
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pressure-dependent kinetic analysis of the N2H3 potential energy surface†
Michal
Keslin
a,
Kfir
Kaplan
a and
Alon
Grinberg Dana
 *ab
*ab
aWolfson Department of Chemical Engineering, Technion – Israel Institute of Technology, Haifa 3200003, Israel. E-mail: alon@technion.ac.il; Tel: +972-73-378-2117
bGrand Technion Energy Program (GTEP), Technion – Israel Institute of Technology, Haifa 3200003, Israel
First published on 9th January 2025
Abstract
The pressure-dependent reactions on the N2H3 potential energy surface (PES) have been investigated using CCSD(T)-F12/aug-cc-pVTZ-F12//B2PLYP-D3/aug-cc-pVTZ. This study expands the N2H3 PES beyond the previous literature by incorporating a newly identified isomer, NH3N, along with additional bimolecular reaction channels associated with this isomer, namely NNH + H2 and H2NN(S) + H. Rate coefficients for all relevant pressure-dependent reactions, including well-skipping pathways, are predicted using a combination of ab initio transition state theory and master equation simulations. The dominant product of the NH2 + NH(T) recombination is N2H2 + H, while at high pressures and low temperatures, N2H3 formation becomes significant. Similarly, collisions involving H2NN(S) + H predominantly produce N2H2 + H. Secondary reactions such as H2NN(S) + H ⇌ NNH + H2 and H2NN(S) + H ⇌ NH2 + NH(T) are found to play a significant role at high temperatures across all examined pressures, while H2NN(S) + H ⇌ NH3N becomes prominent only at high pressures. Notably, none of these four H2NN(S) reactions have been included with pressure-dependent rate coefficients in previous NH3 oxidation models. The rate coefficients reported here provide valuable insights for modeling the combustion of ammonia, hydrazine, and their derivatives in diverse environments.
1. Introduction
Nitrogen-based fuels, including ammonia (NH3),1 hydrazine (N2H4),2,3 hydrazine derivatives such as monomethylhydrazine (MMH, CH3NHNH2),2 and aqueous formulations,4,5 form a versatile class of energy carriers. Hydrazine and its derivatives are widely used as satellite and spacecraft propellants, while ammonia receives an increasing interest as a potential energy storage vector.6 In the envisioned future nitrogen economy,7 ammonia is expected to play a key role. Despite differences in their specific applications, the chemical kinetic modeling of these nitrogen-based fuels relies on a shared core chemistry and a set of fundamental reactions.8Hydrazine, a principal component of diamine-based rocket propellants, stands out as an efficient mono- and bi-propellant. Paired with oxygen as an oxidizer, it is second only to hydrogen in terms of specific impulse—the thrust generated per unit mass of fuel over time.3 Hydrazine can be used as a monopropellant, typically undergoing decomposition using a catalyst,3 leading to autoignition and detonation.9–11 Furthermore, hydrazine and hydrazine-based fuels, including MMH, function as bipropellants when combined with nitrogen tetroxide (N2O4) as the oxidizer. These fuels are ubiquitous as thruster engine propellants for satellite and spacecraft attitude control and in-orbit maneuvers,3 where they are stored and used at a maximum pressure of ∼25 bar.12–14
Nitrogen-based alternative fuels are particularly attractive due to the abundance of molecular atmospheric nitrogen as a feedstock. Among them, ammonia is the simplest molecule and it shows significant promise as an energy storage vector.1,6,15 The advantages of ammonia as a fuel include a relatively high power-to-fuel-to-power (PFP) efficiency,7 a large-scale distribution infrastructure that is already in place,16,17 a high octane rating,18 and a narrow flammability range that makes it relatively safe in terms of explosion risks. Although kinetic modeling of NH3 oxidation received significant attention lately,19–35 there is still substantial disagreements on thermodynamic data and rate coefficients between different authors in this relatively small chemical system.8,36–39
Konnov and De Ruyck40 suggested a chemical kinetic model for hydrazine decomposition, and Izato et al.41 suggested a model for the initial hypergolic reaction of liquid hydrazine with nitrogen tetroxide mixtures. Hydrazine decomposition was also studied by Asatryan et al.,42 and its key pathways in hypergolic ignition with nitrogen dioxide and nitrogen tetroxide were suggested by Raghunath et al.43 and by Daimon et al.,44 respectively. The thermal decomposition of hydrazine was studied using a reactive force field.45 Hydrazine decomposition on catalytic surfaces was also studied theoretically.46,47 Sun and Law48 developed a model for thermal decomposition of MMH, and Zhang et al.49 performed a theoretical analysis of the kinetics of barrierless recombination reactions in the MMH system as well as secondary channels.50 Although chemical kinetic models were previously suggested to predict the reactivity of hydrazine and its derivative, the research on the combustion of these fuels has been based on incomplete kinetic mechanisms, which may not fully capture all the relevant reaction pathways. Despite the development of detailed kinetic models, there is a demand for more advanced modeling approaches.51
The present work focuses on studying the N2H3 potential energy surface (PES) that is relevant for all the chemical systems mentioned above. This PES was previously studied by Klippenstein et al.52 at the CCSD(T)/CBS//CCSD(T)/aug-cc-pVDZ level of theory when exploring reactions originating from the NH2 + NH channel. Klippenstein et al. showed that the reaction proceeds through the abstraction of an H atom on the quartet surface (forming NH3 + N(Q)) or by addition followed by H atom elimination on the doublet surface (forming N2H2 + H), as shown in Fig. 1(A). They computed a rate coefficient for the barrierless initial addition of NH2 + NH(T) using variable reaction-coordinate transition state theory (VRC-TST). The authors stated that a transition state (TS) is likely to exist for H2 elimination from N2H3 but was not found in the cited study.52 They expected such a TS to lie higher in energy than that for H loss, and thus it should make an insignificant contribution to the kinetics. Raghunath et al.43 slightly refined the pressure-dependent network by explicitly considering the cis and trans isomers of N2H2, but did not consider the barrierless addition reaction (Fig. 1(B)). Diévart and Catoire53 used the high-pressure VRC-TST rate coefficient calculated by Klippenstein et al.52 along with an inverse Laplace transform (ILT)54 to suggest pressure-dependent reaction rate coefficients for reactions N2H3 + M ⇌ NH2 + NH + M and N2H3 + M ⇌ N2H2 + H. However, they also only considered the trans isomer of the N2H2 product (Fig. 1(C)) and did not provide a rate coefficient for the well-skipping reaction NH2 + NH(T) ⇌ N2H2 + H.
 | ||
| Fig. 1 Representations of the N2H3 PES from previous literature works in kcal mol−1. (A) CCSD(T)/CBS//CCSD(T)/aug-cc-pVDZ level of theory calculations by Klippenstein et al.52 The solid and dashed lines correspond to the doublet and quartet surfaces, respectively. (B) CCSD(T)/CBS//MP2/6-311++G(3df,2p) level of theory calculations by Raghunath et al.43 on the doublet surface (this inset is not plotted to scale, as in the original manuscript). (C) RCCSD(T)/CBS//M06-2x-D3/aug-cc-pVTZ level of theory calculation by Diévart and Catoire53 on the doublet surface. Usage rights: (A) Reprinted (adapted) with permission from J. Phys. Chem. A 2009, 113, 38, 10241–10259. Copyright 2009 American Chemical Society. (B) Reprinted from Publication P. Raghunath, N. T. Nghia, M.-C. Lin “Chapter Seven – Ab Initio Chemical Kinetics of Key Processes in the Hypergolic Ignition of Hydrazine and Nitrogen Tetroxide”, Adv. Quantum Chem.69, 2014, 253–301, Copyright (2014), with permission from Elsevier. (C) Reprinted (adapted) with permission from J. Phys. Chem. A 2020, 124, 30, 6214–6236. Copyright 2020 American Chemical Society. | ||
Pressure-dependent rate coefficients have not previously been reported for the NH2 + NH(T) ⇌ N2H2 + H and N2H2 + H ⇌ NNH + H2 reactions, nor for any reaction on this PES involving the H2NN(S) + H well, such as the formation of N2H2 + H, NNH + H2, or N2H3. Currently, the literature lacks a comprehensive description of the N2H3 PES, as well as a complete set of pressure-dependent reaction rate coefficients for all relevant pathways. The goal of the present work is to address these gaps and provide kinetic modellers of nitrogen-based fuel systems with high-quality kinetic data for the unimolecular N2H3 system.
