An ab initio study of the rovibronic spectra of CH†
Received
23rd August 2024
, Accepted 21st November 2024
First published on 6th December 2024
Abstract
CH is one of the most spectroscopically studied diatomic molecules. The rovibronic spectra of the methylidyne radical (CH) in adiabatic and diabatic representations are obtained based on ab initio data, including 12 potential energy curves, 38 dipole moment curves, 79 spin–orbit coupling curves, and 18 electronic angular momentum coupling curves. We employed the internally contracted multireference configuration interaction method including the Davidson correction with the aug-cc-pV(5+d)Z basis set for the C atom and the aug-cc-pV5Z basis set for the H atom. The diabatic transformations are performed based on a property-based diabatisation method to remove the avoided crossings for the E 2Π–F 2Π and F 2Π–H 2Π pairs. The coupled nuclear motion Schrödinger equations are then solved using the Duo nuclear motion program to obtain the rovibronic spectra of CH for wavenumbers from 0 to 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 at 5000 K. An overall prediction of the rovibronic spectra of CH is provided in this work. Our results could be beneficial for future calculations of rovibronic spectra of CH and contribute to improving astronomical, chemical, and physical models.
000 cm−1 at 5000 K. An overall prediction of the rovibronic spectra of CH is provided in this work. Our results could be beneficial for future calculations of rovibronic spectra of CH and contribute to improving astronomical, chemical, and physical models.
1. Introduction
The study and analysis of the spectra of the CH radical contribute both to the interpretation of observations and the improvement of astronomical, chemical, and physical models.1,2 CH has a long history in molecular spectroscopy, whose spectra were first identified in the laboratory as early as 1918.3,4 In 1937, the presence of the CH radical in interstellar space was first discovered by Swings et al.5 Subsequently, CH has been detected in various astrophysical environments,6 including the Sun,7 comets,8 stellar atmospheres,9,10 the diffuse interstellar clouds,11 stars,12 and extragalactic galaxies.13 The electronic transition of CH was also detected in solar spectra. CH is one of the most spectroscopically studied diatomic molecules since it has attracted great attention in several applications and has a wide range of thermodynamic conditions. For example, it is used as part of the classification of carbon giants14 as well as a tracer for molecular hydrogen in the interstellar medium,15 and is also an important intermediate in the combustion of hydrocarbons, giving the flame its characteristic blue color.16 Given the importance of the spectra of the CH radical in the field of optical and thermal radiation studies and applications, it has been the subject of numerous spectroscopic investigations in the optical, infrared, far infrared, and microwave regions17 and most of the previous research on CH focused on its spectroscopic properties.18
Molecular spectroscopic data can be obtained by both experimental measurements and theoretical calculations. Previous experimental studies of CH are mainly concerned with transitions between the three low-lying electronic states. The spectra of A 2Δ–X 2Π, B 2Σ−–X 2Π, and C 2Σ+–X 2Π transitions were studied and analyzed by Gerö19 in 1941. Ubachs et al.20 measured the absorption spectra of the C 2Σ+–X 2Π transition of the CH molecule in 1986 by laser-induced fluorescence experiments. In 1996, Kepa et al.21 used conventional spectroscopy to record and analyze the spectral constants of the five (0-0, 0-1, 0-2, 1-0, and 1-1) bands of the B 2Σ−–X 2Π transition of the CH radical using conventional spectroscopy and obtained more precise molecular constants. Bembenek et al.22 obtained emission spectra of the C 2Σ+–X 2Π transition using a water-cooled Geissler-type tube in 1997. Medcraft et al.23 measured in 2019 in a supersonically expanding planar hydrocarbon plasma using a cavity-decay method for the C 2Σ+–X 2Π transition of CH rotationally resolved spectra of the electron leaps of CH and determined the equilibrium bond lengths of the C 2Σ+ state more precisely. However, due to factors such as experimental conditions, measurement environments, and experimental costs, it is difficult to study the high-lying electronic states of molecules in high-temperature environments via experimental measurements. Therefore, theoretical calculations have been widely used and the problems mentioned above can be alleviated using high-quality ab initio data.24 With the development of ab initio calculation methods, not only can the potential energy curves (PECs) of the high-lying electronic states and the coupling effects between different electronic states be obtained without the aid of any semiempirical and experimental parameters, but also the accuracy of the calculation results can be guaranteed. For example, Xiao et al.25,26 used the ab initio method to obtain the electronic structure parameters of the SH+ and PSe molecules and determined the spectroscopic constants of both of their bound states by taking into account the coupling effects between electronic states. Therefore, the ab initio data can be used as a baseline in the absence of spectroscopic experimental data.27
With the increasing demand for computational accuracy, more attention has been paid to the influence of non-adiabatic effects as well as coupling effects. The Born–Oppenheimer approximation for simplified calculations, which are basic approximations of the ab initio calculation method, has some impact on the accuracy of ab initio calculations. The Born–Oppenheimer approximation may lead to the occurrence of avoided crossings between electronic states which are spatially degenerate in energy at some internuclear distances. The phenomenon above may cause the subsequent theoretical calculations to differ significantly from the experimental results. Moreover, previous studies often ignore the coupling effects between electronic states, such as spin–orbit couplings (SOCs) and electronic angular momentum couplings (EAMCs), during the calculation of molecular spectroscopic data.6 For example, Brady et al.27 used an ab initio method to obtain the electronic structure parameters of the SO molecule and determined the absorption spectra generated by the bound-bound transitions, taking into account the coupling effects and nonadiabatic effects. The results show that the influence of both effects can make a significant difference in the obtained results. Semenov et al.28 obtained the rovibronic absorption spectra of PN based on ab initio data, including 9 PECs, 14 SOCs, 7 EAMCs, 9 permanent dipole moments (PDMs), and 8 transition dipole moments (TDMs).
