
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of thorium dioxide powder synthesis methods on conventional and spark plasma sintering†
Sorin-Octavian
Vălu
*,
Walter
Bonani
 ,
Jacobus
Boshoven
,
Karin
Popa
* and
Marco
Cologna
,
Jacobus
Boshoven
,
Karin
Popa
* and
Marco
Cologna
European Commission, Joint Research Centre (JRC), Karlsruhe, Germany. E-mail: octavian.valu@ec.europa.eu; karin.popa@ec.europa.eu
First published on 4th September 2025
Abstract
We investigated the influence of the synthesis method on the properties of ThO2 powders and on their sinterability in conventional and spark plasma sintering (SPS). The powders were obtained by precipitation of thorium oxalate or thorium hydroxide, and further processed by low-temperature calcination at 600 °C or hydrothermal conversion at 250 °C. All powders had nanocrystallites <10 nm and a high surface area, which allow the onset of sintering at reduced temperatures. However, the presence of larger voids and non-ideal particle packing hinder full densification in the final sintering step. The powder obtained by hydrothermal decomposition of thorium hydroxide shows the highest specific surface area among all (SSA > 100 m2 g−1) and reaches a relative density of ∼90% in conventional pressure-less sintering at 1100 °C.
Introduction
Thorium dioxide (ThO2) shows favourable properties (e.g., high melting point, high thermal conductivity, and strong radiation shielding) that makes it valuable in high-tech applications, particularly in the fields of nuclear energy, catalysis, high-temperature applications (crucibles, coatings, and refractory materials), optical glasses (cameras and telescope lenses), aerospace industry (paints), radiation protection, catalysis, etc.1The thorium-based nuclear fuel cycle is of significant interest in nuclear energy due to its potential as an alternative to uranium (and plutonium) in nuclear reactors.2,3 Oxide fuels such as (U,Th)O2 or (Pu,Th)O2 for fuelling nuclear reactors (e.g. light- or heavy-water reactors, gas-cooled fast reactors, etc.) would be typically used in the form of dense pellets. They can be produced by starting from a powder, which is pressed at room temperature and then sintered at high temperature for several hours, in a process called conventional or pressure-less sintering. The high melting point of ThO2 (Tm = 3378 ± 17 °C)4 makes it an excellent candidate for high-temperature applications, but at the same time it also poses challenges for the processing of ThO2 ceramics.5,6 The temperatures for densification of ThO2 powders in conventional sintering often exceed 1700 °C.7 The SPS technique (which employs pressure and pulsed direct current to heat and compact powders, allowing for high heating rates and reduced sintering temperatures and times) is increasingly utilized in the nuclear materials field8 and has been successfully applied to ThO2 as well.9,10
High-energy ball-milling has been used to increase the sinterability of commercial ThO2 powders.11 An alternative and effective way to enhance the sintering of such materials is by using nanoparticles.12 In a larger context, research into actinide chemistry at the nanoscale has become increasingly important due to its potential to enhance safety and efficiency by reducing the grain size in the production of nuclear fuels. Over the past decades, numerous bottom-up methods have been employed to generate nanocrystalline ThO2 and solid solutions.13 The properties, such as size, shape, degree of agglomeration, chemical purity, etc., are determined by the synthesis methods used, among which chemical precipitation, sol–gel, molten salts, hydro- and solvothermal processes, combustion synthesis, microwave-assisted synthesis, sonochemical synthesis, and laser ablation in liquid are some.14–35 The precipitation of thorium ions (Th4+) using hydroxides or oxalic acid, followed by thermal decomposition, are efficient and reproducible methods to fabricate agglomerates or aggregates of thorium dioxide (ThO2) nanoparticles. Hydrothermal treatment of the resulting hydroxide or thorium oxalate can create nanoparticles of various sizes and shapes within the agglomerates. These characteristics influence the sintering behaviour of the powders.
The goal of this study is to examine how the preparation method influences the properties of nanocrystalline ThO2 powders during both conventional and spark plasma sintering.
Experimental
Powder preparation
All preparations were done in gloveboxes under an air atmosphere, while the thermal treatments were performed in a glovebox operating under nitrogen.A stock Th-solution was prepared by dissolution of Th(NO3)4·5H2O (Merck) in Milli-Q® water and acidified with nitric acid 65% (pH < 1). The concentration of this solution (as measured by ICP-MS) was 456 g Th l−1.
Hydroxide route
A volume of 8 ml of this Th4+–nitrate solution was added dropwise to 100 ml of 6 M ammonium hydroxide (Merck Millipore) at room temperature under stirring. The formed precipitate was left in the original solution (pH 12) overnight. 6.0 g of Th-hydroxide was separated after being filtered, washed with Milli-Q® water and dried over the weekend.Half of the precipitate (3.0 g) underwent a 6 hour treatment at 600 °C under an air flow (1 l min−1) with heating and cooling rates of 100 and 200 °C h−1, respectively. For this purpose, a Linn Elektronik tubular (quartz) furnace and quartz crucible were used to obtain approximately 1.9 g of ThO2 after the thermal treatment. This powder is hereinafter referred to as HT (hydroxide–thermal).
Another 2.9 g Th-hydroxide was mixed with 20 ml of Milli-Q® water and placed in a 100 ml autoclave (Parr Instruments model 4793, p–T controlled) with a glass insert that provides a free volume of about 80 ml. The mixture was then exposed to heat treatment for 4 hours at 250 °C at an autogenic pressure of circa 40 bar, from an initial nitrogen atmosphere. After cooling, the pressure in the autoclave returned to atmospheric pressure. After separation from the solution and subsequent washing with water, ethanol and acetone, an amount of approximately 1.9 g of ThO2 was obtained. We will refer to this powder as HH, which stands for hydroxide–hydrothermal.
