
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational investigation of uracil dimers in water and the role of classical potentials†
Tea
Ostojić
 a,
Juraj
Ovčar
ab,
Ali
Hassanali
c and
Luca
Grisanti
*ab
a,
Juraj
Ovčar
ab,
Ali
Hassanali
c and
Luca
Grisanti
*ab
aRuđer Bošković Institute, Divison of Theoretical Physics, Bijenička cesta 54, 10 000 Zagreb, Croatia
bCNR-IOM, Consiglio Nazionale delle ricerche – Istituto Officina dei Materiali, c/o SISSA, via Bonomea 265, 34136 Trieste, Italy. E-mail: grisanti@iom.cnr.it
cCondensed Matter and Statistical Physics, The Abdus Salam International Centre for Theoretical Physics, strada Costiera 11, 34151 Trieste, Italy
First published on 23rd May 2025
Abstract
Nucleobases possess a marked tendency to establish hydrogen bonds – a structural trait at the base of the storage of biological information. It is crucial to understand what is the impact on this intermolecular interaction when free nucleobases are moved to aqueous media. In this work, we present a systematic investigation of the thermodynamics of uracil dimers in explicit water solvent. We employ well-tempered metadynamics simulations to generate a reliable free energy surface and classify all low-energy states. We perform detailed structural analysis of the relevant dimer configurations. The results obtained with different force fields are compared and a methodological assessment of the force fields accuracies are presented.
1 Introduction
Canonical nucleobases serve as the fundamental components of ribonucleic and deoxyribonucleic acids (RNA and DNA), forming respective nucleosides when bonded with sugar and phosphate groups.1 Within these biological macrostructures, nucleobases are the building blocks with a crucial structural role because of their involvement in the formation of hydrogen bonds (HBs). Hydrogen bonding and the consequent creation of complementary pairs (A:T(U), G:C) are at the base of the storage and transmission of information in living beings. However, it remains an open question whether these fundamental interactions are able to endure if nucleobases transition into aqueous solutions and to which degree the complementary pairs survive against the formation of HBs with water.2,3 The question about the fate of HBs when moving paired nucleobases in solution may be better understood by investigating the energetic preference of intermolecular nucleobase interactions in the presence of water, an effective hydrogen-bond competitor.As the mechanisms underlying the selection of the five canonical nucleobases from a broader pool, particularly in the context of prebiotic chemistry, remain incompletely understood,4,5 determining nucleobase tendency to aggregate is also deeply relevant for understanding the origin of life. Indeed, nucleobases exhibit interesting photochemistry, with various processes depending on the configurations of the formed supramolecular states, including photo-induced dimerization as observed in uracil and thymine.6–11 Their resistance to degradation and their evolution into more complex structures have been intrinsically related to their capability to (self-)organize and how their properties are affected by the formation of supramolecular structures.12–16
Uracil is an excellent model system and the subject of several investigations under fundamental and applied aspects crossing chemistry, physics, biology and medicine. As shown by some of us recently,17 together with other nucleobases, it has a capability of forming interactions in a supramolecular context and has applications as a building block for functional molecular materials. π-Stacking interactions, together with HBs, have been shown to be crucial in the formation of assemblies, interfaces and substrates formed of nucleic acids.18–20 Accurately modelling these weak interactions to reliably describe the thermodynamics of nucleobases in solution and being able to connect their structure and properties are challenging. It requires us to have full control on how sensitive the force field and the water models are to these subtle interactions.
From the computational point of view, several works in the literature have investigated uracil dimer systems using both empirical and more accurate potentials.21–25 Kratochvíl et al. have classified all the minima obtained by scanning the configurational space of a uracil dimer in vacuum employing a classical potential, focusing in particular on H-bonded structures.21 In the work by Morgado et al.,22 the accuracy of models based on a classical potential (AMBER force field) or density functional theory (DFT) with dispersion corrections was systematically evaluated for many geometric configurations against high-level quantum-chemistry reference methods. The classical force field was found to overestimate the repulsive component of the potential for both stacked and the H-bonded structures. However, the authors heuristically assumed that this error would not have a large qualitative impact on explicit solvent simulations of nucleic acids. To the best of our knowledge, a more systematic exploration in explicit water solvent is missing. Milovanović et al. conducted several computational investigations of stacked uracil dimers (with solvated water molecules) for the purpose of spectroscopy, photophysics and photochemistry.23–26 In one work, they focused on only two significant configurations, face-2-face (F2F) and face-2-back (F2B) stacked uracil dimers in a cluster of 10 water molecules, revealing that the photodynamics of F2F-stacked uracil–water clusters exhibits a greater propensity to form cyclobutane-type photoproducts.23 Remarkably, it was also concluded that the photophysical and photochemical pathways of stacked uracil bases are controlled, to a large extent, by the initial configuration of the bases. However, the general propensity for a specific stacked configurations in water was not investigated. Another potential photodegradation pathway involves an oxetane-like path, which includes a “close to T-shaped” configuration as a relevant intermediate.27 In this work, we provide a systematic computational investigation of the microscopic configurations of uracil dimers in bulk water and fill this knowledge gap.
Generally, nucleobase self-aggregation (pairing) in a solution is not a spontaneous process, indicating that interactions between water and nucleobases are statistically more probable. However, as evidenced by the experimentally observed photodimerization in uracil,11 nucleobase aggregation becomes relevant, especially at low temperatures or higher concentrations. Therefore, to properly study these systems and provide relevant sampling of microscopic configurations, one must rely on enhanced sampling techniques.28–30 By employing molecular dynamics (MD) with enhanced sampling techniques, we rank and classify the possible stable uracil dimer structures encountered in water and explore the impact on experimental properties such as optical absorption.
Although nucleobases appear to be simple molecules, the selection of an accurate force field for such systems involving free nucleobases in water is not straightforward. Most force fields are optimized for simulating nucleic acids, with dedicated parametrizations to account for changes in phosphates or backbones not present in nucleobases/water systems.31 However, the force field parametrization by Chen and Garcia, derived from the AMBER99 force field and the TIP3P water model, focused more specifically on non-bonding parameters by fitting high-level quantum-chemical calculations specifically on nucleobase dimers.32 Since this specific parametrization has faced criticism within the scientific community for its inability to predict certain experimentally observed structures33 and for being primarily tested on a small set of tetraloops and not having a completely satisfactory agreement with the experimental results,34 we evaluate the accuracy of the Chen&Garcia force field against the widely accepted ff99 force field. We show that the force field obtained by the Chen&Garcia parametrization is a more accurate model of the uracil dimer/water system compared to the force field obtained using the more popular ff99.31,35,36
2 Methods
Enhanced sampling MD
We have run enhanced sampling MD simulations using well-tempered metadynamics (wt-MetaD).37 The wt-MetaD simulations were run using two different force fields: the force field introduced by Chen and Garcia32 with water being modelled using TIP3P (abbreviated as Chen & Garcia) and the AMBER99 force field36 with water being modelled using OPC (abbreviated as ff99). Additionally, to verify that the results obtained using wt-MetaD are independent of the sampling scheme, we have run MD with walls (i.e. adding a restraining constant potential to limit the phase space accessible during the simulation) using the Chen & Garcia force field. For all runs, the simulation time was equal to 1 μs.We proceed to describe the force field parametrization protocol in detail. As a first step, density functional theory (DFT) geometry optimizations of the nucleobase and corresponding nucleoside were performed using Gaussian 0938 with the B3LYP exchange–correlation functional and the 6-311G basis set.39–44 On these geometries, single point HF 6-31G* basis set calculations were combined with ANTECHAMBER45 to determine restrained electrostatic potential (RESP) charges. The topologies for the corresponding nucleobase and nucleoside were generated using the ANTECHAMBER package,45 employing the ff9936 force field and converted to the GROMACS46 format using the acpypi47 Python script. Since the uracil molecule contains one extra H atom as compared to the nucleoside, the parameters corresponding to this atom were obtained using the generalized AMBER force field (GAFF).36 To obtain the parameters for the Chen & Garcia force field, we rescaled the ff99 parameters following the ESI† data provided by Chen and Garcia.32
To facilitate effective sampling using wt-MetaD, a set of collective variables (CVs) was chosen. Both non-covalent interactions, namely π-stacking (s) and hydrogen bonding (h), were quantified using coordination numbers (equivalently number of contacts). We note that our hydrogen bonding descriptor (h) is only distance-based and does not include any directionality. The coordination number as CV was already integrated into the PLUMED plugin,48–50 which was applied in conjunction with the GROMACS simulation package.46 The s or h numbers of contacts for a given configuration are computed through the following sum of switching functions:51
 | (1) |
| CV | n | m | r 0 (nm) | d 0 (nm) |
|---|---|---|---|---|
| s | 12 | 24 | 0.35 | 0.05 |
| h | 12 | 18 | 0.30 | 0.02 |
The equations of motion were integrated using a 0.5 fs time step and the md algorithm.46 Periodic boundary conditions were used. Forthe thermostat, velocity rescaling was used (tau_t = 0.2 ps; ref_t = 298.15 K). As barostat, Berendsen coupling was employed (tau_p = 0.2 ps, ref_p = 1 bar).52 Long-range electrostatic interactions between periodic images were computed utilizing the particle mesh Ewald method,53 employing a grid spacing of 0.12 nm, cubic interpolation of the fourth order and a tolerance threshold of 10−5. Neighbor lists were updated at intervals of every 5 time steps. A cutoff distance of 10 Å was imposed for managing van der Waals interactions and real-space Coulomb interactions. The CVs’ range for sampling was set to s: −0.5, 36 and h: −0.5, 8, with σ (the widths of the Gaussian hills) 0.1 and 0.025 respectively. Pace of hills addition was set at 250 steps, τ at 100 and temperature at 298.15 K. τ is a parameter controlling the height of added Gaussian hills.48–50 The simulation lengths were 1000 ns. The size of the simulated systems were 2 uracil molecules and 4046 water molecules (Chen & Garcia) and 2 uracil molecules and 4127 water molecules (ff99).
When biasing MD with walls using Chen&Garcia, two lower walls were defined, both for s and h CVs. The bias due to the wall is defined as:48–50Uwall = k·(x − a)2, where x = {s,h}, a = 0.3 and k = 1000 kJ mol−1. This choice of parameters leads to walls acting as high barriers, restricting the system from entering regions with s < 0.3 or h < 0.3.
Force field validation
To select configurations which we will use to validate force fields, we disregard scenarios in which two nucleobases interact solely with the solvent and not with each other, which corresponds to the minima basin at (s,h) ≈ (0,0). From each of the minima basins of interest (i.e. (s,h) ≠ (0,0)) in which two uracil molecules are interacting with each other, a randomly selected subset of ten configurations was chosen to validate the accuracy of the employed force fields. Three nontrivial basins were defined using the following ranges of s and h: HT (s ∈ [0.9,2]; h ∈ [1,1.15]); 2H (s ∈ [2.6,3.7]; h ∈ [2,2.20]) and ST (s ∈ [12,20], h ∈ [0.8,1.4]). The analysis specifically retained water molecules within a contact radius of 2.5 Å of the nucleobase dimers to reduce the computational workload.Subsequently, the selected configurations underwent an energy minimization process, employing the same force field and water model as in the originating simulations. The DFT energies of each optimized configuration were then computed at the PBE0/def2-TZVP D3BJ level of theory54–56 using the Orca 5.0.2 software package.57 To be able to compare the energies of the sampled configurations with differing numbers of water molecules calculated using DFT and the classical force fields on a linear scale, we performed a linear regression of the energy dependence on the number of water molecules. The slopes of the lines obtained by the linear regression correspond to a constant contribution to the total energy per water molecule, which is then accordingly subtracted from each sampled configuration. The validation procedure is outlined on Fig. 1 with more details being given in the ESI.†
 | ||
| Fig. 1 Force field validation procedure (see textand ESI†). | ||
For each combination of the force field and water model, a linear regression line was fitted to E(DFT) vs. E(classical). The determination of the appropriateness of the force field was made through an assessment of key statistical parameters, such as the R2 factor and mean average error (MAE), for both sets of data.
Geometry variables for the free energy surface analysis
Number of contacts (NC) between atoms involved in HB was defined as the sum of contributions due to intermolecular contacts (short distance) between atoms X and Y′ (where primed labels stress that the atom belongs to different molecules): | (2) |
Simulated optical absorption
For all four minima, a set of 40 clusters formed by uracil (monomer – if taken from the (s,h) = (0,0) minimum, or dimer otherwise) and a number of water molecules within a 2.5 Å radius of the nucleobase, was selected to undergo a preoptimization using the semi-empirical HF3c exchange–correlation functional58 available in Orca,57 followed by a full optimization at PBE0-D3BJ/def2-TZVP level of theory54–56 in implicit solvent (SMD model59). Such structures were then employed to run time-dependent density functional theory (TDDFT) calculations employing both PBE054 and ωB97X60 functionals in Orca package57 with the above basis set. To calculate the absorption spectra, individual transitions were convoluted with a Gaussian profile with a width of 0.06 eV. For the HT and 2H basins it was necessary to exclude up to 45% of the structures from the spectra calculation due to a change of the state during the optimisation.3 Results and discussion
Well-tempered metadynamics
In the course of our investigation, two distinct force fields have been employed for metadynamics simulations, each paired with different water models: Chen&Garcia32 and ff99.36 The reparametrization by Chen and Garcia was introduced to mitigate the potential overestimation of base stacking interactions. Moreover, specific interactions between carbon, oxygen and nitrogen atoms of nucleobases with water's oxygen atoms were subject to rescaling.32 For these reasons, this has been our initial choice and our results are first presented employing this force field. Below, we present an evaluation of Chen&Garcia and ff99 against DFT and compare the free energy surfaces obtained with the two force fields.The selection of CVs was inspired by research on the association of nucleobases in hydrated ionic liquid.51 The Gibbs energy surface was obtained from wt-MetaD simulations, involving the summation of the negative bias along the trajectory in the space of two CVs (s,h). The uncertainties of energies were assessed by analyzing the energy changes in the last 300 ns of the simulation within the step of 1 ns (ESI,† Fig. S1–S6). The full free-energy surface is shown in Fig. 2. The deepest basin corresponds to the (s,h) ≈ (0,0) configuration, in which uracil molecules interact solely with the surrounding water, so there is no uracil aggregation.
 | ||
| Fig. 2 The Gibbs free energy surface of a uracil dimer in water, obtained using the Chen&Garcia force field. Basins are marked on the free energy surface. | ||
The other three minima basins in Fig. 2 correspond to configurations where two uracil molecules are in contact with each other, with each basin involving different types of interactions. Among these, the lowest energy basin represents configurations where two uracil molecules interact through two hydrogen bonds (2H). The next lowest energy basin is found for configurations with 1 hydrogen-bond and T-shaped (HT) interactions, while the highest energy basin corresponds to configurations where two uracil molecules are in a (π–)π stacking (ST) arrangement. Due to different orientations and arrangements of the (π–)π stacking configurations, this is the widest minimum basin. The obtained results align with existing literature concerning the configurations of the uracil dimer in a vacuum and in solution – see below for extensive discussion. Moreover, both 2H and ST configurations are consistent with the crystal structure of uracil.61 Detailed information on the relative energies of the minima basins is provided in Table 2.
