
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational and structural insights into the zwitterionic and neutral forms of N-substituted hydroxyformamidines in the solid state
David O.
Juma
 a,
Sizwe J.
Zamisa
*a,
Anamika
Sharma
b,
Unathi
Bongoza
a,
Eric M.
Njogu
c,
Fernando
Albericio
bd and
Bernard
Omondi
*a
a,
Sizwe J.
Zamisa
*a,
Anamika
Sharma
b,
Unathi
Bongoza
a,
Eric M.
Njogu
c,
Fernando
Albericio
bd and
Bernard
Omondi
*a
aSchool of Chemistry and Physics, University of KwaZulu Natal, Private Bag X54001, Westville, Durban, 4000, South Africa. E-mail: owaga@ukzn.ac.za; zamisas@ukzn.ac.za
bPeptide Science Laboratory, School of Chemistry and Physics, University of KwaZulu-Natal, Durban 4001, South Africa
cMultimedia University of Kenya, PO Box 15653-00503, Nairobi, Kenya
dCIBER-BBN (Networking Centre on Bioengineering, Biomaterials and Nanomedicine) and Department of Organic Chemistry, University of Barcelona, 08028 Barcelona, Spain
First published on 15th September 2025
Abstract
N-Hydroxyformamidines with the general formula [N-(Ar), N′(OH)-(Ar′)] were synthesized and characterized using NMR, IR and mass spectrometry. Analysis of single-crystal X-ray diffraction data of two of the compounds, along with previously reported structures of N-hydroxyformamidine derivatives, revealed that symmetrical hydroxyformamidines can exist as either zwitterionic or neutral species in the solid state. In contrast, unsymmetrical hydroxyformamidines are exclusively zwitterionic. The zwitterionic forms adopt Zanti isomerism, while the neutral forms exhibit Eanti isomerism. Both symmetrical compounds and unsymmetrical compounds bearing relatively less bulky substituents, exhibit intermolecular interactions that result in the formation of dimeric molecular units characterized by R22(10) graph-set descriptor. In contrast, unsymmetrical compounds with bulkier substituents form extended chain-like structures. N–H⋯O, N–H⋯N, O–H⋯O and O–H⋯N classical hydrogen bonding patterns were observed to support the crystal lattices in dimeric units, while chains were formed by relatively weak C–H⋯O intermolecular interactions. Molecular pairwise interaction energy calculations indicated that electrostatic energy (Eele) contributes more to the total energy of interaction of compounds forming dimeric units, whereas dispersion (Edis) energy is the primary contributor in molecules adopting chain-like structures. Natural Bond order analysis indicate that electron-donating substituents enhance the basicity of the imine and the amine nitrogen atoms, facilitating cooperative resonance-driven proton transfer. Consequently, both zwitterionic and neutral hydroxy species are present in the solid state. This study offers insight into the role of substituents in modulating hydrogen-bonding patterns and charge distribution, providing valuable strategies for controlling crystal packing and polymorphism, which are critical factors in pharmaceutical and materials science.
1 Introduction
Formamidines are versatile chemical compounds with the general formula R2N–CR![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR, where R can be variously substituted with H, alkyl, or aryl groups.1–3 Interest in these compounds advanced in 1950s following the discovery of their pesticidal properties4 along with their negligible toxicity to vertebrates.5 Their diverse structures have since prompted investigation to pharmacological potential, where they exhibit, antimicrobial,6 bactericidal7 and anticancer activities.8
NR, where R can be variously substituted with H, alkyl, or aryl groups.1–3 Interest in these compounds advanced in 1950s following the discovery of their pesticidal properties4 along with their negligible toxicity to vertebrates.5 Their diverse structures have since prompted investigation to pharmacological potential, where they exhibit, antimicrobial,6 bactericidal7 and anticancer activities.8
In addition to biological applications, formamidines serve as a key intermediate in various chemical transformations9 including synthesis of valuable compounds such as imidazole salts.10 The formamidine backbone NC(H)–N contains two nitrogen atoms offering bidentate coordination sites that enable their use as ligands in metal complexes.11 Recently, zinc(II) and copper(II) formamidine metal complexes have been applied in ring opening polymerization reactions as catalysts.11–13
The practical applications of these compounds are greatly influenced by the substituents on their backbone, as well as their overall structure and stereochemistry.11,14 Consequently, extensive research has been conducted to investigate their stereochemistry in both the liquid and solid states. The N–C single bond within the backbone exhibits partial double-bond character, enabling multiple conformations in solution, as supported by data from various spectroscopic techniques.15,16 Both anti and syn conformers, along with varying hydrogen bond association patterns in solution have been reported. However, only the anti-conformers have been observed to form cyclic hydrogen-bonded dimers (analogous to carboxylic acid dimers), stabilized by rapid double proton transfer.17 On the other hand, the CN double bond introduces a rotational barrier around the C–N bond, leading to E and Z conformational isomers with varying activation energies based on the steric and electronic properties of the N-substituents.18
Rapid double proton transfer in formamidine dimers in solution has been shown to be dependent on factors such as temperature, concentration of the dimeric species and the nature of the solvent.17,19 Recent studies by our group have examined the preferred isomerism of both symmetrical and unsymmetrical diarylformamidines in the solid state. We demonstrated that these compounds adopt either Esyn or Eanti isomers, with the steric and electronic properties of the aryl substituents significantly influencing molecular geometry, classical hydrogen-bonding patterns, and isomeric preference.20 However, the influence of N-substituent electronic effects on the formamidine backbone, and their potential role in modulating proton transfer, remains unexplored.
In this study, we report the modification of select diarylformamidines into N-hydroxy-N,N-diarylformamidines and their characterization. Our objectives include investigating solid-state molecular isomerism, molecular interactions, and Hirshfeld surfaces. Additionally, we aim to elucidate how the electronic and steric properties of various substituents influence molecular isomerism, intermolecular interactions, and potentially proton transfer.
2 Experimental section
2.1 Material and instrumentation
All solvents (ACS reagent grade, ≥99.5%) were purchased from Sigma-Aldrich and used as received without further purification. Aniline (99.5%), 2,6-difluoroaniline (98%), 2,6-diisopropylaniline (97%), 2,6-dimethylaniline (99%), 2,4,6-trimethylaniline (98%), triethyl orthoformate (99%), and 3-chloroperoxybenzoic acid were also obtained from the same supplier.The melting points were measured using Stuart Scientific SMP3 melting point apparatus. NMR spectra were recorded on a Bruker 600 MHz spectrometer using DMSO-d6 and CDCl3 as solvents. Chemical shift values (δ) are reported in parts per million (ppm) relative to the residual solvent peak at 2.50 ppm and 7.26 ppm, respectively for 1H NMR, and 39.5 ppm and 77.0 ppm respectively for 13C NMR. Infrared (IR) spectra were acquired using a PerkinElmer Universal ATR Spectrum 100 FTIR spectrometer. High-resolution mass spectra (HRMS) were obtained using a Shimadzu LCMS-2020 with electrospray ionization (ESI) in positive mode.