Oxalate route
In a similar manner, a volume of 8 ml Th4+–nitrate stock solution was added dropwise at room temperature under stirring into 100 ml of oxalic acid 0.7 M (the as-called “reverse strike” approach); the oxalic acid solution was obtained by dissolution of oxalic acid dihydrate (VWR Chemicals) in Milli-Q® water and has an initial pH of 3. The formed precipitate was left ageing in the original solution overnight (pH does not change significantly after precipitation). 9.4 g of Th(C2O4)2·xH2O (as proved by XRD) was separated after filtration, washed with Milli-Q® water and dried over the weekend.An amount of 4.7 g of the oxalate was subjected to a 6 hour treatment at 600 °C under an air atmosphere (heating and cooling rates of 100 and 200 °C h−1, respectively). This temperature was selected as the minimum temperature needed to complete the thermal decomposition process.36 Approximately, 2.3 g of ThO2 was recovered after this heat treatment. This powder will be referred to as OT (oxalate – thermal) from this point forward.
Another portion of Th-oxalate weighing 4.5 g was added to 20 ml of Milli-Q® water, and the resulting mixture was placed in the 100 ml Parr autoclave with a glass insert, as described above. The system was then subjected to heat treatment at 250 °C and approximately 55 bar pressure for 4 hours under an initial nitrogen atmosphere (a peak temperature of 260 °C was initially applied for about 10 minutes in order to initiate the reaction). After cooling, the pressure in the autoclave was measured to be around 15 bar at room temperature, indicating that the decomposition reaction had been completed. Note that the atmosphere in the autoclave changes from an inert atmosphere to a reducing atmosphere due to the formation of CO (and CO2) as a reaction product of the oxalate decomposition. However, the oxidation state of the thorium remains unaffected during the conversion from oxalate to oxide. After separation from the solution, it was washed with water, ethanol and acetone, and approximately 3.0 g of ThO2 was obtained. In the following, this powder will be referred to as OH (oxalate – hydrothermal).
For conventional or spark plasma sintering, all powders were used as-prepared, with no milling or grinding.
Pellet preparation
The disks were placed in a molybdenum container and sintered under reductive gas in a cold wall sinter furnace (Degussa VSL10/18). The temperature of the furnace was measured with a type C thermocouple, placed close to the samples. Two different kinds of experiments were conducted, referred to as step sintering and direct sintering.
Step sintering: one disc of each powder (the only exception is OT powder, where three discs were used) was heat treated for four hours at 1100, 1300, 1500, 1600, and 1700 °C with a heating rate of 200 °C h−1. After cooling at room temperature, the dimensions of the pellets were taken after each heat treatment and the samples were placed back into the furnace for the next thermal treatment.
Direct sintering: one disc of each powder was sintered for four hours directly at 1600 °C, with the same heating rate.
The geometrical densities of all pellets were calculated by measuring the weight (Mettler-Toledo AG204), the diameter and the height (Mitutoyo QuickMike). The dimensions were measured several times under different orientations. The resulting average values were determined and registered. The relative densities of the discs were calculated taking a crystallographic density of ThO2 of 10.0 g cm−3 at room temperature.37,38
Characterisation methods
Results and discussion
Thorium hydroxide and thorium oxalate precursors
The two precursors were characterised by XRD, SEM and FT-IR.The XRD pattern (Fig. S1†) indicates that Th-hydroxide is almost amorphous, in agreement with the findings reported in ref. 42, and the baseline being very little deformed around the main diffraction lines of ThO2. The two peaks at 3600 and 3100 cm−1 of the FT-IR spectrum indicate the presence of ThO2·xH2O as well (Fig. S2†). After hydrothermal treatment, Th(OH)4 de-hydrated and/or reorganised in as ThO2·xH2O (x = 0–2), as proved by XRD. A similar behaviour was observed for ceric hydroxide.43 The micrographs (Fig. 1) show very fine particles within larger aggregates of irregular shape. Some of these aggregates show a layer that looks dense at the highest SEM magnification, which could have formed during the filtration of the precipitate.
 | ||
| Fig. 1 SEM images of the Th-hydroxide ((a), (c), (e) and (g)) and Th-oxalate hydrate ((b), (d), (f) and (h)) powders. | ||
For the second precursor, the XRD indicates a well crystallised Th(C2O4)2·xH2O monoclinic (C2/c) compound. SEM provides very important information regarding the morphology of the powder: we have applied the “reverse strike” approach, meaning addition of a highly concentrated Th-solution (pH < 1) in a highly concentrated oxalic acid solution; the oxalate platelets (Fig. 1) are, however, cavitated, similar to those obtained by36 using the “direct strike” method (addition of oxalic acid solution into a Th-solution).
The FT-IR spectrum (Fig. S2†) shows a typical pattern of the absorption peak of crystallised Th(C2O4)2·xH2O.31 Several non-resolved adsorption bands in the range 3000–3500 cm−1 can be attributed to the different stretching modes of the O–H bond of hydrate oxalate groups. The sharp adsorption peaks at 1630, 1350, 1310 and 795 cm−1 are assigned to the symmetric stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, asymmetric and symmetric C–O stretching and O–CO asymmetric stretching vibration modes, respectively.
O, asymmetric and symmetric C–O stretching and O–CO asymmetric stretching vibration modes, respectively.
