DOI:
10.1039/D5CE00465A
(Paper)
CrystEngComm, 2025, Advance Article
Zn(II), Cd(II) and Hg(II) halide coordination polymers supported by bis-pyridyl-bis-amide: structural diversity and structural transformation†
Received
4th May 2025
, Accepted 21st May 2025
First published on 22nd May 2025
Abstract
Reactions of metal halide with N,N′-bis(3-pyridylmethyl)oxalamide (L1), N,N′-bis(4-methylpyridyl)oxalamide (L2) or N,N′-bis(3-methylpyridyl)adipamide (L3) afforded {[ZnI2(L1)]·2DMF}n, 1, [ZnI2(L1)]n, 2, [CdI2(L1)]n, 3, [HgI2(L1)]n, 4, [HgI2(L2)]n, 5, [Hg2I4(L3)], 6, [CdI2(L3)]n, 7, 1D-[CdBr2(L1)2]n, 8, 2D-[CdBr2(L1)2]n, 9, and [Cd2Br2(ox)(L1)2]n (ox− = oxalate), 10, whereas the reaction of Cd(CH3COO)2·2H2O with L1 gave {[Cd(ox)(L1)]·4H2O}n, 11, which have been structurally characterized by using single-crystal X-ray diffraction. Complexes 1 and 2 form a pair of supramolecular isomers, giving a 1D concavo-convex chain and a 1D zigzag chain, respectively, whereas 3, 4, 5 and 7 are 1D zigzag chains and 6 is a dinuclear complex, demonstrating that the solvent identity and the length of the spacer ligand are important in determining the structural diversity. Complexes 8 and 9 are another pair of supramolecular isomers, adopting a 1D looped-chain and a 2D layer with the (44·62)-sql topology, respectively. Complex 10 is a 3D framework with the (66)-dia topology and 11 is a 2D layer with the (44·62)-sql topology. Moreover, complexes 1 and 2 display irreversible structural transformation upon solvent removal, whereas temperature-dependent structural transformations from 8 to 9, 8 to 10 and 9 to 10 are observed.
Introduction
The rational design and synthesis of coordination polymers (CPs) have been rapidly developing, due to their fascinating structural diversity and potential applications in magnetism, gas storage and separation, drug delivery, catalysis, luminescence, sensing, and so on.1–8 The properties of CPs are mostly dominated by their structures and compositions and it is well known that the structural type of CPs is affected by many factors, including metal ions, counterions, temperature and identity of the organic linker. Metal-to-ligand ratios and solvent systems are also significant in the preparation of CPs.9–12 Therefore, controlling the appropriate factors to construct suitable CPs has become an exciting topic in the crystal engineering of CPs. On the other hand, structural transformations of CPs that lead to the formation of intriguing structures have attracted great attention of the researchers.13–21
One-dimensional (1D) CPs based on Zn(II), Cd(II) and Hg(II) halide have been reported, however, it remains a challenge to elucidate their structure–ligand relationship and thereby the intrinsic properties.22–30 We have investigated the structural diversity and properties of the CPs constructed from the bis-pyridyl-bis-amide ligand (bpba) and Hg(II) that exhibited structural transformations. Reversible structural transformation between [Hg(1,3-pbpa)X2]n [X = Br and I; 1,3-pbpa = 2,2′-(1,3-phenylene)-bis(N-(pyridin-3-yl)acetamide)] and [Hg(1,3-pbpa)X2·MeCN]n was ascribed to the formation and breaking of the N–H⋯N hydrogen bonds to the acetonitrile molecules.23 On the other hand, 1D mercury(II) chloride CPs synthesized by using a semi-rigid N-donor ligand, 2,2′-(1,4-phenylene)-bis(N-(pyridin-3-yl)acetamide) (1,4-pbpa), [Hg(1,4-pbpa)Cl2·CH3OH]n and [Hg(1,4-pbpa)Cl2]n underwent reversible structural transformation upon removal and uptake of CH3OH. Pyridyl ring rotation of the 1,4-pbpa ligand that results in the change of the ligand conformation was proposed for the initiation of the structural transformation.24
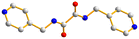
To investigate the effect of metal identity and ligand flexibility on the structural diversity of the metal halide CPs and to elucidate the factors that may govern the structural transformations, ten new CPs and a dinuclear complex containing bpba, N,N′-bis(3-pyridylmethyl)oxalamide (L1), N,N′-bis(4-methylpyridyl)oxalamide (L2) or N,N′-bis(3-methylpyridyl)adipamide (L3), namely {[ZnI2(L1)]·2DMF}n, 1, [ZnI2(L1)]n, 2, [CdI2(L1)]n, 3, [HgI2(L1)]n, 4, [HgI2(L2)]n, 5, [Hg2I4(L3)], 6, [CdI2(L3)]n, 7, 1D-[CdBr2(L1)2]n, 8, 2D-[CdBr2(L1)2]n, 9, [Cd2Br2(ox)(L1)2]n (ox = oxalate), 10, and {[Cd(ox)(L1)]·4H2O}n, 11, are prepared. Fig. 1 depicts the structures of L1, L2 and L3. The synthesis and structural characterization of these complexes form the subject of this report. Irreversible structural transformation from complex 1 to complex 2 upon DMF removal and the temperature-dependent structural transformations in 8–10 are also discussed.
 |
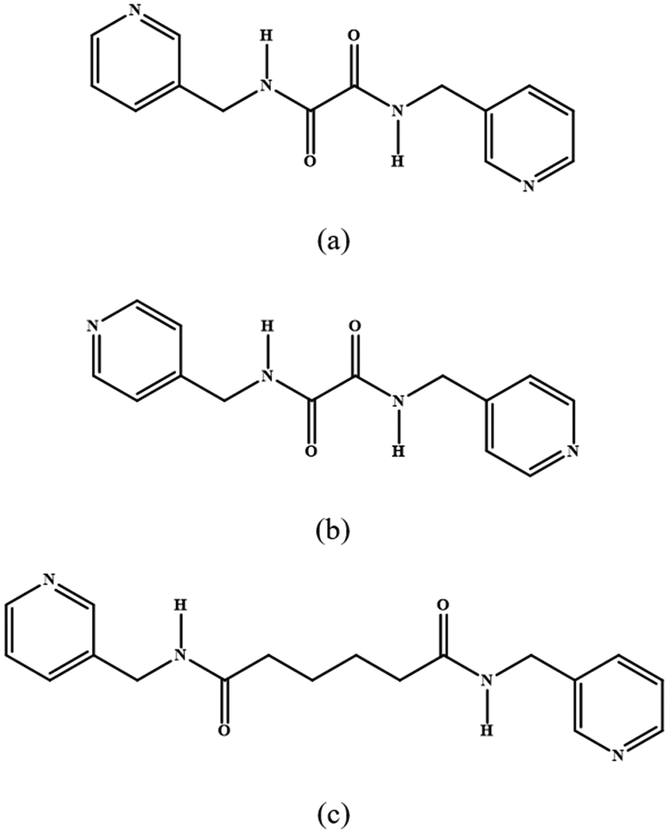
| | Fig. 1 Structures of (a) L1, (b) L2 and (c) L3. | |
Experimental details
General procedures
IR spectra (KBr disk) were obtained from a JASCO FT/IR-4200 FT-IR spectrometer. Elemental analyses were performed on an Elementar vario EL III analyzer. Powder X-ray diffraction was carried out on a Bruker D2 PHASER diffractometer with CuKα (λα = 1.54 Å) radiation.