2.2 General procedure for preparing N-hydroxyformamidines 1–5
The hydroxyformamidines were synthesized following a method outlined in literature.11 Generally, 1 mmol of the formamidine was dissolved in dichloromethane, and 1 mmol solid sodium hydrogen carbonate added. The mixture was cooled to 0 °C. A slight excess of meta-chloroperoxybenzoic acid (1.2 mmol) in dichloromethane was then added dropwise, allowing the reaction mixture to warm to room temperature with stirring over an hour. The mixture was then washed twice with 25 mL of 5% potassium carbonate solution. The combined organic layers were dried over anhydrous sodium sulphate, filtered, and the solvent removed by evaporation to obtain the products as oils or solids.2.3 Single crystal X-ray diffraction
Crystal structure elucidation and data collection for compounds 4 and 5 was performed using a Bruker Smart APEXII diffractometer with Mo Kα radiation, equipped with an Oxford Cryostream low-temperature apparatus operating at 296 K. Reflections were collected from various starting angles, and the APEXII program suite was employed to index the reflections.21 Data reduction was processed using SAINT21 software, with scaling and absorption corrections applied via the SADABS22 multi-scan technique. The structures were solved by intrinsic phasing using the SHELXT23 program and refined with SHELXL.24 Crystal structure graphics were generated using OLEX2.25 Non-hydrogen atoms were initially refined isotropically, followed by anisotropic refinement using the full-matrix least squares method based on F2 with SHELXL.21 X-ray crystal data and structure refinement details are described in the Table 1. Crystallographic data for the structures has been deposited with the Cambridge Crystallographic Data Centre numbers 2452448 and 2452449 for compound 4 and 5, respectively.| 4 | 5 | |
|---|---|---|
| CCDC number | 2452448 | 2452449 |
| Empirical formula | C51H74Cl2N4O2 | C13H8F4N2O |
| Formula weight | 846.087 | 284.21 |
| Crystal system | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
| a/Å | 12.6939(3) | 7.5760(3) |
| b/Å | 13.0267(3) | 23.9624(8) |
| c/Å | 18.5039(5) | 13.8352(4) |
| α/° | 84.192(1) | 90 |
| β/° | 76.836(1) | 100.932(2) |
| γ/° | 61.861(1) | 90 |
| λ/Å | 0.71073 | 0.71073 |
| V/Å3 | 2627.18(12) | 2466.05(15) |
| Z | 2 | 8 |
| Z′ | 1 | 2 |
| ρ calc/g cm−3 | 1.070 | 1.531 |
| μ/mm−1 | 0.162 | 0.140 |
| F(000) | 917.0 | 1152.0 |
| Crystal size/mm3 | 0.21 × 0.16 × 0.13 | 0.25 × 0.17 × 0.12 |
| 2θ range for data collection/° | 3.72 to 52.04 | 3.4 to 52.746 |
| Index ranges | −15 ≤ h ≤ 15 | −9 ≤ h ≤ 9 |
| −16 ≤ k ≤ 12 | −29 ≤ k ≤ 29 | |
| −19 ≤ l ≤ 22 | −17 ≤ l ≤ 17 | |
| Reflections collected | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 842 842 |
95598 |
| Independent reflections | 10046 |
5045 |
| R int/−Rsigma | 0.0345/0.0413 | 0.0312/0.0099 |
| Data/restraints/parameters | 10046/0/549 |
5045/0/363 |
| Goodness-of-fit on F2 | 1.055 | 1.053 |
| Final R indexes [I ≥ 2σ(I)] | ||
| R 1 | 0.0782 | 0.0416 |
| wR2 | 0.2280 | 0.1062 |
| Final R indexes [all data] | ||
| R 1 | 0.1136 | 0.0505 |
| wR2 | 0.2677 | 0.1146 |
| Largest diff. peak/hole/e Å−3 | 0.65/−0.73 | 0.18/−0.25 |
2.4 Hirschfeld surface analysis
Hirshfeld surface analysis and fingerprint plots26,27 for compounds 4 and 5, as well as for compounds with Cambridge Structural Database reference codes QUCMUX28 and GIKFUB,29 and compounds with CCDC reference numbers 2389686 (ref. 30) and 2389590 (ref. 31) (identified as 6 and 7 respectively in this manuscript) were created using CrystalExplorer21.32 This analysis was performed on the crystal structures within the asymmetric units of each compound. All bond lengths involving hydrogen were automatically adjusted to standard neutron values: C–H = 1.083 Å, O–H = 0.983 Å, and N–H = 1.009 Å.33 The fingerprint plot was generated using a standard view range of 0.6 to 2.6 Å, with the distances to the nearest atoms outside (de) and inside (di) the surface displayed on the graph.2.5 Theoretical calculations
For theoretical calculations, a single unit cell was selected as the initial structure from the crystals of compounds 4, 5, as well as the previously reported structures GIKFUB and 6. The structures were optimized using the DFT-B3LYP/6-311G++(d,p) method in Gaussian 09 at a default temperature of 298.15 K. Transition state (TS) calculations were carried out to investigate the conversion from the neutral to the zwitterionic form. The identified TS was validated through intrinsic reaction coordinate (IRC) analysis. Natural bond orbital (NBO) calculations were also performed using the same basis set.3 Results and discussion
3.1 Synthetic consideration of compounds 1–5
