
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural tuning of tetrazole-BODIPY Ag(I) coordination compounds via co-ligand addition and counterion variation†
Matthias
Schöbinger‡
 *a,
Martin
Huber‡
a,
Berthold
Stöger
b,
Christian
Hametner
a and
Peter
Weinberger
*a
*a,
Martin
Huber‡
a,
Berthold
Stöger
b,
Christian
Hametner
a and
Peter
Weinberger
*a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9, 1060, Vienna, Austria. E-mail: matthias.schoebinger@tuwien.ac.at; peter.e163.weinberger@tuwien.ac.at
bX-Ray Center, TU Wien, Getreidemarkt 9/164, 1060, Vienna, Austria
First published on 20th March 2025
Abstract
The coordination properties of a previously described fluorescence active ligand (L), consisting of a coordinating unit (1H-tetrazol-1-yl) and a fluorophore (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) derivative) towards Ag(I) were investigated. Additionally, the influence of different anions (BF4−, PF6−, PF2O2−, ClO4−, ReO4− and NO3−) and a co-ligand (CH3CN) on the crystal structure formation and intramolecular interactions of the Ag(I) coordination compounds was studied. Beside structural investigations via single crystal X-ray diffraction, bulk characterization of the coordination compounds was conducted in both solution and solid-state, including NMR (1H, 11B, 19F, 31P and 13C), ATR-IR, UV-vis and photoluminescence spectroscopy as well as PXRD. Eleven distinct coordination compounds are reported, each falling into one of four classes: the first group (I) comprises of a mononuclear complex, whereas group (II) consists of dinuclear complexes with ligand bridged metal centers (Ag(I)) and weak intermetallic interactions (∼4 Å). Group (III) likewise includes dinuclear complexes, but the bridging mode was prevented and the Ag–Ag distance was reduced (∼3.2 Å) upon the addition of a co-ligand. Group (IV), a structurally diverse category consists of coordination polymers, which in some cases show even shorter intermetallic contacts (<3.1 Å). All investigated coordination compounds exhibit photoluminescence in the solid state, with structurally dependent emission maxima distinct from those of the ligand.
Introduction
Silver (Ag), one of the three coinage metals (Cu, Ag, Au), exhibits diverse coordination behavior in its singly charged cationic state (Ag(I)), enabled by its filled-shell d10 electron configuration. The absence of any crystal field stabilization energy does not lead to a preferred coordination geometry,1 enabling coordination numbers from two to nine.2 Additionally, Ag may establish coordination bonds to N, S, and O based versatile mono- or multitopic ligand systems.3 If such multitopic ligands and/or counterions are used, not only classical (0D) coordination compounds can be formed, but also coordination polymers (CPs) like chains (1D), sheets (2D) and 3D networks can be built.4Further, Ag(I) exhibits the ability to form intermetallic (argentophilic, Ag(I)–Ag(I)) interactions at sub-van der Waals distances (∑rvdW(Ag,Ag) = 3.44 Å), either in a ligand-supported2,5,6 or unsupported7–9 manner. These interactions are comparable to hydrogen bonding in terms of binding energy (5–15 kcal mol−1), the ability to form multiple interactions and lack of strict directionality.10 The primary impact of Ag(I)–Ag(I) interactions is structural, facilitating cross-linking between monomeric units or sheets, which promotes the formation of CPs,2,7 and polynuclear metal clusters.2,10 Additionally, these interactions significantly influence the physicochemical properties of a compound or material. In particular, they can alter fluorescence emission behavior, which may even be quenched upon the insertion of additional molecules, enabling sensor applications.10,11 In some cases, partially covalent interactions can form between Ag(I) (acceptor) and an adjacent H–C (donor).12,13 Depending on the distance between donor and acceptor one can differentiate between agostic (1.8–2.3 Å) and anagostic (2.3–2.9 Å) interactions.14
Ag(I) CPs are increasingly being investigated due to their potential applications e.g., as antimicrobial agents,15–17 as novel materials,18 in catalysis,19,20 in photoluminescence (PL) applications,21–23 as sensors11,24,25 and as anion exchangers.20
Ligand systems consisting of heterocyclic aromatic azoles are frequently used when designing multidimensional CPs.13 Besides diazoles26–28 and triazoles,29 particularly tetrazoles3,7,9,30 have the ability to function as a bridging ligand through multiple N-donors. Tetrazole (tz) exists in three isomeric forms (1H-tz, 2H-tz, 5H-tz), each capable of mono- or di-functionalization (1,5-; 2,5- and 5,5-disubstituted derivatives).31 The most frequently used tz are C- or 1,5-substituted, due to their application in medicine.32 However, also as ligands for Ag(I) coordination compounds C-substituted tz2,3,9,33,34 are more often used than N-substituted tz.15,35,36 Keeping in mind that four different nitrogen atoms can independently function as a donor for the complex bond depending on the substitution pattern, tz derivatives represent a multifaceted ligand system. This versatility was first shown by Bodner37 in 1972 and Carlucci30 in 1999 by crystallizing tz bridged dimeric and 3D coordination compounds, respectively. In addition to multitopic ligands, counterions with multiple donor atoms can facilitate the formation of Ag(I) CPs (vide supra).15,16,38,39 The structural influence of the counterion on the CP is mainly dependent on its geometry and its donor properties as shown in literature.39–41
In this contribution, we report on the structural and PL properties of a novel family of Ag(I) coordination compounds formed with a recently disclosed tz-based ligand (4,4-difluoro-1,3,5,7-tetramethyl-8-[(1H-tetrazol-1-yl)methyl]-4-bora-3a,4a-diaza-s-indacene, L, Chart 1).42 The ligand carries a BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) moiety as the photoactive component, chosen for its remarkable PL characteristics, including high fluorescence quantum yields,43 strong absorption,44 excellent thermal and photochemical stability,45 and sharp fluorescence emission bands spanning the visible to near-infrared (NIR) spectrum.46
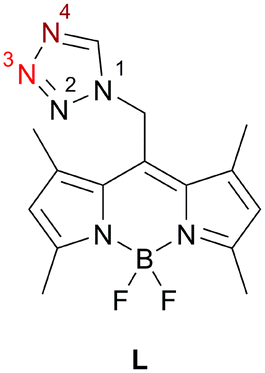 | ||
| Chart 1 Photoluminescence active ligand (L), observed coordination site for monocoordinating mode dark red (N4); for bridging mode dark (N4) and light red (N3). | ||
In order to gain greater insight into the coordination behavior of these compounds, variations of the geometry and the donor properties of the counterion (BF4−, PF6−, PF2O2−, ClO4−, ReO4− and NO3−) as well as the addition of a co-ligand (CH3CN) were performed and the resulting impact on crystal structure, intramolecular bonding (Ag(I)–Ag(I), C–H⋯Ag) and PL properties investigated.