ThO2 powder characterisation
Table 1 summarises several physical properties of the powders obtained by different methods. The temperatures of the processes were selected as the minimum needed for the thermal36 and hydrothermal21,25 decomposition of the oxalate. As a matter of coincidence, the crystallite sizes are similar (∼6.5 and ∼8 nm for the two pairs of thermal and hydrothermal processes, respectively). The crystallite sizes produced from hydroxides are smaller than those obtained from oxalates.43–45 The lattice parameters are larger than the one reported for bulk ThO2 (0.5598 nm) and pairing as a function of the type of the method (thermal or hydrothermal), with larger values obtained for the hydrothermal processes, possibly due to adsorbed water (Fig. S3†).| Powder preparation method | ||||
|---|---|---|---|---|
| Reaction conditions | Hydroxide thermal | Hydroxide hydrothermal | Oxalate thermal | Oxalate hydrothermal |
| HT | HH | OT | OH | |
| T, °C | 600 | 250 | 600 | 250 |
| p, bar | 1 | 40 | 1 | 55 |
| Atmosphere | Oxidising (air) | Inert (N2) | Oxidising (air) | Inert (N2) + CO/CO2 formed in situ |
| ThO2 powder characteristics | ||||
|---|---|---|---|---|
| a, nm | 0.5605(2) | 0.5615(1) | 0.5605(1) | 0.5614(2) |
| d from XRD, nm | 6.6 ± 1.1 | 6.2 ± 0.8 | 7.8 ± 0.9 | 8.1 ± 1.2 |
| SSA, m2 g−1 | 17.3 ± 0.3 | 109.7 ± 0.3 | 12.9 ± 0.1 | 35.0 ± 0.7 |
| Equivalent spherical particle size, dBET, nm | 34.7 | 5.5 | 46.5 | 17.1 |
| Morphology from SEM | 100–500 μm agglomerates of nanometric powders | 100–500 μm agglomerates of nanometric powders | Cavitated plate-like aggregates of 0.5–1 μm | Core shell spherical agglomerates of ∼0.3 μm |
The synthesis method has a significant effect on the SSA. While the thermal method gives the smallest values (17 and 13 m2 g−1 for HT and OT, respectively), these values are far larger for the hydrothermal treatment (110 and 35 m2 g−1 for HH and OH, respectively), in line with the milder conditions of the processes. An equivalent particle size, dBET, was calculated from the SSA, assuming the ideal case of spherical and isolated primary particles, using the formula dBET = 6/(SSA·ρ), with ρ = 10.0 g cm−3 (Table 1). The particle size calculated from the SSA is generally higher than the crystallite size calculated by XRD, indicating that a part of the surface of the sample is inaccessible. This could be due to agglomeration/aggregation or partial sintering of the crystallites. Interestingly, only the SSA for the powder obtained by hydrothermal methods give a calculated particle size in fair agreement with the one obtained from XRD. In the case of HH powder, the close similarity in value suggests that the primary particles and crystallites are the same.
SEM (Fig. 2) and FTIR (Fig. S4†) techniques were used to elucidate this behaviour. All FTIR measurements indicate the presence of water and organic compounds, and the general features of the IR spectra are closer to the one of the precursors (Fig. S4†). The thermal method gives almost pure ThO2 powders. Several features that might be assigned to carbon pollution are present in the powders obtained from oxalate, as the signals at 1510 cm−1 and 1360 cm−1 (νas CO), and 1100 cm−1 (νs CO).46 The two vibrations present at 980 and 870 cm−1 in the powders obtained by hydrothermal treatments could not be assigned but are likely related to the presence of an impurity in the autoclave.
 | ||
| Fig. 2 SEM images of the ThO2 powders obtained by different methods: HT (a) and (b); HH (c) and (d); OT (e) and (f); OH (g) and (h). | ||
Both powders obtained from the hydroxide (HT and HH) show large agglomerates, which are pseudomorphic for the powder obtained by thermal decomposition (Fig. 2 and Fig. 1, respectively). Such agglomerates are composed of nanometric particles, as shown by XRD and BET (Table 1). The apparently dense layer observed in the precursor is still present in the HT powder. Thermal oxalate-to-oxide conversion (OT) is also pseudomorphic and results in agglomerates of nanocrystallites preserving the original size and shape of the oxalate. On the other hand, the hydrothermal treatment (OH) results in almost independent and fairly monodispersed cavitated spheroids, corresponding to agglomerated ThO2 nanoparticles.