Materials
The reagent zinc iodide was purchased from Nova, cadmium iodide from Alfa Aesar, mercury iodide from Sigma-Aldrich and cadmium bromide from Strem Chemicals, Inc. The ligands N,N′-bis(3-pyridylmethyl)oxalamide (L1), N,N′-bis(4-methylpyridyl)oxalamide (L2) and N,N′-bis(3-methylpyridyl)adipamide (L3) were prepared according to published procedures.31
Preparation of {[ZnI2(L1)]·2DMF}n, 1. L1 (0.24 g, 1.0 mmol) was placed in a flask containing 15 mL of DMF, and ZnI2 (0.32 g, 1.0 mmol) was added. The mixture was then heated at reflux for 24 h to afford a yellow solution with some white solids. Colorless crystals suitable for single crystal X-ray diffraction were obtained by slow diffusion of diethyl ether into the DMF solution for several weeks. Yield: 0.36 g (49%). Anal. calcd for C20H28ZnI2N6O4 (MW = 735.65 g mol−1): C, 32.62; N, 11.42; H, 3.81%. Found: C, 28.78; N, 9.78; H, 2.40%. Anal. calcd for C20H28ZnI2N6O4 – 2 DMF (MW = 589.48): C, 28.49; N, 9.49; H, 2.37%.
Preparation of [ZnI2(L1)]n, 2. Complex 2 was prepared by following the same procedures as 1. When the crystals were exposed to air, the DMF solvents evaporated to give 2 and several crystals were suitable for X-ray crystallography. Yield: 0.36 g (61%). Anal. calcd for C14H14ZnI2N4O2 (MW = 589.48 g mol−1): C, 28.49; N, 9.49; H, 2.37%. Found: C, 28.90; N, 9.82; H, 2.43%. FT-IR (cm−1): 3318(s), 3048(s), 2931(s), 1663(s), 1515(s), 1438(s), 1212(w), 1111(w), 1050(s), 971(s), 831(w), 672(w), 511(s).
Preparation of [CdI2(L1)]n, 3. A mixture of CdI2 (0.037 g, 0.10 mmol) and L1 (0.027 g, 0.10 mmol) in 1.25 mL of EtOH and 1.25 mL of H2O was sealed in a 23 mL Teflon-lined steel autoclave, which was heated under autogenous pressure to 120 °C for two days, and then cooled down to room temperature for two days. Colorless crystals were obtained. Yield: 0.059 g (93%). Anal. calcd for C14H14CdI2N4O2 (MW = 636.49 g mol−1): C, 26.39; N, 8.79; H, 2.20%. Found: C, 25.89; N, 8.85; H, 2.18%. FT-IR (cm−1): 3316(s), 3046(m), 2929(s), 1862(m), 1660(s), 1512(s), 1432(s), 1350(w), 1209(w), 1104(w), 1040(s), 965(s), 825(w), 698(s), 618(w), 506(s).
Preparation of [HgI2(L1)]n, 4. Complex 4 was prepared by following the same procedures as 3, except that a mixture of HgI2 (0.045 g, 0.1 mmol) and L1 (0.027 g, 0.1 mmol) was used. Colorless crystals were obtained. Yield: 0.066 g (91%). Anal. calcd for C14H14HgI2N4O2 (MW = 724.68 g mol−1): C, 23.18; N, 7.70; H, 1.93%. Found: C, 23.04; N, 8.20; H, 1.67%. FT-IR (cm−1): 3318(s), 3045(s), 2929(s), 1922(m), 1616(w), 1460(w), 1352(w), 1212(w), 1194(m), 1070(w), 804(m), 700(s), 622(w), 509(s).
Preparation of [HgI2(L2)]n, 5. Complex 5 was prepared by following the same procedures as 3, except that a mixture of HgI2 (0.045 g, 0.10 mmol) and L2 (0.027 g, 0.10 mmol) in 10 ml EtOH was used. Colorless crystals were obtained. Yield: 0.028 g (39%). Anal. calcd for C14H14HgI2N4O2 (MW = 724.68 g mol−1): C, 23.20; H, 1.95; N,7.73%. Found: C, 23.00; H, 1.70; N, 8.03%. FT-IR (cm−1): 3430(s), 2817(w), 2727(w), 2375(w), 2343(w), 1597(s), 1385(m), 1352(m), 1124(w), 768(w), 617(m).
Preparation of [Hg2I4(L3)], 6. Complex 6 was prepared by following the same procedures as 3, except that a mixture HgI2 (0.045 g, 0.10 mmol) and L3 (0.033 g, 0.10 mmol) in 1.25 mL H2O and 1.25 mL EtOH was used. Colorless crystals were obtained. Yield: 0.018 g (29%). Anal. calcd for C18H22Hg2I4N4O2 (MW = 1235.17): C, 17.50; N, 4.54; H, 1.80%. Found: C, 17.49; N, 4.99; H, 1.55%. FT-IR (cm−1): 3440(s), 3240(s), 3073(m), 2926(w), 2857(w), 2820(w), 2370(w), 2344(w), 1647(s), 1599(s), 1478(w), 1439(w), 1386(m), 1351(m), 1268(w), 1191(w), 1134(w), 1048(w), 698(w).
Preparation of [CdI2(L3)]n, 7. Complex 7 was prepared by following the same procedures as 3, except that a mixture of CdI2 (0.037 g, 0.10 mmol) and L3 (0.033 g, 0.10 mmol) was used. Colorless crystals were obtained. Yield: 0.026 g (38%). Anal. calcd for C18H22CdI2N4O2 (MW = 692.61): C, 31.21; N, 8.09; H, 3.20%. Found: C, 31.54; N, 8.43; H, 2.91%. FT-IR (cm−1): 3440(s), 2820(w), 2370(w), 2340(w), 1600(s), 1380(m), 1350(m), 768(m), 610(m), 521(m).
Preparation of 1D-[CdBr2(L1)2]n, 8. A mixture of CdBr2 (0.054 g, 0.20 mmol) and L1 (0.027 g, 0.10 mmol) were placed in a 23 mL Teflon reaction flask containing 10 mL H2O, which was sealed and heated at 100 °C for 48 hours under autogenous pressure and then the reaction system was cooled to room temperature at a rate of 2 °C per hour. Transparent crystals suitable for single-crystal X-ray diffraction were obtained. Yield: 0.013 g (8%). Anal. calcd for C28H28Br2CdN8O4 (MW = 812.79): C, 41.37; H, 3.47; N, 13.79%. Found: C, 41.15; H, 3.18; N, 13.84%. FT-IR (cm−1): 3303(s), 3198(w), 3051(w), 2921(w), 1661(s), 1573(w), 1510(s), 1426(w), 1326(w), 1187(m), 1048(m), 918(w), 758(m), 704(s), 633(m), 523(w).
