
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive quantum chemical and mass spectrometric analysis of the McLafferty rearrangement of methyl valerate†
Mitsuo
Takayama
 *a,
Masahiro
Hashimoto
b,
Keijiro
Ohshimo
c,
Fuminori
Misaizu
c,
Masaaki
Ubukata
b and
Kenji
Nagatomo
b
*a,
Masahiro
Hashimoto
b,
Keijiro
Ohshimo
c,
Fuminori
Misaizu
c,
Masaaki
Ubukata
b and
Kenji
Nagatomo
b
aGraduate School in Nanobioscience, Yokohama City University, 22-2 Seto, Kanazawa-ku, Yokohama 236-0027, Japan. E-mail: takayama@yokohama-cu.ac.jp
bJEOL Ltd, Akishima, Tokyo 196-8558, Japan
cDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aoba, Aramaki, Aoba-ku, Sendai 980-8578, Japan
First published on 26th November 2024
Abstract
The McLafferty rearrangement (McLR) of the methyl valerate molecular ion has been comprehensively studied from the standpoints of the timescale for the keto–enol transformation and the change of the configuration of intermediates and transition state (TS), using mass spectrometry with electron ionization, strong-field tunnel ionization and collision-induced dissociation methods, and the global reaction route mapping (GRRM) program with quantum chemical calculations (QCCs). The timescales estimated from mass spectrometric results suggested that the McLR starts at 100 fs after ionization and is completed at least within 100 ns in the ion source. Whereas the timescales are consistent with a stepwise mechanism of fast (100 fs) and slow (10 ps) steps presented by Stamm et al., the QCCs put forth the possibility that an unanticipated, rapid, concerted process may be involved in completing the McLR reaction. It is worth noting that there appears to be a concerted process as a potential direct route to form the McLR fragments, while the slower step seems to involve a dynamic rearrangement in the configuration of the propene moiety in the TS of the molecular ion.
1. Introduction
McLafferty rearrangement (McLR) is a classical research topic and has long been the subject of a large number of studies on intramolecular hydrogen atom transfer (intraHAT) via a six-membered ring intermediate of radical cations M+˙.1–7 A key controversial or questionable issue of the McLR is that it is unclear whether the reaction is a stepwise or concerted,8–14 although a stepwise process can be assumed to be more likely than a concerted process.5,13,14 It would seem that there is another specific issue of the McLR, namely the disappearance of McLR ions such as m/z 74 and 87 from the collision-induced dissociation (CID) spectra of the molecular ion M+˙ of fatty acid methyl esters (FAMES).15–17 It seems fair to conclude that the disappearance of the McLR ions from the CID spectra is due to the methylene chain migration to the radical site on the carbonyl oxygen of the molecular ion M+˙ of FAMEs, which occurs at the electron ionization (EI) source within 10−6 s before CID. It seems that the methylene chain migration might result in a change in the original structure of molecular ions via radical-initiated migration (RIM) (see upper part in Fig. 1).15–17 It would be beneficial to gain a deeper understanding of both intraHAT and RIM reactions related to the McLR, particularly with regard to the lifetime of the ions and their residence times in the apparatus of mass spectrometers. | ||
| Fig. 1 Overall McLafferty rearrangement reactions of the molecular ions of keto compounds. | ||
While it is challenging to quantify the timescale and to determine the ion structure of each step of the McLR reaction, Fagerquist et al. have estimated using EI that the time interval between intraHAT (d in Fig. 1) and Cα–Cβ bond cleavage involving the McLR and complementary McLR (compMcLR) (f and g in Fig. 1) of FAMEs is 0.1 ns within the ion residence time of 1.0–10 μs in the ionizing cell.18 Stamm et al. reported using a strong-field tunnel ionization (SFTI) method that the McLR of 2-pentanone and substituted 2-pentanones occurs by two timescale steps, namely the fast timescale with molecular internal rotation and intraHAT within 100 fs (10−13 s), which involves the formation of a six-membered ring (c in Fig. 1), and the slow timescale with a π-bond rearrangement and Cα–Cβ bond cleavage (f and g in Fig. 1) for 10 ps (10−11 s).19 The report of Stamm et al. strongly supports a stepwise mechanism with fast and slow steps, and it is worth noting that the second step has a time scale about one hundred times slower than the first step, which involves the formation of the six-membered ring intermediate and γ-hydrogen transfer (d in Fig. 1). While there is undoubtedly great interest in gaining insights into the slower time scale events, it does seem to present a significant challenge to elucidate the gas-phase ion structures.