ThO2 pellet sintering and characterisation
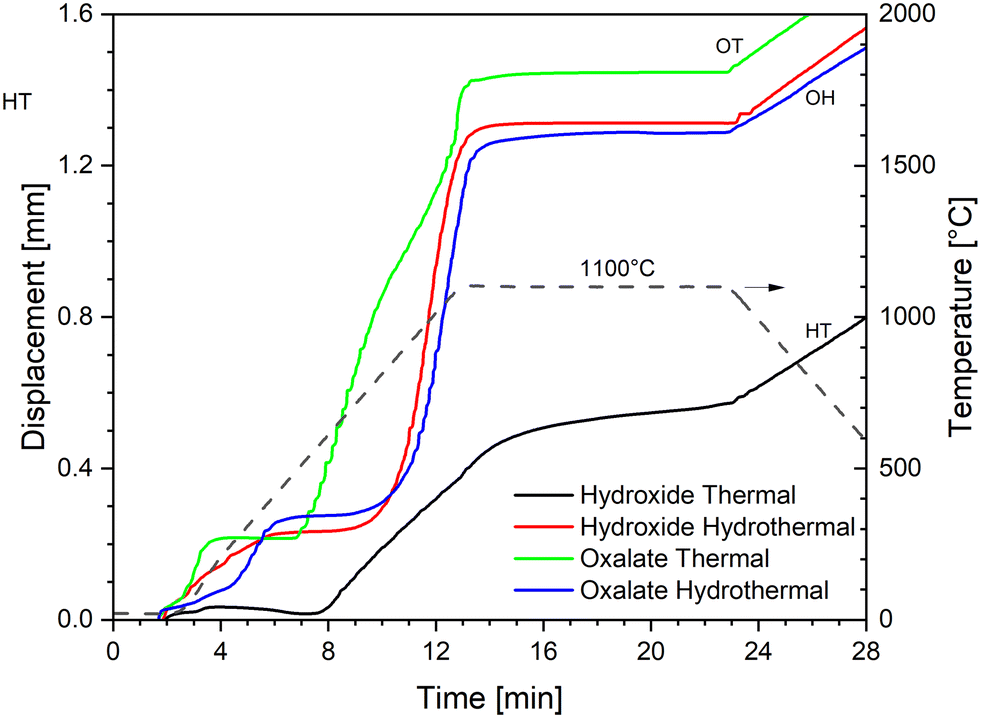 | ||
| Fig. 3 SPS densification curves at a maximum temperature of 1100 °C with a constant pressure of 70 MPa. | ||
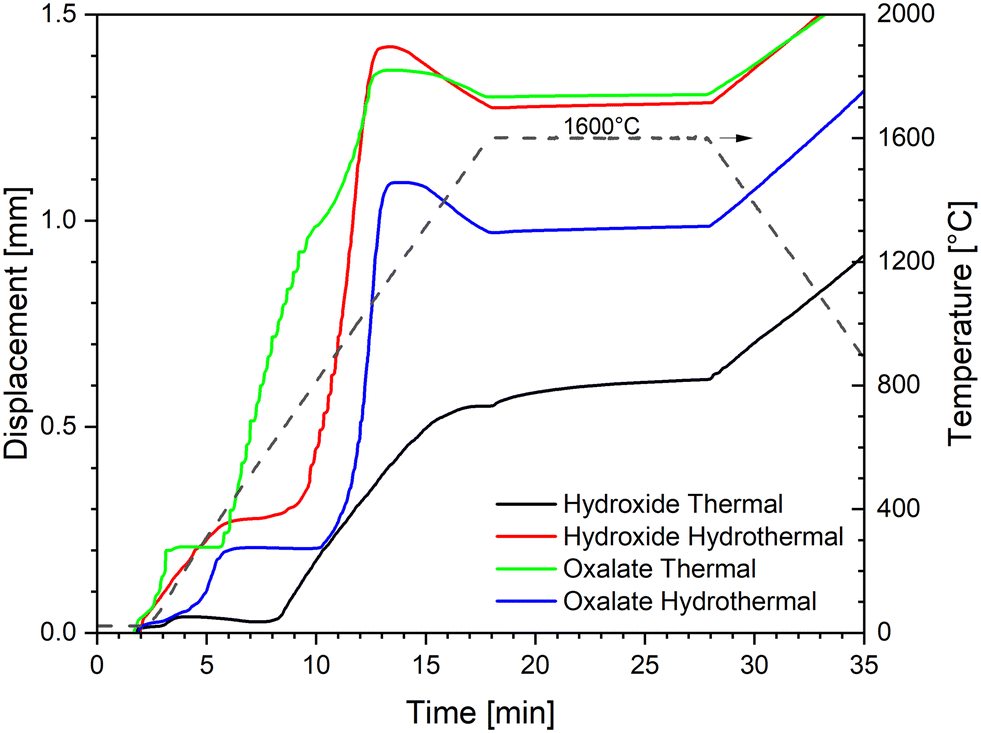 | ||
| Fig. 4 SPS densification curves at a maximum temperature of 1600 °C with a constant pressure of 70 MPa. | ||
The two powders obtained by hydrothermal decomposition (HH and OH) show a very similar behaviour, with the HH curve slightly shifted to lower temperatures (∼50 °C): (i) an initial low-temperature shrinkage up to ∼400 °C, (ii) a steep sintering curve between ∼800 °C and ∼1300 °C, and (iii) the isothermal dwell, where densification continues at a much lower rate.
The first low-temperature shrinkage has been previously observed with nanometric powders and was attributed to degassing of the adsorbed species and particles and agglomerate rearrangement.47 Most of the densification happens in the second step, up to ∼1300 °C, due to the sintering of the primary nanometric particles and the elimination of the nanoporosity. The negative slope of the displacement above 1300 °C indicates that the thermal expansion of the system (sample and pistons) is larger than the sintering shrinkage. Since the slope is almost linear and opposite to the slope of the displacement during cooling, we can conclude that the sintering rate is negligible in comparison to the thermal expansion above 1300 °C. Finally, densification continues very slowly also during the isothermal dwell at 1600 °C. This last phase likely represents the elimination of the larger porosity, which is present between the agglomerates. The similar sintering curves of the HH and OH powders suggest that the hydrothermal treatment gives similar products in terms of the morphological features influencing the densification rate (e.g. the degree of agglomeration, packing, shape and size of the primary particles), and thus the final oxides show little memory of their precursor's shape (oxalate or hydroxide).
