
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acceleration of scintillation response in Sc,Ca,Mg-codoped YAG:Ce crystals†
Ia.
Gerasymov
 a,
S.
Tkachenko
a,
D.
Kurtsev
a,
D.
Kofanov
a,
O.
Viahin
a,
P.
Maksimchuk
a,
I.
Rybalka
a,
B.
Grynyov
a,
J.
Delenne
b,
L.
Martinazzoli
bc,
L.
Roux
bd,
E.
Auffray
b,
A.
Padmanaban
e and
O.
Sidletskiy
*ae
a,
S.
Tkachenko
a,
D.
Kurtsev
a,
D.
Kofanov
a,
O.
Viahin
a,
P.
Maksimchuk
a,
I.
Rybalka
a,
B.
Grynyov
a,
J.
Delenne
b,
L.
Martinazzoli
bc,
L.
Roux
bd,
E.
Auffray
b,
A.
Padmanaban
e and
O.
Sidletskiy
*ae
aInstitute for Scintillation Materials NAS of Ukraine, Kharkiv, Ukraine. E-mail: sidletskiy@isma.kharkiv.ua
bEuropean Organization for Nuclear Research (CERN), Geneva, Switzerland
cINFN & Università degli Studi di Milano-Bicocca, Milan, Italy
dUniversité Lyon, Université Claude Bernard Lyon 1, Institute Lumière Matière UMR 5306, CNRS, Villeurbanne, 69100, France
eCentre of Excellence ENSEMBLE3 Sp. z o. o, Warsaw, 01-919, Poland. E-mail: oleg.sidletskiy@ensemble3.eu
First published on 11th June 2025
Abstract
This study explores fast-timing garnet scintillators as potential candidates for future high-energy physics detectors. Sc-codoped YAG:Ce crystals were successfully grown for the first time using the Czochralski method in a reducing atmosphere from tungsten crucibles. Scintillation decay times were accelerated through codoping of Y3Al5O12:Ce (YAG:Ce) with Sc3+ and divalent alkaline earth metal cations (Ca2+, Mg2+). The resulting codoped YAG:Ce exhibited a decay time of 21 ns and a high light yield of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 photons per MeV, making it a strong candidate for future detectors at the HL-LHC, where particle collisions occur every 25 ns. The study also examines correlations between cation substitutions and the optical and scintillation performance of Ce-doped Al3+/Sc3+-substituted garnets, comparing them with known counterparts.
000 photons per MeV, making it a strong candidate for future detectors at the HL-LHC, where particle collisions occur every 25 ns. The study also examines correlations between cation substitutions and the optical and scintillation performance of Ce-doped Al3+/Sc3+-substituted garnets, comparing them with known counterparts.
1. Introduction
Yttrium aluminum garnet doped with cerium (Y3Al5O12:Ce, or YAG:Ce) is a well-established phosphor and scintillation material that has recently regained attention due to advancements in scintillation performance. These improvements include an increased light yield exceeding 33000 photons per MeV,1–3 high radiation tolerance,4,5 and cost-effective crystal fabrication technologies.6–8
Despite these advantages, YAG:Ce has limitations for fast-timing applications, such as next-generation high-energy physics experiments at colliders9 and time-of-flight positron emission tomography (TOF-PET).10,11 Its primary drawback is a relatively slow luminescence decay, with the main component ranging from 55 to 120 ns, alongside slower decay components and afterglow effects.11–13 Meanwhile, the high-luminosity large hadron collider (HL-LHC) at CERN operates at a 25 ns−1 collision frequency, requiring scintillation signals to be registered within a <25 ns window to prevent pile-up.14
In Ce-doped garnets, slow luminescence decay is primarily attributed to the intrinsically long Ce3+ lifetime of 50–60 ns in garnet hosts. Additionally, intrinsic lattice defects—such as cationic antisites (A3+ and B3+ substitutions in the A3B5O12 lattice), oxygen and Al3+ vacancies, and their combinations—contribute to delayed scintillation.15,16 Furthermore, garnets grown in CO-containing atmospheres using cost-effective W or Mo crucibles often incorporate carbon-related defects. Interestingly, these defects can enhance scintillation performance by creating negatively charged centers that suppress electron trapping and the formation of color centers associated with positively charged defects.17–19
Reducing luminescence decay time below the intrinsic Ce3+ lifetime is only possible by introducing non-radiative processes, which inevitably compromise light output. This can be achieved by substituting Al3+ with Ga3+ or Sc3+ and/or by doping with divalent alkaline earth metals. These modifications influence competing mechanisms of electron ionization from Ce3+ 5d excited states into the conduction band, as detailed in.20–22 While Ga incorporation reduces decay time, it significantly increases production costs, as gallium-containing garnets (e.g., Gd3Al5−xGaxO12:Ce, GAGG:Ce) require high-cost Ir crucibles and an inert or weakly oxidizing atmosphere for large-scale growth.9 In contrast, Sc3+ functions similarly to Ga3+ by forming conduction band states via its 3d orbitals, thereby reducing the energy gap between the Ce 5d1 level and the conduction band bottom and inducing non-radiative electron transitions.22 However, unlike Ga2O3, scandium oxide remains stable under reducing conditions, making it suitable for crystal growth in W and Mo crucibles.
Recent experiments on the crystal growth of Sc-doped garnets have employed various methods, including micro-pulling-down,23–25 liquid phase epitaxy (LPE),26,27 optical floating zone (OFZ),28 and Bridgman.29 However, each of these techniques has intrinsic limitations that can degrade crystal/film quality and scintillation performance. For instance, the OFZ method introduces high thermal gradients, leading to crystal cracking; the micro-pulling-down technique suffers from radial activator segregation; the Bridgman method often results in compositional inhomogeneities due to poor melt mixing; and LPE-grown films exhibit very low Ce distribution coefficients. These factors likely contribute to the slow scintillation decay and moderate-to-poor light yield observed in these crystals,23–29 as discussed in detail in the Discussion section.
The Czochralski method, by contrast, avoids these drawbacks and remains the primary technique for industrial-scale production of many dielectric and semiconductor crystals. Growth of Y3Al5−xScxO12 (YSAG) with Sc contents ranging from 0 to 2 f.u. in the melt using this method has been reported in ref. 30 and 31. The Y2O3–Sc2O3–Al2O3 system supports a broad range of garnet-structured solid solutions, though no ternary garnet has been found to melt congruently.31 The segregation coefficients KSc = 0.83, KAl = 1.16, and KY = 0.96 (ref. 31) close to unity suggest a relatively uniform distribution of host components along the ingots.