2D-[CdBr2(L1)2]n, 9. Complex 9 was prepared by following the same procedures as 8, except that the mixture was heated at 120 °C for 48 hours. Suitable white crystals were obtained. Yield: 0.018 g (11%). Anal. calcd for C28H28Br2CdN8O4 (MW = 812.79): C, 41.37; H, 3.47; N, 13.79%. Found: C, 41.27; H, 3.19; N, 13.87%. FT-IR (cm−1): 3374(s), 3261(s), 3059(w), 2942(w), 1665(s), 1586(s), 1494(s), 1426(m), 1372(w), 1208(w), 1036(w), 935(w), 771(m), 700(s), 611(m), 536(w).
[Cd2Br2(ox)(L1)2]n, 10. Complex 10 was prepared by following the same procedures as 8, except that the mixture was heated at 140 °C for 48 hours. Transparent crystals were obtained. Yield: 0.012 g (12%). Anal. calcd for C30H28Br2Cd2N8O8 (MW = 1013.22): C, 35.56; H, 2.78; N, 11.06%. Found: C, 35.74; H, 2.67; N, 11.41%. FT-IR (cm−1): 3354(s), 3269(w), 3043(w), 2938(w), 1674(s), 1620(s), 1502(s), 1435(w), 1300(w), 1174(m), 1023(w), 947(w), 788(m), 704(s), 620(m), 502(w).
{[Cd(ox)(L1)]·4H2O}n, 11. A mixture of Cd(CH3COO)2·2H2O (0.027 g, 0.10 mmol) and L1 (0.027 g, 0.10 mmol) in 5 mL of H2O was sealed in a 23 mL Teflon-lined steel autoclave, which was heated under autogenous pressure to 120 °C for two days, and then cooled down to room temperature for two days. Colorless crystals were obtained. Yield: 0.0062 g (11%). Anal. calcd for C16H22CdN4O10 (MW = 542.78): C, 35.41; N, 10.32; H, 4.09%. Found: C, 35.51; N, 10.59; H, 3.85%. FT-IR (cm−1): 3286(s), 3049(m), 1652(s), 1520(s), 1432(s), 1306(m), 1191(m), 1106(w), 1035(w), 947(w), 793(m), 708(m), 647(m), 496(w), 409(w).
X-ray crystallography
The diffraction data for complexes 1–11 were collected on a Bruker AXS SMART APEX II CCD diffractometer, which was equipped with a graphite-monochromated Mo Kα (λα = 0.71073 Å) radiation. Data reduction was carried out by standard methods with the use of well-established computational procedures.32 The structure factors were obtained after Lorentz and polarization corrections. An empirical absorption correction based on “multi-scan” was applied to the data. The position of some of the heavier atoms were located by the direct or Patterson method. The remaining atoms were found in a series of alternating difference Fourier maps and least-squares refinements, while the hydrogen atoms except those of the water molecules were added by using the HADD command in SHELXTL 6.1012.33 Table S1† lists the crystal data for 1–11. The alerts A and B appealing in the checkcif of complex 2 are presumably due to the poor crystallinity of the crystal used for the measurement. The crystals of complex 2 were obtained fortunately by exposing the crystals of 1 to air, as shown in the Experimental section.
Results and discussion
Structure of {[ZnI2(L1)]·2DMF}n, 1
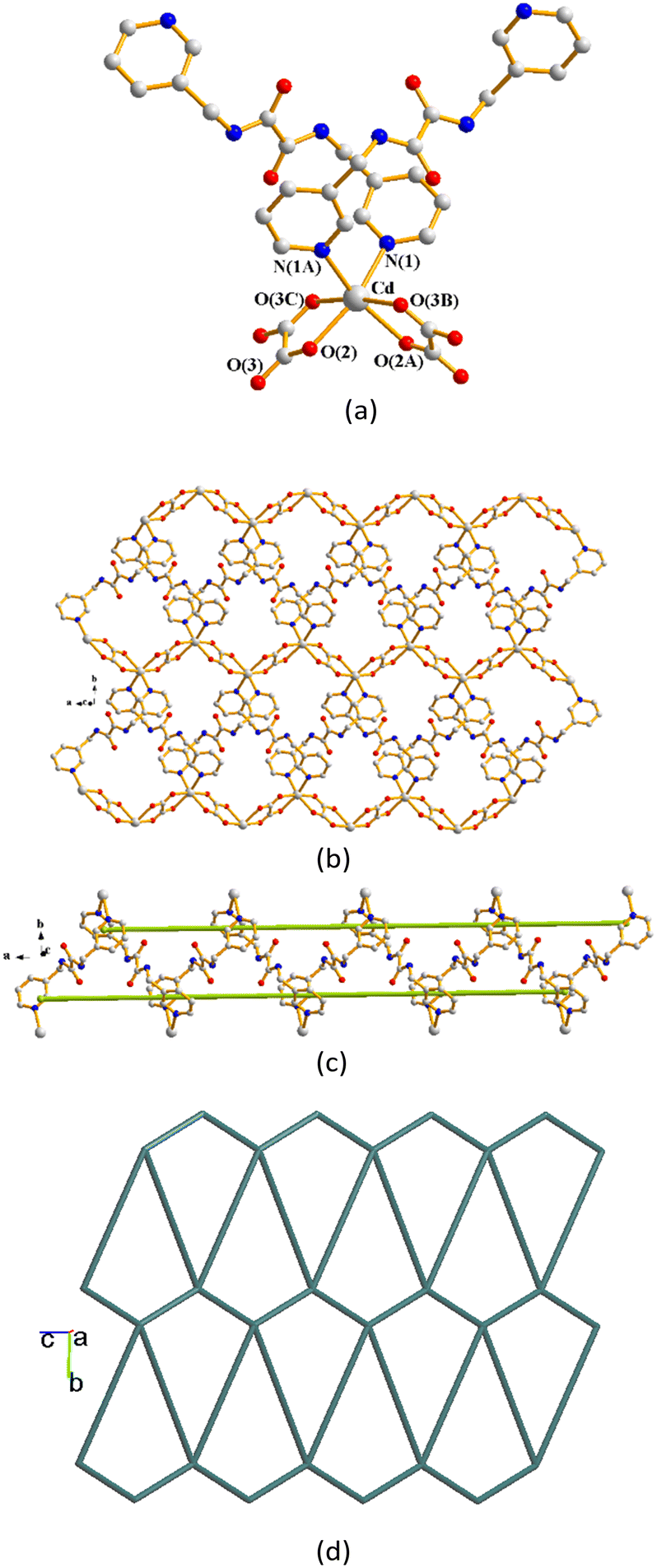
Single-crystal X-ray diffraction analysis shows that complex 1 crystallizes in the monoclinic space group P21/n and each asymmetric unit consists of one Zn(II) cation, one L1 ligand, two iodide anions and two coordinated DMF molecules. The Zn(II) metal center is four-coordinated by two pyridyl nitrogen atoms from two L1 ligands [Zn–N = 2.056(3)–2.058(3) Å] and two iodine atoms [Zn–I = 2.5466(6)–2.5579(6) Å], forming a distorted tetrahedral geometry (Fig. 2(a)). The Zn(II) ions are linked together by L ligands to afford a 1D concavo-convex chain (Fig. 2(b) top). Looking down the 1D chain, it is seen that the ZnI2 groups are located on the right and left sides alternately (Fig. 2(b) bottom). Moreover, the 1D concavo-convex chain is reinforced by the N–H⋯O hydrogen bonds from the amine hydrogen atoms of L1 to the oxygen atoms of the adjacent L1 (H⋯O = 2.423 Å; ∠N–H⋯O = 136.1) and DMF (H⋯O = 2.090 Å; ∠N–H⋯O = 144.1°) (Fig. 2(c)).