Given the above consideration, this work aims to use the state-of-the-art ab initio method to calculate the electronic structures of CH, including the higher-lying electronic states. Duo29,30 and ExoCross31 are used to obtain the rovibronic spectra of CH with the consideration of the coupling effects between electronic states and the non-adiabatic effects. This work is organized as follows. Section 2 provides the theory and methods for computing the electronic structures of CH, as well as the diabatic transformation. Section 3 discusses the corresponding results. Finally, a conclusion is drawn in Section 4.
2. Theory and methods
2.1.
Ab initio calculations
For the CH radical, the electronic structures correlating with 12 electronic states, including the X 2Π, A 2Δ, E 2Π, F 2Π, H 2Π, C 2Σ+, D 2Σ+, B 2Σ−, c 4Σ−, b 4Π, a 4Σ− and d 6Σ− states, were calculated using the MOLPRO 2015 quantum chemistry package.32,33 The PECs, TDMs, PDMs, SOCs, and EAMCs are obtained using the state-averaged complete active space self-consistent field (SA-CASSCF) approach,34,35 followed by internally contracted multireference configuration interaction with the Davidson correction (icMRCI + Q),36 which is widely used to study electronic structures of diatomic molecules.37,38 The calculation is performed in its largest Abelian subgroup (C2v) because MOLPRO cannot deal with the non-Abelian (such as C∞v) symmetry. The irreducible representation of the C2v point group is (A1, B1, B2, and A2) and its corresponding relationship with the C∞v point group can be described as follows: Σ+ → A1, Σ− → A2, Π → (B1, B2), and Δ → (A1, A2). The aug-cc-pV(5+d)Z basis set is selected for the C atom and aug-cc-pV5Z basis set is selected for the H atom and both of them are augmented with diffuse functions. The ab initio data were computed at the internuclear distances from 0.5 to 9.5 Å with step sizes of 0.05 Å for 0.5–1.2 Å, 0.02 Å for 1.2–2 Å, 0.05 Å for 2–4 Å, 0.1 Å for 4–5 Å, and 0.5 Å for 5–9.5 Å.
2.2. Diabatisation
Born–Oppenheimer approximation may lead to the occurrence of avoided crossings between electronic states, which can cause the subsequent theoretical calculations to differ significantly from the experimental results. The inclusion of non-adiabatic couplings (NACs) within numerical dynamics calculations is computationally costly when using analytical forms for the PECs and couplings since both the cusp-like nature of PECs and the NACs have singularities at the region of spatial degeneracy.27,39,40 In this work, the NACs were used to diabatise the electronic structures of CH, which have been studied in depth by Brady et al.,27,41 which can lessen both numerical problems encountered in calculations within the adiabatic representation and the computational cost of calculating NACs. Based on the NACs, the kinetic energy terms are introduced to the molecular Hamiltonian. Then, the Born–Huang expansion42 considering NACs are capable of restoring the non-adiabatic effect at avoided crossing points. The smooth molecular properties can be obtained using the property-based diabatisation method, including PECs, TDMs, SOCs, and EAMCs in this work. First-order nondiagonal derivative couplings will vanish since the smoothness condition of the electronic structures uniquely defines the unitary transformation to the diabatic representation. The smooth results are favorable for nuclear motion calculations because no quantities within the molecular model are cusped. More information about the diabatisation method can be obtained from the published articles.41,43–46
The transformation from the adiabatic to diabatic basis can be described by a unitary matrix U:
| |  | (1) |
where
β(
R) is the mixing angle obtained by integrating the function of the non-adiabatic derivative coupling:
| | 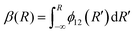 | (2) |
where
ϕ12(
R′) can be expressed as
47–49| | 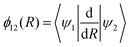 | (3) |
where |
ψ1〉 and |
ψ2〉 are the wavefunctions with the adiabatic representations of the lower and upper electronic states, respectively.
The mixing angle β(R) should be equal to π/4 at the internuclear distance of the crossing point Rc and the path integral of non-adiabatic coupling (NAC) over each side of the crossing point should be π/4 (and the overall integral should be π/2). Therefore, the non-adiabatic coupling function is normalized as follows:
| |  | (4) |
The two-state electronic Hamiltonian can be expressed from the adiabatic potential energy curves Va1(R) and Va2(R):
| |  | (5) |
where the subscripts 1 and 2 refer to the two PECs taken into account, respectively, and
R is the internuclear distance.
Based on the unitary transformation U(β(R)), the diabatic Hamiltonian can be obtained:
| |  | (6) |
where the
Vd(
R) is the diabatic potential energy curve.
The NACs can be obtained from the electronic wave functions using quantum chemical methods or modeled using the Lorentzian function forms, and the latter can reasonably describe the ab initio NACs in the vicinity of the crossing points,48–51 given as follows:
| |  | (7) |
where
α represents the inverse of half-width-at-half-maximum (HWHM). In this work, the absorption cross-sections of CH radical are modeled using the Lorentzian profile with a HWHM of 1 cm
−1.
The aforementioned method, called ‘property-based diabatisation’,27,39 is employed to construct the diabatic PECs. Minimizing the second derivatives of diabatic PECs in the vicinity of the crossing point Rc can obtain the smoothest PECs Vd1(R) and Vd2(R).
| |  | (8) |
where
Vdi(
R) means the
ith diabatic PEC.
3. Results and discussion
3.1.