A major advantage of YSAG over Gd3Al5−xScxO12 (GSAG) is its tunable host composition and electronic structure through Sc3+ doping. In contrast, GSAG garnet formation is restricted to compositions near the congruently melting Gd2.88Sc1.89Al3.23O12.31 As a result, prior studies22–26,28–30,32 predominantly focus on compositions close to Gd3Sc2Al3O12, where Sc3+ substitutes Al3+ almost exclusively in octahedral sites. The only reported study on Ce-doped GSAG grown by the Czochralski method, dating back to 1994,32 found a decay time of 120 ns but a relatively high light yield under γ-ray excitation—approximately 30% that of NaI:Tl. Additionally, the effect of Sc3+ doping on the energy barrier height for Ce3+ 5d1 ionization in Gd2.97Ce0.03Ga2.5Sc1Al1.5O12 (GASGG:Ce) was demonstrated in ref. 22.
The acceleration of rise and decay times in Ce-doped rare-earth garnets through the introduction of divalent alkaline-earth metal ions has been extensively studied.33–40 When alkaline-earth metals substitute Y3+ or Al3+ sites, they introduce additional negative charge into the lattice, which is compensated by the partial oxidation of Ce3+ to Ce4+. Moreover, Mg2+ ions are believed to form complexes with Ce3+/Ce4+, facilitating hole transfer to luminescence centers and thereby enhancing scintillation efficiency.18,19
Promising results in YAG:Ce have been obtained through dual Ca2+–Mg2+ codoping.3 In these studies, Ce3+ absorption intensity decreased nearly to zero, while the shortest scintillation decay time of 26 ns was observed in a sample where approximately 4% of Ce remained in the trivalent state. Based on ionic radii r(Ca2+) = 0.112 nm (8-fold coordination) and r(Mg2+) = 0.072 nm (6-fold coordination)—Ca2+ is expected to preferentially occupy dodecahedral Y3+ sites (r(Y3+) = 0.102 nm), while Mg2+ is likely to replace octahedral Al3+ sites (r(Al3+) = 0.054 nm). This selective incorporation enables precise control over defect structure and concentration in these sublattices.
Despite these advances, further reduction of the effective decay time below 26 ns has proven challenging.3 However, a decay time of 21 ns was reported for Al/Ga-substituted YAGG:Ce,Ca,41 while ultrafast decay times below 1 ns were achieved in Al/Ga-substituted GAGG:Ce,Mg (ref. 42) through a combination of bandgap engineering and divalent cation doping. These findings highlight the potential of Sc-doped YAG:Ce as an alternative to Ga doping, particularly when combined with Ca2+–Mg2+ codoping. Further investigation into the interplay between Sc3+ substitution and divalent cation doping is necessary to optimize the scintillation properties of these materials.
This work presents, for the first time, the crystal growth and characterization of Y3Al1−xScxA3O12:Ce with Sc concentrations of x = 0.05, 0.25, and 1.25. Additionally, YSAG:Ce was codoped with Ca2+ and Mg2+ to regulate the charge states of Ce ions and enhance carrier transfer to Ce3+/Ce4+ luminescence centers. All crystals were grown using the Czochralski method under a reducing Ar + CO atmosphere in tungsten (W) crucibles.
2. Experiment
2.1 Crystal growth and sample preparation
Three sets of crystals were grown by the Czochralski method from 50 mm diameter W crucibles in Ar + CO reducing gas atmosphere. Details of the Czochralski process under these conditions may be found elsewhere.7,8 The melt compositions were (Y1−y−zCe0.01CayMgz)3Al2−xScxAl3O12 (x = 0.01, 0.05, 0.25; y = 0, 0.0075, 0.01; z = 0, 0.0025), corresponding to 1 at%, 5 at%, 25 at% Sc3+ in respect to the overall content of Al3+ ions (Table 1). The first set included YSAG:Ce crystals with the three mentioned Sc3+ concentrations. The second set involved crystals with the same Sc3+ concentrations codoped with 1 at% Ca2+. In the third set, crystals with the same Sc concentrations as in the previous sets were codoped with 0.75 at% Ca2+ and 0.25 at% Mg2+, as these dopant concentrations provided the best combination of light output and decay time in YAG:Ce,Ca,Mg.3 The pulling rate during growth was 1.7 mm h−1, the crystal rotation speed was 10 rpm. The starting raw materials for growth were yttrium oxide, cerium oxide, magnesium oxide, calcium oxide, aluminum oxide, and scandium oxide of 99.99% purity, without prior solid-phase synthesis. Samples of 5 × 5 × 2 mm3 with polished sides of 5 × 5 mm2 for optical and scintillation measurements were cut from the crystals and annealed in air atmosphere for 48 hours at 1500 °C at atmospheric pressure.| Crystal | Ce, at% | Sc, at% | Ca, at% | Mg, at% |
|---|---|---|---|---|
| YAG:1Ce,1Sc | 1 | 1 | — | — |
| YAG:1Ce,5Sc | 1 | 5 | — | — |
| YAG:1Ce,25Sc | 1 | 25 | — | — |
| YAG:1Ce,1Sc,1Ca | 1 | 1 | 1 | — |
| YAG:1Ce,5Sc,1Ca | 1 | 5 | 1 | — |
| YAG:1Ce,25Sc,1Ca | 1 | 25 | 1 | — |
| YAG:1Ce,1Sc,0.75Ca,0.25Mg | 1 | 1 | 0.75 | 0.25 |
| YAG:1Ce,5Sc,0.75Ca,0.25Mg | 1 | 5 | 0.75 | 0.25 |
| YAG:1Ce,25Sc,0.75Ca,0.25Mg | 1 | 25 | 0.75 | 0.25 |
2.2 X-ray diffraction analysis
X-ray phase analysis of powders was carried out using a SmartLab SE X-ray diffractometer from Rigaku. The diffraction patterns were obtained using Cu-Kα radiation at U = 40 kV, I = 50 mA. Data analysis was performed using the Rietveld method. Further, the crystal structure of Y3Al5−xScxO12 garnet was refined using the Rietveld technique. The refinements employed the following atomic positional coordinates in the cubic unit cell: Y3+ at the 24c site (1/8, 0, 1/4), Al/Sc at the 16a and 24d sites (0, 0, 0 and x, 0, 1/4), and O at the 96 h site (x, 0, z). The profile shape and width (FWHM) were modeled using a pseudo-Voigt function and the Caglioti relationship (FWHM2 = utan2θ + vtanθ + w), while the background was approximated by linear interpolation between a set of background points with definable heights. During refinement, the following parameters were varied: zero shift, scale factor, peak width parameters (u, v, w), lattice parameters, positional coordinates, and thermal parameters. The composition was fixed at the nominal value. Refinements for each composition converged rapidly, typically within a few cycles.