This is not the case for the simply calcined powders: HT and OT show two completely distinct sintering curves. The OT powder shows an initial compaction below 200 °C, then the on-set of sintering at a very low temperature (∼400 °C), after which the densification continues in at least two main steps (indicatively 400–1000 °C and 1000–1600 °C). Such behaviour is typical of powder compacts with small primary particle sizes organised in larger agglomerates, resulting in a microstructure with porosity at two distinct scales, nanometric and micrometric. In particular, it has been shown that ex-oxalate urania and thoria powders produced under similar conditions consist of nanometric primary particles, ordered within larger thin platelets of micrometric size. In this configuration, the nanoparticles influences the low-temperature sintering, while the shape and packing of the platelets dominate the densification kinetics at high temperature.18,48
Lastly, the hydroxide powder thermally calcined (HT) also shows an initial compaction below 200 °C (but much lower than that of all the other powders), then it starts sintering at ∼600 °C, and the sintering range extends up to 1600 °C and continues during the dwell time. It can be inferred that the particle size and porosity distribution for this powder is relatively large, and the residual large porosity between the agglomerates are responsible for the high-temperature densification.
The fracture surfaces after sintering at 1100 °C (Fig. 5) and 1600 °C (Fig. 6) provide further insight into the process. At 1100 °C, the microstructure of HT presents an area of well-sintered grains (0.3–1.2 μm size) within the original agglomerates. These areas are surrounded by large voids, partially filled by zones with less-sintered powder (∼50 nm size), which do appear to have been affected by the external force applied during SPS, as the pressure was likely concentrated on the skeleton made by the larger agglomerates. Raising the temperature to 1600 °C increases and homogenises the grain size and partially removes the large voids, possibly by creep and deformation of the large agglomerates. However, the time and temperature are not sufficient for completely removing the voids.
 | ||
| Fig. 5 SEM micrographs of selected fracture surfaces after SPS for 10 min at 1100 °C (scale bar 5 μm). HT (a); HH (b); OT (c); OH (d). | ||
 | ||
| Fig. 6 SEM micrographs of selected fracture surfaces after SPS for 10 min at 1600 °C (scale bar 20 μm; note the lower magnification compared to Fig. 5). HT (a); HH (b); OT (c); OH (d). | ||
The HH powder at 1100 °C not only shows very fine grains (∼100 nm) surrounded by distributed nano-porosity, but also few areas with larger micrometric-sized grains (not shown here). Some elongated flaws are still visible around the original powder agglomerates but are much less than in the HT sample. It appears that the hydrothermal decomposition is effective in destroying the original agglomerates and producing softer agglomerates that are easily deformed under pressure to fill the gaps during the SPS process. At 1600 °C the grains have grown to a few micrometers in size, but the intergranular porosity is not completely eliminated, and some grains have assumed a peculiar rounded shape that could be caused by the incomplete elimination of some impurity or trapped gas during sintering. This confirms previous results, which have shown that incomplete densification is obtained under similar conditions, but dense and nanostructured (50 nm grain size) pellets can be achieved with an HH powder by rising the pressure to 500 MPa in a special high-pressure SPS set-up using SiC dies, at temperatures of only 915 °C.47
The OT powder at 1100 °C gives a fine microstructure with grains of ca. 800 nm surrounded by residual closed porosity, and some regions with larger grains in the order of a few micrometres, located towards the centre of the pellet. At 1600 °C the porosity is almost completely eliminated, and the mean grain size has grown to 3.5 μm, in agreement with the findings in ref. 10 and 18, where relative densities above 95% and comparable grain sizes under similar processing conditions and powders were obtained.
Lastly, the OH powder at 1100 °C has grown into larger grains of ca. 3 μm in size, some of them with a rounded shape. The microstructure after SPS at 1600 °C looks close to fully dense and the grain size approaches 10 μm. While HH and OT ThO2 powders have been previously sintered in SPS, a comparison with the literature for HT and OH powder is not possible, as no previous results were found.
 | ||
| Fig. 7 Relative density during conventional sintering from 1100 °C to 1700 °C (step-sintering test). | ||
The microstructures are shown in Fig. 8. The HT sample appears poorly sintered, in agreement with its low relative density, and the original agglomerates with the peculiar dense layer observed in the powders are clearly recognisable. Conversely, the particles of the HH sample have sintered and grown to grains of several micrometres in size, which show partially intragranular fracture. The residual porosity is well-distributed and intergranular, while larger elongated flaws between the original agglomerates (visible at lower magnifications, not shown here) also contribute to the overall low relative density. Interestingly, this sample achieves the highest density already at 1100 °C, after which the increasing temperature gives only grain growth but has little effect on the densification and elimination of the larger pores and flaws. The OT sample reaches the highest density, giving a homogeneous microstructure with distributed porosity between the original micrometric-sized platelets. The negative effects of larger powder aggregates are less severe than those in the HT and HH powders. The sintering behaviour of analogously produced powders have been extensively studied in the literature (e.g. ref. 36 and 49). It is typically seen that the densification happens in two main stages: initial removal of the small intra-platelet porosity at lower temperatures, followed by the elimination of the larger intra-platelet porosity at higher temperatures. Very high densities (>95%) can be achieved at higher temperatures (>1700 °C). Finally, the OH sample shows sintered grains of a few micrometres in size, surrounded by large micrometric pores. Analogous to the other hydrothermally-produced powder (OH), increasing the sintering temperature from 1100 °C to 1600 °C does not improve the final density, but instead promotes grain growth.