Results and discussion
Synthesis
Ligand L was synthesized following a protocol previously established by our group.42 The synthesis of the coordination compounds 1–4, 6–8, 10 and 11 was straight forward by mixing L with the Ag(I) salt of different anions (BF4−, PF6−, PF2O2−, ClO4−, ReO4− and NO3−) in acetone (ace) or acetonitrile (CH3CN) and stirring at elevated temperature (40 °C) over night. The coordination compounds were precipitated and washed with diethyl ether (Et2O). The molar ratio L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag(I) was fixed at 2:1 for all syntheses. However, in the case of 4, 6, 10 and 11 excess ligand was removed during the workup process, since less equivalents of the ligand were needed to build these structures (Table 1). It has to be noted that unlike in 1, 3, 7 and 10 in the case of 11 the use of CH3CN as solvent did not lead to a coordination of CH3CN as co-ligand, as has already been reported in the literature.39 All syntheses were monitored by IR and NMR spectroscopy as well as powder X-ray diffraction (PXRD), whereby the shift in the tetrazolic CH signal in 1H-NMR and IR spectroscopy was used as a quick indicator of successful complexation. 5 was found as a side product during the crystallization of 4 and 6, while 9 was found as a side product during the crystallization of 8. Therefore, 5 and 9 could not be characterized using bulk analysis methods. Moreover, 6 was also found during crystallization of 4 in ace due to solvolysis of the PF6− anion.
:Ag(I) was fixed at 2:1 for all syntheses. However, in the case of 4, 6, 10 and 11 excess ligand was removed during the workup process, since less equivalents of the ligand were needed to build these structures (Table 1). It has to be noted that unlike in 1, 3, 7 and 10 in the case of 11 the use of CH3CN as solvent did not lead to a coordination of CH3CN as co-ligand, as has already been reported in the literature.39 All syntheses were monitored by IR and NMR spectroscopy as well as powder X-ray diffraction (PXRD), whereby the shift in the tetrazolic CH signal in 1H-NMR and IR spectroscopy was used as a quick indicator of successful complexation. 5 was found as a side product during the crystallization of 4 and 6, while 9 was found as a side product during the crystallization of 8. Therefore, 5 and 9 could not be characterized using bulk analysis methods. Moreover, 6 was also found during crystallization of 4 in ace due to solvolysis of the PF6− anion.
| Counterion | Compound | Formula | Nuclearity or dimensionality | Crystallographically independent Ag-atoms | Coordination number | Coordination geometry of Ag(I) | Ag environment |
|---|---|---|---|---|---|---|---|
| a — multiple – for details, see section Crystal structures. | |||||||
| BF4− | 1·2CH3CN | [Ag2L4(CH3CN)2](BF4)2·2CH3CN | Dinuclear | 1 | 4 | Pseudo tetrahedral | Ag, 2 × N4 tz, NCCH3 |
| 2·Et2O·ace | [Ag2L4](BF4)2·Et2O·ace | Dinuclear | 1 | 3 | Trigonal planar | 2 × N4 tz, N3 tz | |
| PF6− | 3·2CH3CN | [Ag2L4(CH3CN)2](PF6)2·2CH3CN | Dinuclear | 1 | 4 | Pseudo tetrahedral | Ag, 2 × N4 tz, NCCH3 |
| 4 | [AgL3](PF6) | Mononuclear | 1 | 3 | Trigonal planar | 3 × N4 tz | |
| PF2O2− | 5 | [Ag2L4(PF2O2)2] | Dinuclear | 1 | 4 | Pseudo tetrahedral | 2 × N4 tz, N3 tz, OPF2O |
| 6 | {[Ag5L4(H2O)(PF2O2)5]}∞ | 1D | 5 | 3, 4, 4, 5, 6 | —a | —a | |
| ClO4− | 7·2CH3CN | [Ag2L4(CH3CN)2](ClO4)2·2CH3CN | Dinuclear | 1 | 4 | Pseudo tetrahedral | Ag, 2 × N4 tz, NCCH3 |
| 8 | [Ag2L4(ClO4)2] | Dinuclear | 1 | 4 | Trigonal pyramidal | 2 × N4 tz, N3 tz, OClO3 | |
| 9 | {[Ag5L4(H2O)(ClO4)5]}∞ | 1D | 5 | 3, 4, 4, 5, 6 | —a | —a | |
| ReO4− | 10·CH3CN | {[AgL2(CH3CN)(ReO4)2]·CH3CN}∞ | 1D | 1 | 5 | Distorted square pyramidal | 2 × N4 tz, NCCH3, OReO3 |
| NO3− | 11 | {[Ag2L2(NO3)2]}∞ | 1D | 2 (4) | 4, 5, 5, 6 | —a | Ag, N4 tz, N3 tz, ONO3, HC |
Bulk characterization
The analytical data of bulk samples in solution (NMR (1H, 11B, 19F, 31P and 13C)) and solid state (ATR-IR, PXRD, UV-vis and PL) for the coordination compounds 1–4, 6–8, 10 and 11 are shown in the ESI.† According to PXRD, the bulk powders of the coordination compounds 1–4, 6–8, 10 and 11 are single-phase. The coordination compounds 1–3, 7 and 10 crystallize as solvates (vide infra). For these compounds, the solvate molecules are also present in the bulk. To enhance readability in this chapter, the compound numbers for these five coordination compounds will refer to their solvates (e.g.: 1 ≙ 1·2CH3CN).The main difference between the IR spectra of the uncoordinated ligand L and its Ag(I) coordination compounds is observed in the position of the νCH(tz) band. The coordination compounds 1, 2, 7 and 10 show the biggest shift from 3133 cm−1 in L42 to 3147 cm−1 after complexation. Smaller shifts of the aforementioned band to higher wavenumbers are shown by 3 and 4 (3144 cm−1), 8 (3137 cm−1) and 11 (3141 cm−1). For 6, the νCH(tz) band shift is not clear due to low intensities in the spectral region of interest. Additionally, the IR spectra of the coordination compounds show characteristic bands of each anion. For 1 and 2 only the asymmetric deformation mode of the BF4− anion is visible at 520 cm−1.47 The stretching mode of the PF6− anion in 3 and 4 appears as broad bands at 836 cm−1 and 829 cm−1, respectively. However, the bending mode of the same anion appears as sharp bands at 558 cm−1 and 556 cm−1, respectively.48 The IR spectrum of 6 displays the P–F stretching mode of the PF2O2− anion at 839 cm−1.49 The symmetric stretching mode of the ClO4− anion in 7 and 8 appears as a band at 460 cm−1 with very low intensity. Moreover, in 7 the degenerated asymmetric bending mode of the non-coordinating anion is visible at 621 cm−1. However, in 8 this mode splits up due to reduction in symmetry from Td to C3V geometry after coordination of the ClO4− anion and therefore two bands appear at 618 cm−1 and 624 cm−1.50–52 In 10, the ReO4− anion is also coordinated to an Ag-atom and therefore its ν3(Re–O) vibrational mode is split into two bands which appear at 910 cm−1 and 887 cm−1.41,53 For 11, the various coordination modes of the NO3− anion and the disorder of the metal centers do not allow a precise assignment of the observed bands at 703 cm−1 and 794 cm−1 to specific vibrational modes.