2.3 Optical measurements
The absorption spectra of the samples were measured using a PerkinElmer LAMBDA 650 UV/vis spectrometer. The instrument provided with a deuterium and halogen lamp and covers the wavelength range of 190 nm to 900 nm. The light passes through a monochromator and then split into two branches. One of the monochromatic light beams, whose intensity can be tuned by means of an iris, traverses the crystal sample placed on a moving stage. The other beam is instead employed to monitor instrumentation drift during the acquisition. Both beams were focalized through a photomultiplier measuring their intensity.The photoluminescence spectra as well as the photoluminescence excitation spectra of crystals were measured with Perkin Elmer LS55 automatic spectrofluorometer. The spectrofluorometer emits a tunable monochromatic beam of light onto a sample and measures the intensity of luminescence as a function of its wavelength.
2.4 Light output and scintillation decay
The pulse-height spectra of the samples were measured using a Hamamatsu R2059 PMT, an analogue attenuator, and a DT5720 CAEN digitizer working in charge integration mode. The PMT is biased at 2500 V, providing sufficient gain to resolve the charge of single photoelectron pulses. That allows to convert the total charge of a scintillation event to photoelectrons produced. The number of photons impinging on the PMT is obtained by correcting for the average quantum efficiency of the PMT, computed as the weighted average of the scintillator's emission and the PMT's quantum efficiency. The crystals were wrapped in Teflon on all faces coupled with Rhodosil grease to the PMT window. The 137Cs (662 keV) gamma radioactive source was used for excitation of scintillations. The whole setup was enclosed in a temperature controlled black box.The scintillation decay curves were measured using a time correlated single photon counting (TCSPC) setup.43 A pulse diode laser (Pico-Quant, PDL 800-B) was used as an excitation source of an X-ray tube XRT N5084 from Hamamatsu. It generated an X-ray beam with a continuous energy spectrum between 0 and 40 keV (with a ≃10 keV mean energy). The beam was collimated on the tested sample with a brass plate, and its scintillation light was collected using a hybrid photomultiplier (HPM 100-07 Becker Hickl) working in TCSPC mode. A 420 nm long pass filter was placed in front of the hybrid photomultiplier to suppress air excitation contribution to the emission distributions. The measurements were done hitting the sample on one surface and detecting the emitted light from the same surface (reflection mode). The repetition rate of excitation pulse was 500 kHz, and the time window of registration was 1–2 μs, depending on the sample decay time. The signal of the hybrid PMT was then fed to an amplifier and timing discriminator and was then used as the stop signal of a time to digital converter, while the start was provided by an external trigger of the pulse diode laser. Scintillation time profile for each sample was mathematically described with a multi-exponential function:


3. Results
3.1 Crystal growth
Photos of some as-grown crystals are presented in the Fig. 1. The crystals had diameters of about 18 mm and lengths of up to 60 mm. The sets of Sc,Ce-codoped (Fig. 1a–c) and Sc,Ce,Ca-codoped (Fig. 1d–f) crystals contained few inclusions in the form of bubbles, while cracks were observed only in crystals with a large amount of scandium, see Fig. 1f. | ||
| Fig. 1 As grown crystals from the melts with composition: (a) YAG:1Ce,1Sc, (b) YAG:1Ce,5Sc, (c) YAG:1Ce,25Sc, (d) YAG:1Ce,1Sc,1Ca, (e) YAG:1Ce,5Sc,1Ca, (f) YAG:1Ce,25Sc,1Ca. | ||
Samples cut from the crystals before (Fig. 2a) and after annealing (Fig. 2b) under daylight and UV light are presented below. The Ca2+-containing crystals became almost colorless after annealing, while the well-known yellow-green luminescence of Ce3+-doped garnets was not observed under UV irradiation (Fig. 2a and b).
 | ||
| Fig. 2 Samples fabricated for optical and scintillation measurements (from left to right – YAG:1Ce,1Sc, YAG:1Ce,5Sc, YAG:1Ce,25Sc, YAG:1Ce,1Sc,1Ca, YAG:1Ce,5Sc,1Ca, YAG:1Ce,25Sc,1Ca) before (a) and after (b) annealing under daylight (top) and UV (bottom). | ||
Mg-codoped crystals (Fig. 3) were not homogeneous in the entire bulk, with a large number of gas inclusions, apparently due to the dissociation of magnesium oxide in a reducing atmosphere and temperature of about 2000 °C.
 | ||
| Fig. 3 As grown crystals from the melts with composition: (a) YAG:1Ce,1Sc,0.75Ca,0.25Mg, (b) YAG:1Ce,5Sc,0.75Ca,0.25Mg, (c) YAG:1Ce,25Sc,0.75Ca,0.25Mg. | ||
Nevertheless, samples from transparent parts were produced (Fig. 4). Ca2+–Mg2+-codoped crystals had no coloration, except for the 25 at% Sc sample YAG:1Ce,25Sc,0.75Ca,0.25Mg. This may be due to the high Sc3+ concentrations preventing incorporation of Mg2+ into the crystal lattice and stabilizing Ce in the trivalent state.
 | ||
| Fig. 4 Crystalline samples (from left to right – YAG:1Ce,1Sc,0.75Ca,0.25Mg, YAG:1Ce,5Sc,0.75Ca,0.25Mg, YAG:1Ce,25Sc,0.75Ca,0.25Mg) after annealing under daylight (a) and UV (b). | ||
3.2 Crystal structure and host composition
Fig. 5(a) presents the room temperature powder X-ray diffraction (PXRD) patterns of Y3Al5−xScxO12 garnet with x = 0.05, 0.25, and 1.25. The diffraction peaks in all patterns can be indexed to the cubic Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d space group, indicating a single-phase garnet structure. Notably, no impurity peaks are observed in the PXRD patterns. A shift in the peak positions (see the example of the peak around 33.5° in Fig. 5(b)) towards lower 2θ values is observed with increasing Sc content, consistent with the larger ionic radius of Sc3+ (74.5 pm) compared to Al3+ (53.5 pm). This shift suggests an increase in lattice parameter and unit cell volume with increasing Sc3+ substitution. Sc3+ content was evaluated based on the lattice parameters of 12.01 Å in Y3Al5O12 (ref. 44) and 12.271 Å in Y3Al5−xScxO12 (ref. 45) assuming a linear dependence between lattice parameters and Sc content. The Sc content in crystals is x = 0.030, 0.245, and 1.234 at corresponding x = 0.05, 0.25, 1.25 in the melt, pointing at the segregation coefficient close to unity.
d space group, indicating a single-phase garnet structure. Notably, no impurity peaks are observed in the PXRD patterns. A shift in the peak positions (see the example of the peak around 33.5° in Fig. 5(b)) towards lower 2θ values is observed with increasing Sc content, consistent with the larger ionic radius of Sc3+ (74.5 pm) compared to Al3+ (53.5 pm). This shift suggests an increase in lattice parameter and unit cell volume with increasing Sc3+ substitution. Sc3+ content was evaluated based on the lattice parameters of 12.01 Å in Y3Al5O12 (ref. 44) and 12.271 Å in Y3Al5−xScxO12 (ref. 45) assuming a linear dependence between lattice parameters and Sc content. The Sc content in crystals is x = 0.030, 0.245, and 1.234 at corresponding x = 0.05, 0.25, 1.25 in the melt, pointing at the segregation coefficient close to unity.