 | ||
| Fig. 8 SEM micrographs of the selected fracture surfaces after conventional sintering for 4 h at 1600 °C (same magnification as in Fig. 6). HT (a); HH (b); OT (c); OH (d). | ||
Conclusions
We synthetized four different ThO2 powders, starting from two different precursors (either thorium hydroxide or thorium oxalate), and applying two distinct conversion routes (either low-temperature calcination at 600 °C or hydrothermal conversion at 250 °C). Each method yields powders containing nanocrystallites (<10 nm) but with distinct properties, such as the degree of agglomeration/aggregation, specific surface area and residual water content. Hydrothermal decomposition produces powders with a higher specific surface area compared to those obtained through thermal calcination. Among all the samples, the powder obtained from hydrothermal decomposition of Th hydroxide shows the highest specific surface area (SSA > 100 m2 g−1). On the other hand, the powders obtained by hydrothermal decomposition contain more residual water and show the highest mass loss during sintering.The properties of the powders dictate the sintering behaviour. The nanometric size of the primary particles induce an initial fast sintering at low temperatures in both SPS and conventional sintering. However, the presence of large voids left between the agglomerates/aggregates and the degassing of the adsorbed water hinders further densification at low temperatures unless external pressure is applied during sintering, as in SPS. In particular, the very large surface area powder obtained by hydrothermal decomposition of Th hydroxide shows the highest potential for low-temperature sintering: it reached a relative density of almost 90% already at temperatures as low as 1100 °C in conventional sintering. Any further increase in the temperature up to 1600 °C did not improve the density. Additional processing and optimisation steps, e.g. degassing, and better particle packing by de-agglomeration/de-aggregation and granulation could also improve the final densification step.
Data availability
The data that support the findings of this study are available from the authors upon reasonable request.Author contributions
S.-O. V.: methodology, investigation, data curation, and writing – review and editing; W. B.: methodology, investigation, and writing – review and editing; J. B.: investigation and writing – review and editing; K. P.: conceptualization, methodology, investigation, writing – original draft, review and editing, and supervision; M. C.: conceptualization, methodology, validation, investigation, data curation, writing – original draft, review and editing, and supervision.Conflicts of interest
There are no conflicts to declare.Notes and references
- X.-W. Wang, L. Mei, L.-Y. Yuan, S.-A. Wang, Z.-F. Chaiac and W.-Q. Shi, Size-tunable synthesis of monodisperse thorium dioxide nanoparticles and their performance on the adsorption of dye molecules, CrystEngComm, 2014, 60, 10469–10475 RSC.
- U. Humphrey and M. Khandaker, Viability of thorium-based nuclear fuel cycle for the next generation nuclear reactor: Issues and prospects, Renewable Sustainable Energy Rev., 2018, 97, 259–275 CrossRef CAS.
- M. Du Toit, F. Van Niekerk and S. Amirkhosravi, Review of thorium-containing fuels in LWRs, Prog. Nucl. Energy, 2024, 170, 105136 CrossRef CAS.
- C. Ronchi and J. Hiernaut, Experimental measurement of pre-melting and melting of thorium dioxide, J. Alloys Compd., 1996, 240(1–2), 179–185 CrossRef CAS.
- H. Takiishi, L. Gênova, E. Cavalheira, M. Cotrim, W. Santos and P. Lainetti, Use of dopants for thoria sintering temperature reduction-characterization of ThO2, J. Energy Power Eng., 2016, 10, 740–745 CAS.
- O. Shichalin, R. Frolov, I. Buravlev, I. Tanayev, V. Faizova, S. Azon, N. Andreeva and E. Papynov, Synthesis and Spark Plasma Sintering of microcrystalline thorium dioxide for nuclear fuel products, Russ. J. Inorg. Chem., 2020, 65, 1245–1252 CrossRef CAS.
- S. Qi, F. Guan, D. Peng, X. Zhang and W. Liao, A simple method for preparing ThO2 ceramics with high density, Int. J. Appl. Ceram. Technol., 2023, 20(2), 1194–1204 CrossRef CAS.
- M. Cologna, 5.25 - Use of field assisted sintering for innovation in nuclear ceramics manufacturing, Comprehensive Nuclear Materials, 2nd edn, 2020, pp. 811–839 Search PubMed.
- H. Muta, Y. Murakami, M. Uno, K. Kurosaki and S. Yamanaka, Thermophysical properties of Th1−xUxO2 pellets prepared by spark plasma sintering technique, J. Nucl. Sci. Technol., 2013, 50(2), 181–187 CrossRef CAS.
- M. Linu, A. Prasad, J. Ranasinghe, E. Jossou, D. Oladimeji, B. Szpunar, L. Bichler and J. Szpunar, The effect of SPS processing parameters on the microstructure and thermal conductivity of ThO2, J. Nucl. Mater., 2019, 527, 151811 CrossRef.
- S. Scott, T. Yao, F. Lu, G. Xin, W. Zhu and J. Lian, Fabrication of lanthanum-doped thorium dioxide by high-energy ball milling and spark plasma sintering, J. Nucl. Mater., 2017, 485, 207–215 CrossRef CAS.
- S. Mukerjee, T. Kutty, N. Kumar, R. Pai and A. Kumar, Fabrication technologies for ThO2-based fuel, in Thoria-based Nuclear Fuels. Green Energy and Technology, Springer, London, 2013, pp. 205–277 Search PubMed.
- K. Popa and O. Walter, Actinide dioxide nanoparticles, in Comprehensive Nuclear Materials, 2 edn, Elsevier, Oxford, 2020, vol. 6, pp. 579–592 Search PubMed.