Nitrobenzene-d5 was used as NMR solvent, because it showed the best solvation ability for the coordination compounds and no coordination to Ag(I) was observed. Repeated 1H-NMR measurements showed good stability of the coordination compounds in solution over 48 h. The most prominent difference between the 1H-NMR spectra of L and the described coordination compounds is a downfield shift of the tetrazolic H-atom singlet, which has been previously reported in literature.151 and 2 exhibit a difference of 0.63 ppm from 9.19 ppm in L to 9.82 ppm after complexation. However, 3 and 4 show a smaller downfield shift of 0.53 ppm in the tetrazolic H-signal. 6, 10 and 11 show even smaller shifts in comparison to L of 0.39 ppm, 0.16 ppm and 0.11 ppm, respectively. Interestingly, the coordination compounds with ClO4− (7 and 8) as the anion exhibit different shifts, which contrasts with the compounds containing BF4− (1 and 2) and PF6− (3 and 4), where the shifts are identical for each pair. 7 exhibits a chemical shift of 9.83 ppm, but the tetrazolic H-signal of 8 is even more downfield shifted to 9.92 ppm. We attribute this to the fact that the ClO4− anion is coordinated to Ag(I) in 8. Additionally, all investigated coordination compounds show small downfield shifts in comparison to L of the bridging –CH2– group between the BODIPY core and the tz moiety. Moreover, the co-ligand (CH3CN) is also visible in 1H-NMR for 1, 3, 7 and 10 at around 2.10 ppm.
As expected, the PF6− anion of 3 and 4 shows as a septet at around −143 ppm in the 31P-NMR spectra and the PF2O2− anion of 6 shows as triplet at around −14 ppm in the 31P-NMR spectra. The 11B-NMR spectra of L and all coordination compounds exhibit a triplet at around 0.60 ppm originating from the BODIPY core in L. Additionally, the BF4− anion of 1 and 2 is visible as a singlet at around 0.37 ppm in 11B-NMR spectra. Since the anions of 1–4 and 6 contain F-atoms, the 19F-NMR spectra exhibit characteristic signals for BF4−, PF6− and PF2O2− beside the quartet around 145 ppm which originates from the BODIPY core of L.
The solid-state UV-vis spectra of the coordination compounds 1–4, 6–8, 10 and 11 closely resemble those of ligand L. The spectra exhibit a high absorption plateau between 250 nm and 580 nm, featuring several local maxima and minima, as well as a global maximum that varies only slightly for each coordination compound (Fig. S28†). Above 580 nm, the absorption decreases to a low level without any specific features. At 424 nm, L and 1–4, 6–8, 10 and 11 share a common local minimum at a high absorption level which was chosen as excitation wavelength for the PL studies. Due to high absorption levels from the ligand at wavelengths <580 nm the Ag(I)–Ag(I) interaction at around 260 nm could not be observed.35
The solid-state PL spectra of coordination compounds 1–4, 7, 8, 10 and 11 exhibit a bathochromic shift of the emission maxima to varying extents compared to the uncoordinated ligand L (Fig. 1) at room temperature. 1, 3, 7 and 8 show the smallest bathochromic shift of their emission maxima from 632 nm (uncoordinated L) to 647 nm, 645 nm, 643 nm and 641 nm, respectively. Bigger shifts of the emission maxima are displayed by 2, 4 and 10 with emission maxima of 665 nm, 659 nm and 704 nm, respectively. Besides the global emission maximum at 725 nm, 11 exhibits also a local maximum at 623 nm which is slightly hypsochromically shifted compared to the uncoordinated ligand L. 6 also shows a hypsochromic shift of its emission maximum at 621 nm (Fig. 1).