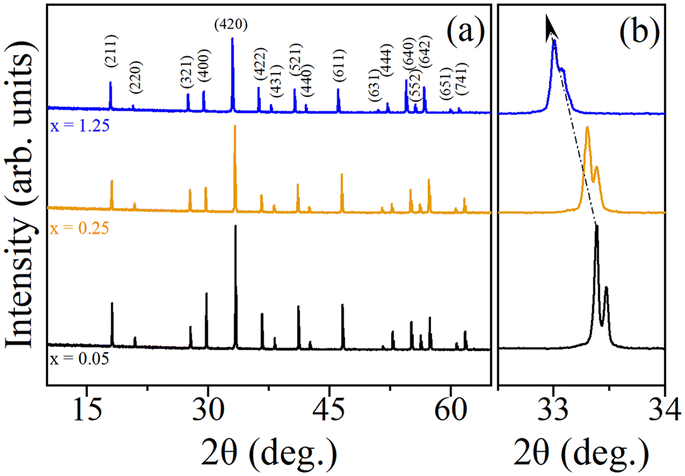 | ||
| Fig. 5 (a) Room temperature powder X-ray diffraction patterns of Y3Al5−xScxO12 garnet x = 0.05, 0.25, and 1.25. (b) Magnified view of peak shifting to low 2θ side with increasing x. | ||
The excellent fit between observed and calculated profiles is evident in Fig. S1,† displaying nearly flat difference profiles, which also confirms no impurity phases present in analyzed compositions. The refined unit cell parameters, positional coordinates, bond lengths, and agreement factors (goodness of fit, χ2) are summarized in Table S1.† These results confirm that Sc3+ substitution does not induce a structural phase transition, maintaining a monophase cubic structure in the Iad space group for all the compositions.
3.3 Optical and luminescent properties
The absorption spectra of YAG:Ce,Sc crystals (Fig. 6) contain three bands with maxima around 460 nm, 340 nm, 225 nm, attributed to 4f–5d1, 4f–5d2, 4f–5d3,4,5 transitions in trivalent cerium, respectively.1 In the absorption spectra of the YAG:Ce,Sc,Ca crystals, only one Ce3+ band at 460 nm is observed, while a broad unresolved band is also present in the UV region. The increase in the absorption intensity in the UV region in Ca2+-codoped crystals is attributed to the Ce3+ transfer into the tetravalent state and the formation of the Ce4+–O2− charge transfer state (CTS).33 The change of Ce valency is confirmed by the weakening absorption band at 460 nm associated with the 4f–5d1 transition in Ce3+. This is consistent with the discoloration of the samples (see Fig. 4). With increasing Sc concentration, the long-wavelength edge of the absorption band shifts to the blue region, both for Ca-free and Ca-containing crystals. The opposite effect, a red shift, is observed for the short-wavelength absorption bands due to a change in the crystal field strength with its subsequent influence on the splitting of cerium levels.46 | ||
| Fig. 6 Absorption spectra of YAG:Ce,Sc and YAG:Ce,Sc,Ca crystals, as well as YAG:Ce for reference. | ||
The luminescence excitation and emission spectra of YAG:Ce,Sc and YAG:Ce,Sc,Ca (Fig. 7) involve the characteristic 5d–4f Ce3+ luminescence band peaked at about 540 nm at excitation wavelengths of 350 nm and 440 nm. Sc3+ induces the blue spectral shift of the luminescence and first excitation bands. In contrast, the band peaked at about 340 nm, corresponding to the 4f–5d2 transition of Ce3+ ion, undergoes a red shift, consistently with the absorption spectra of the crystals, see Fig. 6. Similar shifts were observed in the Lu2Y(Al5−xScx)O12,27 YAGG:Ce,47 and GAGG:Ce (ref. 33) mixed garnets.
 | ||
| Fig. 7 Normalized photoluminescence excitation and emission spectra of crystals annealed in air: (a) YAG:Ce,Sc, λex = 350 nm, λem = 540 nm, (b) YAG:Ce,Sc,Ca, λex = 440 nm, λem = 540 nm. | ||
The band of unidentified nature peaked at 390 nm is observed in the excitation spectrum of the YAG:1Ce,5Sc,1Ca crystal (left red curve in Fig. 7b), which may be related to contamination with an unidentified impurity. Remarkably, the 340 nm band corresponding to the 4f–5d2 transition in Ce3+ weakens in Ca2+-containing crystals because of the increased absorption at λ < 350 nm attributed to Ce4+–O2− charge transfer complex.
The absorption and photoluminescence spectra (Fig. 8) of the third set of crystals additionally codoped with magnesium (YAG:Ce,Sc,Ca,Mg) qualitatively does not differ from the spectra of the Mg-free ones in the first two sets (see Fig. 6 and 7). No Ce3+ absorption was detected in the Ca2+–Mg2+-codoped crystals with 1 and 5 at% of scandium, alongside with the strong absorption at λ < 350 nm attributed to the Ce4+–O2− charge transfer complex (Fig. 8a). The behavior of luminescence emission and excitation curves of Ca–Mg-codoped crystals with 1 and 5 at% Sc is identical to those of the Mg-free crystals (see Fig. 7). In the luminescence excitation spectrum of the 25 at% Sc crystal, the band peaked at about 350 nm attributed to 4f–5d2 transition in Ce3+ is observed, which is typical for YAG:Ce without codoping. Also, in the excitation spectra of samples with 1 and 5 at% of scandium (left black and red curves in Fig. 7b) there is the band of unknown nature in the region of 380 nm, which may be associated with impurities or defects.