- J. Spino, H. Santa Cruz, R. Jovani-Abril, R. Birtcher and C. Ferrero, Bulk-nanocrystalline oxide nuclear fuels – An innovative material option for increasing fission gas retention, plasticity and radiation-tolerance, J. Nucl. Mater., 2012, 422(1–3), 27–44 CrossRef CAS.
- D. Hudry, C. Apostolidis, O. Walter, T. Gouder, E. Courtois, C. Kübel and D. Meyer, Controlled synthesis of thorium and uranium oxide nanocrystals, Chem. – Eur. J., 2013, 19(17), 5297–5305 CrossRef CAS PubMed.
- R. Zhao, L. Wang, Z.-F. Chai and W.-Q. Shi, Synthesis of ThO2 nanostructures through a hydrothermal approach: influence of hexamethylenetetramine (HMTA) and sodium dodecyl sulfate (SDS), RSC Adv., 2014, 4(94), 52209–52214 RSC.
- V. Tyrpekl, J.-F. Vigier, D. Manara, T. Wiss, O. Dieste Blanco and J. Somers, Low temperature decomposition of U(IV) and Th(IV) oxalates to nanograined oxide powders, J. Nucl. Mater., 2015, 460, 200–208 CrossRef CAS.
- V. Tyrpekl, M. Cologna, D. Robba and J. Somers, Sintering behaviour of nanocrystalline ThO2 powder using spark plasma sintering, J. Eur. Ceram. Soc., 2016, 36(3), 767–772 CrossRef CAS.
- N. Clavier, G. Nkou Bouala, J. Léchelle, J. Martinez, N. Dacheux and N. Podor, Novel approaches for the in situ study of the sintering of nuclear oxide fuel materials and their surrogates, Radiochim. Acta, 2016, 105(11), 879–892 CrossRef.
- M. Brykala and M. Rogowski, The complex sol–gel process for producing small ThO2 microspheres, J. Nucl. Mater., 2016, 473, 249–255 CrossRef CAS.
- O. Walter, K. Popa and O. Dieste Blanco, Hydrothermal decomposition of actinide(IV) oxalates: a new aqueous route towards reactive actinide oxide nanocrystals, Open Chem., 2016, 14(1), 170–174 CAS.
- K. Kamali, K. Ananthasivan, T. Ravindran and D. Sanjay Kumar, High pressure Raman spectroscopic studies on nanocrystalline ThO2, J. Nucl. Mater., 2017, 493, 77–83 CrossRef CAS.
- N. Clavier, J. Maynadié, A. Mesbah, J. Hidalgo, R. Lauwerier, G. Nkou Bouala, S. Parrès-Maynadié, D. Meyer, N. Dacheux and R. Podor, Thorium aspartate tetrahydrate precursor to ThO2: Comparison of hydrothermal and thermal conversions, J. Nucl. Mater., 2017, 487, 331–342 CrossRef CAS.
- L. Balice, D. Bouëxière, M. Cologna, A. Cambriani, J.-F. Vigier, E. De Bona, G. Sorarù, C. Kübel, O. Walter and K. Popa, Nano and micro U1-xThxO2 solid solutions: From powders to pellets, J. Nucl. Mater., 2018, 498, 307–313 CrossRef CAS.
- K. Popa, O. Walter, O. Dieste Blanco, A. Guiot, D. Bouëxière, J.-Y. Colle, L. Martel, M. Naji and D. Manara, A low-temperature synthesis method for AnO2 nanocrystals (An = Th, U, Np, and Pu) and associate solid solutions, CrystEngComm, 2018, 20(32), 4614–4622 RSC.
- T. Plakhova, A. Romanchuk, D. Likhosherstova, A. Baranchikov, P. Dorovatovskii, R. Svetogorov, T. Shatalova, T. Egorova, A. Trigub, K. Kvashnina, V. Ivanov and S. Kalmykov, Size effects in nanocrystalline thoria, J. Phys. Chem. C, 2019, 123(37), 23167–23176 CrossRef CAS.
- S. Valu, E. De Bona, K. Popa, J.-C. Griveau, E. Colineau and R. Konings, The effect of lattice disorder on the low-temperature heat capacity of (U1−yThy)O2 and 238Pu-doped UO2, Sci. Rep., 2019, 9, 15082 CrossRef PubMed.
- L. Amidani, G. Vaughan, T. Plakhova, A. Romanchuk, E. Gerber, R. Svetogorov, S. Weiss, Y. Joly, S. Kalmykov and K. Kvashnina, The application of HEXS and HERFD XANES for accurate structural characterisation of actinide nanomaterials: The case of ThO2, Chem. – Eur. J., 2020, 27(1), 252–263 CrossRef PubMed.
- L. Bonato, M. Virot, X. Le Goff, P. Moisy and S. Nikitenko, Sonochemical dissolution of nanoscale ThO2 and partial conversion into a thorium peroxo sulfate, Ultrason. Sonochem., 2020, 69, 105235 CrossRef CAS PubMed.
- L. Bonato, M. Virot, T. Dumas, A. Mesbah, E. Dalodière, O. Dieste Blanco, T. Wiss, X. Le Goff and M. Odorico, Probing the local structure of nanoscale actinide oxides: a comparison between PuO2 and ThO2 nanoparticles rules out PuO2+x hypothesis, Nanoscale Adv., 2020, 2(1), 214–224 RSC.
- J. Manaud, J. Maynadié, A. Mesbah, O. Myrtille, P. Martin, M. Zunino, N. Dacheux and N. Clavier, Hydrothermal conversion of thorium oxalate into ThO2·nH2O oxide, Inorg. Chem., 2020, 59(20), 14954–14966 CrossRef CAS PubMed.