 | ||
| Fig. 1 Normalized emission spectra of 1 (black), 2 (red), 3 (blue), 4 (dark green), 6 (purple), 7 (orange), 8 (turquoise), 10 (olive), 11 (light green), L (brown) as powder sample. λexc. = 424 nm. | ||
Crystal structures
All single crystals (SC) were obtained by vapor diffusion of an antisolvent (Et2O or 2-methoxy-2-methylpropane) into saturated solutions of the coordination compounds 1–4, 6–8, 10 and 11 in CH3CN (1, 3, 7, 10 and 11) or ace (2, 4, 6 and 8) at room temperature (RT).Coordination compound 1 (Fig. 2) crystallizes in the monoclinic P21/n space group, with one crystallographically unique dinuclear complex located on a center of inversion. Two CH3CN molecules and two non-coordinating counterions (BF4−) per complex are located on the general position. Moreover, the BF4− anion is disordered about two positions with an occupancy ratio of 86.8:13.2(3), which can be interconverted by rotation along the B3–F5 axis. Each central atom is coordinated by three N-atoms, two tetrazolic N4-atoms (the italicized numbered N-atoms correspond to the tetrazole numbering system shown in Chart 1) and once by CH3CN (Fig. S1†). The bond distances for N13–Ag1, N12–Ag1 and N6–Ag1 are 2.184(2), 2.389(2) and 2.243(2) Å, respectively. The tetrazolic coordination bond distances are in line with corresponding literature examples (2.1842(16);35 2.328(5) and 2.501(6);36 2.241(5) and 2.264(5) Å15). The distance between the two Ag-atoms of the complex, which are related by inversion, is 3.2427(4) Å. This sub-van der Waals contact indicates ligand-unsupported argentophilic interactions,10 which are consistent with a comparable literature example (3.215–3.242 Å).7 Our preceding structural investigations on L have shown that the tz moiety can rotate almost freely around the N1–CH2 bond and therefore enables different conformers.42 The two crystallographically independent molecules of L in 1 are such conformers mainly differing in the orientation of the tz ring towards the FBF plane (22.9(2)° vs. 39.0(2)°).
 | ||
| Fig. 2 Structure of the coordination compound 1 (ellipsoids: 50% probability level; atom color code: grey – C, blue – N, light green – F, pink – B, light grey – Ag; H-atoms are omitted for clarity) with Ag–Ag interaction, disordered anion and crystal solvate (CH3CN). | ||
Coordination compound 2 (Fig. 3), synthesized in analogy to 1, but using a non-coordinating solvent (ace) instead of CH3CN, crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. 2 likewise shows a crystallographically unique dinuclear complex with one solvate molecule ace, one solvate molecule Et2O and two non-coordinating counterions (BF4−) per complex, which is located on a center of inversion. Moreover, the non-coordinating counterion (BF4−) is disordered in an analogous manner to 1, but with a different occupancy ratio of 75.7:24.3(13). The two Ag-atoms are bridged by two molecules of L, each with their tetrazolic N3- and N4-atom (Fig. 3 and S2†). Beside the two bridging ligands, each Ag-atom is also coordinated by another molecule of L, with its tetrazolic N4-atom. The bond distance between this monocoordinating ligand L and the Ag-atom (N12–Ag1) is rather short with 2.191(4) Å, but well within the range reported in literature.35 Interestingly, the distance between the N3-atom of the bridging ligand and the Ag-atom (N5–Ag1 2.251(4) Å) is shorter than the distance between the N4-atom and the same Ag-atom (N6(1 − x, 1 − y, −z)–Ag1 2.345(3) Å). Generally, it is assumed that the N4-atom of a tz ligand represents the preferred coordination site,37 which was already shown for Ag(I) in 1 and also in 2 for the monocoordinating ligand. Therefore, the N4–Ag(I) bond is expected to be shorter in a bridging coordination mode than the N3–Ag(I) bond.37
space group. 2 likewise shows a crystallographically unique dinuclear complex with one solvate molecule ace, one solvate molecule Et2O and two non-coordinating counterions (BF4−) per complex, which is located on a center of inversion. Moreover, the non-coordinating counterion (BF4−) is disordered in an analogous manner to 1, but with a different occupancy ratio of 75.7:24.3(13). The two Ag-atoms are bridged by two molecules of L, each with their tetrazolic N3- and N4-atom (Fig. 3 and S2†). Beside the two bridging ligands, each Ag-atom is also coordinated by another molecule of L, with its tetrazolic N4-atom. The bond distance between this monocoordinating ligand L and the Ag-atom (N12–Ag1) is rather short with 2.191(4) Å, but well within the range reported in literature.35 Interestingly, the distance between the N3-atom of the bridging ligand and the Ag-atom (N5–Ag1 2.251(4) Å) is shorter than the distance between the N4-atom and the same Ag-atom (N6(1 − x, 1 − y, −z)–Ag1 2.345(3) Å). Generally, it is assumed that the N4-atom of a tz ligand represents the preferred coordination site,37 which was already shown for Ag(I) in 1 and also in 2 for the monocoordinating ligand. Therefore, the N4–Ag(I) bond is expected to be shorter in a bridging coordination mode than the N3–Ag(I) bond.37
 | ||
| Fig. 3 Structure of the coordination compound 2 (ellipsoids: 50% probability level; atom color code: grey – C, blue – N, light green – F, pink – B, light grey – Ag, red – oxygen; H-atoms are omitted for clarity) with bridging L and crystal solvates (ace and Et2O). | ||
The bridging mode of L in 2 leads to a longer distance between the two Ag-atoms of the complex, which are related by inversion (Ag1(1 − x, 1 − y, −z)–Ag1 4.1294(5) Å), in comparison to 1. This is unexpected since short Ag(I)–Ag(I) distances are often promoted by bridging ligands.2,5 Again, there are two crystallographically independent molecules of L in 2 which are conformers. The monocoordinating ligand shows an tz/FBF angle of 42.8(5)°, whereas the bridging ligand shows an angle of 15.6(6)°. In both cases the tetrazolic N2-atom is facing towards the B-atom.
Coordination compound 3 (Fig. 4 and S3†) is structurally closely related to 1, with some noteworthy differences. First of all, the crystallographically unique Ag-atom is disordered about two positions with an occupancy ratio of 53.09:46.91(12), as well as the crystal solvate (CH3CN), where N14 and C33 are disordered with an occupancy ratio of 64.8:35.2(8). In contrast to the BF4− molecule in 1, the octahedral PF6− molecule shows no disorder in 3. The bond distances between the central atom and the tetrazolic N4-atom of the ligands L and also of the coordinating CH3CN molecule are very similar to 1 (N13–Ag1 2.223(3) Å, N12–Ag1 2.362(3) Å and N6–Ag1 2.290(3) Å). The disorder of the central atom allows up to three different Ag–Ag distances (Ag1(−x, 1 − y, 1 − z)–Ag1 2.6028(15) Å, Ag1(−x, 1 − y, 1 − z)[Ag1′(−x, 1 − y, 1 − z)]–Ag1′[Ag1] 3.0946(16) Å, Ag1′(−x, 1 − y, 1 − z)–Ag1′ 3.6073(17) Å) between the two Ag-atoms of the dinuclear complex which are related by inversion. The first two Ag–Ag distances are sub-van der Waals contacts, indicating argentophilic interactions10 like in 1. Similar to 1, there are two crystallographically independent molecules of L in 3, which are conformers (angle between FBF plane and tz: 26.8(3)° and 44.1(3)°).