Ab initio PECs
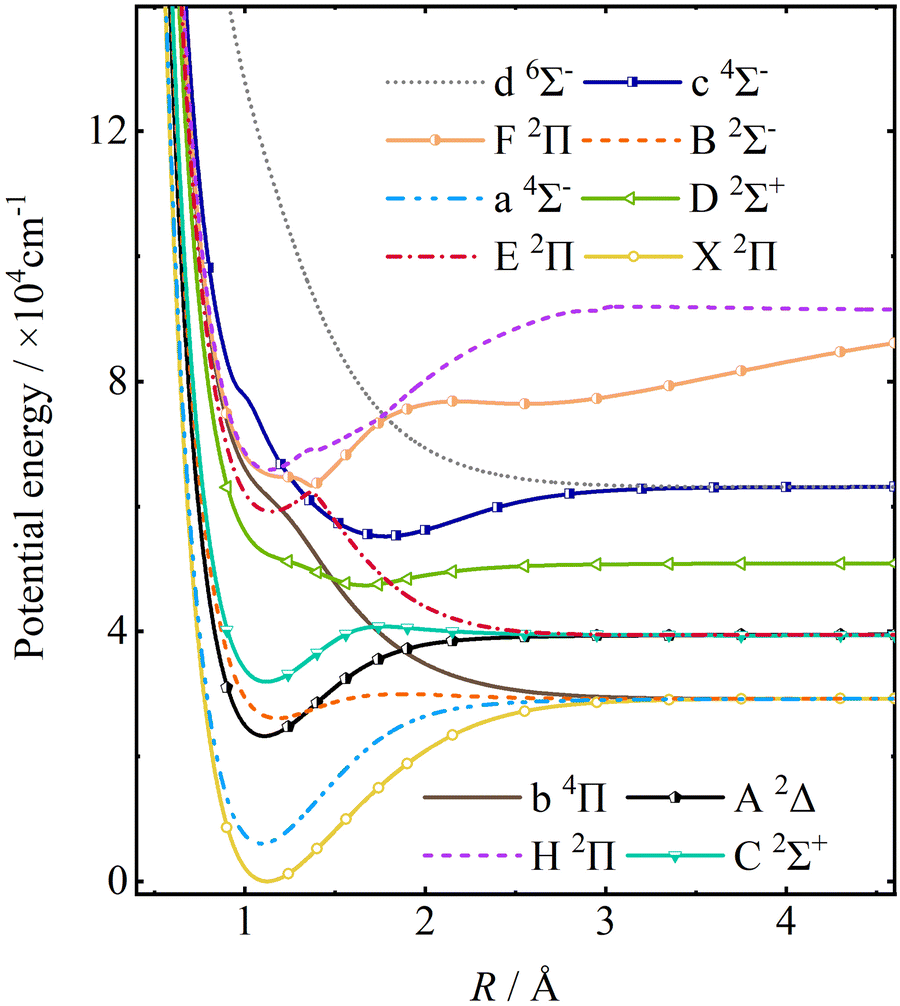
The PECs of twelve electronic states with the minimum energy point of the ground state as the zero-point energy are shown in Fig. 1, including 8 doublet, 3 quartet, and 1 sextet states. Table 1 shows these twelve electronic states of CH and their corresponding dissociation limits. It is worth noting that the spatially degenerate states E 2Π, F 2Π, and H 2Π of CH exhibit avoided crossings due to their shared symmetries, as shown in Fig. 2.
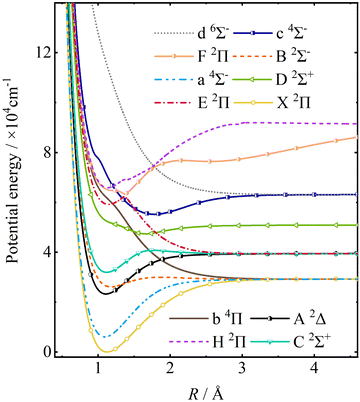 |
| | Fig. 1 PECs of CH calculated by the icMRCI method with the aug-cc-pV(5+d)Z basis set for the C atom and the aug-cc-pV5Z basis set for the H atom in the adiabatic representations. | |
Table 1 Electronic states of CH and their corresponding dissociation limits
| Dissociation limit |
Molecular electronic states |
| C(1P)–H(2S) |
H 2Π |
| C(3P)–H(2S) |
F 2Π |
| C(5S)–H(2S) |
c 4Σ−, d 6Σ− |
| C(1S)–H(2S) |
D 2Σ+ |
| C(1D)–H(2S) |
C 2Σ+, E 2Π, A 2Δ |
| C(3P)–H(2S) |
X 2Π, B 2Σ−, b 4Π, a 4Σ− |
 |
| | Fig. 2 The electronic states of CH molecule with the occurrence of avoided crossings. | |
In order to verify the reliability of our ab initio data, the spectroscopic constants computed in the present work are compared with those of the previous studies17,52–55 and with the experimental values56–62 in Table 2. Our adiabatic excitation energy (Te) and equilibrium internuclear distances (re) for X 2Π, B 2Σ−, a 4Σ−, C 2Σ+, E 2Π and A 2Δ states are close to the experimental values and the previous calculations. There are notable differences between our dissociation energy (De) for the B 2Σ− and C 2Σ+ states between the experimental values56,58–60 and the theoretical calculation results,17,52,54 which are close to those computed by Cui et al.53 The Te for the D 2Σ+ and c 4Σ− states as well as the re for H 2Π state are different from other results,17,55 which may result from the different levels of computational methods and the different basis sets. Overall, our ab initio PECs of CH are reliable and can be used to calculate the corresponding spectroscopic studies.