 |
| | Fig. 2 (a) Coordination environment of the Zn(II) ion in 1. Symmetry transformations used to generate equivalent atoms: (A) 1/2 + x, 3/2 + y, 1/2 + z. (b) a drawing showing the 1D chain of 1, top, and a view looking down the chain, bottom. (c) A drawing showing the N–H⋯O hydrogen bonds. | |
Structures of [MI2(L1)]n (M = Zn, 2; Cd, 3; Hg, 4)
Crystals of complexes 2, 3 and 4 are isomorphous, which conform to the monoclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with the asymmetric unit consisting of one M(II) cation, one L1 ligand, and two iodide anions. The M(II) metal center is four-coordinated by two different pyridyl nitrogen atoms from two different L1 ligands [M–N = 1.991(15) and 2.004(13) Å for 2; 2.290(5) and 2.330(5) Å for 3; 2.396(7) and 2.464(8) Å for 4] and two iodine atoms [M–I = 2.476(5) and 2.484(5) Å for 2; 2.6869(6) and 2.6952(6) for 3; 2.6418(8) and 2.6533(8) for 4], forming a distorted tetrahedral geometry (Fig. 3(a)). It is noted that while the M–N distances increase with the increasing of the size, Zn(II) < Cd(II) < Hg(II), the Cd–I distances are significantly longer than those of Zn–I and Hg–I.
with the asymmetric unit consisting of one M(II) cation, one L1 ligand, and two iodide anions. The M(II) metal center is four-coordinated by two different pyridyl nitrogen atoms from two different L1 ligands [M–N = 1.991(15) and 2.004(13) Å for 2; 2.290(5) and 2.330(5) Å for 3; 2.396(7) and 2.464(8) Å for 4] and two iodine atoms [M–I = 2.476(5) and 2.484(5) Å for 2; 2.6869(6) and 2.6952(6) for 3; 2.6418(8) and 2.6533(8) for 4], forming a distorted tetrahedral geometry (Fig. 3(a)). It is noted that while the M–N distances increase with the increasing of the size, Zn(II) < Cd(II) < Hg(II), the Cd–I distances are significantly longer than those of Zn–I and Hg–I.
 |
| | Fig. 3 (a) A representative drawing showing the coordination environment of the M(II) ion in 2–4. Symmetry transformations used to generate equivalent atoms: (A) 1 + x, 1 + y, 1 + z. (b) A drawing showing the 1D chain of 2–4. (c) A drawing showing the N–H⋯O hydrogen bonds. | |
The M(II) ions are linked together by L1 ligands to afford 1D zigzag chains (Fig. 3(b), top). Looking down the 1D chain, it is shown that the MI2 groups are located in an eclipsed fashion (Fig. 3(b) bottom), which are attributed to the ZnI2 groups in complex 1. Moreover, the 1D zigzag chains are linked by pairs of complementary N–H⋯O hydrogen bonds from the amine hydrogen atoms of L1 to the oxygen atoms of the adjacent L (H⋯O = 2.073, 2.458 Å and ∠N–H⋯O = 131.7, 107.1° for 2; H⋯O = 2.236, 2.523 Å and ∠N–H⋯O = 127.4, 114.6° for 3; H⋯O = 2.223, 2.592 Å and ∠N–H⋯O = 129.3, 109.7° for 4) (Fig. 3(c)).
Structure of [HgI2(L2)]n, 5
Complex 5 crystallizes in the monoclinic space group P2/n and each asymmetric unit consists of a half of a Hg(II) cation, a half of an L2 ligand and one iodide anion. Fig. 4(a) depicts the coordination environment of the Hg(II) ion, which is coordinated by two pyridyl nitrogen atoms from two L2 ligands [Hg–N = 2.466(6) Å] and two iodine anions [Hg–I = 2.6510(6) Å], resulting in a distorted tetrahedral geometry. The HgI2 units are linked by the L2 ligands to form a 1D zigzag chain (Fig. 4(b)). The dinuclear molecules are linked through the extensive self-complementary N–H⋯O (H⋯O = 2.173(4) Å; ∠N–H⋯O = 151.7(4)) to form a 2D layer (Fig. 4(c)). No Hg⋯I interaction can be observed in this complex.
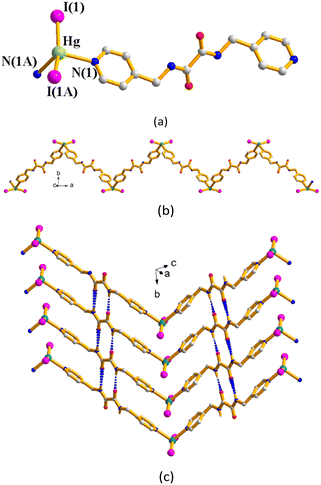 |
| | Fig. 4 (a) Coordination environment of the Hg(II) ion in 5. Symmetry transformations used to generate equivalent atoms: (A) −x + 1/2, y, −z + 1/2. (b) A drawing showing the 1D chain. (c) A drawing showing the 2D layer supported by the self-complementary N–H⋯O hydrogen bonds. | |
Structure of [Hg2I4(L3)], 6
Complex 6 crystallizes in the monoclinic space group C2/c and each asymmetric unit consists of one Hg(II) cation, a half of an L3 ligand and two iodide anions. Complex 6 forms a dinuclear structure and each of the two Hg(II) metal centers is three-coordinated by one pyridyl nitrogen atom from the L3 ligand [Hg–N = 2.370(5) Å] and two iodine atoms [Hg–I = 2.6198(5) and 2.6481(4) Å], forming a distorted triangular planar geometry (Fig. 5(a)). The dinuclear molecules are linked through the Hg⋯I [3.4540(7) Å] interactions and self-complementary N–H⋯O (H⋯O = 1.828(7) Å; ∠N–H⋯O = 169.4(5)) to form a 1D band (Fig. 5(b)).