The synthesis of the compounds followed the general procedure outlined in Scheme 1. The electron-donating properties due to lone pair of electrons on the nitrogen atoms in the formamidine backbone facilitate the oxidation of the compounds using the potent oxidizing agent meta-chloroperoxybenzoic acid. All the compounds were isolated as off-white to brown solids except for compound 1, which was isolated as a brown oil, with good to excellent yields ranging between 50% and 80%. The yields appeared to be influenced by the substituents on the phenyl ring where compounds bearing electron-donating groups afforded relatively higher yields compared to those with electron-withdrawing groups. The electron-donating substituents increase electron density on the imine nitrogen, enhancing its nucleophilicity and reactivity toward the oxidizing agent.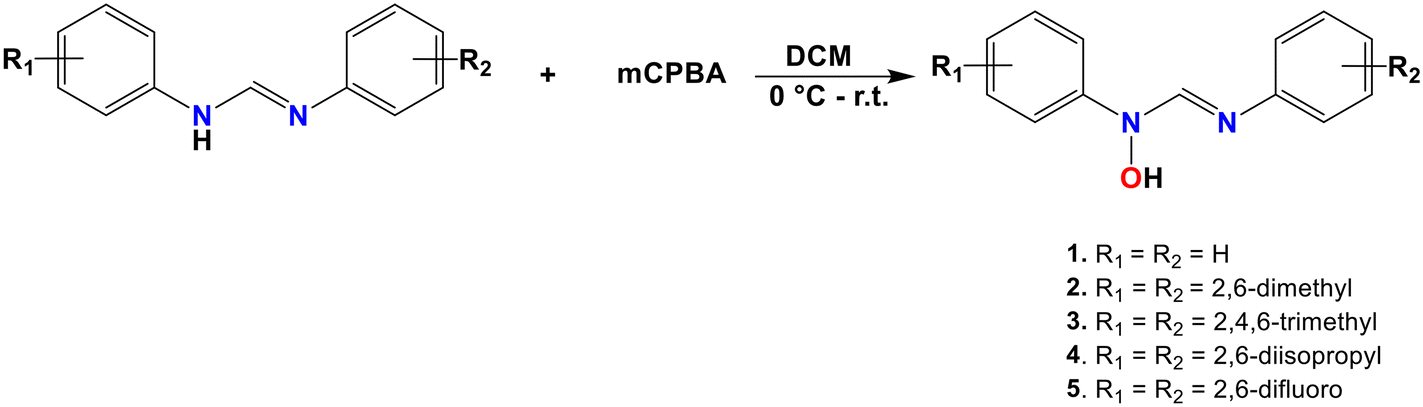 | ||
| Scheme 1 Synthetic route to the formation of N-hydroxyformamidines. | ||
3.2 Spectroscopic analysis of compound 1–5
In the IR spectra, broad O–H and N–H stretching bands overlapping with aliphatic and aromatic C–H stretches are observed. These bands appear significantly broader in the spectra of compounds 1 and 5, spanning the 2700–3400 cm−1 range, compared to compounds 2, 3, and 4, which contain electron-donating substituents and display narrow bands in the 2700–3100 cm−1 region. The broad bands suggest the presence of relatively strong intermolecular hydrogen bonding between the molecules of the compounds. Additionally, the spectra of compounds 1–5 exhibit absorption bands between 2853 and 2886 cm−1 and between 1642 and 1675 cm−1, corresponding to aliphatic C–H and CN stretching vibrations, respectively (Table 1). Compound 5 bearing an electron-withdrawing substituent displays C–H stretching vibrations at relatively higher wavenumbers compared to those with electron-donating groups 2, 3, and 4, and 1 the unsubstituted compound. Notably, 1 shows a CN stretching band at a relatively higher wavenumber compared to compounds 2–5. The NMR spectra of 1–5 demonstrate that chemical shift of the methine proton is shifted to a lower field due to increased electron density around the phenyl rings from electron-donating groups (Table 2). All NMR spectra (Fig. S2–S11) and FTIR (Fig. S12–S16) are provided in the SI.
| Compound | IR bands (cm−1) | 1H NMR (ppm) | |
|---|---|---|---|
| ν(C–H) |
ν(CN) |
δ[NC(H)–N] |
|
| 1 | 2862 | 1675 | 8.79 |
| 2 | 2853 | 1650 | 7.82 |
| 3 | 2856 | 1658 | 7.73 |
| 4 | 2867 | 1665 | 7.74 |
| 5 | 2891 | 1667 | 8.32 |
3.3 Crystal structure description of compounds 4–7
Fig. 1 presents the molecular structures of compounds 4 and 5. Compound 4 crystallized as a dichloromethane hemisolvate, with two crystallographically distinct molecules in the asymmetric unit. Likewise, the crystal structure of compound 5 contains two crystallographically independent molecules. In compound 5, both adopt the neutral hydroxy form as the stable configuration (Fig. 1c). By contrast, compound 4 exhibits one molecule in the neutral form (Fig. 1a) and the other in the zwitterionic form (Fig. 1b). Like their parent formamidines, the presence of an NC double bond enables the formation of E and Z geometric isomers.18Z isomerism is preferred by the zwitterionic forms, whereas E isomerism is prevalent by the neutral molecules. Both isomers can be classified based on their conformation as syn and anti, depending on the orientation of the N–H and C–H hydrogen atoms in the zwitterionic molecules, or the C–H and the N–OH group in the neutral molecules along the C–N single bond. The zwitterionic species adopts Zanti isomerism while the neutral derivatives exhibit Eanti isomerism. This trend is consistent with reported crystal structures of related compounds, such as N-hydroxy-N,N′-bis(2,6-dimethylphenyl)formamidine (QUCMUX)28 and N-hydroxy-N-(4-methoxyphenyl)-N′-phenylformamidine (GIKFUB).29
 | ||
| Fig. 1 Molecular structures of hydroxyformamidines showing atom numbering scheme and displacement ellipsoids drawn at 50% probability level. Only the hydrogen atoms on the formamidine backbone are shown. (a) and (b) show the neutral and the zwitterionic structures of compound 4, respectively, while (c) shows the neutral structure of compound 5. | ||
Molecular overlay of the crystallographically unique molecules in crystal structures of compounds 4 and 5 are shown in Fig. 2(a) and (b) respectively. The overly of molecules in the asymmetric unit of crystal structure of compound 4 have a root mean square deviation (RMSD) of 0.1994 Å resulting primarily from different orientation of the phenyl substituents. The molecules in the asymmetric unit of compound 5 are conformationally near identical with molecular overlay having a RMSD of 0.0491 Å. The observed deviation from similarity in 5 is due to the orientation of the fluorine substituent on the phenyl rings and the hydroxyl group.