 | ||
| Fig. 4 Structure of the coordination compound 3 (ellipsoids: 50% probability level; atom color code: grey – C, blue – N, light green – F, pink – B, light grey – Ag, orange – P; H-atoms are omitted for clarity) with Ag–Ag interaction, disordered metal centers and crystal solvate (CH3CN). | ||
Coordination compound 4, (Fig. S4–S6†) crystallizes in the triclinic P space group with one crystallographically unique mononuclear complex located on the general position. The anion (PF6−) is non-coordinating and also located on the general position. The coordination compound is not disordered and the crystal is free of any incorporated solvent molecules. The Ag-atom is coordinated in a pseudo trigonal planar geometry (N6–Ag1–N12 127.60(11)°; N12–Ag1–N18 140.41(12)°) by three molecules of L, each with the tetrazolic N4-atom. The BODIPY cores of two of the three ligands are located on one side of the plane which is defined by the trigonal planar geometry, whereas the third one points to the other side. The bond distances between the N4-atoms and the central atom consist of one short (Ag1–N12 2.184(4) Å) and two medium (Ag1–N6 2.305(3) Å, Ag1–N18 2.306(3) Å) long contacts. All three coordinating ligands are conformers of L, mainly differing in the orientation of their tz moieties in respect to the FBF plane (53.0(4)°, 55.9(5)° and 32.5(7)°), as it was already discussed earlier. In all three cases, the tetrazolic H-atom faces towards the B-atom.
In contrast to BF4− and PF6−, PF2O2− is acting as a coordinating anion in coordination compounds 5 and 6. 5 (Fig. 5 and S7†) is structurally related to 2, with two main differences. First, 5 crystallizes without any solvate molecules and each PF2O2− is coordinated with one of its O-atoms to one Ag(I) center. Moreover, the crystallographically unique Ag-atom is coordinated by three tetrazolic N-atoms (two N4-atoms and one N3-atom) from three molecules of ligand L. One of these ligand molecules coordinates through a single site, while the other two function as bridging ligands, like in 2. The bond distance between the monocoordinating ligand L and the Ag-atom (N7–Ag1) is longer than in 2 with 2.246(4) Å, yet still in line with literature.15 In contrast to 2, the bond distance between the Ag-atom and the tetrazolic N4-atom (N1–Ag1 2.262(3) Å) is shorter than the distance between the Ag-atom and the tetrazolic N3-atom (N2(1 − x, 2 − y, 1 − z)–Ag1 2.376(3) Å) of the bridging ligand L. This is in good agreement with the already observed coordination behavior of the tz based ligand L and is consistent with literature37 (vide supra). The distance between the two Ag-atoms of the crystallographically unique dinuclear complex, which are related by inversion, in 5 is 4.0191(10) Å, slightly shorter than in 2 (4.1294(5) Å), however it is significantly longer than in 1 (3.2427(4) Å). The distance between the coordinating O-atom of the anion and the Ag-atom (O2–Ag1 2.522(3) Å) is very similar to a literature known Ag–OPF2O bond (2.5166(1) Å).54
 | ||
| Fig. 5 Crystal packing of dinuclear complex 5 (ellipsoids: 50% probability level; atom color code: grey – C, blue – N, light green – F, pink – B, light grey – Ag, red – O, orange – P; H-atoms are omitted for clarity). | ||
In addition to the mentioned differences between 2 and 5, the spatial arrangement of the ligand L is slightly different, which is visualized in Fig. S8.† In 5, the two ligand molecules are two crystallographically independent molecules of L, which are conformers as in 2. In both cases the tetrazolic N2-atom points towards the FBF plane, but the angles between this plane and the tz plane vary: the bridging molecule of L shows 35.8(4)° and the monocoordinating ligand 24.1(4)°.
Coordination compound 6, a 1D coordination polymer, crystallizes in the triclinic P space group and extends along the crystallographic a-axis (Fig. 6). The infinite chain is symmetric only by translation. Besides five crystallographically independent Ag-atoms, the asymmetric unit consists of five anion molecules (PF2O2−), four conformers of L (two monocoordinating and two bridging ligands) and one molecule of water (Fig. S9–S11†). All those molecules are coordinated to at least one Ag-atom. Fig. 7 displays the remarkably complex connectivity graph of coordination compound 6. Before discussing the connectivity of each metal center, two significant structural properties should be noted: first, the Ag5-atom is not part of the chain itself (vide infra). Second, the P3F2O2− molecule coordinates to all metal atoms (Ag1–Ag4) of the infinite chain.
 | ||
| Fig. 6 Crystal packing of coordination polymer 6 viewed along the crystallographic b-axis (ellipsoids: 50% probability level; atom color code: grey – C, blue – N, light green – F, pink – B, light grey – Ag, red – O, orange – P; H-atoms are omitted for clarity). | ||
 | ||
| Fig. 7 Connectivity graph of coordination compound 6 (L – ligand (L), P – PF2O2−, [100] – connection through translation along the crystallographic a-axis, dotted line – short Ag–Ag contact). | ||
The Ag1-atom is coordinated by two molecules of L (L1 and L2 in Fig. 7) each with the tetrazolic N4-atom (N6–Ag1 2.221(13) Å and N12–Ag1 2.188(13) Å) and two O-atoms of two different PF2O2− molecules (P1F2O2−: O1–Ag1 2.588(13) Å and P3(−1 + x, y, z)F2O2−: O5(−1 + x, y, z)–Ag1 2.669(15) Å). Moreover, of the two neighboring Ag-atoms (Ag2–Ag1 3.8354(19) Å and Ag3–Ag1 3.351(2) Å), only the second contact is short enough to indicate intermetallic interactions. Interestingly, as already noted for 2, a bridging tz unit may be detrimental for the formation of shorter Ag–Ag distances.