Table 2 Comparison of spectroscopic constants for electronic states of CH
| State |
Ref. |
T
e (cm−1) |
D
e (eV) |
r
e (Å) |
| X 2Π |
This work |
0.0 |
3.6275 |
1.10 |
| Expt.56,57 |
0.0 |
3.472 |
1.120 |
| Ref. 17 |
0.0 |
3.447 |
1.120 |
| Ref. 52 |
— |
3.413 |
1.123 |
| Ref. 53 |
0.0 |
3.669 |
1.1181 |
| B 2Σ− |
This work |
26524.76 |
0.4764 |
1.20 |
| Expt.58,59 |
26059.52 |
0.305 |
1.164 |
| Ref. 17 |
26140.17 |
0.244 |
1.175 |
| Ref. 52 |
26204.69 |
0.209 |
1.181 |
| Ref. 53 |
26007.57 |
0.438 |
1.1624 |
| a 4Σ− |
This work |
5960.573 |
— |
1.089 |
| Expt.56 |
5844 |
— |
1.085 |
| Ref. 17 |
6024.902 |
2.676 |
1.090 |
| Ref. 52 |
5847.462 |
2.680 |
1.093 |
| Ref. 53 |
6245.28 |
2.894 |
1.0872 |
| C 2Σ+ |
This work |
31883.50 |
1.0950 |
1.116 |
| Expt.56,60 |
31802.13 |
0.769 |
1.114 |
| Ref. 17 |
32205.40 |
0.738 |
1.116 |
| Ref. 52 |
32184.7 |
0.749 |
1.122 |
| Ref. 53 |
31961.97 |
0.930 |
1.1155 |
| Ref. 54 |
32184.7 |
0.749 |
1.118 |
| E 2Π |
This work |
59337.22 |
— |
1.150 |
| Expt.62 |
60394.77 |
— |
1.15 |
| Ref. 17 |
59346.60 |
— |
1.1437 |
| Ref. 55 |
59243.06 |
— |
1.1463 |
| A 2Δ |
This work |
23168.85 |
2.0145 |
1.100 |
| Expt.57,61 |
23147.88 |
1.836 |
1.103 |
| Ref. 17 |
23395.07 |
1.801 |
1.106 |
| Ref. 52 |
23510.83 |
1.760 |
1.109 |
| Ref. 53 |
23280.16 |
2.024 |
1.1032 |
| D 2Σ+ |
This work |
52716.60 |
0.4643 |
1.6500 |
| Ref. 17 |
47606.81 |
0.405 |
1.6635 |
| Ref. 55 |
47385.25 |
— |
1.6611 |
| c 4Σ− |
This work |
72162.93 |
0.9837 |
1.800 |
| Ref. 55 |
55039.22 |
— |
1.7797 |
| Ref. 17 |
55045.96 |
0.967 |
1.7866 |
| F 2Π |
This work |
65395.45 |
3.3264 |
1.3500 |
| Ref. 55 |
63312.55 |
— |
1.3746 |
| Ref. 17 |
63503.92 |
3.285 |
1.3751 |
| H 2Π |
This work |
65863.92 |
— |
1.1270 |
| Ref. 55 |
69895.35 |
— |
1.3434 |
| Ref. 17 |
70350.04 |
2.647 |
1.3762 |
The dipole-allowed DMs for 12 electronic states of CH were calculated using the icMRCI method with the aug-cc-pV(5+d)Z basis set for the C atom and the aug-cc-pV5Z basis set for the H atom. The detailed PDMs and TDMs of CH molecule in the adiabatic representations are given in the ESI.†
Comparisons between our DMs of CH computed in this work and those computed by other researchers18,63–65 are made to verify the reliability of our ab initio data, which are shown in Fig. 3 and 4. As shown in Fig. 3, the TDMs of B 2Σ−–X 2Π and D 2Σ+–X 2Π transitions show reasonable agreement with those calculated by van Dishoeck.64 using ab initio self-consistent-field with configuration-interaction (CI) methods with the Gaussian atomic orbital (AO) basis set for carbon nucleus and the 5s primitive set of Huzinaga contracted to [3s] by Dunning for the H atom. There are notable differences between our TDMs of the B 2Σ−–X 2Π and C 2Σ+–X 2Π and those computed by Kanzler et al.63 in the short-range or long-range regions, who used the correlated, size-consistent, ab initio effective valence-shell dipole operator (μν) method with the basis set consisting of the Dunning triple-zeta Gaussian-type basis with 5s and 3p functions on carbon and 3s functions on hydrogen. The PDMs for the X 2Π–X 2Π transition are in excellent agreement with those calculated by Baluja et al.18 using the R-matrix method and are presented in Fig. 4. Other PDMs for the X 2Π–X 2Π, a 4Σ−–a 4Σ−, A 2Δ–A 2Δ, B 2Σ−–B 2Σ− and C 2Σ+–C 2Σ+ transitions are different from those calculated by Lie et al.65 based the “extended CI” wavefunctions66 and Kanzler et al.63 The above difference between our ab initio data and those computed by other researchers18,63–65 may result from the different levels of computational methods or different basis sets adopted. Overall, our ab initio data of CH are reliable and can be used to calculate the corresponding rovibronic spectra.
 |
| | Fig. 3 Comparison of the TDMs of CH obtained in this work with those calculated by Kanzler et al.63 and van Dishoeck et al.64 | |
 |
| | Fig. 4 Comparison of PDMs for the electronic states of CH obtained in this work with those calculated by Lie et al.,65 Baluja et al.18 and Kanzler et al.63 | |
All results of the ab initio EAMCs and SOCs for CH molecule are shown in the ESI.† SO and L represent spin–orbit and electronic angular moment matrix elements, respectively. The subscripts, X, Y, and Z, mean the component of the parameter on the corresponding axis. The values of these curves have been multiplied by (−i) for better exhibition, except for the Y-component. Table 3 provides values for the optimized NAC parameters α and Rc used to diabatise the energy degenerate pairs, which are obtained using the method mentioned in Section 2.2. The crossing points for F 2Π–E 2Π and H 2Π–F 2Π pairs are 1.37 Å and 1.74 Å, respectively. The Lorentzian parameters are 48.17 and 16.06 for the above pairs, respectively. In addition, the mixing angles increase from 0 to about 1.56 radians with a surge at Rc for both the F 2Π–E 2Π and H 2Π–F 2Π pairs, which are shown in Fig. 5(b). As shown in Fig. 5 (a), the NACs are symmetrical curves with a cusp in the vicinity of Rc, and the peaks of the NACs for the F 2Π–E 2Π and H 2Π–F 2Π pairs are about 26 Å−1 and 11 Å−1, respectively.