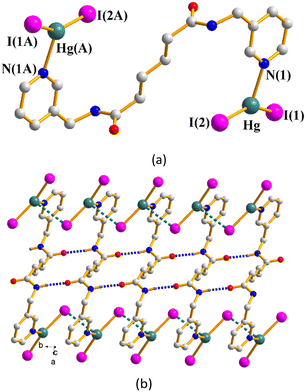 |
| | Fig. 5 (a) A drawing showing the dinuclear structure of 6. Symmetry transformations used to generate equivalent atoms: (A) −x, −y + 2, −z. (b) The dinuclear molecules are linked through Hg⋯I interactions and N–H⋯O hydrogen bonds. | |
Structure of [CdI2(L3)]n, 7
Complex 7 crystallizes in the monoclinic space group Cc and each asymmetric unit consists of one Cd(II) cation, one L3 ligand and two iodide anions. Fig. 6(a) depicts a drawing showing the coordination environment of the Cd(II) ion of 7, which is four-coordinated by two pyridyl nitrogen atoms from two L3 ligands [Cd–N = 2.248(8) and 2.280(8) Å] and two iodine atoms [Cd–I = 2.6783(13) and 2.7095(13) Å], forming a distorted tetrahedral geometry. The Cd(II) ions are linked by the L3 ligands to form a concavo-convex chain (Fig. 6(b)). The 1D chains are linked through N–H⋯O (H⋯O = 1.985(9) and 2.200(12) Å; ∠N–H⋯O = 170.9(6) and 132.4(8)°) hydrogen bonds (Fig. 6(c)) to form a 2D layer.
 |
| | Fig. 6 (a) A drawing showing the coordination environment of the Cd(II) ion of 7. Symmetry transformations used to generate equivalent atoms: (A) −x, −y + 2, −z. (b) A drawing showing the 1D zigzag chain. (c) The 1D chains are linked through N–H⋯O hydrogen bonds. | |
Structure of 1D-[CdBr2(L1)2]n, 8
The single-crystal X-ray diffraction analysis shows that 8 crystallizes in the monoclinic space group Pī. The symmetric unit consists of one Cd(II) cation and two L1 ligands. Fig. 7(a) shows the coordination environment of the Cd(II) metal center, which is six-coordinated by its two original bromine atoms [Cd–Br = 2.6937(13) Å] and four nitrogen atoms from four different ligands [Cd–N = 2.3910(3) and 2.5020(3) Å]. The Cd(II) central metal atom is bridged by two different L1, both horizontally to form a 1D looped-chain (Fig. 7(b)). If the Cd(II) ions are considered as four-connected nodes and the L1 ligands as two-connected nodes, the structure of 8 can be regarded as a 2,4-connected 1D net with the (42)(4)2–2,4C4 topology. Noticeably, the adjacent 1D looped chains are interlinked through extensive N–H⋯O [H⋯O = 2.208(3) and 2.377(3) Å; ∠N–H⋯O = 143.1(2) and 130.9(2)°] hydrogen bonds (Fig. 7(c)).
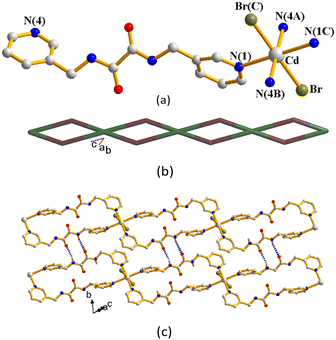 |
| | Fig. 7 (a) Coordination environment of Cd(II) in 8. Symmetry transformations used to generate equivalent atoms: (A) −x + 2, −y − 1, −z + 1; (B) x + 1, y − 1, z + 1; (C) −x + 3, −y 2, −z + 2. (b) A drawing showing the 1D looped chain with the 2,4C4 topology. (c) The adjacent 1D looped chains are interlinked by the N–H⋯O hydrogen bonds. | |
Structure of 2D-[CdBr2(L1)2]n, 9
The single-crystal X-ray diffraction analysis shows that 9 crystallizes in the monoclinic space group P21/c. The symmetric unit consists of one Cd(II) cation and two L1 ligands. Fig. 8(a) shows the coordination environment of the Cd(II) metal center, which is six-coordinated by its two original bromine atoms [Cd–Br = 2.7090(3) Å] and four nitrogen atoms from four different ligands [Cd–N = 2.4045(19) and 2.4685(19) Å]. If the Cd(II) ions are considered as four-connected nodes and the L1 ligands as linkers, the structure of 9 can be simplified as a 4-connected 2D net with the (44·62)-sql topology as illustrated in Fig. 8(b). The Cd(II) metal atoms are bridged by L1 ligands with 2D layers, which are supported by the N–H⋯O [H⋯O = 2.056(3) Å; ∠N–H⋯O = 146.6(1)°] hydrogen bonds (Fig. S1†).
 |
| | Fig. 8 (a) Coordination environment of Cd(II) in 9. Symmetry transformations used to generate equivalent atoms: (A) −x + 1, −y + 2, −z + 2; (B) x − 1, −y + 3/2, z + 1/2; (C) −x + 2, y + 1/2, −z + 3/2. (b) A drawing showing the structure of 9 with the sql topology. | |
Structure of [Cd2Br2(ox)(L1)2]n, 10
The single-crystal X-ray diffraction analysis shows that 10 crystallizes in the monoclinic space group P21/c. The asymmetric unit consists of one Cd(II) cation, two oxalate ligands, one L1 ligand, and one bromine atom. Fig. 9(a) shows the coordination environment of the Cd(II) metal center, which is six-coordinated by two bromine atoms [Cd–Br = 2.6681(4) and 2.9233(4) Å], two oxygen atoms from two decomposed L1 ligands [Cd–O = 2.265(2) and 2.301(2) Å] and two nitrogen atoms from two different L ligands [Cd–N = 2.279(2) and 2.404(3) Å]. The Cd(II) metal atoms are bridged together by two decomposed L, bonded to another Cd(II) by one L, and bonded to another Cd(II) via two bromine atoms. If the Cd(II) ions are considered as four-connected nodes while the other ligands as linkers, the structure of 10 can be simplified as a 4-connected 3D net with the (66)-dia topology as illustrated in Fig. 9(b), determined using ToposPro.34 The 3D framework is supported by the N–H⋯O [H⋯O = 2.223(2) Å; ∠N–H⋯O = 142.3(2)°] hydrogen bonds (Fig. S2†).
 |
| | Fig. 9 (a) Coordination environment of Cd(II) in 10. Symmetry transformations used to generate equivalent atoms: (A) −x, −y, −z + 1; (B) x − 1, −y + 1/2, z + 1/2. (C) −x + 1, −y, −z + 1. (b) A drawing showing the 3D topological structure of 10 with the dia topology. | |
Structure of {[Cd(ox)(L1)]·4H2O}n, 11
Complex 11 crystallizes in the monoclinic space group C2/c. The symmetric unit consists of a half of a Cd(II) cation, a half of an L1 ligand, a half of an oxalate anion and two co-crystallized water molecules. Fig. 10(a) shows the coordination environment of the Cd(II) metal center, which is six-coordinated by two nitrogen atoms from two L1 ligands [Cd–N = 2.3252(19) Å] and four oxygen atoms from two oxalate anions [Cd–O = 2.2959(15) and 2.3185(16) Å]. The Cd(II) ions are linked by the L1 ligands and oxalate anions to form a 2D layer (Fig. 10(b)) supported by the double helical chains (Fig. 10(c)). If the Cd(II) ions are considered as four-connected nodes and the L1 ligands and oxalate anions as linkers, the structure of 11 can be simplified as a 4-connected 2D net with the (44·62)-sql topology as illustrated in Fig. 10(d). The 2D layers are supported by the O–H⋯O hydrogen bonds from the water hydrogen atoms to the oxalate oxygen atoms [H⋯O = 2.035(2) Å; ∠O–H⋯O = 155.9(1)°], the amide oxygen atoms [H⋯O = 1.904(2) Å; ∠O–H⋯O = 168.4(1)°] and the water oxygen atoms [H⋯O = 1.983(2) and 2.055(2) Å; ∠O–H⋯O = 163.9(2) and 144.9(2)°] (Fig. S3†).