 | ||
| Fig. 2 Molecular overlays of the crystallographically unique molecules in the asymmetric units in crystal structures of compounds 4 (a) and 5 (b). | ||
The planes of the two pendant rings and the formamidine backbone are not coplanar. The isomers can thus be described by the dihedral angles between planes A, B, and C as shown in Fig. 4. The dihedral angles are listed in Table 3, along with those of closely related hydroxyformamidine compounds that have been structurally characterized in the literature. These include N-hydroxy-N,N′-bis(2,6-dimethylphenyl)formamidine (QUCMUX),28N-hydroxy-N-(4-methoxyphenyl)-N′-phenylformamidine (GIKFUB),29N-hydroxy-N-(2-methoxyphenyl)-N′-(2,4-dimethylphenyl)hydroxyformamidine (CCDC reference number 2389686)30 (6) and N-hydroxy-N-(2-mehoxyphenyl)-N′-(2,4-diisopropylphenyl) hydroxyformamidines (2389590)31 (7) (Fig. 3). In Eanti isomers the dihedral angle between planes A and B increases with the steric bulk of the substituents, indicating a sterically driven distortion of the molecular geometry. For example, compound 4, with bulky isopropyl groups causing significant steric effects, exhibits the largest dihedral angle of 74.40(2)°. QUCMUX, with slightly smaller methyl substituents, follows with an angle of 68.70(3)°, while compound 5, which has even smaller fluorine substituents, shows the smallest angle, 55.82(19)°.
| Compound | Dihedral angle/° | ||
|---|---|---|---|
| A–C | B–C | A–B | |
| E anti isomerism | |||
| 4 | 74.40(2) | 86.60(2) | 61.91(11) |
| QUCMUX | 68.70(3) | 98.30(3) | 62.12(11) |
| 5 | 55.82(19) | 33.21(18) | 29.45(7) |
| Z anti isomerism | |||
| 4 | 86.30(3) | 74.90(3) | 63.94(12) |
| QUCMUX | 87.60(3) | 87.70(3) | 49.70(11) |
| GIKFUB | 34.90(3) | 21.80(3) | 30.80(13) |
| 6 | 14.84(11) | 81.34(12) | 89.25(5) |
| 7 | 7.88(15) | 81.17(15) | 78.17(15) |
 | ||
| Fig. 3 Illustration of the molecular structures of QUCMUX, GIKFUB, 2389686, and 2389590. QUCMUX is shown in both its neutral (a) and zwitterionic (b) forms. | ||
 | ||
| Fig. 4 The three planes identified within the hydroxy formamidine molecules. Plane A, corresponding to the phenyl ring attached to the amine nitrogen; plane B, corresponding to the phenyl ring attached to the imine nitrogen; and plane C, corresponding to the formamidine backbone (–N–CHN–). | ||
In the Zanti isomers, larger steric substituents result in smaller dihedral angles. For instance, compound 4 the angle is 86.30(3)°, smaller than 87.60(3)° observed in QUCMUX. Similarly, GIKFUB, which has the smallest steric load, shows the largest angle of 34.90(3)°. This is followed by compound 6 with 14.84(11)°, and compound 7, which has the largest steric load but the smallest angle of 7.88(15)°.
3.4 Evaluation of intermolecular interactions
In the crystal structure of compound 4, packing is stabilized by N–H⋯N (H⋯N = 2.120(3) Å, ∠N–H⋯N = 168.12(8)°) and O–H⋯O (H⋯O = 1.727(3) Å, ∠O–H⋯O = 168.50(3)°) hydrogen bonds (Table 4). These interactions occur between Zanti and Eanti isomers, resulting in hydrogen-bonded cyclic dimers with an R22(10) graph-set motif (Fig. 5). A similar hydrogen-bonding pattern is observed in the structure of QUCMUX, which features electron-donating methyl substituents.28 The structure of compound 5, which contains an electron-withdrawing group, also forms R22(10) dimers via O–H⋯N hydrogen bonds (H⋯N = 1.975(14) Å, ∠O–H⋯N = 160.10(9)°) (Fig. 6). In contrast to the crystal structure of compound 4, the dimers in the structure of 5 are formed between two Eanti isomers.| D–H⋯A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|
| Compound 4 | ||||
| O2–H2⋯O1 | 0.819(19) | 1.727(3) | 2.536(3) | 168.5(5) |
| N4–H4a⋯N1 | 0.860(17) | 2.120(3) | 2.967(3) | 168.12(8) |
| Compound 5 | ||||
|---|---|---|---|---|
| O2–H2⋯N2 | 0.820(13) | 1.975(14) | 2.763(19) | 169.10(9) |
 | ||
| Fig. 5 Representation of the hydrogen bonding pattern in the crystal structure of compound 4. For clarity, only the hydrogen atoms on the formamidine backbone are displayed. Hydrogen bonds are indicated by blue and red dotted lines. | ||
 | ||
| Fig. 6 Hydrogen bonding pattern in the crystal structure of compound 5 hydrogen bonds are indicated by dotted blue lines. | ||
The hydrogen bonding patterns in the crystal structures of the unsymmetrical GIKFUB, 6, and 7, all of which adopt Zanti isomerism, have been reported. In the structure of GIKFUB cyclic hydrogen bonded dimers with an R22(10) graph-set descriptor were formed via N–H⋯O hydrogen bonding interactions. In contrast, the crystal parking of compound 6, is driven by C–H⋯O and O–H⋯O interactions leading to chain running along a crystallographic axis. The chains are supported by C–H⋯π intermolecular interactions.30 For compound 7, C–H⋯O interactions result in hydrogen-bonded cycles with an R34(30) graph-set descriptor, creating two-dimensional layers extending along the ac plane.31
3.5 Non-covalent interaction plots (NCI)
NCI analysis is a qualitative approach used to visualize and characterize regions of attractive and repulsive non-covalent interactions. Hydroxyformamidines exhibit both zwitterionic and neutral species unlike formamidines. Hydroxyformamidine derivatives are capable of forming three distinct types of hydrogen-bonded pairs, as detailed in section 3.4. NCI analysis was employed to elucidate the interaction patterns among these pairs, with emphasis placed on the formamidine backbone.Fig. 7 presents the NCI plots mapped as isosurfaces and the plot of the reduced density versus the electron density multiplied by the sign of the second Hessian Eigenvalue,34,35 for the two-molecular aggregates that forms hydrogen bonded dimers. The calculated isosurfaces use a red-blue-green colour scheme: blue for strong attractive interactions (sign(λ2)ρ), red for strong repulsion, and green for weak interactions.35
 | ||
| Fig. 7 Three-dimensional and two-dimensional non-covalent interaction (NCI) plots for the two-molecular aggregate of hydroxy formamidine hydrogen bonded dimers, stabilized by O–H⋯N, N–H⋯N, N–H⋯O, and O–H⋯O interactions. | ||
The hydrogen-bonded dimers of compound 5, formed through O–H⋯N interactions between two Eanti isomers, exhibit relatively symmetrical attractive interaction, as evidenced by the NCI plots shown in Fig. 6. A similar trend is observed in GIKFUB, which forms dimers through N–H⋯O hydrogen bonds between two Zanti isomers. In contrast, the NCI plot of compound 4, which forms cyclic dimers through interactions between Zanti and Eanti isomers stabilized by O–H⋯O and N–H⋯N hydrogen bonds, reveal asymmetrical interaction strengths within the dimeric structure with O–H⋯O having stronger interaction than N–H⋯N. The NCI plots for compounds 6, 7 and 8 are provided in the supplementary information (Fig. S21).