2. Methods
Ab initio calculations were carried out using the open source Automated Rate Calculator (ARC) software suite,55 which automates the calculation of species thermochemistry and reaction rate coefficients. Conformer geometry searches were performed based on a random set of force field conformers generated via RDKit56 using MMFF94s.57 The force field conformers were optimized at the ωB97X-D/Def2-SVP58,59 level of theory. The lowest energy DFT (density functional theory) conformer was further used and optimized at the double-hybrid B2PLYP functional60 with Grimme's dispersion correction, D3(BJ),61 coupled with Dunning's correlation consistent basis set62 augmented with diffuse functions, i.e., B2PLYP-D3/aug-cc-pVTZ. Ro-vibrational analyses were subsequently computed via ARC at the same level of theory. ARC calculated a frequency scaling factor of 0.995 for this functional and basis set combination using the method recommended by Truhlar et al.63 Quantum tunneling effects were incorporated using the Eckart correction.64 TS verification was performed by intrinsic reaction coordinate (IRC) computations65 and automated inspection of the normal mode displacement.Torsional modes were automatically identified as rotatable single bonds (accounting for relevant resonance structures66) and treated with continuous constrained optimizations with all other internal coordinates relaxed using the B2PLYP-D3/aug-cc-pVTZ level of theory at a dihedral angle resolution of 10°. The 1D potential energy torsional scans were fitted to truncated Fourier series that were used as input to compute energy levels and the partition function of anharmonic modes using Arkane,67 an open source software distributed within the RMG software suite.68 All DFT calculations were performed in Gaussian 09.69
Single-point energies were computed using three methods: 1. CCSD(T)-F1270–72 with the correlation-consistent aug-cc-pVTZ-F12 base set,62,73 2. MRCI-F12+Q (internal contracted multiconfiguration reference configuration-interaction with explicit correlation F12 and Davidson correction)74–76 with the same basis set, and 3. CCSDT(Q),77 which approximates full CCSDTQ calculations78 by extending coupled-cluster theory to include full triples excitations (CCSDT) with a perturbative treatment of connected quadruple excitations, using the aug-cc-pVTZ basis set. The MRCI-F12+Q calculations were performed with the full valence active space that included 13 valence electrons and 11 active orbitals for the isomers and TSs on the N2H3 PES. The CCSD(T)-F12 and MRCI-F12+Q calculations were performed in Molpro 2021.2,79 and the CCSDT(Q) calculations were performed in Psi4 v. 1.9.1.80 The frozen core approximation was applied to the CCSDT(Q) computations, wherein the 1s orbitals of nitrogen atoms were kept frozen while the valence and virtual orbitals were fully correlated. This approach effectively reduces computational cost without significantly compromising the accuracy of energy calculations. Empirical systematic errors in atomization energies were corrected using atom energy corrections implemented in Arkane67,81 for the respective levels of theory.
Statistical mechanics processing of all electronic structure calculations were done by Arkane67 to calculate the thermochemical partition functions and macroscopic parameters, including reaction rate coefficients of interest. Pressure-dependent reaction networks were modeled using a master equation (ME) describing molecular processes on a collisional timescale.82 The Modified Strong Collision (MSC) approximation83 was used to solve the ME since the more accurate Chemically Significant Eigenvalues (CSE) method84 did not converge for this case. Collision energy transfer rates for deactivating collisions were modeled using the “single exponential down” expression, 〈ΔEd〉, with nitrogen as the bath gas. Nitrogen-based systems exhibit a somewhat different trend of 〈ΔEd〉 vs. temperature than carbon-based systems.67 Since both N4H6 + N2 and N3H5 + N2 were shown to have relatively similar values,67 the parameters computed for the N3H5 + N2 system are assumed to be representative of the present N2H3 + N2 system as well, and were adopted here: 〈ΔEd〉 = 175·(T/298 K)0.52 cm−1. Lennard-Jones parameters were taken from RMG's “OneDMinN2” transport library85 computed using OneDMin,86 and from Glarborg et al.19
An ILT,54 a method of deriving microcanonical transition state theory rate coefficients as a function of the total energy from high-pressure limit rate coefficients, was used as implemented in Arkane67 for the high-pressure limit recombination rate coefficient of the entrance channel NH2 + NH(T) as determined by Klippenstein et al.,52 and of the recombination channel H2NN(S) + H as estimated by RMG.85 Pressure-dependent rate coefficients were fitted to Chebyshev polynomials87 over a temperature range of 300–3000 K and a pressure range of 0.01–100 bar. Pressure-dependent network sensitivity analysis was performed using Arkane67 by perturbing the E0 values of each well and TS in the PES and determining the effect on the calculated pressure-dependent rate coefficients.
3. Results and discussion
The doublet N2H3 PES (Fig. 2) involves pressure-dependent reactions among five bimolecular channels interconnected via two isomers, N2H3 and NH3N (Fig. 3). Fig. S2 (ESI†) compares the present computations with the literature values shown in Fig. 1. The N2H3 radical is relatively stable, with an endothermic dissociation energy of ∼200 kJ mol−1 (Fig. 2), significantly higher than similar beta-scission reactions.88 The association of NH2 + NH(T) (amino radical and triplet imidogen) without further isomerization can result in the formation of N2H3 or diazene (N2H2) and a hydrogen radical, as suggested by previous works.43,52,53 However, the previous studies did not consider additional isomers in this PES other than N2H3 (Fig. 1). The NH3N isomer (Fig. 2 and 3(B), SMILES89 representation: [NH3+][N-]) is proposed as a conformation that facilitates further transformations on this PES. Thermodynamic properties for this isomer, calculated at the CCSD(T)-F12/cc-pVTZ-F12//B2PLYP-D3/aug-cc-pVTZ level of theory, are provided in Tables S2 and S3 (ESI†). The NH3N isomer, a stable well in this PES, allows the pressure-dependent network to access two additional bimolecular wells: NNH + H2 and H2NN(S) + H. The N2H3 isomer can also produce barrierlessly H2NN(S) + H (Fig. 2 and Fig. S8, S9, ESI†).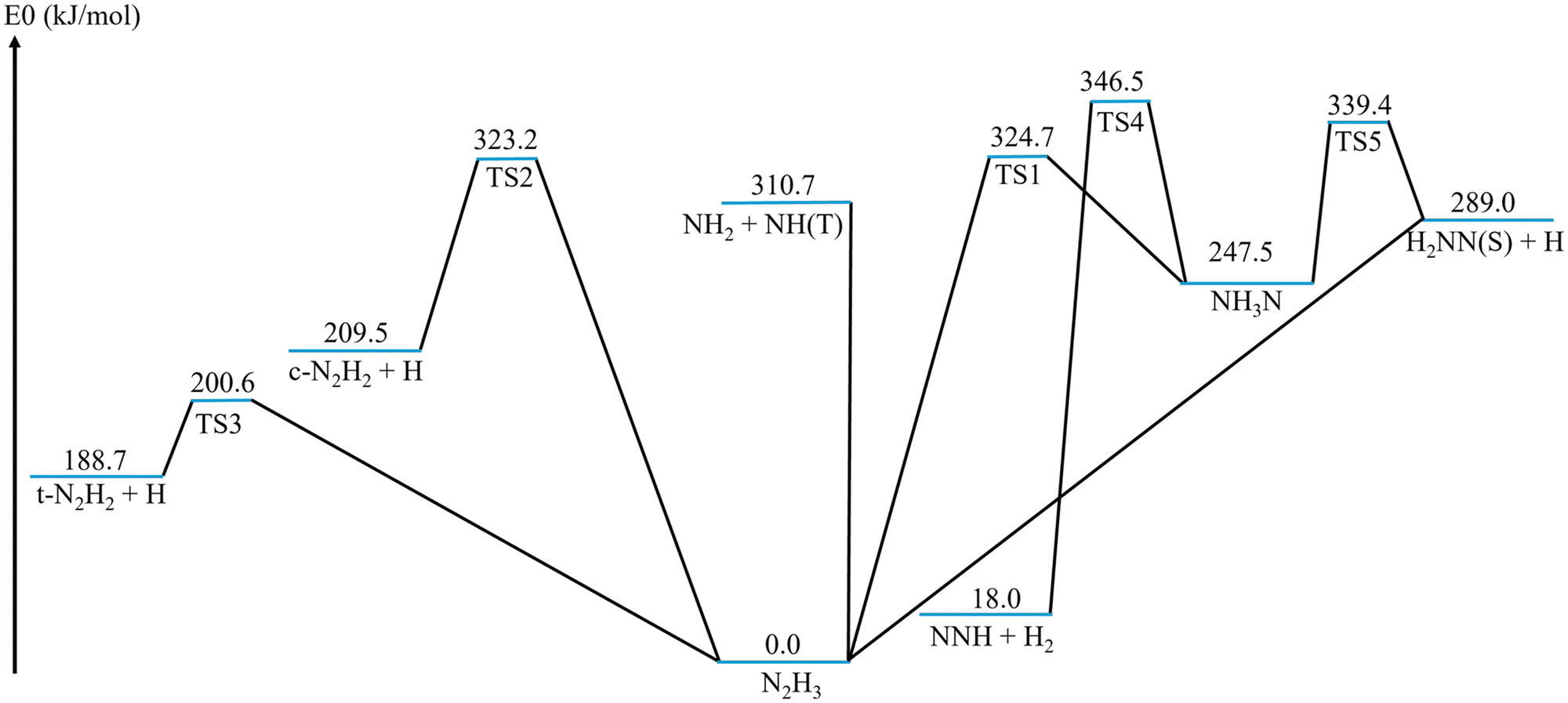 | ||