Ag2 is coordinated by four O-atoms (O4(−1 + x, y, z)–Ag2 2.511(10) Å, O5(−1 + x, y, z)–Ag2 2.696(13) Å, O8–Ag2 2.354(10) Å and O9(−1 + x, y, z)–Ag2 2.517(12) Å) of four different PF2O2− molecules and one tetrazolic N3-atom (N5–Ag2 2.361(13) Å) of the ligand L (L1 in Fig. 7) bridging via N6 to Ag1. Beside the already discussed interaction of Ag2 with Ag1, Ag2 has another interaction with Ag4(−1 + x, y, z) (3.4770(18) Å). However, the distances to Ag3 and Ag5(−1 + x, y, z) are with 3.9329(18) and 4.7529(19) Å, respectively, too long for any interaction.
Ag3 is, like Ag2, coordinated by four different O-atoms (O1–Ag3 2.314(13) Å, O3–Ag3 2.367(13) Å, O5(−1 + x, y, z)–Ag3 2.366(13) Å and O7–Ag3 2.387(14) Å) and it is the only Ag-atom in 6 which is not coordinated by any L molecule. In addition to the already discussed proximity of Ag3 to Ag1 and Ag2, there are two more neighboring Ag-atoms: Ag4 and Ag5. The distance between Ag3 and Ag5 is rather long with 3.860(2) Å, however the Ag4-atom is in close proximity to Ag3 and shows the strongest intermetallic interaction in 6 with a distance of 3.1140(18) Å.
Ag4 is coordinated by four different PF2O2− molecules (O2–Ag4 2.461(15) Å, O4–Ag4 2.509(13) Å, O6–Ag4 2.421(12) Å, O9–Ag4 2.491(12) Å) and once by the tetrazolic N3-atom of a molecule of L (L4 in Fig. 7, N23–Ag4 2.385(14) Å), which bridges to Ag5 with its tetrazolic N4-atom. The remaining Ag–Ag contact of Ag4, which was not yet discussed, is the Ag4–Ag5 contact with a distance of 3.579(2) Å.
Ag5 has a coordination number of only three and therefore the smallest of all metal centers in 6. It is coordinated by two tetrazolic N4-atoms of two different molecules of L (L3 and L4 in Fig. 7, N18–Ag5 2.169(13) Å and N24–Ag5 2.163(13) Å), however, only the second one bridges to the polymeric chain (Ag4). Additionally, Ag5 is coordinated once by water (OW1–Ag5 2.689(14) Å).
All three coordination compounds containing perchlorate as anion (7–9) show structures very similar to already described coordination compounds and are not discussed in detail. 7 is isostructural to 1, but the ClO4− molecule shows no disorder in contrast to the BF4− molecule (Fig. S12†). 8 is isostructural to 5 with minor deviations in the spatial arrangement of the anions (ClO4−vs. PF2O2−) (Fig. S13†). Coordination compound 9 is isostructural to coordination polymer 6.
Coordination compound 10 crystallizes as coordination polymer in the orthorhombic space group Pnma extending along the crystallographic a-axis (Fig. 8). The infinite cation is symmetric by the 21 screw rotation in [100] direction, the m[010] reflection and the a[001] glide reflection of the Pnma space group. The asymmetric unit of 10 (Fig. S14†) consists of one Ag-atom, one molecule of L, two molecules of CH3CN and a perrhenate anion. The crystallographically unique Ag-atom is coordinated by two tetrazolic N4-atoms (N6/N6(x, 1/2 − y, z)–Ag1 2.298(5) Å) of two molecules of L in a monocoordinating fashion. Moreover, one molecule of CH3CN is coordinated to the central atom (N7–Ag1 2.248(10) Å), whereas the other one is embedded as solvate. The ReO4− counterion coordinates with its O1-atom to two symmetry related Ag-atoms: the Ag1-atom (2.520(7) Å) and the Ag1(1/2 + x, 1/2 − y, 3/2 − z)-atom (2.709(8) Å) (Fig. S15†). The ReO–Ag distances are consistent with literature (2.397(2)–2.894(3) Å).29
 | ||
| Fig. 8 Crystal packing of coordination polymer 10 with CH3CN as solvate viewed along the crystallographic c-axis (ellipsoids: 50% probability level; atom color code: grey – C, blue – N, light green – F, pink – B, light grey – Ag, red – O, petrol blue – Re; H-atoms are omitted for clarity). | ||
The ReO4− molecule is disordered about the m[010] reflection plane of the Pnma space group. Overall, the Ag1-atom has a coordination number of five with a distorted square pyramidal geometry.
11, the only coordination compound discussed in this work with NO3− as counterion, crystallizes as coordination polymer (Fig. 9) in the monoclinic P21/c space group extending along the crystallographic c-axis. The infinite chain is located on the c[010] glide reflection plane. Both crystallographically independent Ag-atoms are disordered about two positions with an occupancy ratio of 97.20:2.80(12) and coordinated by two molecules of L in a bridging mode via the tetrazolic N3 and N4-atoms (N5–Ag1 2.222(5), N12–Ag1 2.301(5), N6–Ag2 2.354(4), N11–Ag2 2.301(5)) (Fig. S16†). The distance between the major positions of the two Ag-atoms is 3.6221(7) Å, significantly shorter compared to the equivalent distance of the bridged dimers 2, 5 and 8 (>4 Å). The metal centers are also coordinated each by one NO3− molecule. In the case of Ag1, the anion molecule functions as a bidentate ligand (O1–Ag1 2.417(6) Å, O2–Ag1 2.412(5) Å). For Ag2, the case is more complicated since the N14O3− molecule coordinates once with its O4-atom (O4–Ag2 2.463(6) Å) and the N14O3− molecule, which is related by glide reflection, twice (O5(x, 3/2 − y, 1/2 + z)–Ag2 2.558(5) Å, O6(x, 3/2 − y, 1/2 + z)–Ag2 2.508(7) Å). There are two additional interesting interactions: first a close intermetallic interaction of Ag1′ with Ag2′(x, 3/2 − y, −1/2 + z) (3.06(3) Å) and second an anagostic interaction of the tetrazolic H15 with the Ag1′(x, 3/2 − y, 1/2 + z)-atom (2.44 Å) (Fig. S17†). The unit cell contains two distinct chains related by a 21 screw rotation.