3.6 Hirshfeld surface analysis
Electrostatic potential mappings on Hirshfeld surfaces of molecules of compounds 4, 5, QUCMUX, GIKFUB, 6 and 7 have been created to provide an understanding of charge distribution, which influences various types of intermolecular interactions, including hydrogen bonding.36 In these mappings, electronegative areas are represented in red, while electropositive areas are shown in blue. Fig. 8 illustrates the front and back views of the different isomers of the molecules. | ||
| Fig. 8 Electrostatic potential mapping of Eanti and/or Zanti isomers of 4, 5, QUCMUX, GIKFUB, 6 and 7 showing electropositive region in blue and electronegative region blue. (a) The front view is taken to be surface where the functional groups –OH in Eanti isomers and N–O− in Zanti isomers points while (b) the back view is where the methine proton points. All electrostatic mappings are generated using same scale. | ||
Both the Zanti and Eanti isomers of symmetrical formamidines, such as compound 4 and QUCMUX, exhibit both electronegative and electropositive regions positioned on the same side. A similar pattern is observed in the Eanti isomer of compound 5. In unsymmetrical hydroxy formamidines, the Zanti isomer of GIKFUB, with a methoxy group on the para position of one of its phenyl rings, similarly has both electronegative and electropositive regions on the same side. In contrast, compounds 6 and 7, with increased steric bulk due to methyl and isopropyl substituents on one of their phenyl rings and a methoxy group on the ortho position of the other, show electronegative and electropositive regions on both the front and back of the molecules. This electron distribution accounts for the observed hydrogen-bonding patterns, where compounds 4, 5, QUCMUX, and GIKFUB form hydrogen-bonded cyclic dimers, while compounds 6 and 7 favour chain-like hydrogen-bonded structure.
In our previous work,37 we investigated the close contacts and intermolecular interactions of formamidine using dnorm Hirshfeld surfaces. Our findings indicated that the primary interactions were reciprocal H⋯H and H⋯X/X⋯H (where X represents fluorine or chlorine), with variations dependent on the substituents on the phenyl rings. The crystal packing of these compounds is predominantly influenced by the N⋯H/H⋯N interactions, leading to significant N⋯H hydrogen bonding.20 Crystal parking in hydroxyformamidines on the other hand is influenced by N⋯H/H⋯N and O⋯H/H⋯O interactions, which give rise to N–H⋯O, O–H⋯N, N–H⋯N and O–H⋯O hydrogen bonding patterns The fingerprint plot in Fig. 9 demonstrates that in Eanti isomerism the substituents on the phenyl rings significantly affect the likelihood of N⋯H/H⋯N reciprocal interactions. For instance, the introduction of electron-withdrawing groups, such as fluorine in compound 5 in Eanti isomerism, enhances this interaction from 1.7% in QUCMUX and 1.2% in compound 4 to 5.6%. A similar trend is observed in the O⋯H/H⋯O reciprocal interactions, where the percentage increases from 4.0% in compound 4 and 5.8% in QUCMUX to 7.9% in compound 5.
 | ||
| Fig. 9 Hirschfeld surfaces mapped over dnorm and selected fingerprint plots with respective reciprocal contact contributions of various N-hydroxy formamidines. | ||
A similar trend is observed in the Zanti isomerism. In the unsymmetrical hydroxyformamidines, the introduction of electron-withdrawing groups in compounds 6 and 7 decreases the N⋯H reciprocal interactions in GIKFUB from 4.8% to 1.6% and 2.2%, respectively. However, this trend does not hold for the O⋯H reciprocal interactions due to the solvation effects in compound 7, which increase the H⋯O de interactions and, consequently, the overall reciprocal interactions. In the symmetrical compounds, replacing the methyl group in QUCMUX with a bulkier isopropyl group in compound 4 also tends to reduce both the N⋯H and O⋯H reciprocal interactions.
3.7 Molecular pairwise interaction energies and energy framework
The varying patterns of intermolecular hydrogen bonding in crystal packing are influenced by the electronics and steric effects due to the substituents on the phenyl groups. Therefore, it is valuable to study the interaction energies between molecules. These interaction energies are typically expressed in terms of electrostatic (Eele), polarization (Epol), dispersion (Edis), and repulsion components, all of which contribute to the total energy (Etot).38 The calculated interaction energies are summarized in Table 5.| Compounds | Interaction energy/kJ mol−1 |
E
dis:Eele ratio |
Interaction type | ||||
|---|---|---|---|---|---|---|---|
| E ele | E pol | E dis | E rep | E tot | |||
| QUCMUX | −163.1 | −67.3 | −59.0 | 157.5 | −135.4 | 0.3617 | O–H⋯O |
| N–H⋯N | |||||||
| 4 | −145.1 | −60.5 | −119.8 | 171.9 | −155.8 | 0.8256 | O–H⋯O |
| N–H⋯N | |||||||
| 5 | −145.3 | −48.9 | −79.9 | 141.3 | −137.4 | 0.5499 | O–H⋯O |
| GIKFUB | −90.8 | −33.1 | −29.5 | 60.8 | −91.3 | 0.3249 | N–H⋯O |
| 6 | −11.6 | −8.5 | −33.4 | 18.8 | −32.2 | 2.8793 | C–H⋯O |
| 7 | −5.9 | −6.3 | −16.5 | 5.8 | −20 | 2.7966 | C–H⋯O |
In compounds QUCMUX, 4, 5, and GIKFUB, the Edis:Eele ratios are less than one, indicating that electrostatic energy (Eele) makes the most significant contribution to the total energy (Etot). In contrast, for compounds 6 and 7, where the Edis:Eele ratios exceed one, dispersion energy (Edis) is the dominant factor. This suggests that crystal packing in the latter compounds is primarily driven by classical hydrogen bonding, while in the former compounds, it is mainly influenced by the dispersive forces. Notably, the ratio in compound 4 is nearly equal to one, and is attributed to the bulky isopropyl substituents that stabilize the dispersion energy.