| Configuration | G r (kJ mol−1) | G r (kJ mol−1) |
|---|---|---|
| Chen&Garcia | ff99 | |
| 2H | 0.00 ± 0.11 | 1.84 ± 0.08 |
| HT | 2.26 ± 0.07 | 0.00 ± 0.09 |
| ST | 8.52 ± 0.05 | 2.41 ± 0.06 |
Force field comparison
As mentioned above, our initial decision was to employ the reparameterized AMBER force field by Chen and Garcia (Chen&Garcia, see Methods for the detailed force field description), as it contains rescaled non-bonding parameters obtained by benchmarking against accurate calculations of interacting nucleobases.32 The more widely accepted parametrization schemes, including standard AMBER, primarily focus on altering parameters related to the RNA backbone or phosphate,31 which are not directly relevant to the system under investigation in this study. Therefore, we assumed that the choice of Chen&Garcia ensures a targeted and context-specific approach to force field selection. However, given the common criticisms regarding the parametrization of Chen&Garcia,32 particularly its reported limitations in accurately predicting certain structures,33,62 we evaluate the accuracy of the Chen&Garcia force field against the widely accepted ff99 force field.36 The details of this evaluation are presented in the Method section.The Gibbs energy surface resulting from wt-MetaD simulations using ff99 exhibits a similar shape to the one depicted in Fig. 2. However, as detailed in Table 2, the distinction between the two lies in the obtained energies, particularly in their ordering. Using ff99 (Fig. 3), the deepest minimum remains in (0,0), aligning with the expectation that uracil aggregation is a rare event. Notably, the three minima of primary interest exhibit variations. The deepest of these is HT, followed by 2H and ST. Utilizing ff99 also results in 2H and ST minima being closely matched in energy, in contrast to the proximity of 2H and HT minima obtained with Chen&Garcia.
 | ||
| Fig. 3 The Gibbs free energy surface of a uracil dimer in water, obtained using the ff99 force field. Basins are marked on the free energy surface. | ||
The results of the evaluation of the force fields against DFT are shown on Fig. 4. It is apparent that both force fields perform well in describing the investigated system. However, a closer analysis of the goodness-of-fit coefficient and linear regression line slope reveals that the Chen&Garcia parametrization offers a slightly better description of nucleobases in aqueous solutions. The mean average errors for Chen&Garcia32 and ff9935 were 10.33 kJ mol−1 and 10.45 kJ mol−1, respectively. Furthermore, the wt-MetaD simulations using the Chen&Garcia parametrization converge faster compared to the one employing ff99,35 which can likely be attributed to the simpler description of water molecules and the softened non-bonding interactions.
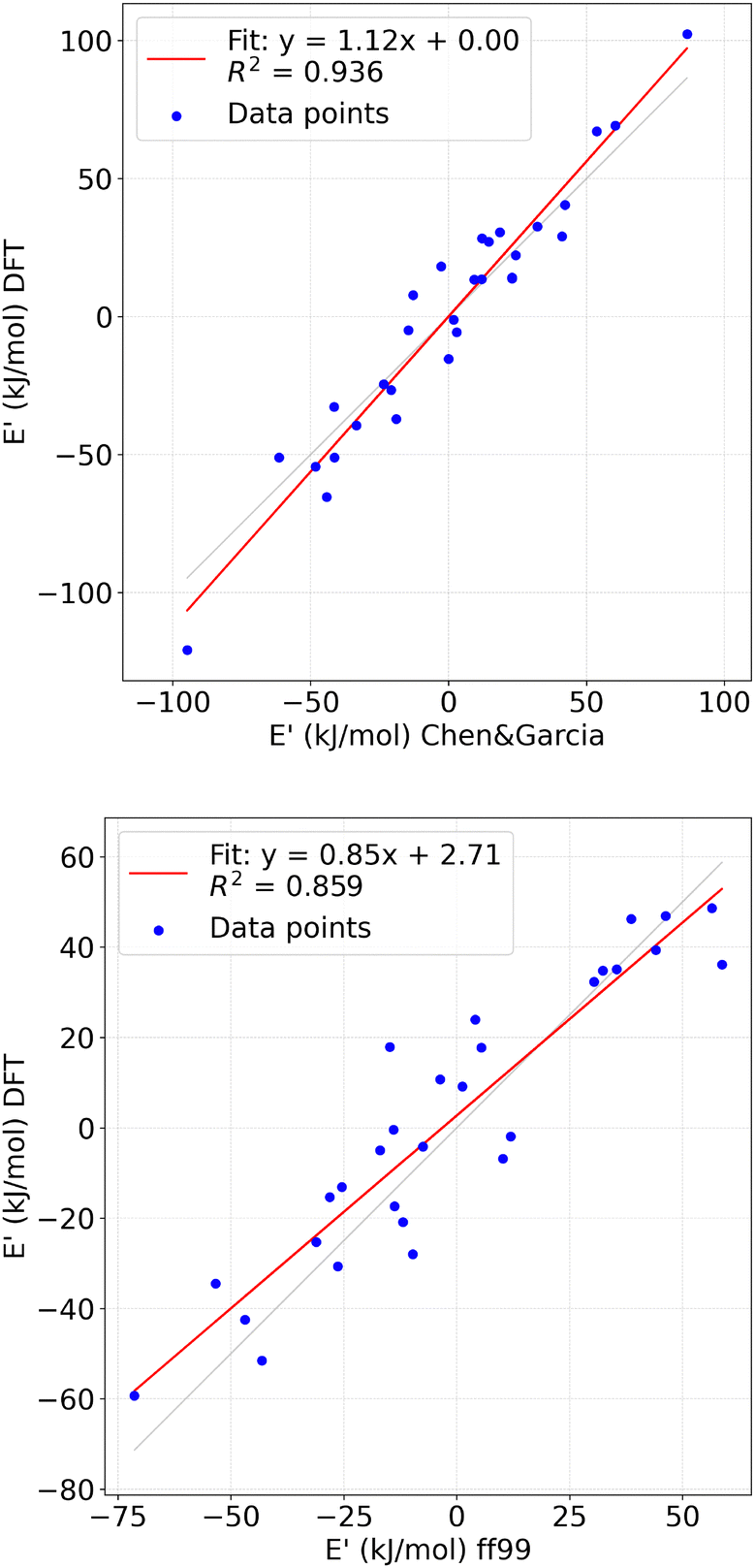 | ||
| Fig. 4 Top panel: transformed DFT energies (E′DFT) vs. transformed classical energies (E′cl.), using Chen&Garcia data. Bottom panel: transformed DFT energies (E′DFT) vs. transformed classical energies (E′cl.), using ff99 data. | ||
The primary challenge in simulating nucleobase systems in water lies in overcoming the infrequency of association. Specifically, escaping the (0,0) minimum basin to explore other minima in the configurational space and the difficulties of reaching convergence, motivated us to perform additional validation of the wt-MetaD suitability. The same system was subjected to biased molecular dynamics simulations with walls, employing the Chen&Garcia force field.32 By placing walls at s = 0.3 and h = 0.3, the system was enhanced to explore the desired configurational space where two uracil molecules interact with each other. The results of the MD simulation biased with walls (Fig. S7 in ESI†) show the same energy ranking and differences obtained by wt-MetaD, except for a small overstabilisation of the HT state. Importantly, this confirms that wt-MetaD can be effectively employed as proposed.