 | ||
| Fig. 8 (a) Absorption spectra of YAG:Ce,Sc,Ca,Mg crystals, (b) photoluminescence excitation spectra (left) and spectra (right) of YAG:Ce,Sc,Ca,Mg crystals annealed in air. | ||
3.4 Scintillation properties
Pulse-height spectra of YAG:Ce,Sc and YAG:Ce,Sc,Ca crystals presented in Fig. 9 display a clear trend to decrease in light output with Sc3+ concentration. The photopeak is clearly distinguishable in all the Ca-containing crystals and YAG:1Ce,1Sc, while it is difficult to determine the precise photopeak position in the rest (Fig. 9a). The values of light output are summarized in Table 2. The highest light output of 28900 photons per MeV is registered in the YAG:1Ce,1Sc crystal. This value almost coincides with the light output of the YAG:Ce crystal,1 grown from a tungsten crucible under the same conditions. The lowest light output of approximately 3900 photons per MeV was registered for YAG:1Ce,25Sc,1Ca sample.
 | ||
| Fig. 9 Pulse-height spectra of the crystals: (a) YAG:Ce,Sc, (b) YAG:Ce,Sc,Ca under gamma quanta excitation by 137Cs source with energy of 662 keV. | ||
| Sample | LO, photon per MeV | τ 1, ns | A 1, % | τ 2, ns | A 2, % | τ 3, ns | A 3, % | τ eff, ns | (τeff/LO)1/2 |
|---|---|---|---|---|---|---|---|---|---|
| YAG:0.9Ce | 28900 |
15.2 | 159.5 | 114.6 | 58.9 | 251.4 | 26.0 | 90.0 | 0.056 |
| YAG:1Ce,1Sc | 28900 |
0.3 | 0.0 | 46.5 | 42.5 | 215.9 | 57.4 | 80.4 | 0.053 |
| YAG:1Ce,5Sc | 15000 |
0.3 | 0.2 | 5.3 | 2.1 | 317.9 | 97.8 | 72.6 | 0.070 |
| YAG:1Ce,25Sc | ND | 0.2 | 0.5 | 4.0 | 3.33 | 253.8 | 96.2 | 21.7 | — |
| YAG:1Ce,1Sc,1Ca |
14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 000 000
|
1.7 | 2.9 | 13.6 | 22.1 | 53.5 | 74.9 | 21.2 | 0.039 |
| YAG:1Ce,5Sc,1Ca | 8700 | 1.4 | 3.1 | 18.5 | 29.7 | 84.3 | 67.1 | 21.4 | 0.050 |
| YAG:1Ce,25Sc,1Ca | 3900 | 1.6 | 5.1 | 15.7 | 33.7 | 72.3 | 61.2 | 16.3 | 0.065 |
| YAG:1Ce,1Sc, 0.75Ca,0.25Mg |
18000
|
2.5 | 2.7 | 16.9 | 22.9 | 54.3 | 74.4 | 25.88 | 0.038 |
| YAG:1Ce,5Sc, 0.75Ca,0.25Mg | 7660 | 2.3 | 4.6 | 20.7 | 34.9 | 84.8 | 60.5 | 22.49 | 0.054 |
| YAG:1Ce,25Sc, 0.75Ca,0.25Mg | 4000 | 1.9 | 3.2 | 16.6 | 28.0 | 72.0 | 68.8 | 23.11 | 0.076 |
The luminescence decay curves under X-ray excitation (Fig. 10) and the values of luminescence decay times given in Table 2 suggest that codoping with both Sc3+ and Ca2+ accelerate scintillation decay. Thus, a shortest τeff of 16.3 ns is achieved in the YAG:1Ce,25Sc,1Ca crystal. It should be noted that although the ultrafast subnanosecond component appears in the decay curves of the Ca-free crystals, they also have a very slow component with a lifetime of few hundred nanoseconds. Ca2+-codoping allows to suppress this slow component significantly.
 | ||
| Fig. 10 Scintillation decay curves under X-ray excitation: (a) YAG:Ce,Sc, (b) YAG:Ce,Sc,Ca. | ||
Among the samples, YAG:1Ce,25Sc and YAG:1Ce,25Sc,1Ca crystals with the addition of 25 at% Sc in solid solution demonstrated the fastest luminescence decay. The light output, however, is remarkably reduced to 14000 and 3900 photons per MeV, respectively, due to the proximity of the 5d1 levels of Ce3+ and Ce4+ to the bottom of the conduction band and subsequent thermal ionization of electrons into the conduction band from these levels. These results are in line with numerous works demonstrated the drop of light output with acceleration of luminescence decay in Ce-doped garnets, see for example.33–40 According to the pulse-height spectra of the YAG:Ce,Sc,Ca,Mg crystals (Fig. 11a), the light output decreases with increasing Sc3+ concentration, like in the two previous sets. Luminescence decay curves and corresponding luminescence decay times of Ca2+–Mg2+ codoped crystals were slightly slower than those in Mg-free crystals (Fig. 11b). Ca–Mg-codoped crystals practically do not differ from Mg-free counterparts in terms of light output, see Table 2.
 | ||
| Fig. 11 (a) Pulse-height spectra of YAG:Ce,Sc,Ca,Mg crystals under excitation by gamma quanta from 137Cs source with energy of 662 keV, (b) scintillation decay curves under X-ray excitation. | ||
Considering the combination of light output and decay time, for the evaluation of photon time density, which is proportional to coincidence time resolution,48 one may use the square root of the decay time (τeff) to the light output (LO) ratio. The lowest (τeff/LO)1/2 parameters, i.e. the best timing resolution were registered in YAG:1Ce,1Sc,1Ca and YAG:1Ce,5Sc,0.75Ca,0.25Mg (appear in bold in Table 2) where a light output of 14000–18000 photons per MeV is combined with the effective decay time of 21–26 ns, nearly reaching the target values for scintillators at future detectors at HL-LHC.1
4. Discussion
The scintillation parameters of the obtained YSAG:Ce crystals were compared with previously studied Sc-doped garnets (Table 3). Y3Al2−xScxAl3O12:Ce with a Sc content of x = 0.05 f.u. exhibited the highest light yield of 14000 photons per MeV along with a relatively fast effective decay time of 21 ns. In contrast, previously reported Sc-doped garnets (Table 3) generally demonstrated lower light yield, likely due to high Sc3+ content (1.89–2 f.u.) in them, which induces strong thermal ionization from Ce3+ 5d levels. However, unlike Ga-admixed garnets, the reduction in light yield in these works23–25,28,29,32 was not accompanied by a significant decrease in decay time. This supports the suggestion in ref. 25 that thermal quenching of Ce3+ at room temperature is not the primary cause of low light yield in the GSAG host. Instead, slow luminescence decay may be attributed to Sc-related traps.
| Material | Sc content, f.u. | Light output, photon per MeV | Decay time, ns | Production method and ref. |
|---|---|---|---|---|
| GSAG:Ce,Mg | 1.89 | 10160 |
120 | μ-PD23 |
| GYSAG:Ce,Mg | 1.9–2 | 3210 | 11.7 | μ-PD24 |
| GYSAG:Ce | 2 | 15100 |
116 (fast) | μ-PD25 |
| GSAG:Ce,Mg | 2 | 555 | 19 | LPE26 |
| Lu2Y(Al5−xScx)O12:Ce,Mg | 1–2 | ≤35% of BGO | 11–17 | LPE27 |
| GSAG:Ce,Mg | 1.89 | 2600–2800 | 94–175 (main) | FZ28 |
| GSAG:Ce | 1.89 | 10240 |
46; 184 | Bridgman29 |
| GSAG:Ce,Mg | 1.89 | 9320 | 27; 92 | Bridgman29 |
| GSAG:Ce | 2 | 30% of NaI(Tl) | 120 | Czochralski32 |
| YSAG:Ce,Ca | 0.05 | 14000 |
21.2 | Czochralski (this work) |
| YSAG:Ce,Ca,Mg | 0.05 | 18000 |
25.9 | Czochralski (this work) |
Fast decay times of 11–19 ns were observed only in LPE-grown films,26,27 likely due to a lower concentration of intrinsic defects introduced during growth.49 However, these films exhibited negligible light output. Sc3+ forms energy levels below the conduction band, reducing the electron ionization barrier from Ce3+ 5d1 levels (which shifts by 0.14 eV under heavy Sc3+ doping in GSAGG:Ce (ref. 22)). A high light yield observed in 1%-Sc-doped YSAG:Ce and YSAG:Ce,Ca in this study may be attributed to the relatively low Sc3+ content, which prevents the formation of a continuous subband merging with the conduction band and thus limits the electron ionization. Furthermore, Sc3+ situated at the dodecahedral site of the garnet lattice serves as a dominant electron trap, creating a bottleneck in the scintillation mechanism of GSAG:Ce.50 Hence, a Sc3+ introduction to the dodecahedral sites should be negligible at a low Sc3+ content in YSAG:Ce, thus decreasing the possibility of such carrier trapping. Both these factors contribute to a relatively high light output of 14000–18000 photons per MeV. This suggests that further optimization of scintillation parameters could be achieved by fine-tuning Sc and Ca concentrations in YSAG:Sc,Ce.
In contrast to YAG:Ce,Ca,Mg,3 Ca–Mg double codoping did not improve the scintillation performance of YSAG:Ce. This could be due to the competition between Mg2+ and Sc3+ ions for octahedral lattice sites, as both cations preferentially occupy these positions based on their ionic radii, thereby limiting Mg2+ incorporation into the lattice. Nevertheless, the Czochralski method appears promising for producing large Ce,Sc-codoped garnets with high optical quality and improved scintillation performance.
5. Conclusions
This study examined the impact of Sc and divalent cation (Ca2+, Mg2+) codoping on the optical and scintillation performance of YAG:Ce crystals. (Y1−x−xCe0.01CayMgz)3(Al1−xScx)2Al3O12 (x = 0.01, 0.05, 0.25; y = 0, 0.0075, 0.01; z = 0, 0.0025) crystals were grown by the Czochralski method using tungsten crucibles in a reducing Ar + CO atmosphere. Unlike gadolinium garnets, where Sc doping is typically constrained to 1.89–2 f.u.,23–26,28,29,32 we explored a broader Sc doping range of 0.05–1.25 f.u.Our results indicate that codoping with Ca2+ and Sc3+ does not compromise crystal growth stability or optical quality under these conditions. However, additional Mg2+ codoping negatively impacted crystal quality. Unlike previous studies on GASG:Ce and GASGG:Ce scintillators, which exhibit low-to-moderate light yield and slow luminescence decay, our findings suggest that low-level Sc3+ doping in YAG:Ce allows fine control over electron thermal ionization from Ce3+ 5d levels while limiting carrier trapping on Sc3+-related traps and preserving light output. Additionally, Ca2+ ions located near Ce3+ promote Ce4+ formation and enhance carrier transport to Ce3+/Ce4+ luminescence centers.
We identified an optimal codopant concentration yielding a high light output of 14000 photons per MeV and a short decay time of 21 ns in YAG:1 at%Ce, 1 at%Sc, 1 at%Ca. These parameters align with the requirements for radiation-hard inorganic scintillators in future HL-LHC detectors, where 25 ns particle collision intervals necessitate minimized pile-up. Further optimization of Sc and Ca content in YSAG:Ce,Ca may enhance scintillation performance, offering a viable alternative to Gd-containing garnets. While YSAG has a lower density (∼2 g cm−3 less than GSAG), this drawback can be mitigated in sampling calorimeters where scintillation fibers are embedded in a W absorber, ensuring most particle energy is attenuated by tungsten. Additionally, low Sc3+ content does not seriously increase crystal production cost, given the high cost of Sc2O3 raw material.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ia. Gerasymov – conceptualization, investigation, writing – original draft, writing – review & editing. S. Tkachenko – investigation, methodology. D. Kurtsev – investigation. D. Kofanov – investigation. O. Viahin – investigation, visualization, writing – review & editing. P. Maksimchuk – investigation, visualization. I. Rybalka – investigation. B. Grynyov – supervision. L. Martinazzoli – investigation, visualization. L. Roux– investigation. E. Auffray – management, writing – review & editing. A. Padmanaban – data curation. O. Sidletskiy – management, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was made in the frame of Horizon Europe ERA Widening Project no. 101078960 “TWISMA”, and supported by the Crystal Clear Collaboration and CERN EP-RD. A. Padmanaban and O. Sidletskiy acknowledge the support from the project “Centre of Excellence in Nanophotonics, Advanced Materials and Technologies Based on Crystal Growth” (ENSEMBLE3) carried out within the International Research Agendas Program of the Foundation for Polish Science and Teaming for Excellence Project under grant MAB/2020/14 and grant 857543.References
- O. Sidletskiy, Ia. Gerasymov, Y. Boyaryntseva, P. Arhipov, S. Tkachenko, O. Zelenskaya, K. Bryleva, K. Belikov, K. Lebbou, C. Dujardin, B. Buchner and B. Grynyov, Cryst. Growth Des., 2021, 21, 3063 CrossRef CAS.
- M. Nikl, V. Babin, J. A. Mares, K. Kamada, S. Kurosawa, A. Yoshikawa, J. Tous, J. Houzvicka and K. Blazek, J. Lumin., 2016, 169, 539 CrossRef CAS.
- O. Sidletskiy, Ia. Gerasymov, D. Kurtsev, O. Viahin, S. Tkachenko, D. Kofanov, S. Sadivnycha, L. Martinazzoli, L. Roux, E. Auffray, V. Kononets and K. Lebbou, Progress in fast-timing Ce-doped garnet scintillators by complex codoping with divalent cations, Mater. Res. Bull., 2025, 184, 113283 CrossRef CAS.
- E. Auffray, A. Fedorov, V. Dormenev, J. Houžvička, M. Korjik, M. T. Lucchini, V. Mechinsky and S. Ochesanu, Optical transmission damage of undoped and Ce doped Y3Al5O12 scintillation crystals under 24 GeV protons high fluence, Nucl. Instrum. Methods Phys. Res., Sect. A, 2017, 856, 7 CrossRef CAS.
- V. Dormenev, K.-T. Brinkmann, A. Borisevich, D. Kazlou, M. Korzhik, M. Moritz, R. W. Novotny, P. Orsich, Ia. Gerasymov, S. Tkachenko, P. Arhipov, O. Sidletskiy and H.-G. Zaunick, Radiation tolerant YAG: Ce scintillation crystals grown under reducing Ar + CO atmosphere, Nucl. Instrum. Methods Phys. Res., Sect. A, 2021, 1015, 165764 CrossRef CAS.
- J. Houžvička and K. Bartoš, Method for the preparation of doped garnet structure single crystals with diameters of up to 500 mm (CRYTUR, SPOL, SRO), US Pat., 9499923B2, 2016 Search PubMed.
- P. Arhipov, S. Tkachenko, S. Vasiukov, K. Hubenko, Ia. Gerasymov, V. Baumer, A. Puzan, P. Mateychenko, K. Lebbou and O. Sidletskiy, J. Cryst. Growth, 2016, 449, 104 CrossRef CAS.
- O. Sidletskiy, P. Arhipov, S. Tkachenko, Ia. Gerasymov, D. Kurtsev, V. Jary, R. Kucerkova, M. Nikl, K. Lebbou and E. Auffray, Springer Proceedings in Physics, 2019, vol. 227, p. 83 Search PubMed.
- DRD 6: Calorimetry, Proposal Team For DRD-on-Calorimetry, CERN, 2024, July 31, https://cds.cern.ch/record/2886494/files/DRD6-cdscern.pdf?version=3.
- P. Lecoq, C. Morel, J. O. Prior, D. Visvikis, S. Gundacker, E. Auffray, P. Križan, R. M. Turtos, D. Thers and E. Charbon, Roadmap toward the 10 ps time-of-flight PET challenge, Phys. Med. Biol., 2020, 65, 21RM01 CrossRef CAS PubMed.
- O. Sidletskiy, P. Arhipov, S. Tkachenko, O. Zelenskaya, S. Vasyukov, F. Moretti and C. Dujardin, Phys. Status Solidi A, 2018, 215, 1800122 CrossRef.
- M. Moszynski, T. Ludziejewski, D. Wolski, W. Klamra and L. O. Norlin, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 345, 461 CrossRef CAS.
- E. Mihokova, M. Nikl, J. A. Mares, A. Beitlerova, A. Vedda, K. Nejezchleb, K. Blazek and C. D'Ambrosio, J. Lumin., 2007, 126, 77 CrossRef CAS.
- C. Parkes and R. Lindner, Framework TDR for the LHCb Upgrade II: Opportunities in flavour physics, and beyond, in the HL-LHC era, CERN-LHCC-2021-012; LHCB-TDR-023.
- M. M. Kuklja, Defects in yttrium aluminium perovskite and garnet crystals: atomistic study, J. Phys.: Condens. Matter, 2000, 12, 2953 CrossRef CAS.
- S. Jiang, T. Lu and J. Chen, Ab initio study the effects of Si and Mg dopants on point defects and Y diffusion in YAG, Comput. Mater. Sci., 2013, 69, 261 CrossRef CAS.
- J. Zhu, O. Sidletskiy, Y. Boyarintseva and B. Grynyov, Opt. Mater., 2021, 111, 110561 CrossRef CAS.
- L. Jia, J. Zhu, Y. Boyarintseva, Ia. Gerasymov, B. Grynyov and O. Sidletskiy, Phys. Status Solidi B, 2021, 258(12), 2100325 CrossRef CAS.
- L. Jia, M. Gu, J. Zhu, B. Grynyov and O. Sidletskiy, Influence of Carbon Doping on Stability of Intrinsic Defects in Y3Al5O12 and Lu3Al5O12, Phys. Status Solidi B, 2024, 2300544 CrossRef CAS.
- V. Babin, P. Bohacek, M. Nikl, L. Vasylechko and S. Zazubovich, Influence of {Ce3+ − Mg2+} pairs on the photo- and thermally stimulated luminescence of Mg2+ co-doped Gd3(Ga,Al)5O12:Ce single crystals, Opt. Mater., 2024, 154, 115691 CrossRef CAS.
- V. Babin, P. Bohacek, A. Krasnikov, M. Nikl, L. Vasylechko and S. Zazubovich, Photoluminescence optical and thermal quenching in heavily doped Gd3(Ga,Al)5O12:Ce, Mg single crystals: Dependence on the Ga3+, Ce3+, and Mg2+ concentration, J. Lumin., 2025, 277, 120945 CrossRef CAS.
- D. Spassky, N. Kozlova, E. Zabelina, V. Kasimova, N. Krutyak, A. Ukhanova, V. A. Morozov, A. V. Morozov, O. Buzanov, K. Chernenko, S. Omelkov and V. Nagirnyi, Influence of the Sc cation substituent on the structural properties and energy transfer processes in GAGG:Ce crystals, CrystEngComm, 2020, 22, 2621 RSC.
- O. Zapadlík, J. Pejchal, R. Kučerková, A. Beitlerová and M. Nikl, Composition-Engineered GSAG Garnet: Single-Crystal Host for Fast Scintillators, Cryst. Growth Des., 2021, 21, 7139 CrossRef.
- O. Zapadlík, J. Pejchal, F. Levchenko, R. Kucerkova, A. Beitlerova, V. Vanecek, K. Jurek and M. Nikl, The Ga-admixed GSAG:Ce single crystal scintillator: Composition tuning, J. Lumin., 2023, 263, 119984 CrossRef.
- O. Zapadlik, J. Pejchal, V. Babin, V. Jary, V. Vanecek, R. Kucerkova, K. Jurek, A. Beitlerova and M. Nikl, GSAG:Ce scintillator: insights from yttrium admixture, RSC Adv., 2025, 15, 2140 RSC.
- K. Wantong, W. Chewpraditkul, W. Chewpraditkul, M. Rathaiah, M. Kucera, S. Danis, A. Beitlerovac, R. Kucerkovac and M. Nikl, Optical, luminescence and scintillation characteristics of Gd3Sc2(Al3-xGax)O12: Ce,Mg (x = 0, 1, 2) single crystalline films, Opt. Mater., 2022, 134, 113240 CrossRef CAS.
- W. Chewpraditkul, K. Wantong, W. Chewpraditkul, N. Pattanaboonmee, R. Kucerkova, A. Beitlerova, M. Nikl, D. Strachotova and M. Kucera, Effects of Sc3+ admixture on luminescence and scintillation properties of Ce3+−doped Lu2Y(Al5-xScx)O12 (x = 1, 1.5, 2) garnet single-crystalline films, Opt. Mater., 2023, 145, 114417 CrossRef CAS.
- F. Zajíc, J. Pospíšil, R. Kučerková, P. Boháček and M. Nikl, Growth of GSAG:Ce scintillator by floating zone method under pressurized oxygen atmosphere, J. Cryst. Growth, 2024, 627, 127479 CrossRef.
- K. L. Hovhannesyan, M. V. Derdzyan, G. Badalyan, G. Kharatyan, J. Pejchal, M. Nikl, C. Dujardin and A. G. Petrosyan, CrystEngComm, 2024, 26, 4812 RSC.
- C. D. Brandle and R. L. Barns, Crystal Stoichiometry and Growth of Rare-earth Garnets Containing Scandium, J. Cryst. Growth, 1973, 20, 1 CrossRef CAS.
- G. B. Lutts, A. L. Denisov, E. V. Zharikov, A. I. Zagumennyi, S. N. Kozlikin, S. V. Lavrishchev and S. A. Samoylova, GSAG and YSAG: A Study on Isomorphism and Crystal Growth, Opt. Quantum Electron., 1990, 22, S269 CrossRef CAS.
- A. Kling, D. Kollewe and D. Mateika, Scintillation properties of cerium-doped gadolinium-scandium-aluminum garnets, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 346, 205 CrossRef CAS.
- M. Nikl, K. Kamada, V. Babin, J. Pejchal, K. Pilarova, E. Mihokova, A. Beitlerova, K. Bartosiewicz, Sh. Kurosawa and A. Yoshikawa, Cryst. Growth Des., 2014, 14, 4827 CrossRef CAS.
- V. Laguta, L. Havlak, V. Babin, J. Barta, J. Pejchal and M. Nikl, Materials, 2023, 12, 4488 CrossRef PubMed.
- M. Tyagi, F. Meng, M. Koschan, S. B. Donnald, H. Rothfuss and C. Melcher, J. Phys. D: Appl. Phys., 2013, 46, 475302 CrossRef.
- W. Chewpraditkul, N. Pattanaboonmee, O. Sakthong, K. Wantong, W. Chewpraditkul, A. Yoshikawa, K. Kamada, S. Kurosawa, T. Szczesniak, M. Moszynski, V. Babin and M. Nikl, Opt. Mater., 2019, 92, 181 CrossRef CAS.
- Ia. Gerasymov, S. Witkiewicz-Lukaszek, T. Zorenko, K. Bartosiewicz, Yu. Zorenko, J. Winiecki, D. Kofanov, Y. Boyarintseva, S. Tkachenko, P. Arhipov, E. Galenen, D. Kurtsev, O. Zelenskaya, V. Alekseev, K. Lebbou and O. Sidletskiy, IEEE Trans. Nucl. Sci., 2023, 70(7), 1362 CAS.
- V. Kononets, K. Lebbou, O. Sidletskiy, Yu. Zorenko, M. Lucchini, K. Pauwels and E. Auffray, Springer Proc. Phys., 2016, 200, 114 Search PubMed.
- S. Liu, J. A. Mares, X. Feng, A. Vedda, M. Fasoli, Y. Shi, H. Kou, A. Beitlerova, L. Wu, C. D'Ambrosio, Y. Pan and M. Nikl, Adv. Opt. Mater., 2016, 4, 731 CrossRef CAS.
- F. Moretti, K. Hovhannesyan, M. Derdzyan, G. A. Bizarri, E. D. Bourret, A. G. Petrosyan and C. Dujardin, ChemPhysChem, 2017, 18(5), 493 CrossRef CAS PubMed.
- O. Sidletskiy, Ia. Gerasymov, D. Kurtsev, V. Kononets, V. Pedash, O. Zelenskaya, V. Tarasov, A. Gektin, B. Grinyov, K. Lebbou, E. Auffray, V. Dormenev, A. Borisevich and M. Korjik, CrystEngComm, 2017, 19(6), 1001 RSC.
- L. Martinazzoli, S. Nargelas, P. Boháček, R. Calá, M. Dušek, J. Rohlíček, G. Tamulaitis, E. Auffray and M. Nikl, Compositional engineering of multicomponent garnet scintillators: towards an ultra-accelerated scintillation response, Mater. Adv., 2022, 3, 6842 RSC.
- F. Pagano, N. Kratochwil, I. Frank, S. Gundacker, M. Paganoni, M. Pizzichemi, M. Salomoni and E. Auffray, A new method to characterize low stopping power and ultra-fast scintillators using pulsed X-rays, Front. Phys., 2022, 10, 1021787, DOI:10.3389/fphy.2022.1021787.
- K. Yasuhiko, K. Suda, N. Ishizawa and T. Yamada, Crystal growth and properties of (Lu, Y) 3Al5O12, J. Cryst. Growth, 2004, 260, 159 CrossRef.
- T. H. Allik, C. A. Morrison, J. B. Gruber and M. R. Kokta, Crystallography, spectroscopic analysis, and lasing properties of Nd3+: Y3Sc2Al3O12, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 21 CrossRef CAS PubMed.
- P. Dorenbos, J. Lumin., 2000, 91(3), 155 CrossRef CAS.
- Yu. Zorenko, T. Zorenko, P. Malinowski, O. Sidletskiy and S. Neicheva, Luminescent properties of Y3Al5−xGaxO12:Ce crystals, J. Lumin., 2014, 156, 102 CrossRef CAS.
- S. Vinogradov, Approximations of coincidence time resolution models of scintillator detectors with leading edge discrimination, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 912, 149 CrossRef CAS.
- Yu. Zorenko, V. Gorbenko, I. Konstankevych, A. Voloshinovskii, G. Stryganyuk, V. Mikhailin, V. Kolobanov and D. Spassky, J. Lumin., 2005, 114, 85 CrossRef CAS.
- M. Nikl, J. Pejchal, J. Jezek, D. Sedmibudsky, V. Laguta, V. Babin, A. Beitlerova and R. Kucerkova, GSAG:Ce scintillator: material optimization and intrinsic bottlenecks, Mater. Adv., 2025 10.1039/d5ma00095e.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ce00373c |
| This journal is © The Royal Society of Chemistry 2025 |