We are pleased to see the recent development of the artificial force-induced reaction method named the global reaction route mapping (GRRM) program with quantum chemical calculations (QCCs). The GRRM enables the elucidation of feasible pathways, including transition state (TS) structures, in an automatic manner. We have preliminarily used the GRRM to explain the formation of the McLR ion at m/z 74 and the formation of compMcLR ions at m/z 224 and m/z 42 of methyl stearate (SteaMe) and methyl valerate (MetVal), respectively.20 The program was able to automatically find a TS structure for the smaller molecule MetVal (Mr 116), but unfortunately the calculations for the larger molecule SteaMe (Mr 298) did not finish within a reasonable time. As will be illustrated and described in more detail in the section of materials and methods in this article, a recently developed orthogonal time-of-flight high-resolution mass spectrometer equipped with an SFTI source has the potential to focus on more detailed time windows and detect resulting fragment ions than an EI source.21
In this study, we aim to estimate the timescales observable for the McLR ion and to gain insight into the TS structure of the enol-form molecular ion by employing mass spectrometers and the GRRM program, respectively. We propose the use of MetVal as an analyte molecule, given that it is a suitable size for QCCs and has fortunately received attention from a wide variety of applications such as the internal rotation and QCCs,22 ultraviolet-induced excitation,23 and the conformation analysis using microwave spectroscopy of methyl alkynoates with short alkyl chains.24
2. Materials and methods
2.1 Reagents and sample preparation
Methyl valerate (99.0%), methyl stearate (99.8%), and n-hexane (96.0%) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Perfluorotributylamine (PFTBA) as a calibrant was purchased from Sigma Aldrich (St. Louis, MO, USA). The analyte was dissolved in n-hexane (Tokyo Kasei) at a concentration of 10 mmol L−1, and 1 μL of the analyte solution was injected into a gas chromatograph (GC) via an autosampler.2.2 Mass spectrometry
EI and SFTI mass spectra were acquired on an AccuTOF GC-Alpha high-resolution time-of-flight mass spectrometer JMS-T2000GC (JEOL, Tokyo, Japan) coupled to a model 8890 gas chromatograph (Agilent Technologies, Santa Clara, CA, USA). The orthogonal time-of-flight (oTOF) mass analyzer used has a resolving power of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (FWHM) at m/z 613.9642 (PFTBA) and sub-mu mass accuracy. Data analysis was performed using msFineAnalysis version 3.2 software (JEOL Ltd) for use with GC/TOF MS with JEOL msAxel software. The electron energies for EI were 15, 20, and 70 eV. The source temperatures for EI and SFTI were 373 and 323 K, respectively. The SFTI ion source was operated at 7.9 × 1010 V m−1 field strength with a carbon emitter whisker (30 nm diameter), a 9.6 kV ion acceleration voltage, and a vacuum pressure of 10−3 Pa. The vacuum pressure of the oTOF mass analyzer was 10−5 Pa. The timescales and schematic illustrations for the SFTI and EI sources of the mass spectrometer used are shown in Fig. S1 in the ESI.† The EI and CID spectra of MetVal and SteaMe were acquired on a triple–quadrupole tandem mass spectrometer JMS-TQ4000GC UltraQuad™ TQ (JEOL, Tokyo, Japan). The electron energy for EI, the source temperature, and ion accelerating voltage were 15 eV, 373 K, and 10 V, respectively. The collision gas was nitrogen and and the collision energy was 10 eV. The timescale and schematic illustration of the TQ4000GC mass spectrometer are shown in Fig. S1 in the ESI.†
000 (FWHM) at m/z 613.9642 (PFTBA) and sub-mu mass accuracy. Data analysis was performed using msFineAnalysis version 3.2 software (JEOL Ltd) for use with GC/TOF MS with JEOL msAxel software. The electron energies for EI were 15, 20, and 70 eV. The source temperatures for EI and SFTI were 373 and 323 K, respectively. The SFTI ion source was operated at 7.9 × 1010 V m−1 field strength with a carbon emitter whisker (30 nm diameter), a 9.6 kV ion acceleration voltage, and a vacuum pressure of 10−3 Pa. The vacuum pressure of the oTOF mass analyzer was 10−5 Pa. The timescales and schematic illustrations for the SFTI and EI sources of the mass spectrometer used are shown in Fig. S1 in the ESI.† The EI and CID spectra of MetVal and SteaMe were acquired on a triple–quadrupole tandem mass spectrometer JMS-TQ4000GC UltraQuad™ TQ (JEOL, Tokyo, Japan). The electron energy for EI, the source temperature, and ion accelerating voltage were 15 eV, 373 K, and 10 V, respectively. The collision gas was nitrogen and and the collision energy was 10 eV. The timescale and schematic illustration of the TQ4000GC mass spectrometer are shown in Fig. S1 in the ESI.†
2.3 Calculations