| Fig. 2 The pressure-dependent kinetic network of the doublet N2H3 PES computed at the CCSD(T)-F12a/aug-cc-pVTZ-F12//B2PLYP-D3/aug-cc-pVTZ level of theory. Zero-point energies (electronic energy plus zero-point energy correction) are in kJ mol−1. | ||
 | ||
| Fig. 3 Geometric representation of 3D structures of the two isomers on the explored PES, (A) N2H3 and (B) NH3N, optimized at the B2PLYP-D3/aug-cc-pVTZ level of theory. Numbers represent internal coordinate values at the B2PLYP-D3/aug-cc-pVTZ level and in parenthesis at the CCSD/aug-cc-pVDZ level for comparison. The Cartesian coordinates are given in Table S1 (ESI†). Additional internal coordinate comparisons of computed geometries are given in Tables S6–S10 (ESI†). | ||
The NH3 + N(D) bimolecular well, which is not shown in Fig. 2, was found to lie more than 400 kJ mol−1 above the N2H3 isomer (Table 1) and was therefore excluded from the ME analysis. This path originates from the NH3N isomer. A TS for this channel was located using DFT, but its energy at the CCSDT(Q)/aug-cc-pVTZ level was lower than that of the corresponding bimolecular products. This reaction is likely barrierless, as previously determined for a similar reaction, CH4 + N(D).90 This channel is not important for the kinetics of N2H3 decomposition due to its high energy, although it might be relevant for plasma-assisted ammonia oxidation systems.91
| Stationary point | T1 diagnostic coefficient(s) | CCSD(T)-F12a | MRCI-F12+Q | CCSDT(Q) | ATcT |
|---|---|---|---|---|---|
| N2H3 | 0.025 | 0.0 | 0.0 | 0.0 | 0.0 |
| NH3N | 0.012 | 247.5 | 234.6 | 243.9 | — |
| NH2 + NH(T) | 0.008, 0.001 | 310.7 | 287.4 | 295.1 | 312.9 |
| c-N2H2 + H | 0.013, 0.000 | 209.5 | 194.9 | 196.3 | 210.1 |
| t-N2H2 + H | 0.013, 0.000 | 188.7 | 170.7 | 175.2 | 188.4 |
| H2NN(S) + H | 0.021, 0.000 | 289.0 | 270.6 | 276.5 | 288.8 |
| NNH + H2 | 0.028, 0.006 | 18.0 | −2.4 | 16.5 | 17.4 |
| NH3 + N(D) | 0.008, 0.006 | 447.4 | 413.8 | 415.3 | 427.2 |
| TS1 | 0.025 | 324.7 | 311.6 | 324.7 | |
| TS2 | 0.020 | 223.2 | 208.7 | 220.3 | |
| TS3 | 0.021 | 200.6 | 190.5 | 197.3 | |
| TS4 | 0.047 | 346.5 | 333.8 | 293.4 | |
| TS5 | 0.088 | 339.4 | 321.6 | 321.9 |
Klippenstein et al.52 commented that a TS which leads to the elimination of H2 from N2H3 likely exists, but was not found in their study. Klippenstein et al. also noted that the missing TS is likely to have a higher energy than TSs for H loss and therefore this pathway should not contribute significantly to the kinetics.52 In the present study, we were also unable to identify a TS corresponding to H2 elimination directly from the N2H3 isomer. However, we show here that the elimination of H2 in this PES stems from the NH3N isomer (TS4, Fig. 2) rather than from N2H3. This TS was indeed found to lie significantly higher in energy than the H loss TSs (TS2 and TS3, Fig. 2). Consequently, this isomerization-mediated H2 elimination channel from N2H3 or from NH2 + NH(T) becomes relevant only at high temperature and low pressure conditions.
The NNH + H2 products are the lowest-lying bimolecular well in this PES, whereas the t-N2H2 + H products' well is the most kinetically accessible from the NH2 + NH(T) direction due to the relatively low energy barrier (TS3, Fig. 2). Another important entry channel into the N2H3 pressure-dependent system is the H2NN(S) + H well that could lead to either of the two isomers or to the NH2 + NH(T), N2H2 + H, or NNH + H2 bimolecular wells (Fig. 2).
The geometries of the TSs identified in this PES are shown in Fig. 4. The computed imaginary frequencies of all TSs are provided in Table S1 (ESI†). Table 1 provides the T1 diagnostic coefficients93–95 for all stationary points on the PES. It also lists the corresponding zero-point energies calculated at three levels of theory: CCSD(T)-F12a/aug-cc-pVTZ-F12, MRCI-F12+Q/aug-cc-pVTZ-F12, and CCSDT(Q)/aug-cc-pVTZ. All calculations were performed using B2PLYP-D3/aug-cc-pVTZ geometries and frequencies.
 | ||
| Fig. 4 Geometric representations of TSs on the N2H3 PES corresponding to the five non-barrierless path reactions, optimized at the B2PLYP-D3/aug-cc-pVTZ level of theory. Blue spheres represent nitrogen atoms, smaller light shaded balls represent hydrogen atoms, bond orders are given for illustration purposes only. Cartesian coordinates for all TS structures are available in Table S1 (ESI†). | ||
The energy deviations between the CCSD(T)-F12a and the CCSDT(Q) levels were approximately −9 kJ mol−1 on average, with a standard deviation of ∼15 kJ mol−1. The energy deviations between the MRCI-F12+Q and the CCSDT(Q) levels averaged 6 kJ mol−1, with a standard deviation of ∼15 kJ mol−1 as well (Table 1). All TSs in this PES, along with N2H3, NNH, and H2NN(S), have T1 diagnostic coefficients exceeding the 0.018–0.020 range, suggesting that a multireference treatment should be used. The MRCI-F12+Q computations account for the system's multireference nature but lack size-consistency, a limitation only partially mitigated by the Davidson (Q) correction. We therefore also computed this PES using the CCSDT(Q) method, which extends coupled-cluster theory to include full triples excitations (CCSDT) with a perturbative treatment of connected quadruple excitations. However, both the MRCI-F12+Q and CCSDT(Q) methods significantly under-predicted the E0 values of nearly all wells relative to the ATcT data (Tables 1 and 2). The CCSD(T)-F12a level, on the other hand, produced highly accurate zero-point energy predictions when compared to ATcT values (Tables 1 and 2).
| Stationary point | ΔE0 CCSD(T)-F12a | ΔE0 MRCI-F12+Q | ΔE0 CCSDT(Q) |
|---|---|---|---|
| NH2 + NH(T) | −2.20 | −25.5 | −17.80 |
| c-N2H2 + H | −0.59 | −15.20 | −13.79 |
| t-N2H2 + H | 0.31 | −17.70 | −13.19 |
| H2NN(S) + H | 0.19 | −18.21 | −12.31 |
| NNH + H2 | 0.62 | −19.78 | −0.88 |
The CCSD(T)-F12a/aug-cc-pVTZ-F12 energies computed here are in excellent agreement with the complete basis set extrapolation value based on CCSD(T) energies reported in the literature (Fig. 5). The MRCI calculations, both with and without the explicit F12 correlation75 or the Davidson correction,76 consistently produced lower energies than the CCSD(T) values (Fig. 5). Similarly, MRCI-F12+Q yielded lower energies than the ATcT values (Tables 1 and 2). The inclusion of the F12 correlation reduced the CCSD(T) energies but increased the MRCI energies. Since the CCSD(T)-F12a/aug-cc-pVTZ-F12 energies showed excellent agreement with both the literature CCSD(T)/CBS values (Fig. 5) and the ATcT values for the wells (Table 2), they were used in this work to compute rate coefficients. The computed pressure-dependent reaction rate coefficients are reported in Tables S4 and S5 (ESI†).