To investigate the topology of pairwise interaction energies for compounds 4, 5, QUCMUX and GIKFUB, 6 and 7 a graphical energy framework was generated using CrystalExplorer21.32 The components Eele, Edis, and Etot are presented in Fig. 10 as cylinders connecting the centroids of molecular pairs. The Coulomb energy cylinders mirror the intermolecular N–H⋯O, N–H⋯N, O–H⋯N, and C–H⋯O hydrogen bonding patterns observed in the crystal packing of the compounds. In QUCMUX, 4, 5, and GIKFUB, the Coulomb energy forms discrete cylinders between molecular pairs, while compounds 6 and 7 exhibit a chain-like, interlinked network. In compound 6, which is a dichloromethane solvate, the cylinders between the compound and solvent molecules are notably larger, attributable to the classical O–H⋯O hydrogen bonds between them.
 | ||
| Fig. 10 Calculated energy frameworks for compounds 4–7, QUCMUX, and GIKFUB, highlighting the contributions of electrostatic and dispersion energies to the total energy. An adjusted scale factor of 100 was applied uniformly to all cylindrical radii, reflecting the strength of intermolecular interactions. | ||
Interestingly the dispersion energy cylinders in compounds QUCMUX, 4, 5, and GIKFUB form a chain-like, interlinked network, with the cylinders between hydrogen-bonded pairs appearing larger and aligned in the same direction as those of the Coulomb energy. Similarly, compounds 6 and 7 exhibit an interlinked network of dispersion energy cylinders. Furthermore, the total energy cylinders of all the compounds correspond to the patterns of the calculated values, with the hydrogen-bonded dimers demonstrating enhanced stability.
4 Theoretical calculation
4.1 Optimized molecular geometries of compounds 4, 5, GIKFUB and 6
Theoretical calculations were carried out using the Gaussian09 program package.39 Geometry optimizations were performed in the gas phase at 298.15 K employing density functional theory (DFT) with the B3LYP functional, which integrates Becke's three-parameter hybrid exchange functional with the gradient-corrected correlation functional of Lee, Yang, and Parr, the 6-311G++(d,p) basis set. The initial geometries were obtained from X-ray crystallographic data except for 1 whose geometry was obtained through modification of the geometry of 5 (The optimized geometry coordinates are included in the supplementary data). The absence of imaginary frequencies in the vibrational analysis verified that the optimized structures correspond to a minimum on the potential energy surface. The optimized molecular geometries in the free state were compared to those observed in the crystalline state. The resulting structures exhibit strong agreement with the experimentally determined crystal structures, as indicated by root mean squared deviation (RMSD) values ranging from 0.3566 to 0.1206 Å for the molecular overlay of crystal and DFT-calculated structures (Fig. 11). This close correlation underscores the reliability of the DFT method employed. The total energies of the optimized structures are presented in Table 6. | ||
| Fig. 11 Molecular overlay analysis comparing DFT-optimized structures with experimental crystal structures for compounds 4 (both the zwitterionic, a and neutral, b molecules), 5 (GIKFUB), and 6. | ||
| E anti isomer | Energies (EO) | Z anti isomers | Energies (EO) | Dimeric units | Energies (EO) |
|---|---|---|---|---|---|
| 4 | −1159.343 | 4 | −1159.342 | 4 | −2169.046 |
| 5 | −1084.514 | 6 | −996.434 | 5 | −3277.793 |
| 1 | −687.459 | 1 | −687.458 | GIKFUB | −1604.046 |
| GIKFUB | −802.0127 |
4.2 Transition state (TS) calculations
A transition state corresponds to a first-order saddle point on the potential energy surface (PES) of a molecular system. Its vibrational spectrum is characterized by a single imaginary frequency, which reflects a negative force constant. This indicates that the energy is maximized along one nuclear coordinate while minimized along all other orthogonal coordinates. TS calculations were performed on the optimized structures of the neutral and zwitterionic forms of compound 1 to examine the conversion between the two isomers. As shown in Fig. 12, the reaction proceeds preferentially toward the neutral form. These calculations were carried out in the gas phase; hence, solvent effects were not considered. The activation energy of the reaction was determined to be 6.17 kcal mol−1. | ||
| Fig. 12 Transition state in case of 1, conversion from neutral to zwitterionic form. | ||
4.3 Natural bond order (NBO)
NBO analysis is a quantum chemistry tool for investigating electron distribution, bonding interactions, and charge delocalization at the atomic level in a molecule. In this study, we utilize NBO analysis to examine the natural atomic charges of both the zwitterionic and neutral hydroxy isomers of the 4, 5, GIKFUB and 6, aiming to elucidate the factors that govern their relative stability in the solid state. Only atoms in the formamidine backbone are considered (Fig. 13). As anticipated, the electron-donating substituents (diisopropyl) in 4 increase the electron density on both the amine and imine nitrogen atoms, yielding calculated charges of −0.206 and −0.616, respectively, for the free (non-hydrogen-bonded) neutral molecule. In contrast, in 5, which contains electron-withdrawing substituents, the imine nitrogen in the free molecule exhibits slightly lower charges of −0.133 and −0.598, respectively. Interestingly, the phenyl substituents in unsubstituted formamidine 1 exhibit greater electron-withdrawing character compared to those in 2,6-difluorophenyl-substituted formamidine 5, as reflected by the calculated charges of −0.126 and −0.585 on the amine and imine nitrogen atoms, respectively. This enhanced electron withdrawal may be attributed to the reduced steric strain in the unsubstituted formamidine, allowing the phenyl groups to adopt a conformation that facilitates π-conjugation with the electronegative nitrogen atoms. The reduced electron density along the molecular backbone of 1 likely account for the observed shift of its CH stretching vibration to a relatively higher wavenumber compared to compounds 2–5.
 | ||
| Fig. 13 Natural atomic charges of the atoms in the formamidine backbone determined based on DFT calculations. These calculations were performed on both the free molecules of the compounds and their hydrogen-bonded forms, as obtained from the experimental crystal structure. | ||
Notably, hydrogen bonding between two neutral molecules and between two zwitterionic molecules alters the atomic charge distribution, increasing the electron density on the imine and amine nitrogen atoms as well as the oxygen atom, while simultaneously enhancing the positive character of the carbon and hydrogen atoms. This effect is evident from the comparison of atomic charges in free and hydrogen-bonded molecules, as observed in compound 5 and GIKFUB. The same trend is observed in the zwitterion hydrogen bonded to the neutral molecule, as seen in compound 4, while the opposite effect is observed for the neutral molecule.
The increased basicity of the imine nitrogen due to electron donating substituents in 4, combined with electron delocalization (tautomerism) on the formamidine backbone likely promote cooperative resonance proton transfer (Fig. 14) contributing to the stabilization of both the zwitterionic and neutral hydroxy species in the solid state. In contrast electron withdrawing substituents in 5 hinders proton transfer and consequently formation of zwitterions. Compound 6 and GIKFUB on the other hand, adopts a solely zwitterionic molecular structure, a behaviour likely driven by asymmetrical charge distribution due to the presence of unequal electron-donating substituents.
 | ||
| Fig. 14 A proposed mechanism illustrating intermolecular (b) and intramolecular (a) proton transfer, coupled with tautomerism due to the position of the CN double bond. | ||
The dipole moments of all structures are summarized in Table 7. In general, the dipole moment increases when going from the neutral to the zwitterionic form. For example, in the fluorine-containing derivative (compound 5), the dipole moment increases from 3.6 D in the neutral Eanti form to 5.3 D in the hydrogen-bonded dimer. For compound 4, three forms were observed: Eanti (2.6 D), dimeric unit (1.8 D), and Zanti (3.2 D). These differences arise from whether the crystal exists in the neutral, hydrogen-bonded, or zwitterionic form. In the unsubstituted compound 1, the dipole moment increases by 1.5 D when going from the neutral Eanti form (2.0 D) to the zwitterionic Zanti form (3.5 D). Overall, the dipole moment tends to increase upon zwitterion formation, although the increase is not very large, likely because the charged atoms are relatively close (<2 Å). In the unsymmetrically substituted derivative GIKFUB, the dipole moments were 5.0 D for the zwitterionic Zanti form and 0.0 D for the hydrogen-bonded dimer, showing a larger difference compared to the symmetrical derivatives.
| E anti isomer | Dipole moment (D) | Z anti isomers | Dipole moment (D) | Dimeric units | Dipole moment (D) |
|---|---|---|---|---|---|
| 4 | 2.6 | 4 | 3.2 | 4 | 1.8 |
| 5 | 3.6 | 6 | 2.8 | 5 | 5.3 |
| 1 | 2.0 | 1 | 3.5 | GIKFUB | 0.0 |
| GIKFUB | 5.0 |
4.4 Second order perturbation theory analysis of Fock matrix in NBO basis
NBO analysis of the dimeric unit of compound 5 identified a strong n → π* interaction from LP(1) N7 to BD(2) N8–C18 (E(2) = 55.62 kcal mol−1), consistent with the short N8–C18 bond (1.2908 Å) and the nonbonded N7–N8 separation (2.326 Å). LP(1) N8 also donates to σ*(O33–H34) (E(2) = 26.67 kcal mol−1), forming an intramolecular N8⋯H–O hydrogen bond [N8⋯H34 = 1.762 Å, N8⋯O33 = 2.758 Å, O–H = 1.0035 Å], and to σ*(C18–H19) (E(2) = 10.89 kcal mol−1), reflecting a strong hyperconjugative effect. In the Eanti isomer of 5, a strong n(N8) → π*(N7–C18) interaction (55.82 kcal mol−1) enhances partial double-bond character and contributes to molecular stabilization. A weaker n(N7) → σ*(C18–H19) interaction (12.44 kcal mol−1) is also observed, indicating a modest hyperconjugative effect compared to the dimeric unit. The dimeric unit of 5 further benefits from additional hydrogen-bond stabilization, absent in the Eanti form. For compound 6, strong conjugation is observed through n(N40) and n(O44) → π*(C13–N42) interactions. The lone pair on N40 donates significantly to the π* orbital of C13–N42 (E(2) = 62.10 kcal mol−1), while O44 contributes similarly (E(2) = 48.59 kcal mol−1), demonstrating cooperative resonance effects that enhance bond delocalization and stability. In the Zanti isomer of GIKFUB, the lone pair on N16 shows strong n → π* donation into the σ*(N1–C18) orbital, imparting partial double-bond character to the CN bond. This interaction is even stronger in the dimeric unit of GIKFUB, likely due to additional hydrogen bonding. Furthermore, O17 donates into the same σ* orbital (E(2) = 45.09 kcal mol−1), reinforcing conjugation and indicating optimal orbital overlap from both N16 and O17. Among derivatives of compound 4, both the Eanti isomer and the dimeric unit display strong n → π* interactions involving nitrogen lone pairs donating into adjacent N–C antibonding orbitals (E(2) ≈ 49–59 kcal mol−1), providing significant resonance stabilization and partial double-bond character. The dimeric unit also exhibits interactions consistent with hydrogen bonding, adding conformational rigidity. In contrast, the Eanti isomer of 4 shows a moderate n(N2) → σ*(N2–H3) interaction (20.69 kcal mol−1), reflecting weaker intramolecular hydrogen bonding or hyperconjugation without substantial π* delocalization.
Both Zanti and Eanti isomers of compound 1 exhibit strong n → π* interactions from nitrogen lone pairs into σ*(C–N), with the Zanti isomer displaying greater stabilization (64.30 vs. 53.09 kcal mol−1), consistent with stronger resonance and partial double-bond character in its zwitterionic form. Additionally, both isomers show weaker hyperconjugative interactions from nitrogen lone pairs into nearby σ*(C–H) orbitals (≈12 kcal mol−1), indicating modest bond polarization effects.
5 Conclusion
This study highlights how substituents modulate the structural and electronic properties of N-hydroxyformamidines, particularly their solid-state behaviour. Electron-donating groups promote the coexistence of neutral and zwitterionic forms, while electron-withdrawing groups stabilize only the neutral hydroxy form. For unsymmetrical derivatives, steric factors are decisive, with smaller substituents allowing classical dimeric hydrogen-bonding motifs, whereas bulkier groups enforce chain-like architectures stabilized primarily by weaker C–H⋯O interactions.Computational analyses have provided deeper insights into these trends. Pairwise interaction energy calculations confirmed that electrostatic forces dominate in dimeric units, while dispersion interactions are more significant in chain structures. NBO analysis further revealed strong n → π* and n → σ* delocalizations that reinforce partial double-bond character and stabilize the zwitterionic form. Importantly, electron-donating substituents enhance nitrogen basicity, enabling resonance-driven proton transfer, which may explain the coexistence of neutral and zwitterionic species in some systems.