 | ||
| Fig. 9 Crystal packing of coordination polymer 11 viewed along the crystallographic b-axis (ellipsoids: 50% probability level; atom color code: grey – C, blue – N, light green – F, pink – B, light grey – Ag, red – O; H-atoms are omitted for clarity). | ||
Conclusion
In this study the versatile coordination behavior of ligand L towards Ag(I) was demonstrated. In addition to the monocoordinating mode involving the tetrazolic N4-atom (1, 3, 4, 7 and 10), a bridging coordination mode utilizing both the tetrazolic N3- and N4-atoms (2, 5, 6, 8, 9 and 11) was observed. Indeed, some examples (2, 5, 6, 8 and 9) display both the monocoordinating and the bridging mode within the same coordination compound. The bridging coordination mode does not inherently result in short Ag–Ag interactions in Ag bridging scenarios (2, 5 and 8) in contrast to the literature. However, the addition of a co-ligand (CH3CN) inhibits such bridging and results in shorter intermetallic distances (1: 3.2427(4) Å and 7: 3.2213(4) Å). Even shorter Ag–Ag interactions (3.1140(18) Å and 3.06(3) Å) were found when the compounds formed CPs (6, (9) and 11). In addition to the bridging mode of L and Ag–Ag interactions, interconnecting bonds in these CPs were formed by multitopic anions (PF2O2−, ClO4−, ReO4−, NO3−).Not only do the geometric differences (Td, D3h and Oh) of the anions result in distinct structures, but geometrically identical anions (BF4−, PF2O2−, ClO4−, ReO4−) also form different structures due to variations in their donor abilities. BF4− exhibits no coordination (1, 2), while PF2O2− and ClO4− can coordinate to the metal center forming similar structures (5vs.8 and 6vs.9) and ReO4− functions as a bridging anion (10).
The strong PL properties of L in the solid state were found to be retained after complexation. However, the emission maxima of all investigated coordination compounds are significantly shifted. The differences in emission wavelength are primarily attributed to structural variations of the coordination compounds, as structurally similar coordination compounds (1, 3 and 7), possessing different anions, exhibit comparable emission maxima (647 nm, 645 nm, 643 nm).
Regarding the PL properties, future studies will focus on solid state quantum yield measurements and in-depth investigations into the mechanisms underlying the differences in emission maxima compared to the uncoordinated ligand L. To complete the structure–property relationship for these investigations, the selective bulk preparation of coordination compounds 5 and 9, which were identified as side products during crystallization, is highly important. Finally, this study highlights the potential of L as a photoluminescence-active ligand, suggesting that coordination compounds of L and its analogues42 with various metal centers could lead to greater structural diversity and new structure–property relationships.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1–11 has been deposited at the CCDC under 2409394–2409399 and 2409407–2409411.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dominik Eder and Alexey Cherevan, Institute of Materials Chemistry, TU Wien for the use of their FluoTime 300 fluorescence lifetime spectrometer. Furthermore, we thankfully acknowledge the support of Werner Artner, X-ray Center, TU Wien. This research was funded in part by the Austrian Science Fund (FWF) [10.55776/P31076]. For open access purposes, P. Weinberger has applied a CC BY public copyright license to any author accepted manuscript version arising from this submission.Notes and references
- A. P. Cote and G. K. Shimizu, Inorg. Chem., 2004, 43, 6663–6673 CAS.
- Z. Y. Shi, J. Peng, Z. Y. Zhang, X. Yu, K. Alimaje and X. Wang, Inorg. Chem. Commun., 2013, 33, 105–108 CAS.
- X. Wang, N. Li, A. Tian, J. Ying, G. Liu, H. Lin, J. Zhang and Y. Yang, Dalton Trans., 2013, 42, 14856–14865 CAS.
- A. Xie, Z. Wang, L. P. Cheng, G. G. Luo, Q. Y. Liu and D. Sun, Eur. J. Inorg. Chem., 2018, 2019, 496–501 CrossRef.
- C. S. Liu, P. Q. Chen, Z. Chany, J. J. Wang, L. F. Yan, H. W. Sun, X. H. Bu, Z. Y. Lin, Z. M. Li and S. R. Batten, Inorg. Chem. Commun., 2008, 11, 159–163 CrossRef CAS.
- Z. Y. Shi, J. Peng, X. Yu, Z. Y. Zhang, X. Wang and W. L. Zhou, Inorg. Chem. Commun., 2014, 41, 84–87 CrossRef CAS.
- D. S. Liu, F. Q. Qiu, Y. Luo, W. T. Chen and Y. Sui, J. Solid State Chem., 2022, 314, 123358 CrossRef CAS.
- M. H. H. Wurzenberger, M. S. Gruhne, M. Lommel, V. Braun, N. Szimhardt and J. Stierstorfer, Inorg. Chem., 2020, 59, 17875–17879 CrossRef CAS.
- X. M. Zhang, Y. F. Zhao, H. S. Wu, S. R. Batten and S. W. Ng, Dalton Trans., 2006, 3170–3178, 10.1039/b518052j.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- J. S. Shen, D. H. Li, M. B. Zhang, J. Zhou, H. Zhang and Y. B. Jiang, Langmuir, 2011, 27, 481–486 CrossRef CAS PubMed.
- E. Hupf, L. A. Malaspina, S. Holsten, F. Kleemiss, A. J. Edwards, J. R. Price, V. Kozich, K. Heyne, S. Mebs, S. Grabowsky and J. Beckmann, Inorg. Chem., 2019, 58, 16372–16378 CAS.
- S. Demir, M. Gür, N. Sener, I. Sener and G. Alpaslan, J. Coord. Chem., 2019, 72, 3071–3087 CAS.
- M. Brookhart, M. L. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 Search PubMed.
- T. P. Andrejević, A. M. Nikolić, B. Đ. Glišić, H. Wadepohl, S. Vojnovic, M. Zlatović, M. Petković, J. Nikodinovic-Runic, I. M. Opsenica and M. I. Djuran, Polyhedron, 2018, 154, 325–333 Search PubMed.
- Q. Gaudillat, A. Krupp, T. Zwingelstein, V. Humblot, C. Strohmann, I. Jourdain, M. Knorr and L. Viau, Dalton Trans., 2023, 52, 5859–5864 Search PubMed.
- T. V. Slenters, I. Hauser-Gerspach, A. U. Daniels and K. M. Fromm, J. Mater. Chem., 2008, 18, 5359–5362 CAS.