Analysis of the Chen & Garcia free energy surface
We take advantage of our complete characterization of the free energy surface to conduct additional analysis. The barriers between the different minima evaluated at the end of our trajectories (simulation time 1000 ns) with Chen&Garcia are 2.04 kT and 3.11 kT at 300 K for 2H-HT and HT-ST, respectively.The three minima were also characterized in term of their relative preferred interactions, both in terms of intermolecular (uracil–uracil) interactions and with respect to the solvent. We calculated the number of contacts between N and O atoms of the uracil molecules and the O atoms of the water molecules (either donor or acceptor of hydrogen bonds). As presented in Fig. 5(a) together with results extracted from monomer states (i.e. (s,h) = (0,0)), the largest number of contacts is developed by the ST dimer, followed by the HT and 2H. This is not surprising based on the decreasing availability of N and O atoms. We proceed by evaluating the distributions of relevant short distances for the HT and 2H basins. In Fig. 5(b), the contacts between pairs of O and N (HB donor or acceptor; labeled as Oi.Nj or Ni.Oj, where i and j are atomic indices) atoms are shown. A few observations can be made with respect to these data. Firstly, in HT, there is a preference for the formation of the HBs between N1 and O4. Such configurations have been already reported in the literature.21 Secondly, while N3.O2 interactions dominate in the 2H configuration, they are almost not encountered in the HT basin. We propose that these two facts are correlated: the formation of the N3.O2 contact immediately triggers the formation of a second HB and therefore it's highly correlated to its transformation to the 2H configuration. These hydrogen-bonding interactions can be compared with the ones encountered in the uracil crystal,61 where O4 has a central role acting as the HB acceptor for the two HBs coming from the nearest in-plane neighboring uracil molecules (through N1 and N3). Additionally, O2 is forming a weak HB with C6.
 | ||
| Fig. 5 (a) Interaction with solvent quantified by the number of contacts developed by HB donor or acceptor atoms in the uracil (N1, N3, O2, O4) and the surrounding water molecules, for the different dimer states (continuous lines) and monomer (dashed lines). (b) Intermolecular interactions quantified as the distribution of specific contact for the HT and 2H states at heavy atom distances. | ||
In ref. 21, 7 minima were reported for the 2H configuration (in vacuum). We relate those results to HB intermolecular connectivity. The details of the analysis are presented in the Methods section. We build a 2D distribution of two quantities: the difference between the NC developed by the N1 atom (NC (N1.O2) minus NC (N1.O4)) and the difference between the contacts developed by the N3 atom (NC (N3.O2) minus NC (N3.O4)). The constructed 2D histogram is reported in Fig. S8 in ESI† and it clearly presents 6 spots that are related to 6 of the 7 configurations previously presented.21 This confirms that all of these states survive in our explicit solvent simulation except the one involving weak HB on the C6 (HB7 in ref. 21). However, the ranking of the different configurations is clearly altered by the change of the environment. A detailed report is given in ESI,† Table S4.
As intermolecular and stacking motifs, in particular, are connected to the propensity for photochemical dimerisation, we have investigated the formation of intramolecular contacts and their preferred orientations. We introduce two supramolecular geometrical descriptors, namely (a) the angle α(n,n′) formed by the two normals of uracil plane n, n′ and (b) the angle τ(N1C4,N1′C4′) formed by the two in-plane vectors connecting N1 to C4 (see Fig. 6(a) and (b)). In Fig. 6(c) and (d), we show the results for the pi-stacking configuration, while the results for the remaining two configurations are shown in Fig. S8 in ESI.†ST state (being the widest basin) is characterized by a larger disorder and the only significant barrier is the one going from the parallel to antiparallel alignment of the uracil rings. Interestingly, the heatmaps computed for the ST configuration suggest the presence of 3 subminima: two for a parallel alignment of uracil rings (or using the nomenclature presented in ref. 23, face-to-back or F2B) and one for an antiparallel alignment of uracil rings (face-to-face or F2F). The definition and the populations of these states are reported in Table S5 in ESI.† When comparing the τ angle (or, equivalently, twist angle) distributions, only 2 out of 3 ST states match previously reported ab initio MD results23 and the F2B states with τ ≈ 160° was not sampled. Hunter and van Mourik63 presented a systematic DFT investigation of all minima both in continuum solvent and in vacuum. They reported 3 minima for the F2F and 3 minima for the F2B configuration. However, their continuum treatment of the solvent does not appear to provide an accurate description of the twist angles distribution and our results better match the results obtained in vacuum. Three minima were also presented for stacking dimers in vacuum,21 but alike ref. 63, there is no complete correspondence with our results since the rotations profile of the two rings is likely to be disturbed by the water bridges participating in the H-bonds with either uracil molecule. In terms of population, we find a small preponderance for the F2B configuration. In an earlier work,23 F2F configurations have proven to be pro-reactive towards photodimerization. Lastly, our ST basin is only capturing two (trans-syn and cys-sin) of the four possible states giving rise to photodimerization.27,64
Several nucleobase homo- and hetero-dimers were investigated by Florián et al. using post-HF and Langevin dipoles solvation model.65 It was found that that free energies for the formation of stacked and hydrogen-bonded complexes fell in a narrow region between 0.3 and −1.9 kcal mol−1. However, for uracil dimer, only stacking configuration were investigated, reporting a positive free energy of +0.3 kcal mol−1. Interestingly, this was the only reported stacking nucleobase dimer with a positive free energy, in agreement with our results that shows that the (0,0) basin is the deepest. While this work reported that the free energy differences between hydrogen-bonded and stacked configurations for investigated bases were smaller than 0.5 kcal mol−1 (consistent with our findings), no conclusions could be drawn for uracil since hydrogen-bonded complexes were not studied. Thus, a direct comparison with our results remains unfeasible. Besides, uracil ribodinucleoside monophosphates (UpU) were investigated along with others dinucleoside monophosphate anions in aqueous solution using MD.66 For UpU the stacking free energy was estimated to be around +0.1 kcal mol−1, in line with previous results. Even in this bound environment, purine dinucleoside were found to have unfavourable stacking (ΔG > 0), again in line with our results.
 | ||
| Fig. 6 (a), (b) sketches used to define the normal to the plane (C2–C4–C6) and the τ angle formed by N1 and C4 atoms. (c) Heatmap describing the number of intramolecular contacts developed by the N1 atom (x axis) and the N3 atom (y axis) in the 2H state. HB1,…, HB6 labels follow the definitions provided by Kratochvíl et al.21 The corresponding contact populations are reported in the Table S5 in the ESI.† (d) Heatmap of the angular vector-based descriptors for the ST states. | ||
Impact on the optical absorption
After having demonstrated the accuracy of the free energy surface obtained with Chen&Garcia, we calculate experimentally relevant quantities, so as to indicate observables capable of differentiating the three dimeric states of uracil in water. Optical properties of uracil have been studied in the past in the presence and absence of solvated water molecules. There are two low-lying excitations: a nπ* dark excitation and a ππ* bright excitation,26,67 whose order is dependent on the environment. The presence of water has been found to redshift the ππ* transition and blue-shift the nπ* one. Additionally, light absorption in pyrimidine nucleobases is intimately connected with its photochemistry.6,16 Here, we establish a relationship between the formation of supramolecular assemblies and the absorption profile.We compare the absorption spectra for several dimer configurations extracted from the three nontrivial minima basins with spectra obtained for monomer configurations from the (0,0) basin. Such water–uracil clusters were optimized by DFT (PBE0-D3BJ functional) in the presence of continuum solvent, followed by a time-dependent DFT (TDDFT) calculation (PBE0 or ωB97X functional) to compute the spectra. Other computational details are given in the Methods section. For the clusters of monomer with water molecules, we obtain absorption peaks at 5.48 eV (for PBE0) and 5.71 eV (ωB97X). As reported in Table 3 and in ESI,† Fig. S9, we found that the formation of dimers leads to small effects on the absorption band, with HT and, to a minor extent, ST being slightly red-shifted, while 2H being only minimally blue-shifted. In spite of minor differences, the shifts are consistent across the two different DFT functionals. The systematic redshift associated with the formation of dimers, and HT in particular, seems to depend on a complex interplay of factors. We found a mild dependence on the elongation of the C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O4 bonds (see ESI,† Fig. S10), which is generally coupled to the different H-bonding patterns occurring for the monomer in water vs. the patterns occurring in the different dimer states. This appears logical as the most common low-lying transitions are the ππ*(C4O4). In particular, we found that HT is often associated with more pronounced elongation of C4O4, which is probably a consequence of a single and stronger hydrogen bond with the other uracil molecule. On the other hand, a shorter distance between the two COMs does not imply larger redshifts – as in the case of ST basin. Only one previous investigation of the dependence of absorption on uracil concentration in water solution exists.68 Although the simulated spectral shifts are small, repetitions of these experiments may present a strategy to validate the proposed free-energy rankings and their spectral consequences.