All calculations were performed using the Gaussian 16 suite of programs,25 and the initial molecule and molecular ion structures of methyl valerate, as well as neutral and ionic fragments, were generated by visual inspection using the GaussView program 6.0.25 The geometry optimization and vibrational frequency analysis of all the mentioned species were performed with the DFT restricted and unrestricted M06-2X26 level of theory and the 6-311++G(d,p), 6-31+G(d′,p′), and 6-31+G(p) basis sets. The vertical and adiabatic ionization energies of MetVal were evaluated from the sum of electronic and zero point energies calculated by the same level of theory and basis sets. The GRRM17 program27,28 was used to calculate the reaction path for the formation of McLR fragment ions from methyl valerate ions. The reaction path was searched by the anharmonic downward distortion following (ADDF) method which can follow reaction paths from local minima to transition states in GRRM17.27 First, the global reaction pathway was mapped by ADDF at the HF/STO-3G level. The resulting equilibrium (EQ) and transition state (TS) structures were reoptimized at the M06-2X/6-31+G(d) level. Zero-point vibrational energies of the EQ and TS structures were corrected at the same level. Energy, gradient and Hessian were calculated using the Gaussian 16 program suite. The reaction path for keto–enol transformation was determined by intrinsic reaction coordinate (IRC) calculations with up to 10 steps. The intermediates were obtained starting from the TS structure, which we believe to be the most appropriate approach. It was thought that the stability and the ion–molecule complex structures of the intermediate and TS ions might be estimated by natural bond orbital (NBO) calculations.3. Results and discussion
3.1 Time scales for ionization and fragmentation estimated from EI and SFTI measurements of methyl valerate
The EI mass spectra of MetVal were obtained at 70, 20, and 15 eV impact energy, as shown in Fig. S2 in the ESI.† The fragment ions at m/z 74, 85, and 87 did not change with decreasing the impact energy in relative intensity, while the aliphatic fragment ions [CnH2n+1]+ in the mass range of m/z 15 to m/z 60 rapidly decreased due to the decrease in the internal energy to cleave the C–C bond of alkyl chains. Here we focus on the fragment ions related to the McLR reaction and low internal energy EI spectra for comparison with the SFTI spectrum, as reported by Shulze and Richter,29 and Levsen and Beckey.30 The EI mass spectrum of MetVal obtained at 15 eV energy is shown in Fig. 2a. The spectrum showed peaks corresponding to the molecular ion M+˙ at m/z 116, the protonated molecule [M + H]+ at m/z 117 and the dehydrated molecule [M − H]+ at m/z 115, which agree with the NIST database.31 The formation of [M + H]+ and [M − H]+ ions may be due to self-chemical ionization in the ionizing cell, occurring due to the high volatility of MetVal.32 The spectra showed major characteristic fragment ions at m/z 74 and 87 resulting from single and double hydrogen rearrangements, respectively,5 while the other fragment ions at m/z 59 and 85 were observed from radical-initiated α-cleavage, as shown in Fig. 3a. The most intense ion at m/z 74 is an McLR ion arising from keto–enol transformation via intraHAT and the Cα–Cβ bond cleavage (c and charge retention in Fig. 3a). The fragment ion at m/z 87 is an McLR related ion arising from the keto–enol transformation and following intraHAT from α-hydrogen to γ-carbon (see Fig. 3a).5 The EI mass spectrum showed aliphatic fragment ions at m/z 29, 43, and 57 originating from σ-cleavage at the C–C bond of the aliphatic chain. The ion peak at m/z 42, assigned by b in the insets of Fig. 2a, represents the compMcLR ion resulting from charge migration of the enol-form molecular ion [Menol]+˙ (Fig. 3a). | ||
| Fig. 2 Mass spectra of methyl valerate (Mr116) obtained by the (a) 15 eV EI and (b) SFTI method. The peaks labeled a and b in the insets represent fragment ions at m/z 42.0101 [C2H2O]+˙ and 42.0464 [C3H6]+˙, respectively, which were separated by a high resolving power of 30000 (FWHM) of the oTOF mass analyzer. | ||
 | ||
| Fig. 3 Time scales for the main fragmentation pathways of the molecular ion of methyl valerate in (a) EI MS and (b) SFTI MS. According to EI MS, the McLR ion at m/z 74 forms via single intraHAT of γ-hydrogen (red) and the ion at m/z 87 forms via double intraHAT of γ-hydrogen (red) and α-hydrogen (blue). | ||
On the other hand, the SFTI mass spectrum showed intense peak for the molecular ion M+˙ at m/z 116 with low internal energy due to a tunneling effect (Fig. 2b), which enables an electron to be removed from analyte molecules at a lower energy than the ionization energy (IE0) without the strong field.33 In the case of MetVal (IE0 = 10.04 eV),21 it is expected that an effective IE in the field strength of 7.9 × 1010 V m−1 is to be about 6 eV by referring to the data for methyl butylate (IE0 = 10.07 eV).33 The spectrum showed SFTI characteristic fragment ions at m/z 29 [C2H5]+ and m/z 87 [M–C2H5]+via field dissociation that occurs within a timescale of 10 fs to 1.0 ps (10−14 to 10−12 s)21,34 and simple α-cleavage ions at m/z 59 and 85, as shown in Fig. 3b. It is known that the SFTI mass spectra show no or weak fragment peaks resulting from rearrangement reactions such as McLR.34 It is important to recognize that the fragment ions observable in the SFTI mass spectra can provide important information about the timescales for fragmentation.29,34–38 That is, the SFTI mass spectrum in Fig. 2b merely showed the molecular and fragment ions produced within 10 fs to 1.0 ps in the ionizing cell, as described in Fig. S1 (ESI†). The reason why the McLR ions at m/z 74 and 87 could not be observed in the SFTI spectrum is that the McLR ions are produced with a longer timescale than 1 ps (10−12 s) in the acceleration region of the ionizing cell. Another reason is that the McLR ions could not be produced in the regions of deceleration and uniform motion in the ion source, because the molecular ions reaching the regions have already changed from the original keto or enol structure of molecular ions to other rearranged ions (see upper part in Fig. 1).15–17 This suggests that the McLR reaction may occur in the time range from 10 ps to 100 ns (10−11 to 10−7 s) in the acceleration and deceleration regions.
To provide evidence for the change in the original structure of the FAMEs, the CID experiments of MetVal and SteaMe were performed using a triple quadrupole tandem mass spectrometer. The CID spectrum of SteaMe showed the product ions at every 14 Da unit pattern resulting from the cleavage at the methylene chain, but hardly showed the McLR ions at m/z 74 and 87 (see Fig. S3, ESI†), as known from previous work.15–17 On the other hand, the CID spectrum of MetVal showed the McLR ions, while the product ions at m/z 59 and 85 disappeared from the spectrum, as shown in Fig. 4. The disappearance of the 59 and 85 ions indicates that the original structure of the MetVal molecular ion was changed within or less than 10 μs in the ion source or the first quadrupole Q1 in the triple quadrupole mass spectrometer. The preferential appearance of the McLR ions at m/z 74 and 87 in the CID spectrum of MetVal is due to the short methylene chain because the short chain length does not enable migration to the radical site on the carbonyl oxygen. It is therefore expected that the MetVal molecular ions would hold the enol form molecular ions until reaching the quadrupole collision cell Q2, while the SteaMe molecular ion could not hold the enol form due to the long chain length.
 | ||
| Fig. 4 (a) 15 eV EI mass spectrum and (b) CID spectrum of the molecular ion at m/z 116 of methyl valerate. | ||
Now, we might apply timescales to the EI fragmentation of MetVal, based on the EI ion source and the organic photochemical knowledge,39 as shown in Fig. 3a. The observed ions correspond to the ions accumulated from the short time scales within 10 ps (10−11 s) from ionization (0.1 fs), internal rotation (0.1 ps), vibronic motion to break the σ-bond (0.1–1.0 ps) and rearrangement reactions (10 ps) in the ionizing cell to the long timescale within 10 μs for metastable decay in the transverse region towards the entrance of the oTOF. In SFTI MS, the observed ions correspond to the ions accumulated by the limited time windows of 10 fs to 1.0 ps in the ionization cell, as shown in Fig. 3b. It is important to recognize that the σ-bond cleavage within 1.0 ps is a non-ergodic event, which makes it difficult to explain the cleavage mechanisms by the quasi-equilibrium theory and the RRKM theory, as pointed out by Stamm et al.19 Consequently, it would be implied that the McLR reaction starts at 100 fs (10−13 s) after ionization (M+˙ formation) and is completed at the least within 100 ns (10−7 s) in the ion source. The timescales evaluated for the McLR are consistent with the results of Stamm et al.19
3.2 Molecular ion formation and spontaneous keto–enol transformation of methyl valerate
As shown in Fig. 3a, the McLR and compMcLR ions at m/z 74 and 42 are formed by an intraHAT from γ-hydrogen to carbonyl oxygen via a six-membered intermediate, which is a keto–enol transformation of the molecular ion, i.e., [Mketo]+˙ → [Menol]+˙. The formation of McLR and compMcLR ions can be classically explained by charge retention and charge migration, respectively.5 The keto–enol transformation is a key process for the McLR reaction after the formation of the molecular ion. The DFT calculations were performed for estimating the ionization energy (IE) of the linear and ring type conformers and the energy of conformational change. The vertical IE of methyl valerate is 10.04 eV.21 The calculations performed for vertical IE suggested that the ring type molecular ion [Mketo,ring]+˙ may undergo a conformational change into the enol-form molecular ion [Menol,ring]+˙ without remaining in the keto-form. This is based on the indications from the calculations of the conformational change of the molecular ion, [Mketo,linear]+˙ ⇄ [Mketo,ring]+˙. This leads us to the tentative conclusion that the six-membered intermediate molecular ion [Mketo,ring]+˙ may be a TS. The DFT calculations suggested that in the case of the molecular ion [Mketo,ring]+˙, the keto–enol transformation occurs by an exothermic reaction (Fig. 5), although the reaction does not occur in the neutral molecule Mketo,ring due to the absence of the unpaired electron on the carbonyl oxygen. Furthermore, the intrinsic reaction coordinate (IRC) analysis was performed for the keto–enol transformation. The IRC plot shows that the transformation occurs spontaneously as shown in Fig. 6. This indicates that the keto–enol transformation of the molecular ions of MetVal occurs within vibronic motion timescales (0.1–1.0 ps) after the formation of the molecular ions and that the keto–enol transformation is an energetically favorable process. | ||
| Fig. 5 The energy profile in eV for the conformational change of the neutral molecule M and the molecular ion M+˙ and the keto–enol transformation at the M06-2X/6-31+G(d) level of theory. | ||
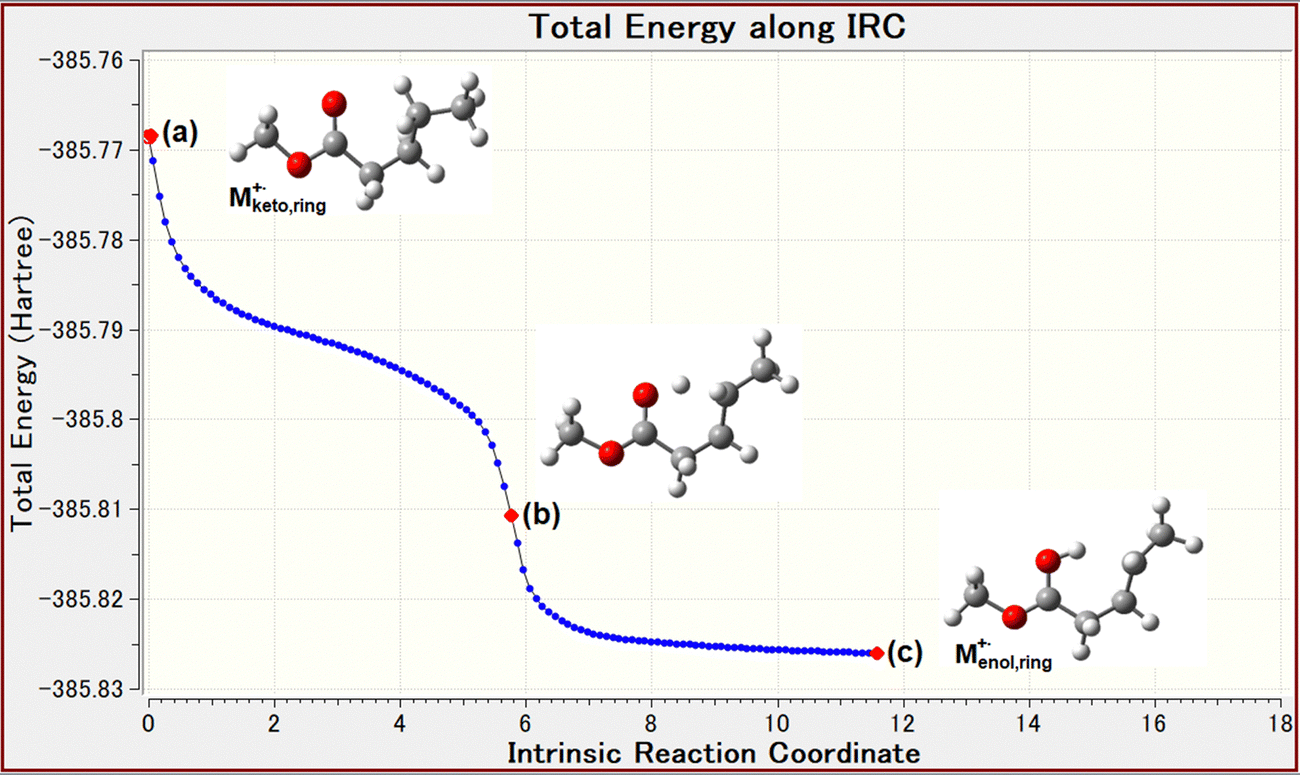 | ||
| Fig. 6 Intrinsic reaction coordinate (IRC) for (a) keto to (c) enol transformation of the molecular ion M+˙ of methyl valerate. Points a, b, and c represent the start, inflection, and end points, respectively. | ||
It is clear that the keto–enol transformation must involve the six-membered ring intermediate involving a specific conformation formed with the rotational frequency on the 1.0 ps timescale. The most provable conformer of neutral MetVal is known to be the conformer Mketo,ring shown in Fig. 3a, and the energy of a ring type conformer Mketo,ring was lower than that of a linear type conformer Mketo,linear.22 We also calculated the energy of the linear and six-membered ring conformers of neutral MetVal molecules. The results indicated that the ring type conformer appeared to be slightly more stable than the linear type (Fig. 5). It might be suggested that the Boltzmann distributions for both initial neutral conformers could be estimated by considering the experimental conditions of the ion source temperatures, 373 K for EI and 323 K for SFTI. The distributions obtained suggest that the amount of the ring type conformer may be approximately 2.7 times larger than that of the linear type under both conditions. From this, we can infer that both neutral conformers remain in the ionizing cell in accordance with the Boltzmann distributions. Furthermore, we calculated the vertical and adiabatic ionization energies (calc.ver. and calc.ad. IE) of the linear and ring conformers of MetVal using the M06-2X method and 6-31+G(d), 6-31+(d′,p′), and 6-311++G(d,p) levels of theory, as summarized in Table 1. The calc.ver. IEs of MetVal were larger than the calc.ad. IEs, regardless of the conformers and basis sets used. In the ring conformer, as shown in Table 1, it should be noted that the calc.ad. IEs of the enol-form molecular ion [Menol,ring]+˙ are smaller than the calc.ver. IEs of the keto-form molecular ion [Mketo,ring]+˙ and that the difference ΔIE could be converted to the internal energy of the enol-form molecular ion via the spontaneous keto–enol transformation, as shown in the IRC in Fig. 6. If so, the internal energy would be used to form fragment ions.