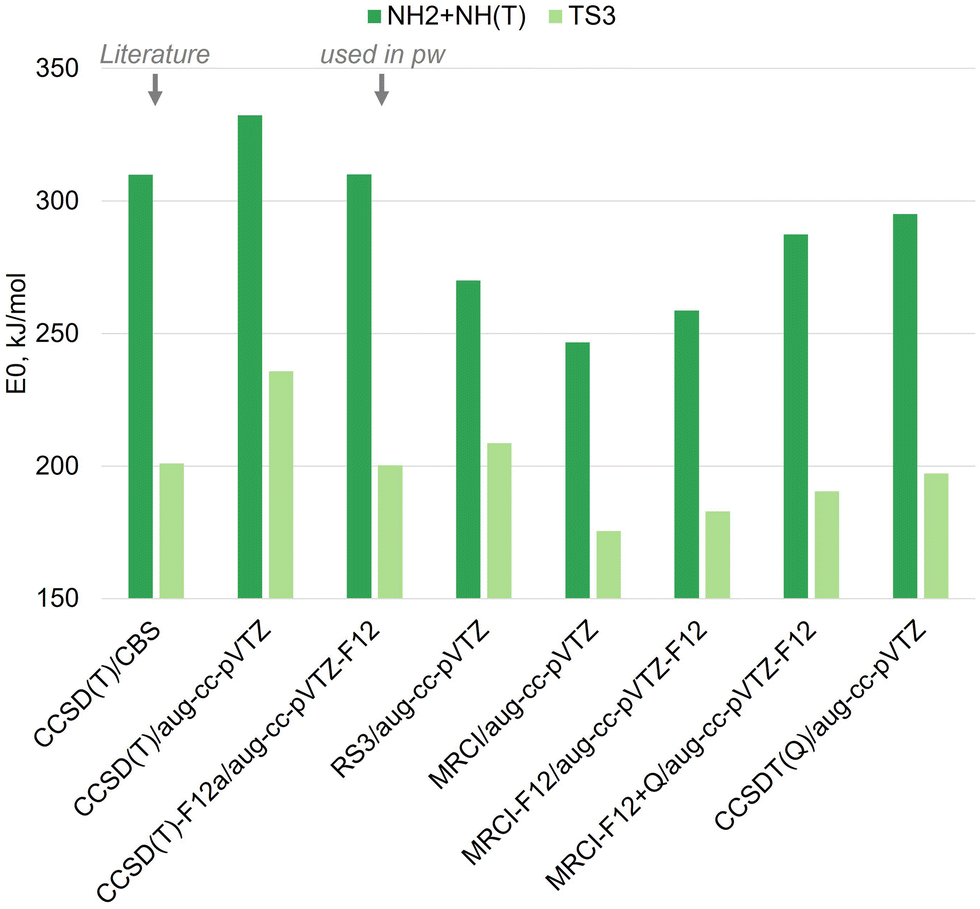 | ||
| Fig. 5 Zero-point energy comparisons of the NH2 + NH(T) well and of TS3, both relative to the zero-point energy of the N2H3 isomer at the respective level. The CCSD(T)/CBS values were taken from Klippenstein et al.52 (annotated as “Literature” in the figure). All other data were computed in the present work, and the CCSD(T)-F12a/aug-cc-pVTZ-F12 level of theory was further used for kinetic computations in the present work (annotated as “used in pw” in the figure). | ||
The primary products of NH2 + NH(T) association at all relevant pressures (1–100 bar) are N2H2 + H, primarily the trans N2H2 isomer due to the lower energy barrier for its formation (Fig. 2 and 6). At high pressures, N2H3 formation becomes relevant but only plays a significant role at temperatures below 1000 K (Fig. 6). The pressure dependence of NH2 + NH(T) reactions was found to be significant only for the isomer products, N2H3 and NH3N; the rate coefficients of bimolecular products that result from well-skipping reactions showed negligible pressure effect (not shown). As expected, the isomers reach thermal equilibrium more rapidly at higher pressures.
 | ||
| Fig. 6 Comparison of computed rate coefficients for unimolecular NH2 + NH(T) reactions at 1 and 100 bar. Rate coefficients that do not exhibit a significant pressure-dependence in the examined range are only plotted at 1 bar. The N2H2 + H product channel shown here accounts for both trans and cis N2H2 isomers. | ||
The association of H2NN(S) + H exhibits more complex pressure-dependent branching (Fig. 7) than the NH2 + NH(T) channel (Fig. 6), and has not been examined in prior studies of this PES. Similarly to the NH2 + NH(T) case, the N2H2 + H products dominate across almost all relevant temperature and pressure ranges (Fig. 7). The formation of NH3N becomes the dominant pathway only at relatively low temperatures and high pressures; however, it remains significant across the entire examined temperature range at high pressures. At high temperatures, NNH + H2 formation becomes important, and NH2 + NH(T) formation is also a significant minor channel. Vibrationally excited NH3N species produced by H2NN(S) + H collisions via TS5 are formed well above the TS1 barrier and near or exceeding the TS4 barrier. From the H2NN(S) + H association direction, once the reactive complex crosses TS5, it can proceed at low pressures past TSs 1, 2, and 3, all of which are submerged relative to TS5. Lower-energy collisions of H2NN(S) + H can also lead to reactivity via a barrierless pathway (Fig. 2), forming N2H3 at high pressures. Since NNH + H2 formation requires a concerted motion of two H atoms (TS4, Fig. 4), it becomes competitive only at high temperatures, while the dominant pathway consists of consecutive single H atom motions, e.g., via TS1. At all examined pressures, the significance of NNH + H2 formation increases with temperature. Consequently, in addition to H2NN(S) + H ⇌ N2H2 + H, the well-skipping H2NN(S) + H ⇌ NNH + H2 reaction should be considered in kinetic models of nitrogen-based fuels that describe high-temperature reactivity.
 | ||
| Fig. 7 Comparison of computed rate coefficients for unimolecular H2NN(S) + H reactions at 1 and 100 bar. Rate coefficients that do not exhibit a significant pressure-dependence in the examined range are only plotted at 1 bar. The N2H2 + H product channel shown here accounts for both trans and cis N2H2 isomers. | ||
The N2H2 + H products are the dominant pathway for NH2 + NH(T) association at all relevant temperature and pressure ranges (Fig. 6). Our calculated pressure-dependent rate coefficient for the NH2 + NH(T) ⇌ N2H2 + H reaction is benchmarked in Fig. 8. All rate coefficients compared in Fig. 8 show good agreement. The calculated rate coefficients for the trans-N2H2 product (by Kippenstein et al.) or the combined trans and cis products (reported in the experimental references) are in excellent agreement with values computed in this work, all within a factor of 1.5×. This strong agreement between the computed rate coefficient and experimental data is seen across both low- and high-temperature regions when validated against different sources (Fig. 6).
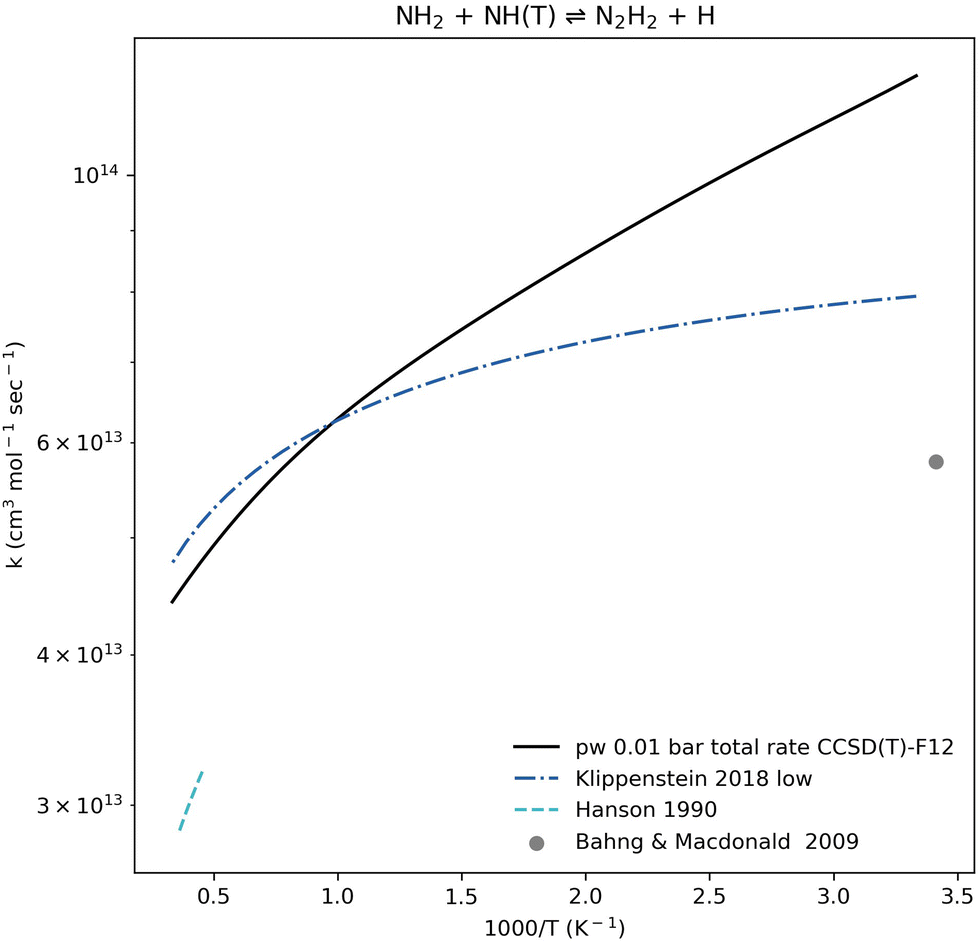 | ||
| Fig. 8 Rate coefficient comparison for the well-skipping reaction NH2 + NH(T) ⇌ N2H2 + H. The present work (pw) computation shown here accounts for both trans and cis N2H2 isomers at 0.01 bar. The computed rate coefficient is benchmarked against the zero-pressure rate coefficient computed by Kippenstein et al.,52 a rate coefficient determined by Hanson et al.96 based on experimental data at 2200–2800 K and 0.8–1.1 bar, and an experimental result by Bahng and Macdonald97 at 293 K and pressures up to 0.013 bar. | ||
The rate coefficient of the NH2 + NH(T) ⇌ N2H3 reaction is in the fall-off regime in the mid- and high-temperature range, even at 100 bar (Fig. 9(A)). Our computed high-pressure limit rate coefficient aligns with the Klippenstein et al.52 high-pressure limit rate coefficient (not shown), since it was directly derived from it via an ILT. The pressure-dependent rate coefficient computed here shows good agreement with the previously computed high-pressure limit rate coefficient. The low temperature and low pressure rate coefficient reported by Pagsberg and Eriksen98 in 1979 (Fig. 9(A)) is based on an outdated kinetic model manually fitted to low-pressure measurements. It is therefore unreliable, and the experimental result that was interpreted using this model cannot be given a significant weight in this analysis.
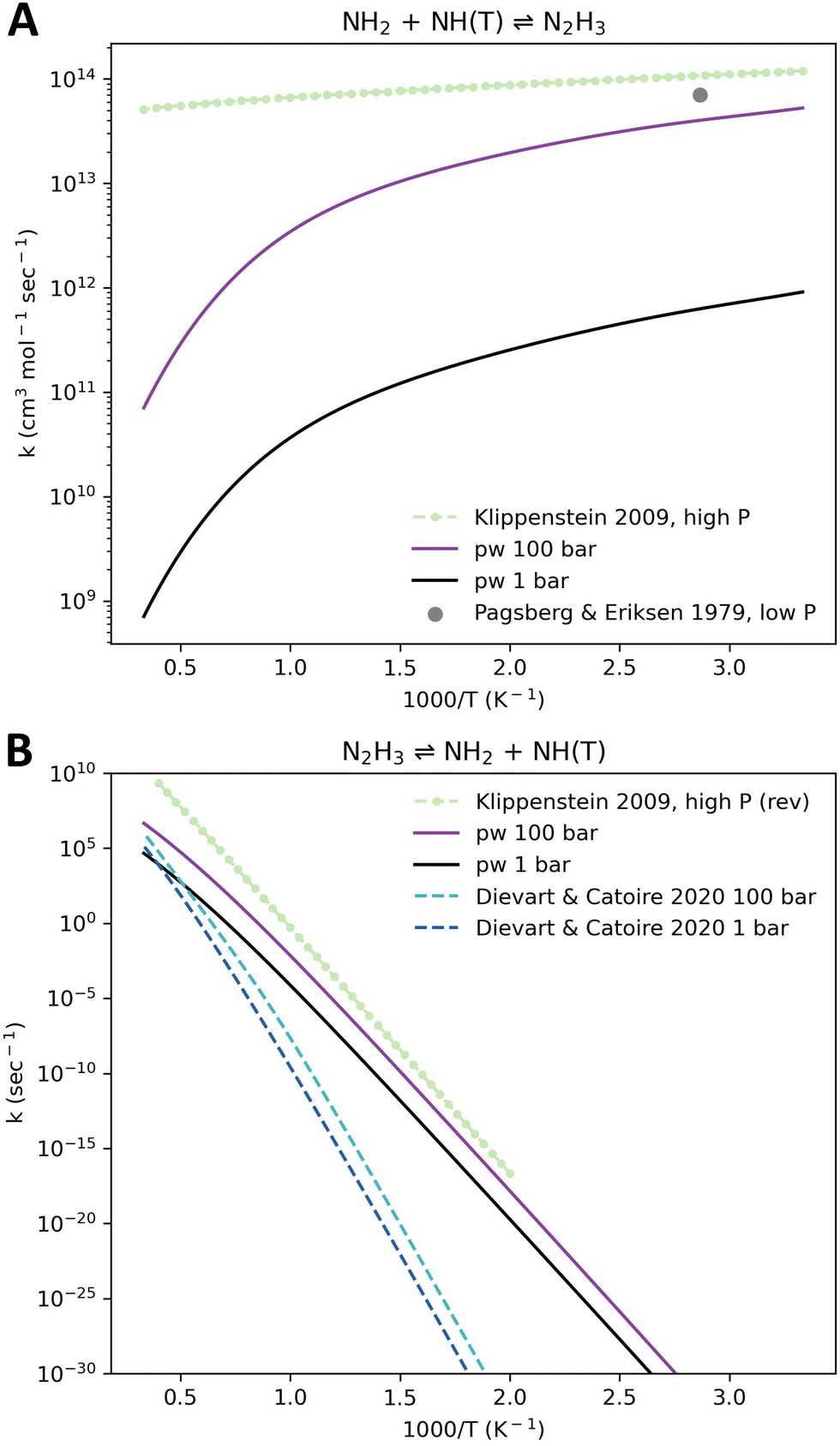 | ||
| Fig. 9 Rate coefficient comparison for the reaction NH2 + NH(T) ⇌ N2H3 (A) in the forward direction and (B) in the reverse direction. The high-pressure limit rate coefficient computed by Klippenstein et al.52 at CCSD(T)/CBS//CCSD(T)/aug-cc-pVDZ is given in (A) in the original reported direction and in (B) in the reverse direction after being reversed here using thermodynamic data computed by Grinberg Dana et al.8 This rate coefficient was also inferred by Pagsberg and Eriksen98 at 349 K and a sub-atmospheric pressure on the basis of indirect measurements combined with their model. Diévart and Catoire53 computed a pressure-dependent rate coefficient in the reverse direction at CCSD(T)/CBS//M062x-D3/aug-cc-pVTZ. | ||
The rate coefficient computed by Diévart and Catoire53 and reported by them in the reverse direction relative to Klippenstein et al.'s high-pressure limit rate coefficient exhibits a different slope compared to the values reported here and compared to the reversed high-pressure limit rate coefficient. At both 1 and 100 bar the Diévart and Catoire rate coefficient is several orders of magnitude lower than the reversed high-pressure limit rate coefficient in most of the explored temperature range (Fig. 9(B)).
The pressure-dependent rate coefficient computed for the N2H3 ⇌ NH2 + NH(T) reaction is highly sensitive to the energy of TS2 and TS3 relative to the N2H3 well (Fig. 10). These transition state barriers dictate the primary competing pathways for N2H2 + H formation (Fig. 2), making their accurate determination critical for reliable rate predictions. Additionally, the rate coefficient is sensitive to the energy difference between the N2H3 isomer and the NH2 + NH(T) well (Fig. 10), as this difference influences the overall reaction kinetics. Uncertainties in these energy values were propagated to estimate their impact on the high-pressure limit rate coefficients, as shown in Fig. S10 (ESI†). As shown above, the system's energy strongly depends on the level of theory used (Fig. 5 and Table 1).
 | ||
| Fig. 10 Normalized sensitivity coefficients of the computed N2H3 ⇌ NH2 + NH(T) rate coefficient evaluated with respect to the E0 values of all wells (the two isomers and bimolecular channels) as well as all TSs on the N2H3 PES at 1000 and 1500 K, both at 1 bar. Only the highest normalized sensitivity coefficients are reported. | ||
The rate coefficient for the N2H3 ⇌ N2H2 + H reaction reported by Dean and Bozzelli88 is lower than that computed in this work at the same pressure (Fig. 11), since Dean and Bozzelli used a significantly higher barrier for this beta-scission reaction. Dean and Bozzelli reported an endothermicity of 226 kJ mol−1 for this reaction. The reaction barrier they used is estimated by us to be at least ∼238 kJ mol−1 based on the reverse energy barrier found here (Fig. 2). This barrier is significantly higher than the E0 barrier of 200.6 kJ mol−1 computed here for the trans-N2H2 isomer (Fig. 2). The rate coefficients calculated in the present work and by Diévart and Catoire53 agree reasonably well (Fig. 11).