- L. Moreau, A. S. M. Herve, D. Russo, R. Abergel, S. Alayoglu, J. Arnold, A. Braun, G. Deblonde, Y. Liu, T. Lohrey, D. Olive, Y. Qiao, J. Rees, D. Shuh, S. Teat, C. Booth and S. Minasian, Structural properties of ultra-small thorium and uranium dioxide nanoparticles embedded in a covalent organic framework, Chem. Sci., 2020, 11, 4648–4668 RSC.
- Q. Zhang, Z. Qian, X. Liu, L. Li, X. Duan, T. Yu, X. Liu and Y. Qiao, Synthesis of thorium dioxide nanocrystals via molten salt thermal decomposition for nuclear energy-related applications, ACS Appl. Nano Mater., 2022, 5(12), 17977–17985 CrossRef CAS.
- A. Zakharanka, L. Gubbels, B. Acevedo, M. Verwerft and V. Tyrpekl, Homogeneous precipitation of thorium oxalate: Structural, kinetic, and morphological aspects, J. Nucl. Mater., 2025, 605, 155574 CrossRef CAS.
- N. Cabanas, V. Manukyan, K. Bauer, P. Burns and A. Aprahamian, ThO2 and Th1–xUxO2 nanoscale materials and thin films for nuclear science applications, ACS Appl. Nano Mater., 2025, 8(7), 3345–3355 CrossRef CAS.
- T. Wangle, V. Tyrpekl, S. Cagno, T. Delloye, O. Larcher, T. Cardinaels, J. Vleugels and M. Verwerft, The effect of precipitation and calcination parameters on oxalate derived ThO2 pellets, J. Nucl. Mater., 2017, 495, 128–137 CrossRef CAS.
- W. Rüdorff and G. Valet, Über das Ceruranblau und Mischkristalle im System CeO2-UO2-U3O8, Z. Anorg. Allg. Chem., 1953, 271, 257–272 CrossRef.
- H. Whitfield, D. Roman and A. Palmer, X-ray study of the system ThO2-CeO2-Ce2O3, J. Inorg. Nucl. Chem., 1966, 28(12), 2817–2825 CrossRef CAS.
- V. Tyrpekl, C. Berkmann, M. Holzhäuser, F. Köpp, M. Cologna, T. Wangle and J. Somers, Implementation of a spark plasma sintering facility in a hermetic glovebox for compaction of toxic, radiotoxic, and air sensitive materials, Rev. Sci. Instrum., 2015, 86, 023904 CrossRef CAS PubMed.
- P. Scherrer, Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen, Göttinger Nachrichten Gesell, 1918, vol. 2, p. 98 Search PubMed.
- S. Brunauer, P. Emmett and E. Teller, Adsorption of gases in multimolecular layers, J. Am. Chem. Soc., 1938, 60(2), 309–3019 CrossRef CAS.
- T. Plakhova, A. Romanchuk, I. Seregina, R. Svetogorov, D. Kozlov, Y. Teterin Jr., A. Kuzenkova, A. Egorov and S. Kalmykov, From X-ray amorphous ThO2 to crystalline nanoparticles through long-term aging at room temperature, J. Phys. Chem. C, 2023, 127(1), 187–195 CrossRef CAS.
- D. Prieur, W. Bonani, K. Popa, O. Walter, K. Kriegsman, M. Engelhard, X. Guo, R. Eloirdi, T. Gouder, A. Beck, T. Vitova, A. Scheinost, K. Kvasnina and P. Martin, Size dependence of lattice parameter and electronic structure in CeO2 nanoparticles, Inorg. Chem., 2020, 59(8), 5760–5767 CrossRef CAS PubMed.
- D. Manara, K. Popa, D. Robba, L. Fongaro, J.-Y. Colle and A. Bulgheroni, Infrared laser absorption and melting behaviour of nano-sized cerium dioxide: A laser heating study, J. Eur. Ceram. Soc., 2021, 41(2), 1384–1390 CrossRef CAS.
- V. Baumann, K. Popa, O. Walter, M. Rivenet, G. Senentz, B. Morel and R. Konings, Synthesis of nanocrystalline PuO2 by hydrothermal and thermal decomposition of Pu(IV) oxalate: A comparative study, Nanomaterials, 2023, 13, 240 CrossRef PubMed.
- L. De Almeida, S. Grandjean, N. Vigier and F. Patisson, Insights into the thermal decomposition of lanthanide(III) and actinide(III) oxalates – from neodymium and cerium to plutonium, Eur. J. Inorg. Chem., 2012, 4986–4999 CrossRef CAS.
- E. De Bona, O. Walter, H. Störmer, T. Wiss, G. Baldinozzi, M. Cologna and K. Popa, Synthesis of nanostructured ThO2 pellets, J. Am. Ceram. Soc., 2019, 102(7), 3814–3818 CrossRef CAS.
- V. Tyrpekl, M. Cologna, J.-F. Vigier, A. Cambriani, W. De Weerd and J. Somers, Preparation of bulk-nanostructured UO2 pellets using high-pressure spark plasma sintering for LWR fuel safety assessment, J. Am. Ceram. Soc., 2017, 100(4), 1269–1274 CrossRef CAS.
- T. Wangle, V. Tyrpekl, J. Pakarinen, T. Cardinaels, T. Delloye and J. V. M. Vleugels, Morphology dependent sintering path of nanocrystalline ThO2, J. Nucl. Mater., 2020,(533), 152081 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ce00459d |
| This journal is © The Royal Society of Chemistry 2025 |