| Basis set | Linear conf. | Ring conf. | |||
|---|---|---|---|---|---|
| calc.ver. IE | calc.ad. IE | ΔIE | calc.ver. IE | calc.ad. IE | |
| 6-31+G(d) | 10.44 | 10.04 | 1.14 | 10.12 | 8.98 |
| 6-31+(d′,p′) | 10.43 | 10.10 | 1.48 | 10.41 | 8.93 |
| 6-311++G(d,p) | 10.45 | 10.06 | 1.91 | 10.80 | 8.89 |
3.3 Two-pathway possibility of the stepwise and concerted processes
As previously reported,20 the McLR and compMcLR ions at m/z 74 and 42 can be formed by charge retention and charge migration of the enol-form molecular ion [Menol,ring]+˙ of MetVal via a transition state (TS). The TS structure of the [Menol,ring]+˙ ion was searched for starting from the most stable enol-form molecular ion (see Fig. 6c) using the ADDF method in GRRM17 at the HF/STO-3G level of theory. After a preliminary ADDF search, we performed a reoptimized and frequency analysis of the TS at the M06-2X/6-31+G(d) level of theory. In a somewhat unexpected turn of events, the ADDF method identified a hitherto unrecognized structural type as a TS, as illustrated in Fig. S4b in the ESI.† The structure estimated as a TS indicated that the dissociation into the ester and propene fragments from the molecular ion may not be a simple cleavage. It is worth noting that the methylene chain (propene) of the MetVal ion is unexpectedly changed in configuration by the transition from the initial state to the TS. As can be seen in Fig. S4b′ (ESI†), the propene moiety (indicated by the dotted circle) wraps around the side of the ester moiety. The refined TS structure and the data for imaginary frequencies including 1 negative sign are given in Fig. S5 and Tables S1 and S2 in the ESI.† Furthermore, the charge and spin density distributions applied to the most stable enol-form molecular ion [Menol]+˙ and the TS ion [MTS]+˙ were estimated at the M06-2X/6-31+G(d) level. The charge and spin of the TS structure were distributed on both parts of the ester [CH3OCOH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2] and propene [CH2CHCH3] fragments (Fig. S4b, ESI†), whereas in the initial state, the charge and spin were mainly localized on the carbonyl carbon (C2) and γ-carbon (C5), respectively (Fig. S4a, ESI†). The ester ion [CH3OCOHCH2]+˙ and the propene ion [CH2CHCH3]+˙ correspond to the McLR and compMcLR ions, respectively, as shown in Fig. 3a. The formation of both ester and propene ions indicates that charge/spin separation occurs with the Cα–Cβ bond cleavage of the TS molecular ion.
CH2] and propene [CH2CHCH3] fragments (Fig. S4b, ESI†), whereas in the initial state, the charge and spin were mainly localized on the carbonyl carbon (C2) and γ-carbon (C5), respectively (Fig. S4a, ESI†). The ester ion [CH3OCOHCH2]+˙ and the propene ion [CH2CHCH3]+˙ correspond to the McLR and compMcLR ions, respectively, as shown in Fig. 3a. The formation of both ester and propene ions indicates that charge/spin separation occurs with the Cα–Cβ bond cleavage of the TS molecular ion.
In order to study the dynamic change in the configuration of the propene moiety, we tried to search for more equilibrium states or intermediates. Starting from the TS structure [MTS]+˙, two intermediates 1 and 2, which were more stable than the TS, could be found by using geometry optimization and vibrational frequency analysis at the M06-2X/6-31+G(d) level of theory, as shown in Fig. 7. Intermediate 1 (Fig. 7a) was almost the same in the TS structure (Fig. 7b), but the bond distance (1.67651 Å) between the enol oxygen (O1) and a propene carbon (C5) is shorter than that of the TS (1.99180 Å). Intermediate 2 (Fig. 7c) is similar in configuration to the propene moiety of the initial molecular ion [Menol]+˙ (Fig. S4a and a′, ESI†). It may be of interest to note that in the structure of intermediate 2, the bond distance (2.11692 Å) between the ester and propene moieties appears to be longer than that of the TS structure. This could indicate that intermediate 2 has been separated into the ester and propene fragments. It seems reasonable to assume that intermediate 2 may be formed directly from the initial molecular ion [Menol]+˙, potentially bypassing the TS. This offers a new perspective on the overall response of the McLR reaction with respect to the issue whether it is a stepwise or concerted process.
 | ||
| Fig. 7 The structure and the stabilization energy of intermediates (a) 1 and (c) 2 and (b) the TS of the enol-form molecular ion of methyl valerate, at the M06-2X/6-31+G(d) level. | ||
In order to gain further insight into the ion structures and stability of the TS and intermediates 1 and 2 (Fig. 7), we conducted a natural bond orbital (NBO) analysis at the M06-2X/6-31+G(d) level of theory. It seems reasonable to suggest that the NBO analysis may provide insight into the stabilization energy through the interactions between orbitals of the ion–molecule complex.40 The NBO analysis of intermediate 2 indicated that the ion–molecule complex could be divided into three units, namely ester (unit 1), propene (unit 2), and hydrogen (unit 3), although the ester and hydrogen were almost one unit from the bond distance (1.01218 Å) between the enol oxygen (O1) and the hydrogen (H11). This suggests that the stabilization energy based on the interaction between units 2 and 3 may be estimated to be 0.89 eV. However, it should be noted that intermediate 1 and TS could not be divided into multiple units.
In addition, the principal stabilization energy, which is thought to arise from the donor–acceptor interactions between the ester and propene moieties, was estimated by the NBO analysis. It seems that the principal donor–acceptor interaction of the TS may be between the O1–C5 of the ester and the C4 of the propene, with a stabilization energy of approximately 1.39 eV (Fig. 7b). It seems that the principal interaction of intermediate 1 was between C4 of the propene and the O1–C5 of the ester, with a stabilization energy of 1.81 eV (Fig. 7a). For intermediate 2, the interaction and stabilization energy were between C4 and O1–C5, with a value of 0.89 eV (Fig. 7c). The order of the stabilization energy is consistent with that of the bond length, as illustrated in Fig. 7. It might be worth reiterating that intermediate 2, which bears resemblance to the initial enol-form molecular ion in terms of configuration, has undergone fragmentation to give rise to the ester and propene ions. This could be a new insight into the McLR reaction. The results estimated above suggest that the McLR reaction may not be fully explained by a single pathway, i.e., a stepwise or concerted process. It may be helpful to view them in the context of at least two processes, as shown in the comprehensive potential profiles of the McLR reactions (Fig. 8).
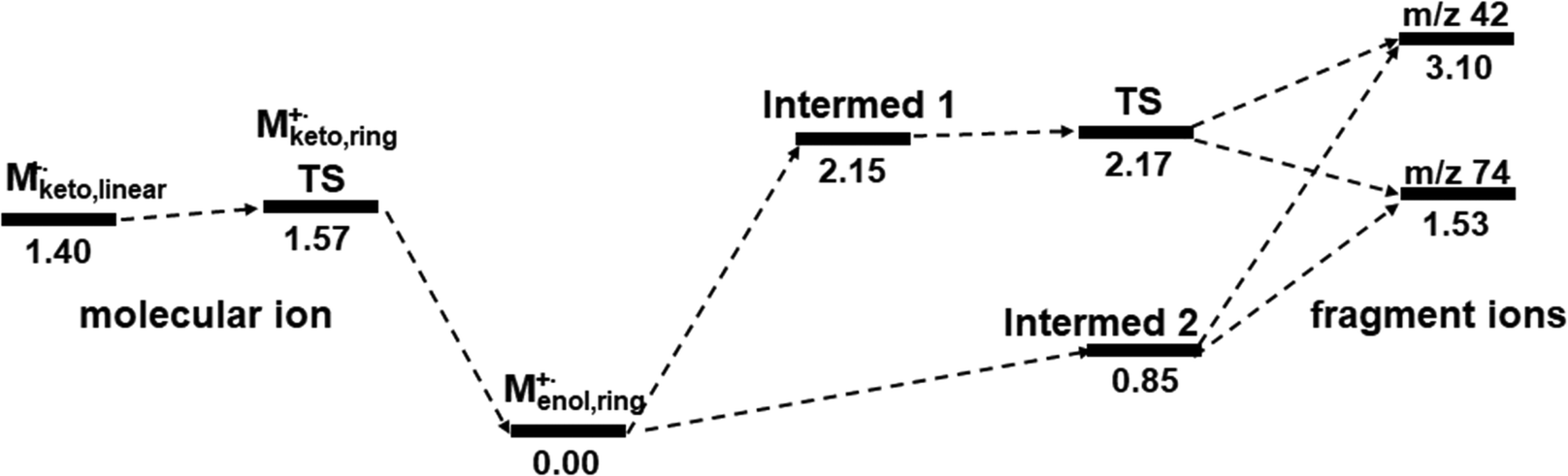 | ||
| Fig. 8 The comprehensive potential energy profiles in eV for the McLafferty rearrangement of methyl valerate, indicating two-pathways to form the fragment ions at m/z 74 and 42 from the enol-form molecular ion at m/z 116. The pathway via intermediate 1 and TS suggests a stepwise process, while the pathway via intermediate 2 suggests the presence of a concerted process. | ||
4. Conclusion
A comprehensive analysis of the McLR reaction of methyl valerate was examined using EI, SFTI, and CID MS methods and QCCs. The results obtained with the MS data indicated the possibility that the McLR reaction may occur and complete in a time range from 10 ps to 100 ns in the ion source. It is worth noting that the GRRM program with QCCs identified a hitherto unrecognized configuration as a TS structure, which could potentially support the second slower step time scale, which is about one hundred times slower than the first step in the McLR reported by Stamm et al.19 One particularly intriguing insight obtained with the QCCs was that the McLR reaction may trace at least two pathways, a slower stepwise process with TS and a fast concerted process without TS. While the precise mechanism of the McLR reaction remains a topic of debate, it seems that further integration of the QCCs and MS approaches may offer insights that could help us move closer to a rational understanding of the process.Author contributions
Mitsuo Takayama: conceptualization, theoretical investigation, formal analysis, visualization, writing – original draft preparation, data curation, and supervision. Masahiro Hashimoto: experimental investigation, formal analysis, and review & editing. Keijiro Ohshimo: theoretical investigation, formal analysis, and review & editing. Fuminori Misaizu: theoretical investigation, formal analysis, and review & editing. Masaaki Ubukata: experimental investigation, formal analysis, and review & editing. Kenji Nagatomo: ion optic calculations, formal analysis, and review & editing.Data availability
Derived data supporting the findings of this study are available from the corresponding author M. T. on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The theoretical calculations with the GRRM17 program were performed using Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C054).References
- F. W. McLafferty, Anal. Chem., 1959, 31, 82 CrossRef CAS.
- J. T. Bursey, M. M. Bursey and G. D. I. Kingston, Chem. Rev., 1973, 73, 191 CrossRef CAS.
- D. G. I. Kingston, J. T. Bursey and M. M. Bursey, Chem. Rev., 1974, 74, 215 CrossRef CAS.
- G. Bouchoux, Mass Spectrom. Rev., 1988, 7, 1 CrossRef CAS; G. Bouchoux, Mass Spectrom. Rev., 1988, 7, 203 CrossRef.