O4 bonds (see ESI,† Fig. S10), which is generally coupled to the different H-bonding patterns occurring for the monomer in water vs. the patterns occurring in the different dimer states. This appears logical as the most common low-lying transitions are the ππ*(C4O4). In particular, we found that HT is often associated with more pronounced elongation of C4O4, which is probably a consequence of a single and stronger hydrogen bond with the other uracil molecule. On the other hand, a shorter distance between the two COMs does not imply larger redshifts – as in the case of ST basin. Only one previous investigation of the dependence of absorption on uracil concentration in water solution exists.68 Although the simulated spectral shifts are small, repetitions of these experiments may present a strategy to validate the proposed free-energy rankings and their spectral consequences.
 , with S(ω) being the first absorption band) with respect to monomer results, using two different DFT functionals
, with S(ω) being the first absorption band) with respect to monomer results, using two different DFT functionals
| Abs. max. shift (eV) | Abs. centroid shift (eV) | |||
|---|---|---|---|---|
| with respect to monomer | ||||
| PBE0 | ωB7X | PBE0 | ωB7X | |
| HT | −0.13 | −0.11 | −0.087 | −0.088 |
| 2H | −0.03 | −0.040 | −0.034 | −0.036 |
| ST | −0.05 | −0.05 | −0.072 | −0.075 |
4 Conclusion
Modeling nucleobase assemblies and aggregates involves the challenging determination of their structural properties through the dominant interaction motifs. This study employs computational techniques to explore the configurational space of uracil dimers in a water solution, overcoming computational challenges by utilizing classical force fields. wt-MetaD simulations were employed to effectively sample the large configuration space of a uracil dimer in water. The results of the wt-MetaD simulations were validated against MD simulations biased with walls. In our study, the CVs were defined to include specific atoms of uracil molecules that are needed to describe (π–)π stacking interactions and hydrogen bonds.51 This approach aimed to capture the key intermolecular interactions in the system. Using configurations extracted from minima basins of the obtained free energy surface, we carried out TDDFT calculations to assess the impact of different dimer arrangements on the optical properties of the investigated system.Additionally, we evaluated the accuracy of the Chen&Garcia32 and ff9935 force fields against DFT. We found that the Chen&Garcia force field32 is the more accurate force field for the investigated system, thereby resolving previous concerns about its predictive power33,62 and showing that in general, it might present the preferred choice as a model of nucleobases in solution.
Data availability
All the methodological details and the relevant information to reproduce our results are available: in the manuscript, under the Methods section in the ESI† as a dataset on Zenodo repository, accessible at: https://doi.org/10.5281/zenodo.15262465.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Croatian Science Foundation (HrZZ) under projects: IP-2020-02-7262 and DOK-2021-02-5899 (HYMO4EXNOMOMA). We thank Tomislav Stolar and Barbara Rossi for the useful discussions concerning the related experimental information on these systems. We thank Sandro Bottaro and Giovanni Bussi for valuable discussions at the preliminary phases of this investigation. We acknowledge ISCRA for awarding access to the LEONARDO supercomputer through BASOP (HP10CBY5GJ) and MASOP (HP10CQ302Z) projects, owned by the EuroHPC Joint Undertaking, hosted by CINECA (Italy). We acknowledge Zagreb University Computing Centre – SRCE for providing the Advanced computing service “Supek”.Notes and references
- D. L. Nelson, A. L. Lehninger and M. M. Cox, Lehninger principles of biochemistry, Macmillan, 2008 Search PubMed.
- P. Cieplak and P. A. Kollman, J. Am. Chem. Soc., 1988, 110, 3734–3739 CrossRef CAS.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2011, 13, 10908–10922 RSC.
- N. V. Hud, S. S. Jain, X. Li and D. G. Lynn, Chem. Biodiversity, 2007, 4, 768–783 CrossRef CAS PubMed.
- R. Szabla, M. Zdrowowicz, P. Spisz, N. J. Green, P. Stadlbauer, H. Kruse, J. Šponer and J. Rak, Nat. Commun., 2021, 12, 3018 CrossRef PubMed.
- W. J. Schreier, P. Gilch and W. Zinth, Annu. Rev. Phys. Chem., 2015, 66, 497–519 CrossRef CAS PubMed.
- R. Saladino, C. Crestini, G. Costanzo and E. DiMauro, Prebiotic Chem., 2005, 29–68 CAS.
- M. Winkler, B. M. Giuliano and P. Caselli, ACS Earth Space Chem., 2020, 4, 2320–2326 CrossRef CAS.
- A. Varghese, Biochemistry, 1971, 10, 4283–4290 CrossRef CAS PubMed.
- R. Improta, F. Santoro and L. Blancafort, Chem. Rev., 2016, 116, 3540–3593 CrossRef CAS PubMed.
- G. Fisher and H. Johns, Photochem. Photobiol. Nucleic Acids, 1976, 1, 225–294 CAS.
- T. Montenay-Garestier, M. Charlier and C. Hélène, Photochemistry and photobiology of nucleic acids, Elsevier, 1976, pp. 381–417 Search PubMed.
- A. A. Beckstead, Y. Zhang, M. S. de Vries and B. Kohler, Phys. Chem. Chem. Phys., 2016, 18, 24228–24238 RSC.
- L. Martínez Fernández, F. Santoro and R. Improta, Acc. Chem. Res., 2022, 55, 2077–2087 CrossRef PubMed.
- S. Boldissar and M. S. de Vries, Phys. Chem. Chem. Phys., 2018, 20, 9701–9716 RSC.
- M. Shukla and J. Leszczynski, J. Biomol. Struct. Dyn., 2007, 25, 93–118 CrossRef CAS PubMed.
- T. Stolar, B. K. Pearce, M. Etter, K.-N. Truong, T. Ostojić, A. Krajnc, G. Mali, B. Rossi, K. Molčanov, I. Lončarić, E. Meštrović, K. Užarević and L. Grisanti, iScience, 2024, 27, 109894 CrossRef CAS PubMed.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC.
- A. Sikder, C. Esen and R. K. OReilly, Acc. Chem. Res., 2022, 55, 1609–1619 CrossRef CAS PubMed.
- F. Pu, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 1285–1306 RSC.
- M. Kratochvíl, O. Engkvist, J. Šponer, P. Jungwirth and P. Hobza, J. Phys. Chem. A, 1998, 102, 6921–6926 CrossRef.
- C. A. Morgado, P. Jurecka, D. Svozil, P. Hobza and J. Sponer, J. Chem. Theory Comput., 2009, 5, 1524–1544 CrossRef CAS PubMed.
- B. Milovanović, J. Novak, M. Etinski, W. Domcke and N. Došlić, Phys. Chem. Chem. Phys., 2022, 24, 14836–14845 RSC.
- B. Milovanović, M. Kojić, M. Petković and M. Etinski, J. Chem. Theory Comput., 2018, 14, 2621–2632 CrossRef PubMed.
- J. Smets, W. J. McCarthy and L. Adamowicz, J. Phys. Chem., 1996, 100, 14655–14660 CrossRef CAS.
- B. Milovanović, J. Novak, M. Etinski, W. Domcke and N. Došlić, Phys. Chem. Chem. Phys., 2021, 23, 2594–2604 RSC.
- V. B. Delchev and W. Domcke, J. Photochem. Photobiol., A, 2013, 271, 1–7 CrossRef CAS.
- R. C. Bernardi, M. C. Melo and K. Schulten, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 872–877 CrossRef CAS PubMed.
- G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 2, 200–212 CrossRef.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- M. Zgarbová, M. Otyepka, J. Sponer, A. Mladek, P. Banas, T. E. Cheatham III and P. Jurecka, J. Chem. Theory Comput., 2011, 7, 2886–2902 CrossRef PubMed.
- A. A. Chen and A. E. García, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16820–16825 CrossRef CAS PubMed.
- K. B. Hall, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16706–16707 CrossRef CAS PubMed.
- J. Sponer, G. Bussi, M. Krepl, P. Banáš, S. Bottaro, R. A. Cunha, A. Gil-Ley, G. Pinamonti, S. Poblete and P. Jurecka, et al. , Chem. Rev., 2018, 118, 4177–4338 CrossRef CAS PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1996, 118, 2309 CrossRef CAS.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- Y. I. Yang, Q. Shao, J. Zhang, L. Yang and Y. Q. Gao, J. Chem. Phys., 2019, 151, 070902 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. B. B. M. G. Scalmani, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. N. Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. McLean and G. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- J.-P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016–5021 CrossRef CAS.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- P. Bauer, B. Hess and E. Lindahl, GROMACS 2022.3 Manual DOI:10.5281/zenodo.6103567.
- A. W. Sousa da Silva and W. F. Vranken, BMC Res. notes, 2012, 5, 1–8 CrossRef PubMed.
- The PLUMED consortium, Nat. Methods, 2019, 16, 670–673 CrossRef PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci and R. A. Broglia, et al. , Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- S. Dasari and B. S. Mallik, J. Phys. Chem. B, 2018, 122, 9635–9645 CrossRef CAS PubMed.
- H. J. Berendsen, J. v Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- R. Sure and S. Grimme, J. Comput. Chem., 2013, 34, 1672–1685 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- R. F. Stewart and L. H. Jensen, Acta Crystallogr., Sect. A, 1967, 23, 1102–1105 CrossRef CAS.
- C. Bergonzo, N. M. Henriksen, D. R. Roe and T. E. Cheatham, RNA, 2015, 21, 1578–1590 CrossRef CAS PubMed.
- R. S. Hunter and T. Van Mourik, J. Comput. Chem., 2012, 33, 2161–2172 CrossRef CAS PubMed.
- P. B. Kancheva and V. B. Delchev, Acta Chim. Slov., 2018, 65, 521–530 CrossRef CAS PubMed.
- J. Florian, J. Šponer and A. Warshel, J. Phys. Chem. B, 1999, 103, 884–892 CrossRef CAS.
- S. Jafilan, L. Klein, C. Hyun and J. Florián, J. Phys. Chem. B, 2012, 116, 3613–3618 CrossRef CAS PubMed.
- T. Gustavsson, A. Bányász, E. Lazzarotto, D. Markovitsi, G. Scalmani, M. J. Frisch, V. Barone and R. Improta, J. Am. Chem. Soc., 2006, 128, 607–619 CrossRef CAS PubMed.
- N. Iza, M. Gil, J. Montero and J. Morcillo, J. Mol. Struct., 1988, 175, 19–24 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04238g |
| This journal is © the Owner Societies 2025 |