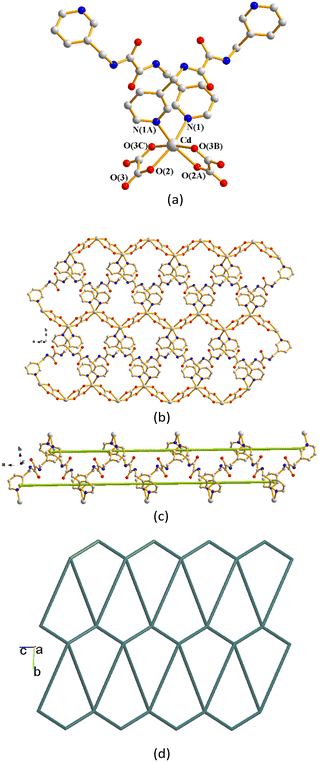 |
| | Fig. 10 (a) Coordination environment of Cd(II) in 11. Symmetry transformations used to generate equivalent atoms: (A) −x + 1, y, −z + 3/2; (B) x, −y + 1, z + 1/2; (C) −x + 1, −y + 1, −z + 1. (b) A drawing showing the 2D layer structure. (c) A drawing showing the double helical structure. (d) A drawing showing the 2D net with the sql topology. | |
Ligand conformations and bonding modes
L3 can be arranged in A and G conformations when the C–C–C–C torsion angles (θ) are 180 ≥ θ > 90° and 0 ≤ θ ≤ 90°, respectively, and based on the relative orientation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, each conformation can adopt cis or trans arrangement.35 Due to the difference in the orientations of the pyridyl nitrogen atom positions, three more orientations, anti–anti, syn–anti and syn–syn, are possible for the ligands L1–L3. Accordingly, the ligand conformations of 1–11 are assigned and listed in Table 1.
O groups, each conformation can adopt cis or trans arrangement.35 Due to the difference in the orientations of the pyridyl nitrogen atom positions, three more orientations, anti–anti, syn–anti and syn–syn, are possible for the ligands L1–L3. Accordingly, the ligand conformations of 1–11 are assigned and listed in Table 1.
Table 1 Ligand conformations of 1–11
| 1 |
 |
2 |
 |
| trans syn–anti |
trans syn–syn |
| 3 |
 |
4 |
 |
| trans syn–syn |
trans syn–syn |
| 5 |
 |
6 |
 |
| trans anti–anti |
AAA trans syn–syn |
| 7 |
 |
8 |
 |
| AAG cis anti–syn |
trans syn–anti |
| 9 |
 |
10 |
 |
| trans syn–syn |
trans syn–syn |
| 11 |
 |
|
|
| trans syn–syn |
Structural comparisons
Structural comparisons of complexes 1–11 indicate that the subtle structural difference in the 1D zigzag chains of 4 and 5 is due to the different donor atom positions of the bpba ligands, whereas the difference between 5 and 6 can be ascribed to the ligand flexibility. Moreover, the structural difference between 6 and 7 is attributable to the metal identity. Table 2 lists the structures of the reported d10-metal halide complexes containing bpba ligands. It is interesting to note that regardless of the flexibility and the shapes of the bpba ligands, only 0D and 1D structures can be obtained for the Zn(II) and Hg(II) halide complexes, whereas higher dimensionality can be achieved for the Cd(II) halide ones. The combined effect of the sizes of the metal centers and halide anions may thus play an important role in determining the dimensionality of the d10-metal halide complexes containing bpba ligands. Complex 10 represents a rare example that a Cd(II) halide CP is supported by three different ligands.
Table 2 d10-Metal halide complexes containing bpba ligands
| Complex |
Structure |
References |
| L4 = N,N′-di(4-pyridyl)adipoamide; L5 = N,N′-bis-(2-pyrimidinyl)-1,4-benzenedicarboxamide; L6 = N,N′-di(3-pyridyl)oxamide; L7 = N,N′-di(3-pyridyl)adipoamide; L8 = 2,2′-(1,2-phenylene)-bis(N-pyridin-3-yl)acetamide; L9 = 2,2′-(1,3-phenylene)-bis(N-pyridin-3-yl)acetamide; L10 = 2,2′-(1,4-phenylene)-bis(N-pyridin-3-yl)acetamide; L11 = N,N′-di(pyridin-3-yl)naphthalene-1,4-dicarboxamide; L12 = N,N′-bis(3-pyridyl)bicyclo(2,2,2,)oct-7-ene-2,3,5,6-tetracarboxylic diamide; L13 = N,N′-bis(4-pyridylmethyl)bicyclo(2,2,2,)oct-7-ene-2,3,5,6-tetracarboxylic diamide; L14 = 4,4′-oxybis(N-(pyridine-3-yl)benzamide). |
| {[ZnCl2(L4)]·H2O}n |
1D double-stranded helical chain |
36 |
| {[ZnBr2(L4)]·H2O}n |
1D double-stranded helical chain |
36 |
| {[ZnI2(L4)]·H2O}n |
1D sinusoidal chain |
36 |
| [Zn2Cl4(L4)2]·2DMF |
Dinuclear metallocycle |
36 |
| [Zn2Br4(L4)2]·2DMF |
Dinuclear metallocycle |
36 |
| [Zn2I4(L4)2]·4DMF·C4H10O |
Dinuclear metallocycle |
36 |
| {[HgBr2(L5)]·H2O}n |
1D zigzag chain |
37 |
| {[ZnCl2(L6)]·2DMF·C4H10O}n |
1D zigzag chain |
38 |
| {[ZnBr2(L6)]·2DMF}n |
1D zigzag chain |
38 |
| {[ZnI2(L6)]·2DMF}n |
1D concavo-convex chain |
38 |
| [HgCl2(L6)]n |
1D zigzag chain |
38 |
| {[HgBr2(L6)]·2DMF}n |
1D zigzag chain |
38 |
| Hg2I4(L6)2 |
Dinuclear metallocycle |
38 |
| [HgBr2(GAG-L7)]n |
1D double helical chain |
28 |
| [HgBr2(AAA-L7)]n |
1D helical chain |
28 |
| [HgI2(GAG-L7)]n |
1D sinusoidal chain. |
28 |
| [HgI2(AAA-L7)]n |
1D helical chain |
28 |
| [HgCl2(L8)]n |
1D zigzag chain |
23 |
| [HgBr2(L8)]n |
1D zigzag chain |
23 |
| [HgI2(L8)]n |
1D zigzag chain |
23 |
| [HgI2(L8)·MeOH]n |
1D helical chain |
23 |
| [HgI2(L8)·MeCN]n |
1D helical chain |
23 |
| [HgCl2(L9)]n |
1D helical chain |
24 |
| [HgBr2(L9)]n |
1D helical chain |
24 |
| [HgI2(L9)]n |
1D helical chain |
24 |
| {[HgBr2(L9)]·MeCN}n |
1D double helical chain |
24 |
| {[HgI2(L9)]·MeCN}n |
1D double helical chain |
24 |
| {[HgCl2(L10)]·CH3OH}n |
1D sinusoidal chain |
39 |
| [HgCl2(L10)]n |
1D helical chain |
39 |
| [HgBr2(L10)]n |
1D helical chain |
39 |
| [HgI2(L10)]n |
1D helical chain |
39 |
| [HgCl2(L11)]n |
1D helical chain |
40 |
| [HgBr2(L11)]n |
1D helical chain |
40 |
| [HgI2(L11)]n |
1D helical chain |
40 |
| [HgBr2(L12)]n |
1D zigzag chains |
30 |
| [HgI2(L12)] |
Mononuclear complex |
30 |
| [Hg2Cl4(L13)2] |
Dinuclear metallocycle |
30 |
| [Hg2Br4(L13)2] |
Dinuclear metallocycle |
30 |
| [Hg2I4(L13)2] |
Dinuclear metallocycle |
30 |
| {[HgCl2(L14)]·H2O}n |
1D zigzag chain |
30 |
| {[HgBr2(L14)]·H2O}n |
1D zigzag chain |
30 |
| {[HgI2(L14)]·H2O}n |
1D zigzag chain |
30 |
| {[ZnI2(L1)]·2DMF}n, 1, |
1D concavo-convex chain |
This work |
| [ZnI2(L1)]n, 2 |
1D zigzag chain |
This work |
| [CdI2(L1)]n, 3 |
1D zigzag chain |
This work |
| [HgI2(L1)]n, 4 |
1D zigzag chain |
This work |
| [HgI2(L2)]n, 5 |
1D zigzag chain |
This work |
| [Hg2I4(L3)], 6 |
Dinuclear complex |
This work |
| [CdI2(L3)]n, 7 |
1D zigzag chain |
This work |
| 1D-[CdBr2(L1)2]n, 8 |
1D looped-chain |
This work |
| 2D-[CdBr2(L1)2]n, 9 |
2D layer with (44·62)-sql topology |
This work |
| [Cd2Br2(ox)(L1)2]n, 10 |
3D framework with (66)-dia topology |
This work |
Powder X-ray analysis
In order to check the phase purity of the products, powder X-ray diffraction (PXRD) experiments have been carried out for complexes 1–11. As shown in Fig. S4–S14,† the peak positions of the experimental and simulated PXRD patterns are in agreement with each other, except complex 1, which has been transformed into complex 2 upon solvent removal, vide infra.
Supramolecular isomerism and structural transformation
Supramolecular isomers are network structures comprising identical chemical compositions but differ from one another in their structures.41 Complexes 1 and 2 as well as 8 and 9 thus form two pairs of supramolecular isomers. Importantly, supramolecular isomers prepared under various experimental conditions may be considered as potential candidates for structural transformation. Consequently, the investigation of both supramolecular isomerism and structural transformation may prove crucial for advancing the crystal engineering of CPs.
Structural transformation in complexes 1 and 2
Complexes 1 and 2 provide an opportunity to investigate the structural transformation due to the removal of DMF molecules in the zinc(II) CPs. The structure of the as-synthesized complex 1 which contains DMF was not stable. When it was exposed to air for 5 minutes, the experimental PXRD pattern (Fig. 11(b)) shows a similar pattern to that of complex 2 (Fig. 11(d)), indicating structural transformation from 1 to 2 upon the removal of the DMF molecules. When the desolvated sample of 1 was immersed in DMF, the PXRD pattern (Fig. 11(c)) reveals no significant change. The irreversible structural transformation from 1 to 2 probably indicates that the self-complementary N–H⋯O hydrogen bonds in 2 that link the 1D chains may be much stronger than the N–H⋯O hydrogen bonds in 1 that link the 1D chains and DMF solvents.
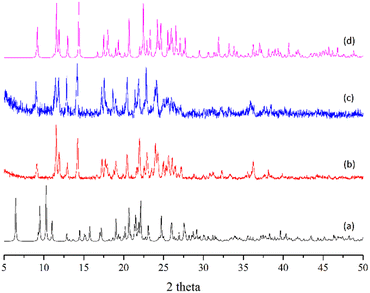 |
| | Fig. 11 (a) Simulated PXRD pattern of 1, (b) PXRD pattern of the as-synthesized 1 exposed to air for five minutes, (c) PXRD pattern of the sample of (b) immersed in DMF and (d) simulated PXRD pattern of 2. | |
Structural transformation in complexes 8–10
In order to achieve the structural transformations, complex 8 was hydrothermally heated at 100 and 120 °C, respectively, for 48 hours. The PXRD patterns (Fig. 12 and S15†) indicate that the structure of 8, originally a 1D structure, has been changed into a 2D layer precisely alike 9, and a 3D framework of 10, respectively. On the other hand, heating 9 hydrothermally at 140 °C leads to the formation of 10, as demonstrated by the PXRD patterns shown in Fig. S16.† Fig. 13 depicts a drawing showing the formation pathways of complexes 8–10 and their corresponding structural transformations. Noticeably, the independent structural transformations from 8 to 10 and 9 to 10 have led to the decomposition of the L1 ligands and subsequently the formation of the oxalate anions (ox−). The ox− anion can also be observed in the structure of complex 11, which was prepared at 120 °C by using the starting metal salt Cd(CH3COO)2·2H2O, probably indicating that the halide anion is not important in the decomposition of the L1 ligand.
 |
| | Fig. 12 (a) Simulated PXRD pattern of 8, (b) PXRD pattern of the as-synthesized 8, (c) PXRD pattern of the sample of (b) heated in water at 100 °C for 2 days and (d) simulated PXRD pattern of 9. | |
 |
| | Fig. 13 A drawing showing the formation pathways of complexes 8–10 as well as their structural transformations. | |
Conclusions
Ten new CPs and one dinuclear complex supported by the bpba ligands have been successfully obtained, in which complexes 1–10 are d10-metal halides. Their structural types are susceptible to the changes of the ligand isomerism and flexibility as well as the metal identity. Complexes 1–4 represent a unique example demonstrating that the cocrystallized DMF molecules play a more important structure-directing role than the metal identity. Irreversible structural transformation from complex 1 to complex 2 upon DMF removal has been demonstrated, presumably owing to the conformational change of the L1 ligand. The formation of 8–10 is temperature-dependent, affording a 1D chain, a 2D layer and a 3D framework, respectively. Complexes 10 and 11 were obtained on account of the decomposition of the L1 ligand, leading to the formation of the ox− anion. Structural transformations in 8–10 are observed, resulting in the conformational change of L1 in 8 to 9 and ligand decomposition in 8 to 10 and 9 to 10. Careful evaluation of the reaction temperature may thus lead to the observation of unique structural alterations. Structural comparisons of the reported bpba-based Zn(II), Cd(II) and Hg(II) halide complexes indicate that only 0D and 1D structures can be obtained for the Zn(II) and Hg(II) halide complexes, while higher dimensionality can be achieved for the Cd(II) halide ones, irrespective of the increasing size Zn(II) < Cd(II) < Hg(II).
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for complexes 1–11 have been deposited at the CCDC with no. 2445340–2445350.
Author contributions
Investigation, A. R., H.-C. Z., Y.-S. L., Y.-H. Y., Y.-T. K. and Y.-F. L.; data curation, Z.-L. C. and S.-W. W.; review and supervision, J.-D. C. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Science and Technology Council of the Republic of China for support.
References
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers Design, Analysis and Application, The Royal Society of Chemistry, London, UK, 2009 Search PubMed.
- S. Mondal and P. Dastidar, Cryst. Growth Des., 2020, 20, 7411–7420 CrossRef CAS.
- V. Chandrasekhar, C. Mohapatra, R. Banerjee and A. Mallick, Inorg. Chem., 2013, 52, 3579–3581 CrossRef CAS PubMed.
- J. Yu, Y. Cheng, X. Zhang, L. Zhou, Z. Song, A. Nezamzadeh-Ejhieh and Y. Huang, J. Environ. Chem. Eng., 2025, 13, 116870 CrossRef.
- P. Yan, Z. Chen, X. Li, F. Liang, Y. Tan, Y. Lin, K. Yang, C. Xiao, J. Wu and D. Ma, J. Solid State Chem., 2024, 330, 124461 CrossRef CAS.
- F. Liang, D. Ma, L. Qin, Q. Yu, J. Chen, R. Liang, C. Zhong, H. Liao and Z. Peng, Dalton Trans., 2024, 53, 10070–10074 RSC.
- Y. Zhang, H. Tan, J. Zhu, L. Duan, Y. Ding, F. Liang, Y. Li, X. Peng, R. Jiang, J. Yu, J. Fan, Y. Chen, R. Chen and D. Ma, Molecules, 2024, 29, 5903 CrossRef CAS PubMed.
- V. Lakshmanan, Y.-T. Lai, X.-K. Yang, M. Govindaraj, C.-H. Lin and J.-D. Chen, Polymer, 2021, 13, 3018 CAS.
- W.-T. Lee, T.-T. Liao and J.-D. Chen, Int. J. Mol. Sci., 2022, 23, 3603 CrossRef CAS PubMed.
- V. Lakshmanan, C.-Y. Lee, Y.-W. Tseng, Y.-H. Liu, C.-H. Lin and J.-D. Chen, CrystEngComm, 2022, 24, 6076–6086 RSC.
- J. L. Sague, M. Meuwly and K. M. Fromm, CrystEngComm, 2008, 10, 1542–1549 RSC.
- J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef CAS.
- G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755–1775 RSC.
- G. Chakraborty, I.-H. Park, R. Medishetty and J. J. Vittal, Chem. Rev., 2021, 121, 3751–3891 CrossRef CAS PubMed.
- C.-H. Hsu, W.-C. Huang, X.-K. Yang, C.-T. Yang, P. M. Chhetri and J.-D. Chen, Cryst. Growth Des., 2019, 19, 1728–1737 CrossRef CAS.
- X.-K. Yang and J.-D. Chen, CrystEngComm, 2019, 21, 7437–7446 RSC.
- Y.-F. Liu, J.-H. Hu, W.-T. Lee, X.-K. Yang and J.-D. Chen, Cryst. Growth Des., 2020, 20, 7211–7218 CrossRef CAS.
- C.-Y. Lee, Y.-H. Ye, S.-W. Wang and J.-D. Chen, Molecules, 2024, 29, 1748 CrossRef CAS PubMed.
- C.-Y. Lee, M. Usman, S.-W. Wang, K. B. Thapa, T.-R. Chen and J.-D. Chen, CrystEngComm, 2024, 26, 5099–5107 RSC.
- X. Hao, Y.-X. Wang, Y. Yang, Z. Song, X. Dong, S. Wang, Y. Liang, L. Li and P. Cheng, Cryst. Growth Des., 2025, 25, 2792–2797 CrossRef CAS.
- A. Morsali and M. Y. Masoomi, Coord. Chem. Rev., 2009, 253, 1882–1905 CrossRef CAS.
- P. M. Chhetri, X.-K. Yang and J.-D. Chen, Cryst. Growth Des., 2017, 17, 4801–4809 CrossRef CAS.
- P. M. Chhetri, X.-K. Yang and J.-D. Chen, CrystEngComm, 2018, 20, 2126–2134 RSC.
- G. Mahmoudi, J. K. Zareba, A. Bauzá, M. Kubicki, A. Bartyzel, A. D. Keramidas, L. Butusov, B. Mirosławh and A. Frontera, CrystEngComm, 2018, 20, 1065–1076 RSC.
- L. K. Rana, S. Sharma and G. Hundal, Cryst. Growth Des., 2016, 16, 92–107 CrossRef CAS.
- G. Mahmoudi, A. Bauzá, A. V. Gurbanov, F. I. Zubkov, W. Maniukiewicz, A. Rodriguez-Diéguez, E. López-Torres and A. Frontera, CrystEngComm, 2016, 18, 9056–9066 RSC.
- K. B. Thapa, Y.-F. Hsu, H.-C. Lin and J.-D. Chen, CrystEngComm, 2015, 17, 7574–7582 RSC.
- S. M. Mobin, A. K. Srivastava, P. Mathur and G. K. Lahiri, Dalton Trans., 2010, 39, 8698–8705 RSC.
- M. Govindaraj, W.-C. Huang, C.-Y. Lee, V. Lakshmanan, Y.-H. Liu, P. B. So, C.-H. Lin and J.-D. Chen, Int. J. Mol. Sci., 2022, 23, 7861 CrossRef CAS PubMed.
- J.-H. Hu, H.-H. Hsu, Y.-W. Chen, W.-H. Chen, S.-M. Liu and J.-D. Chen, J. Mol. Struct., 2023, 1289, 135896 CrossRef CAS.
- Bruker AXS, APEX2, V2008.6, SADABS V2008/1, SAINT V7.60A, SHELXTL V6.14, Bruker AXS Inc., Madison, WI, USA, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- K. B. Thapa and J.-D. Chen, CrystEngComm, 2015, 17, 4611–4626 RSC.
- Y.-F. Hsu, W. Hsu, C.-J. Wu, P.-C. Cheng, C.-W. Yeh, W.-J. Chang, J.-D. Chen and J.-C. Wang, CrystEngComm, 2010, 12, 702–710 RSC.
- T.-P. Tsai, Y.-T. Huang, U. Ray, Y.-J. Chang, P.-C. Cheng, C.-J. Wu, J.-D. Chen and J.-C. Wang, Polyhedron, 2010, 29, 3081–3088 CrossRef CAS.
- H.-L. Hua, Y.-F. Hsu, C.-J. Wu, C.-W. Yeh, J.-D. Chen and J.-C. Wang, Polyhedron, 2012, 33, 280–288 CrossRef.
- P. M. Chhetri, X.-K. Yang, C.-T. Yang and J.-D. Chen, Polymer, 2019, 11, 436 Search PubMed.
- P. M. Chhetri, X.-K. Yang and J.-D. Chen, J. Mol. Struct., 2021, 1239, 130543 CrossRef.
- J.-P. Zhang, X.-C. Huang and X.-M. Chen, Chem. Soc. Rev., 2009, 38, 2385–2396 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Packing diagrams (Fig. S1–S3). PXRD patterns (Fig. S4–S14). CCDC no. 2445340–2445350 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00465a |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*