X.-H. Gan, D.-Y. Hu and B.-A. Song, Chin. Chem. Lett., 2018, 29, 127–130 CrossRef CAS .
- A. R. Y. Eff, R. D. Pertiwi, I. Rakhmawati and T. P. Utami, J. Young Pharm., 2018, 10, S106 CrossRef CAS .
- Y. Zhang, C. Shang, Y. Cao and C. Sun, J. Mol. Liq., 2023, 372, 121165 CrossRef CAS .
- V. S. Vadivoo, C. V. Mythili, R. Balachander, N. Vijayalakshmi and P. Vijaya, Biointerface Res. Appl. Chem., 2021, 12, 7159–7176 Search PubMed .
- Y. Hu, L. Yue, F. L. Gu and C. Zhu, Phys. Chem. Chem. Phys., 2021, 23, 4300–4310 RSC .
- A. P. de Sousa, H. D. d S. Souza, A. Almeida-Júnior, M. F. R. da Silva, L. V. Cordeiro, E. d O. Lima, G. F. Fiss and P. F. de Athayde-Filho, Synth. Commun., 2024, 54(12), 973–991 CrossRef CAS .
- H. Song, Z. Kuang, X. Wang, Y. Guo, Q. Guo, H. Zhang and A. Xia, J. Phys. Chem. C, 2018, 122, 15108–15117 CrossRef CAS .
- Y. Shinozaki, M. Yamaji and T. Arai, J. Photochem. Photobiol., A, 2018, 350, 17–22 CrossRef CAS .
- R. J. Abdel-Jalil, A. R. Ibrahim and O. K. Abou-Zied, Chem. Phys. Lett., 2023, 813, 140299 CrossRef CAS .
- R. Mathew, S. Kayal and A. L. Yapamanu, Phys. Chem. Chem. Phys., 2019, 21, 22409–22419 RSC .
- C. Ma, C. T.-L. Chan, R. C.-T. Chan, A. K.-W. Wong, B. P.-Y. Chung and W.-M. Kwok, Phys. Chem. Chem. Phys., 2018, 20, 24796–24806 RSC .
- C. Ma, Y.-Q. Ou, C. T.-L. Chan, A. K.-W. Wong, R. C.-T. Chan, B. P.-Y. Chung, C. Jiang, M.-L. Wang and W.-M. Kwok, Phys. Chem. Chem. Phys., 2018, 20, 1240–1251 RSC .
- E. M. Tan, M. Hilbers and W. J. Buma, J. Phys. Chem. Lett., 2014, 5, 2464–2468 CrossRef CAS PubMed .
- Y. Peperstraete, M. Staniforth, L. A. Baker, N. D. Rodrigues, N. C. Cole-Filipiak, W.-D. Quan and V. G. Stavros, Phys. Chem. Chem. Phys., 2016, 18, 28140–28149 RSC .
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS .
-
Origin(Pro), Version 2021, OriginLab Corporation, Northampton, MA, USA Search PubMed .
- R. Mathew, P. Verma, A. Barak and A. L. Yapamanu, J. Phys. Chem. A, 2023, 127, 7419–7428 CAS .
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed .
- R. Ghosh and D. K. Palit, J. Phys. Chem. A, 2015, 119, 11128–11137 CrossRef CAS PubMed .
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed .
- S. Lower and M. El-Sayed, Chem. Rev., 1966, 66, 199–241 CAS .
Footnote |
| † Electronic supplementary information (ESI) available: fs-TA in MeOH, SWK plots, fs-fluorescence upconversion of 4-HC, ns-TA spectra, steady-state measurements in MeOH:KOH and in various pH solutions, excitation spectra, computational plots of NTO and MEPS maps. See DOI: https://doi.org/10.1039/d4cp04525d |
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Reshma
Mathew
a,
Reshma
Mathew