 | ||
| Fig. 11 Rate coefficient comparison for the N2H3 ⇌ N2H2 + H reaction. Calculations done in the present work (pw) are shown in this figure as an overall rate coefficient for the formation of both the trans-N2H2 and the cis-N2H2 isomers at 1 and 100 bar. The Dean and Bozzelli88 rate coefficient is given here only at 1 bar (Dean and Bozzelli also reported 0.1 bar and 10 bar values), and the Diévart and Catoire53 rate coefficient is plotted at both 1 and 100 bar. | ||
4. Conclusions
This study provides a deeper understanding of the N2H3 potential energy surface (PES), building upon prior work at the CCSD(T)/CBS level. This work identified the NH3N isomer as an important well in the N2H3 PES, which introduces three new bimolecular wells: NNH + H2, H2NN + H, and NH3 + N(D). The N2H3 ⇌ NH2 + NH(T) rate coefficient was improved compared to previous calculations, and the computed NH2 + NH(T) ⇌ N2H2 + H rate coefficient shows strong agreement with experimental measurements across both high and low temperatures. The NH2 + NH(T) reaction predominantly yields N2H2 + H on the doublet surface, with N2H3 formation becoming significant at low temperatures and high pressures. For H2NN(S) + H reactions, N2H2 + H remains the primary product, with minor contributions from NNH + H2 and NH2 + NH(T) at high temperatures; NH3N formation becomes significant at elevated pressures.The comprehensive dataset of pressure-dependent rate coefficients presented in this study is valuable for refining models of nitrogen-based fuel combustion and pyrolysis, particularly for ammonia and hydrazine.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that there are no conflicts of interest to disclose.Acknowledgements
Financial support from the Stephen and Nancy Grand Technion Energy Program (GTEP) is gratefully acknowledged.References
- O. Elishav, B. Mosevitzky Lis, E. M. Miller, D. J. Arent, A. Valera-Medina and A. Grinberg Dana, et al., Progress and Prospective of Nitrogen-Based Alternative Fuels, Chem. Rev., 2020, 120(12), 5352–5436 CrossRef CAS PubMed.
- G. P. Sutton and O. Biblarz, Rocket Propulsion Elements, Wiley, 9th edn, 2019 Search PubMed.
- E. F. Rothgery, Hydrazine and Its Derivatives, John Wiley & Sons, Ltd, 2004 Search PubMed.
- A. Grinberg Dana, G. E. Shter and G. S. Grader, Nitrogen-based alternative fuel: an environmentally friendly combustion approach, RSC Adv., 2014, 4, 10051–10059, 10.1039/C3RA47890D.
- A. Grinberg Dana, G. E. Shter and G. S. Grader, Nitrogen-Based Alternative Fuels: Progress and Future Prospects, Energy Technol., 2016, 4(1), 7–18, DOI:10.1002/ente.201500232.
- O. Herbinet, P. Bartocci and A. Grinberg Dana, On the use of ammonia as a fuel – A perspective, Fuel Commun., 2022, 11, 100064 CrossRef.
- A. Grinberg Dana, O. Elishav, A. Bardow, G. E. Ster and G. S. Grader, Nitrogen-Based Fuels: A Power-to-Fuel-to-Power Analysis, Angew. Chem., Int. Ed., 2016, 55(31), 8798–8805 CrossRef CAS.
- A. Grinberg Dana, K. Kaplan, M. Keslin, C. Cao and W. H. Green, Thermodynamic and Chemical Kinetic Parameters in Ammonia Oxidation: A Comparison of Recent Studies and Parameter Recommendations, Energy Fuels, 2024, 38(22), 22482–22500 Search PubMed.
- P. Gray, J. C. Lee and M. Spencer, Combustion, flame and explosion of hydrazine and ammonia I—The spontaneous ignition of pure gaseous hydrazine, Combust. Flame, 1963, 7, 315–321 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/0010218063902062.
- M. Auzanneau and M. Roux, Self-Detonation Range in Inert Atmosphere of Ternary Systems Using Hydrogen Peroxide and Hydrazine or Hydrazine Derivatives, Combust. Sci. Technol., 1990, 73(4–6), 505–520 CrossRef CAS.
- J. C. Leyer, A. A. Borisov, A. L. Kuhl and W. A. Sirignano, Dynamics of Detonations and Explosions: Detonations, American Institute of Aeronautics and Astronautics, Washington DC, 1991, DOI: DOI:10.2514/4.866067.
- M. Rath, H. Schmitz and M. Steenborg In: Development of a 400 N hydrazine thruster for ESA's Atmospheric Reentry Demonstrator; 1996. Available from: DOI:10.2514/6.1996-2866.
- A. S. Yang and T. C. Kuo, Design Analysis of a Satellite Hydrazine Propulsion System, J. Propul. Power, 2002, 18(2), 270–279 CrossRef CAS.
- K. Anflo, R. Mollerberg, K. Neff and P. King In: High Performance Green Propellant for Satellite Applications. Aerospace Research Central; 2009. Available from: DOI:10.2514/6.2009-4878.
- A. M. Elbaz, S. Wang, T. F. Guiberti and W. L. Roberts, Review on the recent advances on ammonia combustion from the fundamentals to the applications, Fuel Commun., 2022, 10, 100053 CrossRef . Available from: https://www.sciencedirect.com/science/article/pii/S266605202200005X.
- P. V. Blarigan Advanced internal combustion engine research. DOE Hydrogen Program Review NREL-CP-570-28890. 2000:1-19.
- C. H. Christensen, T. Johannessen, R. Z. Sørensen and J. K. Nørskov, Towards an ammonia-mediated hydrogen economy?, Catal. Today, 2006, 111(1), 140–144 CrossRef CAS . Frontiers in Catalysis: A Molecular View of Industrial Catalysis. Available from: https://www.sciencedirect.com/science/article/pii/S0920586105007157.
- P. J. Feibelman and R. Stumpf Comments on potential roles of ammonia in a hydrogen economy—a study of issues related to the use of ammonia for on-board vehicular hydrogen storage; 2006. https://www.pc.gov.au/inquiries/completed/climate-change-adaptation/submissions/sub046-attachment7.pdf.
- P. Glarborg, J. A. Miller, B. Ruscic and S. J. Klippenstein, Modeling nitrogen chemistry in combustion, Prog. Energy Combust. Sci., 2018, 67, 31–68 CrossRef.
- K. P. Shrestha, L. Seidel, T. Zeuch and F. Mauss, Detailed Kinetic Mechanism for the Oxidation of Ammonia Including the Formation and Reduction of Nitrogen Oxides, Energy Fuels, 2018, 32(10), 10202–10217 CrossRef CAS.
- B. Mei, X. Zhang, S. Ma, M. Cui, H. Guo and Z. Cao, et al., Experimental and kinetic modeling investigation on the laminar flame propagation of ammonia under oxygen enrichment and elevated pressure conditions, Combust. Flame, 2019, 210, 236–246 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0010218019303979.
- A. Stagni, C. Cavallotti, S. Arunthanayothin, Y. Song, O. Herbinet and F. Battin-Leclerc, et al., An experimental, theoretical and kinetic-modeling study of the gas-phase oxidation of ammonia, React. Chem. Eng., 2020, 5, 696–711, 10.1039/C9RE00429G.
- S. Arunthanayothin, A. Stagni, Y. Song, O. Herbinet, T. Faravelli and F. Battin-Leclerc, Ammonia-methane interaction in jet-stirred and flow reactors: An experimental and kinetic modeling study, Proc. Combust. Inst., 2021, 38(1), 345–353 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S1540748920304934.
- A. Bertolino, M. Fürst, A. Stagni, A. Frassoldati, M. Pelucchi and C. Cavallotti, et al., An evolutionary, data-driven approach for mechanism optimization: theory and application to ammonia combustion, Combust. Flame, 2021, 229, 111366 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0010218021000754.
- K. P. Shrestha, C. Lhuillier, A. A. Barbosa, P. Brequigny, F. Contino and C. Mounaïm-Rousselle, et al., An experimental and modeling study of ammonia with enriched oxygen content and ammonia/hydrogen laminar flame speed at elevated pressure and temperature, Proc. Combust. Inst., 2021, 38(2), 2163–2174 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S1540748920302881.
- K. P. Shrestha, B. R. Giri, A. M. Elbaz, G. Issayev, W. L. Roberts and L. Seidel, et al., A detailed chemical insights into the kinetics of diethyl ether enhancing ammonia combustion and the importance of NOx recycling mechanism, Fuel Commun., 2022, 10, 100051 CrossRef . Available from: https://www.sciencedirect.com/science/article/pii/S2666052022000036.
- J. Zhang, B. Mei, W. Li, J. Fang, Y. Zhang and C. Cao, et al., Unraveling Pressure Effects in Laminar Flame Propagation of Ammonia: A Comparative Study with Hydrogen, Methane, and Ammonia/Hydrogen, Energy Fuels, 2022, 36(15), 8528–8537 CrossRef CAS.