Overall, the work demonstrates that subtle changes in electronic and steric substituent effects can dictate hydrogen-bonding patterns, charge distribution, and crystal packing preferences. These findings not only deepen the understanding of hydroxyformamidine chemistry but also point to broader applications in the deliberate design of supramolecular assemblies, crystal engineering, and the tuning of polymorphic behaviour in materials and pharmaceutical sciences.
Author contributions
David O. Juma: investigation, data curation, formal analysis, validation, and writing original draft. Unathi Bongoza and Anamika Sharma: investigation, data curation, formal analysis, and validation. Sizwe J. Zamisa, Bernard Omondi and Eric M. Njogu: conceptualization, methodology, supervision, and writing – review and editing. Fernando Albericio: supervision, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The IR, ESI-MS, 1H-NMR, 13C-NMR data and atomic coordinates of the geometry optimized structures, have been included in the manuscripts SI. Crystallographic data for the structures has been deposited with the Cambridge Crystallographic Data Centre (CCDC) numbers 2452448 and 2452449 for compound 4 and 5, respectively. Compounds with Cambridge Structural Database (CSD) reference codes QUCMUX and GIKFUB, and compounds with CCDC numbers 23899686 and 2389590 (identified as 6 and 7 respectively) were also used this manuscript). See DOI: https://doi.org/10.1039/D5CE00521C.CCDC 2452448 and 2452449 contains the supplementary crystallographic data for this paper.40a,b
Acknowledgements
We would like to thank ESKOM for paying David's fees through TESP and the University of KwaZulu Natal for creating a supportive environment for this research.References
- J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219–300 Search PubMed.
- M. Cibian, S. Langis-Barsetti and G. S. Hanan, Synlett, 2011, 2011, 405–409 CrossRef.
- G. Jin, C. Jones, P. C. Junk, K.-A. Lippert, R. P. Rose and A. Stasch, New J. Chem., 2009, 33, 64–75 RSC.
- R. Hollingworth, Environ. Health Perspect., 1976, 14, 57–69 CrossRef CAS PubMed.
- T. Farooqui, Neurochem. Int., 2013, 62, 122–136 CrossRef CAS.
- S. D. Oladipo, F. A. Olotu, M. Soliman, C. Mocktar and B. Omondi, J. Mol. Struct., 2020, 1219, 128553 CrossRef CAS.
- C. Mallikarjunaswamy, D. Bhadregowda and L. Mallesha, J. Saudi Chem. Soc., 2016, 20, S606–S614 CrossRef CAS.
- M. Stojak, M. Łukawska, I. Oszczapowicz, M. Opydo-Chanek and L. Mazur, Anticancer Res., 2014, 34, 7151–7158 CAS.
- R. L. Shriner and F. W. Neumann, Chem. Rev., 1944, 35, 351–425 Search PubMed.
- K. Hirano, S. Urban, C. Wang and F. Glorius, Org. Lett., 2009, 11, 1019–1022 Search PubMed.
- W. A. Munzeiwa, B. Omondi and V. O. Nyamori, Polyhedron, 2017, 138, 295–305 CrossRef CAS.
- W. A. Munzeiwa, V. O. Nyamori and B. Omondi, Appl. Organomet. Chem., 2018, 32, e4247 CrossRef.
- E. D. Akpan, S. O. Ojwach, B. Omondi and V. O. Nyamori, Polyhedron, 2016, 110, 63–72 CrossRef CAS.
- R. Anulewicz, T. Marek Krygowski and B. Pniewska, J. Crystallogr. Spectrosc. Res., 1987, 17, 661–670 Search PubMed.
- S. Patai and Z. Rappoport, The Chemistry of Amidines and Iminidates, Wiley, New York, 1991, vol. 2, pp. 547–564 Search PubMed.
- E. D. Raczyńska and C. Laurence, Analyst, 1992, 117, 375–378 RSC.
- L. Meschede and H. H. Limbach, J. Phys. Chem., 1991, 95, 10267–10280 CrossRef CAS.
- L. Xing, C. Wiegert and A. Petitjean, J. Org. Chem., 2009, 74, 9513–9516 Search PubMed.
- R. Anulewicz, I. Wawer, T. M. Krygowski, F. Männle and H.-H. Limbach, J. Am. Chem. Soc., 1997, 119, 12223–12230 CrossRef CAS.
- S. J. Zamisa, U. Bongoza and B. Omondi, CrystEngComm, 2021, 23, 4459–4474 RSC.
- A. S. Bruker and A. Bruker, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 Search PubMed.
- Bruker, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- I. Feddaoui, M. S. Abdelbaky, S. García-Granda, K. Essalah, C. B. Nasr and M. Mrad, J. Mol. Struct., 2019, 1186, 31–38 CrossRef CAS.
- M. Cibian, S. Derossi and G. S. Hanan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2485 CrossRef CAS PubMed.
- A. G. Giumanini, N. Toniutti, G. Verardo and M. Merli, Eur. J. Org. Chem., 1999, 1999, 141–143 CrossRef.
- D. O. Juma, S. J. Zamisa, W. Munzeiwa and B. Omondi, IUCrData, 2024, 9, x240989 CrossRef CAS PubMed.
- D. O. Juma, B. Omondi, S. J. Zamisa and W. Munzeiwa, IUCrData, 2024, 9, x240988 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS.
- S. Wolff, CrystalExplorer, Version 3.1, University of Western Australia, 2012 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- S. L. Tan, M. M. Jotani and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 308–318 CrossRef CAS PubMed.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- U. Bongoza, S. J. Zamisa, W. A. Munzeiwa and B. Omondi, Appl. Organomet. Chem., 2022, 36, e6726 CrossRef CAS.
- M. A. Spackman, P. R. Spackman and S. P. Thomas, Complementary Bonding Analysis, 2021, pp. 329–352 Search PubMed.
- A. Frisch, Gaussian 09W Reference, Wallingford, USA, 2009, vol. 470 Search PubMed.
- (a) D. O. Juma, S. J. Zamisa, A. Sharma, U. Bongoza, E. M. Njogu, F. Albericio and B. Omondi, CCDC 2452448: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2n9z76; (b) D. O. Juma, S. J. Zamisa, A. Sharma, U. Bongoza, E. M. Njogu, F. Albericio and B. Omondi, CCDC 2452449: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2n9z76.
| This journal is © The Royal Society of Chemistry 2025 |