Table 3 Avoided crossing points Rc and Lorentzian parameter α for the F 2Π–E 2Π and H 2Π–F 2Π pairs
| Electronic states |
R
c (Å) |
α
|
| E 2Π F 2Π |
1.37 |
48.17 |
| F 2Π H 2Π |
1.74 |
16.06 |
 |
| | Fig. 5 The NACs (a) and mixing angle (b) for the F 2Π–E 2Π and H 4Π–F 2Π pairs of CH. | |
A diabatic representation for PECs of the F 2Π, E 2Π and H 2Π states was obtained based on the parameters given above and the comparisons of PECs in the adiabatic and diabatic representations are shown in Fig. 6(a). The examples of comparisons of TDMs, SOCs, and EAMCs in the adiabatic and diabatic representations are presented in the ESI.† All examples suggest that the results are more reasonable after diabatisation due to the fact that coupling curves and DMs are smoothed out at the vicinity of avoided crossing points. Finally, all the TDMs, EAMCs, and SOCs in the diabatic representations are also shown in the ESI.†
 |
| | Fig. 6 Illustrations of the diabatic and adiabatic representations of PECs for E 2Π, F 2Π and H 2Π states. | |
3.2. Rovibronic spectra
The solar photosphere at 5800 K and the sunspots above 3000 K show prominent molecular features. The electronic transition of CH was detected in the solar spectra, so we calculated the rovibronic spectra of CH at 5000 K in this work. Based on the adiabatic electronic structures of the CH molecule, we calculate the total absorption cross-sections using the Duo29,30 and ExoCross31 programs in the wavenumber range between 0 and 80000 cm−1 at 5000 K. As shown in Fig. 7, a comparison between our total absorption cross-sections between those obtained using the states file (.states) and the transitions file (.trans) from Masseron et al.6 and Bernath67 was made to verify the reliability of our results. The input files of Masseron et al.6 and Bernath67 are available from the ExoMol database, which only contain part of the data for the X 2Π, B 2Σ−, A 2Δ and C 2Σ+ states. As can be seen from Fig. 7, the trends in our absorption cross-sections show reasonable agreement with the results obtained using the two files of Masseron et al.6 and Bernath,67 but there is a significant difference in the values of the two results in the ranges 5400–13000 and 26000–30000 cm−1. The above difference may be due to the use of different levels of theoretical calculations and basis sets during the calculation of the electronic structure parameters. At wavenumbers above 38000 cm−1 the differences might be attributed to the electronic structure parameters of different numbers of electronic states, which show the importance of high-lying electronic states. In this work, ab initio data of CH are obtained based on the state-of-the-art icMRCI + Q method using suitable basis sets. The obtained electronic structures and spectroscopic constants in the present work are compared with those of the previous theoretical calculations and with the experimental values to verify their reliability, which enables accurate calculation of the rovibronic spectra of CH.
 |
| | Fig. 7 Comparisons of our adiabatic total absorption cross-sections with those calculated based on the states file (.states) and the transitions file (.trans) from Masseron et al.6 and Bernath67 at T = 5000 K (HWHM = 1 cm−1). | |
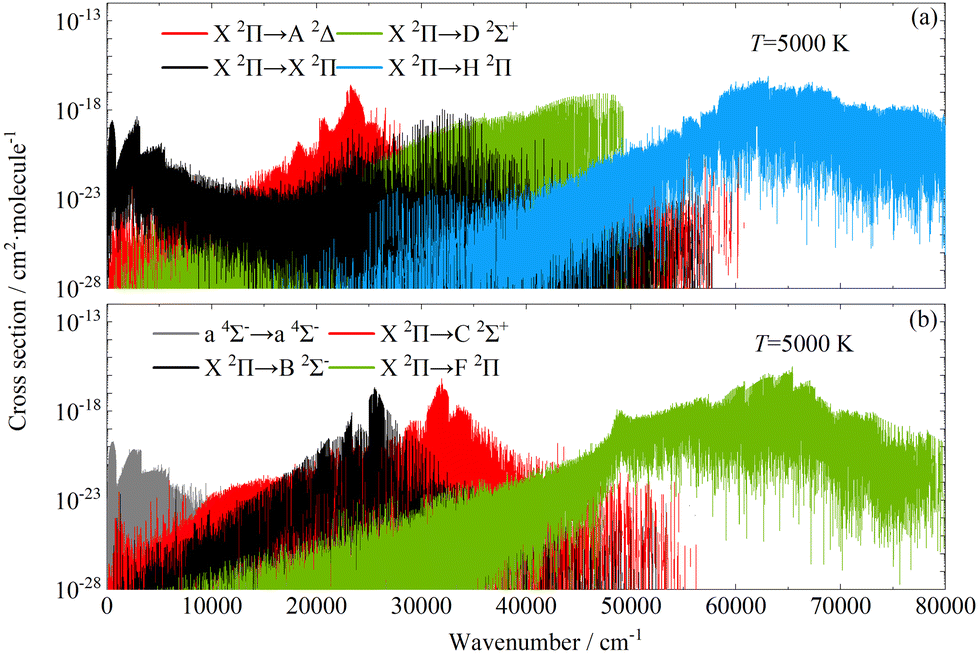
For the CH molecule, the adiabatic absorption cross-sections of eight significant transitions that make the main contribution at temperatures of 5000 K are shown in Fig. 8. It can be seen that a 4Σ− → a 4Σ− and X 2Π → X 2Π transitions contribute more obviously in the wave number range of 0–6000 cm−1. Then, the contribution of the a 4Σ− → a 4Σ− transition decreases with the increasing wavenumbers, while the contribution of the X 2Π → X 2Π transition decreases firstly. The absorption cross-sections of X 2Π → A 2Δ, X 2Π → B 2Σ− and X 2Π → C 2Σ+ transitions tend to rise and then fall with the increasing wavenumbers. The absorption cross-sections of X 2Π → H 2Π and X 2Π → F 2Π transitions exist in a wider range of wave numbers and contribute significantly at wavenumbers above 60000 cm−1.
 |
| | Fig. 8 The adiabatic absorption cross-sections of the CH molecule at 5000 K (HWHM = 1 cm−1). | |
Fig. 9 shows the results for the eight transitions that are the main contributors to the total absorption cross-sections at 5000 K, which are obtained based on the diabatic electronic structure of the CH molecule. Compared with the diabatic results obtained, the adiabatic absorption cross-sections of X 2Π → X 2Π, X 2Π → A 2Δ, X 2Π → B 2Σ−, X 2Π → C 2Σ+, X 2Π → D 2Σ+ and a 4Σ− → a 4Σ− transitions show slight changes. The X 2Π → E′ 2Π transition contributes significantly to the total absorption cross-section in the wavenumbers ranging from 28000 to 35000 cm−1 as well as in the high wavenumber region due to the potential well of E 2Π state becoming deeper after the diabatic transition. The absorption cross-section of the X 2Π → H′ 2Π transition also exhibits large variations in the range of wavenumbers less than 55000 cm−1.