- J. Liu, C. Zou and J. Luo, Experimental and modeling study on the ignition delay times of ammonia/methane mixtures at high dilution and high temperatures, Proc. Combust. Inst., 2022, 4399–4407 Search PubMed . Available from: https://www.sciencedirect.com/science/article/pii/S1540748922005284.
- R. Tang, Q. Xu, J. Pan, J. Gao, Z. Wang and H. Wei, et al., An experimental and modeling study of ammonia oxidation in a jet stirred reactor, Combust. Flame, 2022, 240, 112007 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0010218022000268.
- P. García-Ruiz, M. Uruén, M. Abián and M. U. Alzueta, High pressure ammonia oxidation in a flow reactor, Fuel, 2023, 348, 128302 CrossRef Available from: https://www.sciencedirect.com/science/article/pii/S0016236123009158.
- X. Zhang, K. K. Yalamanchi and S. Mani Sarathy, Combustion chemistry of ammonia/C1 fuels: A comprehensive kinetic modeling study, Fuel, 2023, 341, 127676 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0016236123002892.
- Y. Zhu, H. J. Curran, S. Girhe, Y. Murakami, H. Pitsch and K. Senecal, et al., The combustion chemistry of ammonia and ammonia/hydrogen mixtures: A comprehensive chemical kinetic modeling study, Combust. Flame, 2024, 260, 113239 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0010218023006132.
- A. S. Singh, S. Mohapatra, R. Boyapati, A. M. Elbaz, S. K. Dash and W. L. Roberts, et al., Chemical Kinetic Modeling of the Autoignition Properties of Ammonia at Low-Intermediate Temperature and High Pressure using a Newly Proposed Reaction Mechanism, Energy Fuels, 2021, 35(16), 13506–13522 CrossRef CAS.
- M. Hamdy, S. Nadiri, A. Mohamed, S. Dong, Y. Wu and R. Fernandes, et al. An Updated Comprehensive Chemical Kinetic Mechanism for Ammonia and its Blends with Hydrogen, Methanol, and N-Heptane. In: WCX SAE World Congress Experience. SAE International; 2023. Available from: DOI:10.4271/2023-01-0204.
- S. Nadiri, B. Shu, C. F. Goldsmith and R. Fernandes, Development of comprehensive kinetic models of ammonia/methanol ignition using Reaction Mechanism Generator (RMG), Combust. Flame, 2023, 251, 112710 CrossRef CAS.
- J. Bugler, K. P. Somers, J. M. Simmie, F. Güthe and H. J. Curran, Modeling Nitrogen Species as Pollutants: Thermochemical Influences, J. Phys. Chem. A, 2016, 120(36), 7192–7197 CrossRef CAS PubMed.
- S. J. Klippenstein and P. Glarborg, Theoretical kinetics predictions for NH2 + HO2, Combust. Flame, 2022, 236, 111787 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0010218021005307.
- L. Kawka, G. Juhász, M. Papp, T. Nagy, I. G. Zsély and T. Turányi, Comparison of detailed reaction mechanisms for homogeneous ammonia combustion, Z. Phys. Chem., 2020, 234(7–9), 1329–1357, DOI:10.1515/zpch-2020-1649.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. de Joannon and R. X. Fernandes, et al., Review on Ammonia as a Potential Fuel: From Synthesis to Economics, Energy Fuels, 2021, 35(9), 6964–7029 CrossRef CAS.
- A. A. Konnov and J. De Ruyck, Kinetic modeling of the decomposition and flames of hydrazine, Combust. Flame, 2001, 124(1), 106–126 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S0010218000001875.
- Y. Ichiro Izato, K. Shiota and A. Miyake, A detailed mechanism for the initial hypergolic reaction in liquid hydrazine/nitrogen tetroxide mixtures based on quantum chemistry calculations, Combust. Flame, 2021, 229, 111389 CrossRef . Available from: https://www.sciencedirect.com/science/article/pii/S0010218021001048.
- R. Asatryan, J. W. Bozzelli, G. D. Silva, S. Swinnen and M. T. Nguyen, Formation and Decomposition of Chemically Activated and Stabilized Hydrazine, J. Phys. Chem. A, 2010, 114(21), 6235–6249 CrossRef CAS PubMed.
- P. Raghunath, N. T. Nghia and M. C. Lin, Chapter Seven – Ab Initio Chemical Kinetics of Key Processes in the Hypergolic Ignition of Hydrazine and Nitrogen Tetroxide, in Energetic Materials, ed. J. R. Sabin, Academic Press, 2014, vol. 69 of Advances in Quantum Chemistry, pp. 253–301. Available from: https://www.sciencedirect.com/science/article/pii/B9780128003459000076 Search PubMed.
- Y. Daimon, H. Terashima and M. Koshi, Chemical Kinetics of Hypergolic Ignition in Hydrazine/Nitrogen-dioxide Gas Mixtures, Available from: DOI:10.2514/6.2013-159.
- L. Zhang, A. C. Tv Duin, S. V. Zybin and W. A. Goddard III, Thermal Decomposition of Hydrazines from Reactive Dynamics Using the ReaxFF Reactive Force Field, J. Phys. Chem. B, 2009, 113(31), 10770–10778 CrossRef CAS PubMed.
- M. W. Schmidt and M. S. Gordon, The Decomposition of Hydrazine in the Gas Phase and over an Iridium Catalyst, Z. Phys. Chem., 2013, 227(11), 1301–1336, DOI:10.1524/zpch.2013.0404.
- S. S. Tafreshi, A. Roldan and N. H. de Leeuw, Density functional theory calculations of the hydrazine decomposition mechanism on the planar and stepped Cu(111) surfaces, Phys. Chem. Chem. Phys., 2015, 17, 21533–21546, 10.1039/C5CP03204K.
- H. Sun and C. K. Law, Thermochemical and Kinetic Analysis of the Thermal Decomposition of Monomethylhydrazine: An Elementary Reaction Mechanism, J. Phys. Chem. A, 2007, 111(19), 3748–3760 CrossRef CAS.
- P. Zhang, S. J. Klippenstein, H. Sun and C. K. Law, Ab initio kinetics for the decomposition of monomethylhydrazine (CH3NHNH2), Proc. Combust. Inst., 2011, 33(1), 425–432 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/S1540748910000350.
- P. Zhang, S. J. Klippenstein, L. B. Harding, H. Sun and C. K. Law, Secondary channels in the thermal decomposition of monomethylhydrazine (CH3NHNH2), RSC Adv., 2014, 4, 62951–62964, 10.1039/C4RA13131B.
- J. Wu, F. N. O. Bruce, X. Bai, X. Ren and Y. Li, Insights into the Reaction Kinetics of Hydrazine-Based Fuels: A Comprehensive Review of Theoretical and Experimental Methods, Energies, 2023, 16(16), 6006 CrossRef CAS . Available from: https://www.mdpi.com/1996-1073/16/16/6006.
- S. J. Klippenstein, L. B. Harding, B. Ruscic, R. Sivaramakrishnan, N. K. Srinivasan and M. C. Su, et al., Thermal Decomposition of NH2OH and Subsequent Reactions: Ab Initio Transition State Theory and Reflected Shock Tube Experiments, J. Phys. Chem. A, 2009, 113(38), 10241–10259 CrossRef CAS.
- P. Diévart and L. Catoire, Contributions of Experimental Data Obtained in Concentrated Mixtures to Kinetic Studies: Application to Monomethylhydrazine Pyrolysis, J. Phys. Chem. A, 2020, 124(30), 6214–6236 Search PubMed.
- P. K. Venkatesh, R. W. Carr, M. H. Cohen and A. M. Dean, Microcanonical Transition State Theory Rate Coefficients from Thermal Rate Constants via Inverse Laplace Transformation, J. Phys. Chem. A, 1998, 102, 8104–8115 CrossRef CAS.
- A. Grinberg Dana, D. Ranasinghe, H. Wu, C. Grambow, X. Dong and M. Johnson, et al., ARC – Automated Rate Calculator, version 1.1.0., GitHub, 2019, https://github.com/ReactionMechanismGenerator/ARC Search PubMed.
- G. Landrum RDKit: Open-Source Cheminformatics Software;. https://www.rdkit.org. Available from: https://www.rdkit.org.
- T. A. Halgren, MMFF VI. MMFF94s option for energy minimization studies, J. Comput. Chem., 1999, 20(7), 720–729 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620, 10.1039/B810189B.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/B508541A.
- S. Grimme, Semiempirical hybrid density functional with perturbative second-order correlation, J. Chem. Phys., 2006, 124(3), 034108, DOI:10.1063/1.2148954.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- J. Dunning and H. Thom, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90(2), 1007–1023, DOI:10.1063/1.456153.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries, J. Chem. Theory Comput., 2010, 6(9), 2872–2887 CrossRef CAS PubMed.
- C. Eckart, The Penetration of a Potential Barrier by Electrons, Phys. Rev., 1930, 35(11), 1303–1309 CrossRef CAS.
- K. Fukui, The path of chemical reactions-the IRC approach, Acc. Chem. Res., 1981, 14(12), 363–368 CrossRef CAS.
- A. Grinberg Dana, M. Liu and W. H. Green, Automated chemical resonance generation and structure filtration for kinetic modeling, Int. J. Chem. Kinet., 2019, 51(10), 760–776, DOI:10.1002/kin.21307.
- A. Grinberg Dana, M. S. Johnson, J. W. Allen, S. Sharma, S. Raman and M. Liu, et al., Automated reaction kinetics and network exploration (Arkane): A statistical mechanics, thermodynamics, transition state theory, and master equation software, Int. J. Chem. Kinet., 2023, 55(6), 300–323 CrossRef.
- M. Liu, A. Grinberg Dana, M. S. Johnson, M. J. Goldman, A. Jocher and A. M. Payne, et al., Reaction Mechanism Generator v3.0: Advances in Automatic Mechanism Generation, J. Chem. Inf. Model., 2021, 61(6), 2686–2696 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- T. B. Adler, G. Knizia and H. J. Werner, A simple and efficient CCSD(T)-F12 approximation, J. Chem. Phys., 2007, 127(22), 221106, DOI:10.1063/1.2817618.
- G. Knizia, T. B. Adler and H. J. Werner, Simplified CCSD(T)-F12 methods: Theory and benchmarks, J. Chem. Phys., 2009, 130(5), 054104 CrossRef.
- T. B. Adler and H. J. Werner, An explicitly correlated local coupled cluster method for calculations of large molecules close to the basis set limit, J. Chem. Phys., 2011, 135(14), 144117 CrossRef.
- K. A. Peterson, T. B. Adler and H. J. Werner, Systematically convergent basis sets for explicitly correlated wavefunctions: The atoms H, He, B–Ne, and Al–Ar, J. Chem. Phys., 2008, 128(8), 084102, DOI:10.1063/1.2831537.
- G. Knizia and H. J. Werner, Explicitly correlated RMP2 for high-spin open-shell reference states, J. Chem. Phys., 2008, 128(15), 154103 CrossRef PubMed.
- T. Shiozaki and H. J. Werner, Multireference explicitly correlated F12 theories, Mol. Phys., 2013, 111(5), 607–630 CrossRef CAS.
- E. R. Davidson and D. W. Silver, Size consistency in the dilute helium gas electronic structure, Chem. Phys. Lett., 1977, 52(3), 403–406 CrossRef CAS . Available from: https://www.sciencedirect.com/science/article/pii/0009261477804752.
- S. A. Kucharski and R. J. Bartlett, An efficient way to include connected quadruple contributions into the coupled cluster method, J. Chem. Phys., 1998, 108(22), 9221–9226 CrossRef CAS.
- Y. J. Bomble, J. F. Stanton, M. Kállay and J. Gauss, Coupled-cluster methods including noniterative corrections for quadruple excitations, J. Chem. Phys., 2005, 123(5), 054101 CrossRef.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: a general-purpose quantum chemistry program package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(2), 242–253, DOI:10.1002/wcms.82.
- D. G. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish, M. C. Schieber and R. Galvelis, et al., PSI4 1.4: Open-source software for high-throughput quantum chemistry, J. Chem. Phys., 2020, 152(18), 184108 CrossRef CAS.
- H. Wu, A. M. Payne, H. W. Pang, A. Menon, C. A. Grambow and D. S. Ranasinghe, et al., Toward Accurate Quantum Mechanical Thermochemistry: (1) Extensible Implementation and Comparison of Bond Additivity Corrections and Isodesmic Reactions, J. Phys. Chem. A, 2024, 128(21), 4335–4352 CrossRef CAS.
- M. J. Pilling and S. H. Robertson, Master Equation Models for Chemical Reactions of Importance in Combustion, Annu. Rev. Phys. Chem., 2003, 54, 245–275 CrossRef CAS.
- A. Y. Chang, J. W. Bozzelli and A. M. Dean, Kinetic Analysis of Complex Chemical Activation and Unimolecular Dissociation Reactions using QRRK Theory and the Modified Strong Collision Approximation, Z. Phys. Chem., 2000, 214, 1533–1568 CAS.
- J. A. Miller and S. J. Klippenstein, Determining phenomenological rate coefficients from a time-dependent, multiple-well master equation: “species reduction” at high temperatures, Phys. Chem. Chem. Phys., 2013, 15(13), 4744–4753 RSC.
- M. S. Johnson, X. Dong, A. Grinberg Dana, Y. Chung, D. J. Farina and R. J. Gillis, et al., RMG Database for Chemical Property Prediction, J. Chem. Inf. Model., 2022, 62(20), 4906–4915 CrossRef CAS.
- A. W. Jasper and J. A. Miller, Lennard-Jones parameters for combustion and chemical kinetics modeling from full-dimensional intermolecular potentials, Combust. Flame, 2014, 161(1), 101–110 CrossRef CAS.
- P. K. Venkatesh, A. M. Dean, M. H. Cohen and R. W. Carr, Chebyshev expansions and sensitivity analysis for approximating the temperature-and pressure-dependence of chemically-activated reactions, Rev. Chem. Eng., 1997, 13(1), 1–67 Search PubMed.
- A. M. Dean and J. W. Bozzelli, Combustion chemistry of nitrogen, Gas-phase combustion chemistry, Springer, 2000, pp. 125–341 Search PubMed.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS.
- N. Balucani, A. Bergeat, L. Cartechini, G. G. Volpi, P. Casavecchia and D. Skouteris, et al., Combined crossed molecular beam and theoretical studies of the N (2D) + CH4 reaction and implications for atmospheric models of Titan, J. Phys. Chem. A, 2009, 113(42), 11138–11152 CrossRef CAS PubMed.
- G. Faingold and J. K. Lefkowitz, A numerical investigation of NH3/O2/He ignition limits in a non-thermal plasma, Proc. Combust. Inst., 2021, 38(4), 6661–6669 CrossRef CAS Available from: https://www.sciencedirect.com/science/article/pii/S1540748920306179.
- B. Ruscic Active Thermochemical Tables (ATcT) values based on ver. 1.130 of the Thermochemical Network, 2024;. https://atct.anl.gov/ (Accessed August 2024).
- T. J. Lee and P. R. Taylor, A diagnostic for determining the quality of single-reference electron correlation methods, Int. J. Quantum Chem., 1989, 36(S23), 199–207, DOI:10.1002/qua.560360824.
- D. Jayatilaka and T. J. Lee, Open–shell coupled–cluster theory, J. Chem. Phys., 1993, 98(12), 9734–9747, DOI:10.1063/1.464352.
- T. J. Lee and G. E. Scuseria, in Achieving Chemical Accuracy with Coupled-Cluster Theory, ed. S. R. Langhoff, Springer, Dordrecht, Netherlands, 1995. pp. 47–108. Available from: DOI:10.1007/978-94-011-0193-6_2.
- D. F. Davidson, K. Kohse-Höinghaus, A. Y. Chang and R. K. Hanson, A pyrolysis mechanism for ammonia, Int. J. Chem. Kinet., 1990, 22(5), 513–535 CrossRef CAS.
- M. K. Bahng and R. G. Macdonald, Determination of the rate constants for the radical-radical reactions NH2(X2B1) + NH(X3Σ-) and NH2(X2B1) + H(2S) at 293 K, J. Phys. Chem. A, 2009, 113(11), 2415–2423 CrossRef CAS.
- P. B. Pagsberg, J. Eriksen and H. Christensen, Pulse radiolysis of gaseous ammonia–oxygen mixtures, J. Phys. Chem., 1979, 83(5), 582–590 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Stationary points geometries, T1 diagnostic factors, and imaginary frequencies (Table S1). Torsional scan of N2H3 at a resolution of 10° at the B2PLYP-D3/aug-cc-pVTZ level (Fig. S1). Representation of the N2H3 PES along with energy values from previous studies (Fig. S2). NH3N Thermodynamic properties, Chemkin format (Table S2). NH3N Thermodynamic properties, Cantera format (Table S3). Computed rate coefficient, Chemkin format (Table S4). Computed rate coefficient, Cantera format (Table S5). 3D representations of species (Fig. S3–S7). Species internal coordinate comparisons (Tables S6–S10). An unsuccessful TS search for the N2H3 ⇌ H2NN(S) + H reaction (Fig. S8). A scan of the N– –H distance in the N2H3 ⇌ H2NN(S) + H reaction (Fig. S9). Estimated reaction rate uncertainty based on computed energies (Fig. S10). Comparison of experimental and computed frequencies (Table S11). See DOI: https://doi.org/10.1039/d4cp03837a |
| This journal is © the Owner Societies 2025 |