- F. W. McLafferty and F. Turecek, Interpretation of Mass Spectra, University Science Books, CA, USA, 4th edn, 1993 Search PubMed.
- M. J. Van Stipdonk, D. R. Kerstetter, C. M. Leavitt, G. S. Groenewold, J. Steill and J. Oomens, Phys. Chem. Chem. Phys., 2008, 10, 3209 RSC.
- J. Opitz, A. S. K. Hashmi, B. Miehlich and M. Woelfe, Eur. J. Mass Spectrom., 2019, 26, 3 CrossRef PubMed.
- F. P. Boer, T. W. Shannon and F. W. McLafferty, J. Am. Chem. Soc., 1968, 90, 7239 CrossRef CAS.
- J. S. Smith and F. W. McLafferty, Org. Mass Spectrom., 1971, 5, 483 CrossRef CAS.
- F. Turecek, D. E. Drinkwater and F. W. McLafferty, J. Am. Chem. Soc., 1990, 112, 993 CrossRef CAS.
- T. H. Osterheld and J. I. Brauman, J. Am. Chem. Soc., 1990, 112, 2014 CrossRef CAS.
- M. B. Stringer, D. J. Underwood and J. H. Bowie, Org. Mass Spectrom., 1992, 27, 270 CrossRef CAS.
- D. Norberg and N. Salhi-Benachenhou, J. Comput. Chem., 2007, 29, 392 CrossRef PubMed.
- R. Yasumoto, Y. Matsuda and A. Fujii, Phys. Chem. Chem. Phys., 2020, 22, 19230 RSC.
- J. A. Zirrolli and R. C. Murphy, J. Am. Soc. Mass Spectrom., 1993, 4, 223 CrossRef CAS PubMed.
- I. Vidavsky, R. A. Chorush, P. Longevialle and F. W. McLafferty, J. Am. Chem. Soc., 1994, 116, 5865 CrossRef CAS.
- M. Takayama, Int. J. Mass Spectrom. Ion Processes, 1995, 144, 199 CrossRef CAS.
- C. K. Fagerquist, R. A. Neese and M. K. Hellerstein, J. Am. Soc. Mass Spectrom., 1999, 10, 430 CrossRef CAS PubMed.
- J. Stamm, S. Kwon, S. Sandhu, M. Shaik, R. Das, J. Sandhu, B. Curenton, C. Wicka, B. G. Levine, L. Sun and M. Dantus, J. Phys. Chem. Lett., 2023, 14, 10088 CrossRef CAS PubMed.
- M. Takayama, M. Ubukata, K. Ohshimo, K. Nagatomo and F. Misaizu, Int. J. Mass Spectrom., 2023, 484, 116978 CrossRef CAS.
- M. Takayama, M. Ubukata, K. Nagatomo, J. Tamura and A. Kubota, J. Am. Soc. Mass Spectrom., 2023, 343, 2731 CrossRef PubMed.
- H. V. L. Nguyen, M. Andresen and W. Stahl, Phys. Chem. Chem. Phys., 2021, 23, 2930 RSC.
- M. A. Smialek, D. Duflot, N. C. Jones, S. V. Hoffmann, L. Zuin, M. Macdonald, N. J. Mason and P. Limao-Vieira, Eur. Phys. J. D, 2020, 74, 153 CrossRef CAS.
- N.-N. Dang, H.-N. Pham, I. Kleiner, M. Schwell, J.-U. Grabow and H. V. L. Nguyen, Molecules, 2022, 27, 2639 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683 RSC.
- S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama and Y. Ono, J. Comput. Chem., 2018, 39, 233 CrossRef CAS PubMed.
- P. Schulze and W. J. Richter, Int. J. Mass Spectrom. Ion Phys., 1971, 6, 131 CrossRef CAS.
- K. Levsen and H. D. Beckey, Int. J. Mass Spectrom. Ion Phys., 1972, 9, 63 CrossRef CAS.
- Methyl valerate (nist.gov).
- M. Takayama, K. Hiraoka and H. Nakata, J. Mass Spectrom. Soc. Jpn., 1996, 44, 531 CrossRef CAS.
- N. Davari, P.-O. Astrand, M. Unge, L. E. Lundgaard and D. Linhjell, AIP Adv., 2014, 4, 037117 CrossRef.
- H. D. Becky, Angew. Chem., Int. Ed. Engl., 1969, 8, 623 CrossRef.
- K. Levsen and H. D. Beckey, Int. J. Mass Spectrom. Ion Phys., 1969, 15, 333 CrossRef.
- H. D. Beckey, Int. J. Mass Spectrom. Ion Phys., 1970, 5, 182 CrossRef CAS.
- K. Levsen and H. D. Beckey, Int. J. Mass Spectrom. Ion Phys., 1974, 15, 333 CrossRef CAS.
- F. Borchers and K. Levsen, Int. J. Mass Spectrom. Ion Phys., 1979, 31, 247 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, The Benjamin/Cummings, CA, USA, 1978 Search PubMed.
- I. V. Alabugin, G. dos P. Gomes, N. V. Krivoshapov, P. Mehaffy and M. G. Medvede, Chem. Soc. Rev., 2021, 50, 10212 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03577a |
| This journal is © the Owner Societies 2025 |