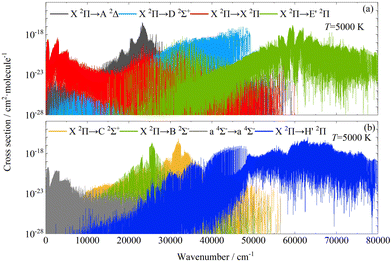 |
| | Fig. 9 The diabatic absorption cross-sections of the CH molecule at 5000 K (HWHM = 1 cm−1). | |
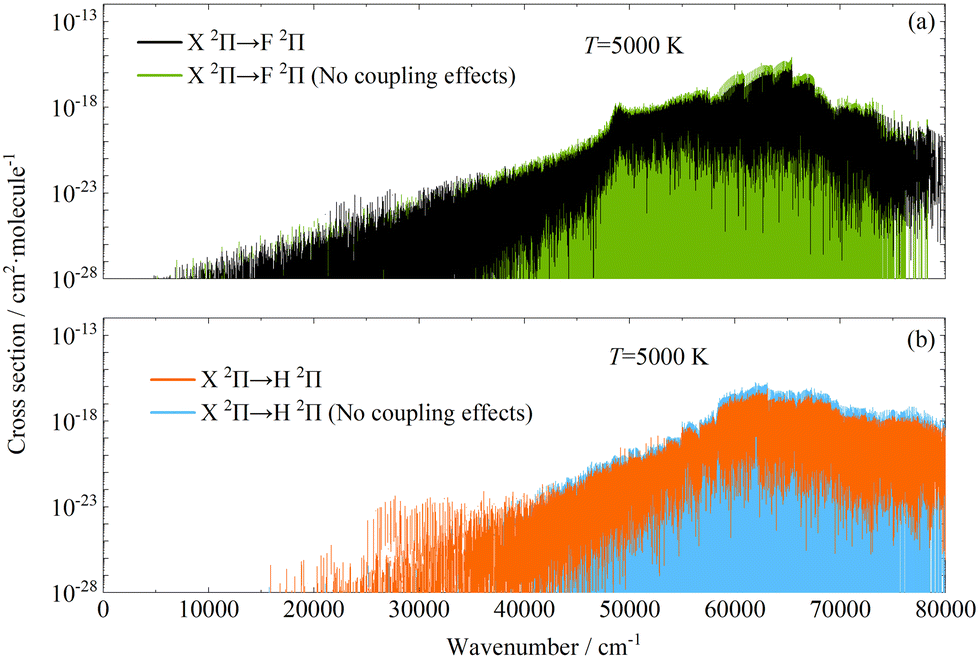
Fig. 10 shows the comparison of the adiabatic and diabatic absorption cross-sections of X 2Π → H 2Π and X 2Π → E 2Π transitions at 5000 K. Comparison of the adiabatic and diabatic PECs of E 2Π and H 2Π states is shown in Fig. 11. In the vibrational energy levels and dissociation limits of the diabatic H 2Π and E 2Π there appeared significant changes. Especially for the E 2Π state, the number of the vibrational energy levels and the dissociation energy increased. The absorption cross-sections of X 2Π → H 2Π and X 2Π → H′ 2Π transitions have huge differences at wavenumbers below about 58000 cm−1, since the vibrational energy levels and the dissociation energy of H 2Π state changed after the diabatisation. The absorption cross-sections of the X 2Π → E 2Π transition are mainly concentrated in the range 0–45000 cm−1. For the X 2Π → E′ 2Π transition, the absorption cross-sections are distributed over the whole wavenumber range. To present the influence of the coupling effects and the non-adiabatic effects on the rovibronic spectra of CH, a comparison of the absorption cross-sections for X 2Π → E 2Π transition was made at 5000 K and is shown in Fig. 12. Differences of the absorption cross-sections for the X 2Π → E 2Π transition are visible at wavenumbers below about 44000 cm−1, when only the coupling effect is considered. The absorption cross-sections differ widely over the entire range of wavenumbers considered in this work. Fig. 13 exhibits the comparison of the adiabatic absorption cross-sections for X 2Π → H 2Π and X 2Π → F 2Π transitions with/without the consideration of the coupling effects between electronic states at 5000 K. As shown in Fig. 13(a) and (b), the absorption cross-sections for the above two transitions in the adiabatic representations with/without the consideration of the coupling effects have similar trends over the whole wavenumber range. At the wavenumbers below about 40000 cm−1, the absorption cross-sections with the consideration of the coupling effects are larger than those with no coupling effects, and there is an opposite pattern in the range 40000–80000 cm−1. The above differences between the absorption cross-sections certainly show that consideration of the non-adiabatic effects and the coupling effects is necessary to calculate the rovibronic spectra of CH.
 |
| | Fig. 10 Comparison of the adiabatic and diabatic absorption cross-sections for X 2Π → E 2Π and X 2Π → H 2Π transitions at 5000 K (HWHM = 1 cm−1). | |
 |
| | Fig. 11 Comparison of the adiabatic and diabatic PECs of E 2Π and H 2Π states. | |
 |
| | Fig. 12 Comparison of the absorption cross-sections for X 2Π → E 2Π transition with/without the consideration of the coupling effects between electronic states and the non-adiabatic effects at 5000 K (HWHM = 1 cm−1). | |
 |
| | Fig. 13 Comparison of the adiabatic absorption cross-sections for X 2Π → H 2Π and X 2Π → F 2Π transitions with/without the consideration of the coupling effects between electronic states at 5000 K (HWHM = 1 cm−1). | |
4. Conclusions
In this work, we have obtained the rovibronic spectra for the CH molecule based on the ab initio data. The electronic structures of CH are obtained using the icMRCI + Q method with the aug-cc-pV(5+d)Z basis set for the C atom and the aug-cc-pV5Z basis set for the H atom, including 12 PECs, 38 DMs, 79 SOCs, and 18 EAMCs, which are also compared with those computed by other researchers to verify the reliability of our ab initio data. Moreover, the non-adiabatic and coupling effects were considered for accurately calculating vibrational spectra. In the wavenumber range between 0 and 80000 cm−1, the total absorption cross-sections and absorption cross-sections of the significant transitions involving high-lying electronic states of CH in adiabatic and diabatic representations are obtained using the Duo and ExoCross programs at 5000 K. At wavenumbers below about 50000 cm−1, the adiabatic absorption cross-sections of X 2Π → X 2Π, X 2Π → A 2Δ and X 2Π → D 2Σ+ transitions make the main contribution, while at wavenumbers above 50000 cm−1, the X 2Π → H 2Π, X 2Π → F 2Π, X 2Π → E′ 2Π and X 2Π → H′ 2Π transitions are significant and include the high-lying electronic states. Overall, our ab initio data of CH are reliable. The resulting vibrational spectra could be used as a benchmark for future calculations for CH and contribute to planetary exploration studies. In future work, more electronic structures of other diatomic molecules, including the higher-lying electronic states, can be obtained using the state-of-the-art ab initio method. Based on the ab initio data, a comprehensive and accurate theoretical investigation of the rovibronic spectra can be carried out by considering the coupling effects between electronic states and the non-adiabatic effects.
Author contributions
Zhenlu Hou: data curation, formal analysis, methodology, software, writing – original draft. Linhua Liu: funding acquisition, supervision, writing – review & editing.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
This work was financed by the National Natural Science Foundation of China (Grant no. 52076123).
Notes and references
- S. A. Davidson, K. M. Evenson and J. M. Brown, Astrophys. J., 2001, 546, 330 CrossRef CAS.
- M. Jackson, L. R. Zink, M. C. McCarthy, L. Perez and J. M. Brown, J. Mol. Spectrosc., 2008, 247, 128–139 CrossRef CAS.
-
T. Heurlinger, PhD thesis, University of Lund, 1918.
- T. Heurlinger and E. Hulthen, Z. Wiss. Photogr. Photophys. Photochem., 1919, 18, 241 CAS.
- P. Swings and L. Rosenfeld, Astrophys. J., 1937, 86, 483–486 CrossRef CAS.
- T. Masseron, B. Plez, S. Van Eck, R. Colin, I. Daoutidis, M. Godefroid, P.-F. Coheur, P. Bernath, A. Jorissen and N. Christlieb, Astron. Astrophys., 2014, 571, A47 CrossRef.
- A. Sauval and N. Grevesse, Astron. Express, 1985, 1, 153–158 Search PubMed.
- C. Arpigny, Annu. Rev. Astron. Astrophys., 1965, 3, 351–376 CrossRef CAS.
- D. L. Lambert, B. Gustafsson, K. Eriksson and K. H. Hinkle, Astrophys. J., Suppl. Ser., 1986, 62, 373–425 CrossRef CAS.
- P. Bernath, J. Chem. Phys., 1987, 86, 4838–4842 CrossRef CAS.
- A. Danks, S. Federman and D. Lambert, Astron. Astrophys., 1984, 130, 62–66 CAS.
- I. Crawford, Mon. Not. R. Astron. Soc., 1995, 277, 458–470 CrossRef CAS.
- J. Whiteoak, F. Gardner and B. Höglund, Mon. Not. R. Astron. Soc., 1980, 190, 17P–22P CrossRef CAS.
- A. Goswami, Mon. Not. R. Astron. Soc., 2005, 359, 531–544 CrossRef CAS.
- M. Gerin, D. A. Neufeld and J. R. Goicoechea, Annu. Rev. Astron. Astrophys., 2016, 54, 181–225 CrossRef CAS.
- P. Versailles, G. M. Watson, A. C. Lipardi and J. M. Bergthorson, Combust. Flame, 2016, 165, 109–124 CrossRef CAS.
- A. Kalemos, A. Mavridis and A. Metropoulos, J. Chem. Phys., 1999, 111, 9536–9548 CrossRef CAS.
- K. Baluja and A. Msezane, J. Phys. B: At., Mol. Opt. Phys., 2001, 34, 3157 CrossRef CAS.
- L. Gerö, Z. Phys., 1941, 118, 27–36 CrossRef.
- W. Ubachs, G. Meyer, J. Ter Meulen and A. Dymanus, J. Chem. Phys., 1986, 84, 3032–3041 CrossRef CAS.
- R. Kepa, A. Para, M. Rytel and M. Zachwieja, J. Mol. Spectrosc., 1996, 178, 189–193 CrossRef CAS.
- Z. Bembenek, R. Ke and M. Rytel, J. Mol. Spectrosc., 1997, 183, 1–5 CrossRef CAS.
- C. Medcraft, H. Linnartz and W. Ubachs, J. Mol. Spectrosc., 2019, 360, 15–23 CrossRef CAS.
- A. Mahfouf, P. André, G. Faure and M.-F. Elchinger, Chem. Phys., 2017, 491, 1–10 CrossRef CAS.
- Z. Xiao, X. Ren, Y. Liu and B. Yan, J. Quant. Spectrosc. Radiat. Transfer, 2021, 267, 107624 CrossRef CAS.
- L. Xiao, Y. Liu, X. Ren and B. Yan, J. Quant. Spectrosc. Radiat. Transfer, 2021, 259, 107422 CrossRef CAS.
- R. P. Brady, S. N. Yurchenko, G.-S. Kim, W. Somogyi and J. Tennyson, Phys. Chem. Chem. Phys., 2022, 24, 24076–24088 RSC.
- M. Semenov, N. El-Kork, S. N. Yurchenko and J. Tennyson, Phys. Chem. Chem. Phys., 2021, 23, 22057–22066 RSC.
- S. N. Yurchenko, L. Lodi, J. Tennyson and A. V. Stolyarov, Comput. Phys. Commun., 2016, 202, 262–275 CrossRef CAS.
- J. Tennyson and S. N. Yurchenko, Int. J. Quantum Chem., 2017, 117, 92–103 CrossRef CAS.
- S. N. Yurchenko, A. F. Al-Refaie and J. Tennyson, Astron. Astrophys., 2018, 614, A131 CrossRef.
-
H. Werner, P. Knowles, G. Knizia, F. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona and R. Lindh, MOLPRO, version 2015.1, a package of ab initio programs, see https://www.molpro.net.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona and D. A. Kreplin, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053–5063 CrossRef CAS.
- P. J. Knowles and H.-J. Werner, Chem. Phys. Lett., 1985, 115, 259–267 CrossRef CAS.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1988, 89, 5803–5814 CrossRef CAS.
- Z. Hou, Z. Qin and L. Liu, Astron. Astrophys., 2023, 672, A25 CrossRef CAS.
- Z. Ding, Z. Qin and L. Liu, Phys. Fluids, 2023, 35, 027127 CrossRef CAS.
- T. Karman, M. Besemer, A. van der Avoird and G. C. Groenenboom, J. Chem. Phys., 2018, 148, 094105 CrossRef.
- M. Desouter-Lecomte, J.-C. Leclerc and J.-C. Lorquet, Chem. Phys., 1975, 9, 147–156 CrossRef CAS.
- R. P. Brady, C. Drury, S. N. Yurchenko and J. Tennyson, J. Chem. Theory Comput., 2024, 20, 2127–2139 CrossRef CAS PubMed.
-
M. Born and K. Huang, Dynamical Theory of Crystal Lattices, Clarendon Press, Oxford, Revised edition, 1988 Search PubMed.
- C. A. Mead and D. G. Truhlar, J. Chem. Phys., 1982, 77, 6090–6098 CrossRef CAS.
- J. B. Delos, Rev. Mod. Phys., 1981, 53, 287 CrossRef CAS.
-
A. W. Jasper, B. K. Kendrick, C. Alden Mead and D. G. Truhlar, Non-Born-Oppenheimer chemistry: Potential surfaces, couplings, and dynamics, World Scientific, Singapore, 2004, ch. 8, pp. 329–391 Search PubMed.
- M. Baer, Phys. Rep., 2002, 358, 75–142 CrossRef CAS.
- D. Simah, B. Hartke and H.-J. Werner, J. Chem. Phys., 1999, 111, 4523–4534 CrossRef CAS.
- H. An and K. K. Baeck, J. Chem. Phys., 2015, 143, 4102 CrossRef.
- K. K. Baeck and H. An, J. Chem. Phys., 2017, 146, 4107 CrossRef.
- T. Mondal and A. Varandas, J. Chem. Phys., 2011, 135, 4304 Search PubMed.
- H. J. Werner and W. Meyer, J. Chem. Phys., 1981, 74, 5802–5807 CrossRef CAS.
- M. Kleinschmidt, T. Fleig and C. M. Marian, J. Mol. Spectrosc., 2002, 211, 179–188 CrossRef CAS.
- J. Cui, J.-G. Xu, J.-X. Qi, G. Dou and Y.-G. Zhang, Chin. Phys. B, 2018, 27, 276–282 Search PubMed.
- H. Hettema and D. R. Yarkony, J. Chem. Phys., 1994, 100, 8991–8998 CrossRef CAS.
- G. Vázquez, J. Amero, H. Liebermann, R. Buenker and H. Lefebvre-Brion, J. Chem. Phys., 2007, 126, 164302 CrossRef.
- C. DL and W. G. Richards, J. Phys. B: Atom. Mol. Phys., 1981, L131 Search PubMed.
- M. Zachwieja, J. Mol. Spectrosc., 1995, 170, 285–309 CrossRef CAS.
- R. Kepa, A. Para, M. Rytel and M. Zachwieja, J. Mol. Spectrosc., 1996, 178, 189–193 CrossRef CAS.
- A. Kumar, C.-C. Hsiao, W.-C. Hung and Y.-P. Lee, J. Chem. Phys., 1998, 109, 3824–3830 CrossRef CAS.
- G. Herzberg and J. Johns, Astrophys. J., 1969, 158, 399–418 CrossRef CAS.
- A. Kasdan, E. Herbst and W. Lineberger, Chem. Phys. Lett., 1975, 31, 78–82 CrossRef CAS.
- X. Li and Y.-P. Lee, J. Chem. Phys., 1999, 111, 4942–4947 CrossRef CAS.
- A. W. Kanzler, H. Sun and K. F. Freed, Int. J. Quantum Chem., 1991, 39, 269–286 CrossRef CAS.
- E. F. van Dishoeck, J. Chem. Phys., 1987, 86, 196–214 CrossRef CAS.
- G. C. Lie, J. Hinze and B. Liu, J. Chem. Phys., 1973, 59, 1887–1898 CrossRef CAS.
- G. C. Lie, J. Hinze and B. Liu, J. Chem. Phys., 1973, 59, 1872–1886 CrossRef CAS.
- P. F. Bernath, J. Quant. Spectrosc. Radiat. Transfer, 2020, 240, 106687 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The Duo input files consisting of the adiabatic data, and diabatic data with optimized PECs, respectively. The DMs, SOCs, and EAMCs of CH and the data of absorption cross-sections in Fig. 8 and 9. See DOI: https://doi.org/10.1039/d4cp03298e |
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b
*b