- J. Armstrong, P. Shea, C. C. Cornell, T. Bryson, H. E. Mason, K. D. Morrison, M. Tofanelli, J. P. Lewicki, B. C. Wood, B. F. Guilliams, W. S. Compel and C. J. Ackerson, Cell Rep. Phys. Sci., 2023, 4, 101738 CAS.
- R. M. Wang, H. Haspel, A. Pustovarenko, A. Dikhtiarenko, A. Russkikh, G. Shterk, D. Osadchii, S. Ould-Chikh, M. Ma, W. A. Smith, K. Takanabe, F. Kapteijn and J. Gascon, ACS Energy Lett., 2019, 4, 2024–2031 CAS.
- H. Fei, D. L. Rogow and S. R. Oliver, J. Am. Chem. Soc., 2010, 132, 7202–7209 CAS.
- C. J. Yang, Z. W. Zhang, J. Q. Chen, X. Y. Zhang, Y. K. Dai, X. Y. Li, Y. Y. Chen, J. Q. Xu and L. Y. Feng, New J. Chem., 2023, 47, 4472–4477 Search PubMed.
- G.-P. Yang, Y.-Y. Wang, P. Liu, A.-Y. Fu, Y.-N. Zhang, J.-C. Jin and Q.-Z. Shi, Cryst. Growth Des., 2010, 10, 1443–1450 CAS.
- M. Gutiérrez, C. Martín, B. E. Souza, M. Van der Auweraer, J. Hofkens and J.-C. Tan, Appl. Mater. Today, 2020, 21, 100817 CrossRef.
- D. H. Li, J. S. Shen, N. Chen, Y. B. Ruan and Y. B. Jiang, Chem. Commun., 2011, 47, 5900–5902 RSC.
- H. Tan and Y. Chen, Chem. Commun., 2011, 47, 12373–12375 RSC.
- L. M. T. Frija, A. J. L. Pombeiro and M. N. Kopylovich, Coord. Chem. Rev., 2016, 308, 32–55 CrossRef CAS.
- G.-G. Hou, Y. Wu, J.-P. Ma and Y.-B. Dong, CrystEngComm, 2011, 13, 6850 CAS.
- M. Maekawa, M. Munakata, T. Kuroda-Sowa, Y. Suenaga and K. Sugimoto, Inorg. Chim. Acta, 1999, 290, 153–158 CrossRef CAS.
- H. Lin, X. Wu and P. A. Maggard, Inorg. Chem., 2009, 48, 11265–11276 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani and D. M. Proserpio, Angew. Chem., Int. Ed., 1999, 38, 3488–3492 CAS.
- H. R. Meier and H. Heimgartner, in Hetarene III – Teil 4, ed. E. Schaumann, Georg Thieme Verlag, 8 edn, 1994, ch. Tetrazole, p. 664, DOI:10.1055/b-0035–110361.
- C. X. Wei, M. Bian and G. H. Gong, Molecules, 2015, 20, 5528–5553 CAS.
- J. Xiang, Y. Luo and J. S. Wu, Z. Anorg. Allg. Chem., 2013, 639, 1279–1283 CrossRef CAS.
- T. Wu, R. Zhou and D. Li, Inorg. Chem. Commun., 2006, 9, 341–345 CAS.
- W. W. Dong, J. Zhao and L. Xu, J. Solid State Chem., 2008, 181, 1149–1154 CAS.
- T.-J. Ni, Z.-J. Yang, G.-J. He, Y.-L. Wang, Q.-Q. Huang and Q.-T. Yuan, Asian J. Chem., 2015, 27, 3117–3120 CrossRef CAS.
- R. L. Bodner and A. I. Popov, Inorg. Chem., 1972, 11, 1410–1414 CrossRef CAS.
- B. Yan, M. D. Capracotta and P. A. Maggard, Inorg. Chem., 2005, 44, 6509–6511 CrossRef CAS PubMed.
- C. Q. Wan, H. J. Yan, Z. J. Wang and J. Yang, Polyhedron, 2014, 83, 116–121 CrossRef CAS.
- Z. Y. Zhang, Z. P. Deng, L. H. Huo, H. Zhao and S. Gao, Inorg. Chem., 2013, 52, 5914–5923 CrossRef CAS PubMed.
- F. Deiser, F. Kraus and H. Schmidbaur, Chem. Commun., 2015, 51, 6746–6748 RSC.
- M. Huber, M. Schöbinger, J. Cirera, B. Stöger and P. Weinberger, Eur. J. Org. Chem., 2024, e202401239, DOI:10.1002/ejoc.202401239.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- J. Banuelos, Chem. Rec., 2016, 16, 335–348 CrossRef CAS.
- B. Hinkeldey, A. Schmitt and G. Jung, ChemPhysChem, 2008, 9, 2019–2027 CrossRef CAS PubMed.
- N. Boens, B. Verbelen, M. J. Ortiz, L. J. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- R. L. Hunt and B. S. Ault, Spectrochim. Acta, Part A, 1981, 37, 63–69 CrossRef.
- A. M. Heyns, Spectrochim. Acta, Part A, 1977, 33, 315–322 CrossRef.
- R. C. Thompson and W. Reed, Inorg. Nucl. Chem. Lett., 1969, 5, 581–585 CAS.
- H. Siebert, Z. Anorg. Allg. Chem., 2004, 275, 225–240 Search PubMed.
- D. L. Lewis, E. D. Estes and D. J. Hodgson, J. Cryst. Mol. Struct., 1975, 5, 67–74 CAS.
- J.-L. Pascal and F. Favier, Coord. Chem. Rev., 1998, 178–180, 865–902 CrossRef CAS.
- M. R. Mohammad and W. F. Sherman, J. Phys. C: Solid State Phys., 1981, 14, 283–288 CrossRef CAS.
- G. Lamming, O. El-Zubir, J. Kolokotroni, C. McGurk, P. G. Waddell, M. R. Probert and A. Houlton, Inorg. Chem., 2016, 55, 9644–9652 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental protocols, crystal structure, PXRD spectra, UV/vis and PL spectra, NMR (1H, 13C, 19F, 11B, 31P) and IR spectra. CCDC 2409394–2409399 and 2409407–2409411. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00197h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |