
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural variety in calcium metal–organic frameworks with a tetratopic carboxylate ligand†
Baiwen
Zhao
a,
Guy J.
Clarkson
 a,
Jie
Liu
b,
Thi Huong
Le
cd,
Jérôme
Marrot
d,
Franck
Millange
e,
Michel
Frigoli
d and
Richard I.
Walton
*a
a,
Jie
Liu
b,
Thi Huong
Le
cd,
Jérôme
Marrot
d,
Franck
Millange
e,
Michel
Frigoli
d and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
cDepartment of Advanced Materials Science and Nanotechnology, University of Science and Technology of Hanoi (USTH), Vietnam Academy of Science and Technology (VAST), 18 Hoang Quoc Viet, Ha Noi 11307, Vietnam
dUMR CNRS 8180, UVSQ, Institut Lavoisier de Versailles, Université Paris-Saclay, 45 Avenue des Etats-Unis, 78035 Versailles Cedex, France
eDépartement de Chimie, UVSQ, Université Paris-Saclay, 45 Avenue des Etats-Unis, 78035 Versailles Cedex, France
First published on 31st March 2025
Abstract
The solvothermal synthesis of three new calcium-based metal–organic frameworks (MOFs) employing the tetratopic carboxylate ligand, 4,4′,4′′,4′′′-(naphthalene-2,3,6,7-tetrayl)tetrabenzoate (NTTB) is reported. 1: Ca(H2NTTB)(DMA)2, 2: Ca5(H2NTTB)(NTTB)2(H2O)8 and 3: Ca5(H2NTTB)(NTTB)2(H2O)5 (DMA = N,N-dimethylacetamide). These have distinct structures and compositions depending on the solvent used in synthesis. The crystal structures of the materials were elucidated through single-crystal X-ray diffraction (SC-XRD) analysis, along with a solvate of the acid form of the ligand. Sample purity was confirmed via powder XRD (PXRD), and thermal stability was assessed using thermogravimetric analysis (TGA). The resulting crystal architectures were analysed in detail with respect to the ligand binding modes, the connectivity of the calcium centres, the ligand conformation and hydrogen bonding. Notably, a solvent-dependent structural connectivity was observed: while the structures all contain six-coordinate Ca, substituting DMA with ethanol in synthesis induced a change in the MOF building units from discrete calcium nodes in 1 to infinite chains in 2 and interrupted chains in 3.
Introduction
Metal–organic frameworks (MOFs), a class of coordination polymers, are distinguished by their exceptional porosity and structural diversity.1,2 Comprising metal ions or clusters linked by organic ligands in three-dimensional networks, MOFs allow for extensive functionalisation, enabling the development of materials with precisely tailored properties. Their porosities are widely explored for potential applications in molecular capture, separation, and energy storage.3,4 Additionally, MOFs hold promise in heterogeneous catalysis, where solid-state acidity, redox activity, and unique surface properties can be leveraged.5,6 MOFs based on s-block metals, have been less studied compared to those containing transition or p-block metals, such as Cu, Fe, Al, Zn and Zr. The unpredictable coordination geometry of s-block metals, arising from their more ionic bonding interactions with carboxylate ligands, presents both challenges and opportunities for novel framework discovery and prediction.7,8 The coordination environment and resulting topology of such MOFs are highly influenced by the organic ligand and synthetic conditions.Calcium-based MOFs present several possible advantages over transition metal-based frameworks. Their strong ionic bonds can result in high thermal stability,8 while calcium's abundance (3.4% of the earth's crust), non-toxic nature, and cost-effectiveness make these frameworks sustainable and economical. Additionally, the lightweight nature of calcium offers a gravimetric advantage for gas storage and adsorption applications.9,10 Furthermore, the non-toxicity and biocompatibility of calcium-based MOFs make them promising candidates for drug delivery systems.11
The structure of a MOF is largely determined by the geometry and connectivity of its metal secondary building units (SBUs) and organic linkers. In calcium-based MOFs, calcium typically exhibits higher coordination numbers (6, 7 or 8) compared to most transition metal cations, owing to its larger ionic size. Calcium is commonly coordinated to oxygen atoms from carboxylate or phosphonate linkers, solvent molecules, or nitrogen atoms from organic ligands. SBUs in Ca-MOFs exhibit significant diversity. These include isolated octahedral units like [CaO6], where calcium is coordinated to a combination of carboxylates and phosphates, as demonstrated by Janczak et al.12 Another example is the CaO4N4 SBU, featuring a calcium centre coordinated to oxygen and nitrogen atoms from carboxylate and nitrogen-containing ligands.13 The SBUs in Ca-MOFs also frequently consist of multinuclear clusters or 1D Ca–O chains. For example, dimeric SBUs involve two calcium centres coordinated to ligands and terminal water molecules,14 while trimeric15 or tetrameric16 clusters are interconnected by carboxylates and bridging oxygens. Notably, 1D chains are a recurring feature in Ca MOFs, often stabilised by carboxylate ligands and bridging solvents, as seen in an ultra-microporous framework constructed with squaric acid, where each calcium centre is 8-coordinated.17
Tetratopic linkers with carboxylate arms exhibiting significant rotational freedom have been shown to produce MOFs with diverse topologies. For example, a series of copper-based MOFs with the formula [Cu2(L)(H2O)2] (L = tetracarboxylate linker) were synthesised by coordinating the tetracarboxylate linkers with varying extendibility to dicopper SBUs, resulting in distinct porosities.18,19 Similarly, research has focused on Zr-MOFs constructed from tetratopic linkers and Zr6 SBUs.20–22 Notably, substitutional modifications to the backbone of 1,2,4,5-tetrakis(4-carboxyphenyl)benzene (TCPB) linker to influence linker conformation, resulted in Zr-MOFs with varied topologies and porosities under same reaction conditions.23 Additionally, the linker H4TCPB–Br2 has been used to synthesise Zr-based MOFs with four distinct topologies.24 These structural variations have been ascribed to geometrical adjustments of the linker influenced by different combinations of solvents, such as N,N-dimethylformamide (DMF) and N,N-diethylformamide (DEF), and modulators, including formic acid and acetic acid, used during synthesis. These findings underscore the critical role of linker flexibility and conformation in determining the structural diversity of MOFs.
Calcium-based MOFs incorporating tetracarboxylate linkers have also attracted interest due to their structural diversity.8 Notable examples include Ca(H2TCPB) composed of isolated [CaO6] octahedra connected by a half-deprotonated TCPB linkers;25 and Ca2(AZBZ)(H2O)(DMF) (AZBZ = 3,3′,5,5′-azobenzenetetracarboxylate) featuring Ca tetramers with 1-D channels;26 and Ca2(BIPA-TC)(DMF)4 (BIPA-TC = 5,5′-(1,3,6,8 tetraoxobenzo[lmn][3,8] phenanthroline-2-7-diyl)bis-1,3-benzene dicarboxylate), which consists of infinite Ca chains.27 Despite these advances, the synthesis of calcium-based MOFs using tetratopic carboxylate ligands remains relatively under-investigated compared to Cu- and Zr-based systems, highlighting a significant opportunity for further exploration.
In this work, we investigate the synthesis and structural characterisation of Ca-MOFs using the tetratopic carboxylic acid ligand 4,4′,4′′,4′′′-(naphthalene-2,3,6,7-tetrayl)tetrabenzoic acid (H4NTTB) (Fig. 1) previously used to prepare a covalent organic framework, but to our knowledge not yet used in the crystallisation of MOFs.28 We report the structures of three novel materials, which emphasise the relationship between coordination modes, framework structures, and synthetic conditions, contributing to the advancement of s-block MOF chemistry.
 | ||
| Fig. 1 Molecular structure of 4,4′,4′′,4′′′-(naphthalene-2,3,6,7-tetrayl)tetrabenzoic acid (H4NTTB). | ||
Results
Synthesis and structure of Ca(H2NTTB)(DMA)2 (1)
Compound 1, Ca(H2NTTB)(DMA)2, was synthesised via a solvothermal method using a solvent mixture of N,N-dimethylacetamide (DMA) and water in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. Calcium nitrate was employed as the calcium precursor, with a molar ratio of 2:1 between the precursor and the ligand, H4NTTB. The synthesis was conducted at 125 °C for 6 hours.
:1 volume ratio. Calcium nitrate was employed as the calcium precursor, with a molar ratio of 2:1 between the precursor and the ligand, H4NTTB. The synthesis was conducted at 125 °C for 6 hours.
Compound 1 crystallises in the monoclinic space group C2/c. The structure consists of mononuclear SBUs featuring six-coordinate calcium centres. Each calcium centre is coordinated in an octahedral geometry, forming [CaO6] polyhedra that act as 4-connected nodes with the trans position occupied by two DMA molecules. The [CaO6] octahedron is slightly distorted, as evidenced by the variation in Ca–O bond distances. The bond length for calcium coordinated to DMA (2.30 Å) is slightly shorter than that for calcium coordinated to carboxylate groups (2.32 Å), which might be due to steric effects. The asymmetric unit consists of half of the H2NTTB2− ligand. The complete ligand is generated by a twofold rotation axis parallel to the b-axis at coordinates (0, y, 1/4), combined with translations along the a and c axes, corresponding to the operation (1 − x, y, 1.5 − z). The single Ca SBUs are linked by H2NTTB2− ligands, with two DMA molecules from the solvent coordinating to each calcium centre, as depicted in Fig. 2a.
 | ||
| Fig. 2 Crystal structure of compound 1, Ca(H2NTTB)(DMA)2. (a) Coordination environment of the [CaO6] centre (blue). (b) Overall framework structure with isolated Ca2+ ions depicted as blue polyhedra, viewed along the crystallographic a-axis. (c) Framework structure viewed along the crystallographic b-axis. (d) Framework structure viewed along the crystallographic c-axis, highlighting the presence of channels within the framework. The channel width is approximately 17.5 Å, measured as the atom-to-atom distance. | ||
The framework, viewed along the c-axis (Fig. 2d), reveals well-defined 1-D channels with a width of 17.5 Å, measured as the atom-to-atom distance, which can potentially accommodate guest molecules or ions. These voids are filled with coordinated DMA molecules. The coordination mode of the H2NTTB2− ligand is shown in Fig. 3a. The tetratopic ligands bind to the Ca centre in an η1 mode. Two of the four carboxylic groups (one crystallographically distinct carboxylic group since the ligand is symmetrical) are deprotonated and directly coordinated to calcium, contributing to the formation of the Ca–O clusters. The remaining two carboxylic groups, located on neighbouring phenyl rings (those at positions 2,3), remain protonated as carboxylic acid groups, stabilising the framework, Fig. 3a. Evidence for the protonation of two of the carboxylic groups comes from analysis of possible hydrogen bonding interactions: the distance O2⋯O4 between neighbouring ligand molecules is 2.45 Å within the reasonable range of hydrogen bonded distances found in carboxylic acids.29 Furthermore, the intramolecular distances C16–O2 = 1.26 Å, and C17–O4 = 1.28 Å reveal that the proton is likely present on O4, such that the two equivalent O3–C17–O4 groups are carboxylic while the O1–C16–O2 are carboxylate.
 | ||
| Fig. 3 (a) The coordination mode of H2NTTB2− in compound 1. (b) Definition of the dihedral angle φ between the planes of the carboxylphenyl rings on the naphthalene ring for the acid form of the ligand. | ||
The geometry of the ligand within the MOF structure can be described by the dihedral angle φ between the planes of the carboxylphenyl rings attached at adjacent positions on the naphthalene ring (i.e. at positions 2,3 and at 6,7) (Fig. 3b). In the crystal structure of the ligand (crystallised as a solvate with dimethyl sulfoxide (DMSO) via intermolecular hydrogen bonding: see Fig. S1, ESI†), the dihedral angle between the carboxylphenyl rings is 64.97°. In comparison, within the crystal structure of compound 1, the dihedral angles are reduced to 52.31° for the deprotonated carboxylphenyl groups and 55.13° for the protonated carboxylphenyl groups, reflecting the influence of ligand and framework constraints necessary to connect to the calcium ions and establish the octahedral Ca centres.
Powder XRD analysis of compound 1 (Fig. 4a) confirmed the material's phase purity. A Pawley fit was applied to the powder pattern using the crystal structure determined from single-crystal XRD, demonstrating excellent agreement with the bulk sample. The observed preferred orientation in the PXRD pattern (Fig. S2†) is attributed to the highly crystalline nature of the material. The refined lattice parameters are shown in Table S1.† TGA (Fig. 4b) in air of compound 1 showed an initial mass loss of 28% up to 300 °C, attributed to the removal of coordinated DMA in Ca(H2NTTB)(DMA)2 and adsorbed species in the channels and surface such as moisture and volatile gases, exceeding the expected 18% from the single crystal structure which may be due to extra disordered solvent not located crystallographically, or surface solvent. At approximately 350 °C, the thermal decomposition of the organic linker results in a total mass loss of 61%, in agreement with the theoretical value expected for the formation of CaCO3.30 Above 600 °C, the decomposition of CaCO3 yields CaO as the final residue.
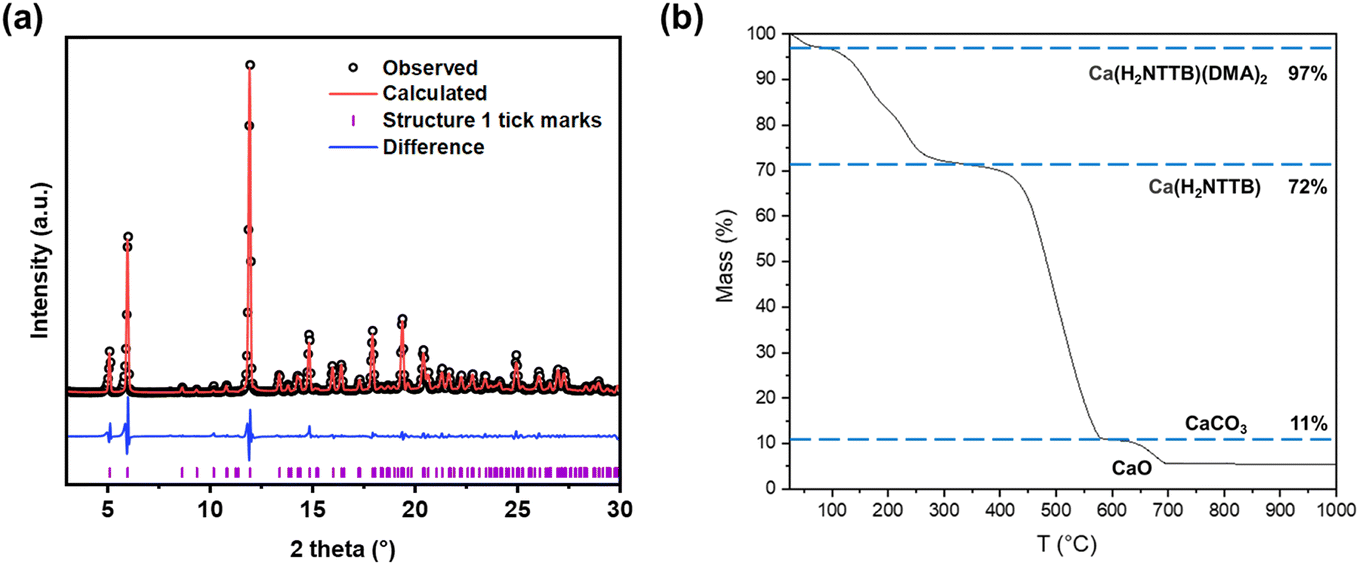 | ||
| Fig. 4 (a) Pawley fit of powder XRD pattern of 1 (Ca(H2NTTB)(DMA)2). The data points are shown in black, the fitted pattern in red, the difference curve in blue and the allowed tick positions in purple. The refined lattice parameters are shown in Table S1.† (b) Thermogravimetric analysis of compound 1 in air. | ||
Synthesis and structure of Ca5(H2NTTB)(NTTB)2(H2O)8 (2)
In comparison to compound 1, the synthesis of compound 2 used calcium chloride as the metal precursor instead of nitrate salt, with the solvent system replaced by a mixture of ethanol and water in a ∼10:1 volume ratio. The reaction was carried out at 130 °C and extended to 18 hours.
Single crystal X-ray analysis revealed that compound 2 crystallises in a triclinic symmetry, with the space group as P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and chemical formula Ca5(H2NTTB)(NTTB)2(H2O)8. The crystal structure features calcium atoms arranged in an infinite chain-like motif, as shown in Fig. 5a. These parallel Ca chains are interconnected and bridged by H2NTTB2− and NTTB4− ligands. A simplified wireframe representation of the structure is provided in Fig. 5d. Viewed along the b-axis (Fig. 5b), the structure exhibits channels with a diameter of 10.6 Å in atom-to-atom distance, between the calcium chains within the framework.
and chemical formula Ca5(H2NTTB)(NTTB)2(H2O)8. The crystal structure features calcium atoms arranged in an infinite chain-like motif, as shown in Fig. 5a. These parallel Ca chains are interconnected and bridged by H2NTTB2− and NTTB4− ligands. A simplified wireframe representation of the structure is provided in Fig. 5d. Viewed along the b-axis (Fig. 5b), the structure exhibits channels with a diameter of 10.6 Å in atom-to-atom distance, between the calcium chains within the framework.
 | ||
| Fig. 5 Crystal structure of compound 2, Ca5(H2NTTB)(NTTB)2(H2O)8, viewed along a (a), b (b) and c (c) crystallographic axes. (d) Shows the simplified wireframe structure viewing along a axis. | ||
In Fig. 6, the building units of the Ca chain within the crystal structure compound 2 are depicted, revealing the arrangement and coordination of calcium atoms in the Ca chain with Ca–O–Ca connectivity. To simplify visualisation, Fig. 6a omits bonded water molecules, focusing instead on the bridging carboxylate oxygens.
 | ||
| Fig. 6 (a) Local coordination of the Ca chain in compound 2. (b) Coordination mode of the fully deprotonated NTTB4− ligand. (c) Coordination mode of the partially deprotonated H2NTTB2− ligand. | ||
The asymmetric unit consists of a [Ca2(H2O)2]4+ dimer at the core (Fig. 6a), formed by two six-coordinate calcium atoms (Ca1) bridged by two water molecules. This water-bridging motif has been previously observed in other Ca MOFs, such as Ca-BTC (BTC = 1,3,5-benzenetricarboxylate).31 The dimer is further stabilised by intramolecular H-bonding interactions.32 Specifically, the bridging water molecules form H-bonds with neighbouring carboxylate oxygen atoms, with an O4⋯O10 distance of 2.72 Å. Each Ca1 is further connected to a six-coordinate calcium atom (Ca3) through two carboxylate groups. The Ca3 atoms coordinate to two water molecules, which engage in potential intramolecular hydrogen bonding with adjacent carboxylate groups, with O2⋯O15 and O1⋯O20 distances of 2.85 Å and 2.97 Å, respectively. This completes the six-coordinate geometry of Ca3. Additionally, each Ca3 connects to a distinct six-coordinate calcium atom (Ca2) via three carboxylate groups. The repeating sequence of the building unit within the chain can be described as Ca3–Ca1–Ca1–Ca3–Ca2, forming the continuous chain structure in the crystal lattice of compound 2.
Fig. 6b and c illustrate the coordination of two distinct types of carboxylate ligands to the calcium chain, each contributing unique geometric and binding characteristics to the structure. One linker, depicted in Fig. 6c, is half-deprotonated, with its carboxylate groups adopting a μ2-η1:η1 binding mode. The remaining protonated carboxylic acid groups do not participate in coordination but instead form intermolecular hydrogen bonds with uncoordinated water molecules in the channels. The dihedral angle between the planes of the carboxylphenyl rings attached at adjacent positions on the naphthalene core is 56.75°, compared to 64.97° in the acid form of the ligand (DMSO solvate). The other type of linker, NTTB4−, shown in Fig. 6b, is fully deprotonated and exhibit asymmetric coordination modes to the calcium chains. One carboxylate adopts a μ3-η1:η2 bridging mode, linking three calcium centres. Another carboxylate shows an η2 chelating mode, while two additional carboxylates coordinate through μ2-η1:η1 binding modes. The dihedral angle between the carboxylphenyl planes in the linker with μ3-η1:η2 and μ2-η1:η1 modes is 55.19°, while the angle between the carboxylphenyl planes in the linker with η2 and μ2-η1:η1 modes is 57.45°. These subtle variations in dihedral angles suggest that coordination interactions and framework constraints induce slight adjustments in ligand geometry.
Powder XRD analysis of compound 2 (Fig. 7a) confirmed the material's phase purity. A Pawley fit was made to the pattern using the crystal structure determined from single-crystal X-ray diffraction, demonstrating excellent agreement with the bulk sample. Refined lattice parameters are summarised in Table S1.† The observed preferred orientation in the PXRD pattern (Fig. S3†) is attributed to the highly crystalline nature of the material.
 | ||
| Fig. 7 (a) Pawley fit of powder XRD patterns of as-made sample of compound 2 (Ca5(H2NTTB)(NTTB)2(H2O)8). The data points are shown in black, the fitted pattern in red, the difference curve in blue and the allowed tick positions in purple. The refined lattice parameters are shown in Table S1.† (b) Thermogravimetric analysis of compound 2 in air. | ||
The TG curve of compound 2 measured in air is presented in Fig. 7b, indicating that the framework is stable in air up to 300 °C. An initial mass loss is observed between 25 °C and 100 °C, likely due to the desorption water. Between 100 °C and 300 °C, further mass loss occurs, corresponding to the removal of coordinated water molecules. Above 300 °C, the TG curve reveals a two-step decomposition of the NTTB ligands, consistent with an expected total mass loss of approximately 60%. The observed mass losses occur in a ratio of approximately 1:2, suggesting sequential decomposition, where the half-deprotonated H2NTTB2− ligand decomposes first, followed by the fully deprotonated form. The residual components decompose further to form CaCO3, which ultimately decomposes to CaO at 650 °C. FTIR spectra (Fig. S9†) indicate differences in carboxylate binding modes between compounds 1 and 2. The bands for compound 2 are shifted to higher wavenumbers compared to 1, suggesting changes in the carboxylate environment. This shift, along with a more complex band structure, is consistent with the unsymmetrical binding mode of the ligands in compound 2.
Synthesis and structure of Ca5(H2NTTB)(NTTB)2(H2O)5 (3)
The synthesis of compound 3 used the same ethanol/water solvent system as compound 2. However, 1 M HCl was added to adjust the pH of the reaction mixture to approximately 2. Additionally, the reaction temperature was reduced to 110 °C, and the reaction time was extended to 48 hours.The crystal structure of compound 3 is shown in Fig. 8, with the chemical formula Ca5(H2NTTB)(NTTB)2(H2O)5. The material crystallises in the triclinic system, with space group P. The calcium coordination structure in compound 3 deviates from the continuous chain arrangement in compound 2, exhibiting a ‘disrupted’ chain configuration. As shown in Fig. 8a, this motif consists of a sequence of four-calcium centres interconnected via oxygen atoms from carboxylate groups, forming an associated unit with Ca–O–Ca connectivity. In contrast, a single, spatially separated calcium cation is connected to this four-calcium unit through two carboxylate oxygen bridges via μ2-η1:η1 coordination mode. This isolated calcium cation is further coordinated to two water molecules, completing its coordination environment. The disrupted chain configuration creates a distinctly different framework structure.
 | ||
| Fig. 8 Crystal structure of compound 3, Ca5(H2NTTB)(NTTB)2(H2O)5. (a): [(Ca5)10+] building unit. Structure viewed along a (b), b (c) and c (d) crystallographic axes. | ||
Fig. 9a shows the coordination unit of the calcium centres through carboxylate oxygen bridges from the NTTB ligands. Each calcium atom exhibits a six-coordinated environment, with Ca–O bond distances ranging from 2.27 to 2.53 Å. Within this unit, the single cation unit Ca3 is capped by two molecules of water. These water molecules engage in H-bonding interactions with carboxylate groups in the framework, as indicated by O3A–O135 and O3B–O227 distances of 2.80 Å and 2.82 Å, respectively. Ca1, Ca2 and Ca5 each coordinate to one water molecule, which serves as a hydrogen bonding donor, interacting with multiple neighbouring carboxylate groups. The O⋯O distances range from 2.90 Å to 3.40 Å, suggesting weak H-bonding interactions. These water molecules are oriented toward the void spaces within the framework and could therefore potentially be removed to create open metal sites on the Ca ions.
 | ||
| Fig. 9 (a): Coordination unit of the disrupted Ca chain in compound 3. (b) and (c): Coordination mode of the fully deprotonated NTTB4− ligand. (d): Coordination mode of the partially deprotonated H2NTTB2− ligand. | ||
This structure incorporates three distinct ligand types, each displaying unique coordination modes. The NTTB4− ligand, as shown in Fig. 9b, adopts four different carboxylate binding modes with calcium centres: η1 mono-donor coordination, μ2-η1:η1 bridging, μ3-η1:η2 bridging, and μ2-η2:η1 chelating and bridging. Notably, the latter coordination mode is exclusive to this material and not observed in the other two structures. The dihedral angles between the carboxylphenyl planes in the linker vary depending on the binding modes: 60.73° for the μ3-η1:η2 and μ2-η2:η1 modes, and 59.62° for the η1 and μ2-η1:η1 modes. Another ligand, depicted in Fig. 9c, adopts a centrosymmetric coordination mode involving μ2-η1:η1 bridging and μ2-η2:η1 chelating and bridging modes, with a dihedral angle of 51.53° between the carboxylphenyl planes. This value represents a significant deviation from the pristine H4NTTB ligand's dihedral angle of 64.97°, highlighting the structural constraints imposed by the ‘disrupted’ chain configuration. Lastly, the partially deprotonated H2NTTB2− ligand, shown in Fig. 9d, features μ2-η1:η1 and η1 binding modes, while retaining uncoordinated protonated oxygens (O20). These uncoordinated oxygens engage in hydrogen bonds linking the nearby carboxylate groups, with an O20⋯O301 distance of 3.07 Å. The dihedral angle between the carboxylphenyl planes in this ligand is 54.39°.
The phase purity of compound 3 was determined through PXRD analysis (Fig. 10a). Pawley refinement against the PXRD data, based on the single-crystal X-ray structure, showed strong consistency in lattice parameters with the bulk material, confirming its crystalline uniformity. Refined lattice parameters are summarised in Table S1.† The FTIR spectrum (Fig. S9†) of compound 3 is similar to compound 2, consistent with their similar carboxylate binding modes. The TG curve (Fig. 10b) of the as-synthesised compound 3 is similar to that of compound 2. Decomposition begins at approximately 300 °C, displaying a two-step ligand decomposition in a mass loss ratio of approximately 1:2. This indicates the sequential decomposition: the half-deprotonated H2NTTB2− ligand decomposes first, followed by the fully deprotonated NTTB4− ligand. Notably, the total mass loss attributed to ligand decomposition is 61%, lower than the theoretical value of 74% calculated for the idealised chemical formula. This discrepancy is likely due to missing ligand defects,33 which is commonly observed in other MOFs, such as UiO-66,34 where structural imperfections can exist while maintaining the overall framework integrity.
 | ||
| Fig. 10 (a) Pawley fit of powder XRD patterns of as-made sample of compound 3 (Ca5(H2NTTB)(NTTB)2(H2O)5). The data points are shown in black, the fitted pattern in red, the difference curve in blue and the allowed tick positions in purple. (b) Thermogravimetric analysis in air of compound 3. | ||
Discussion
The structural diversity of calcium-based MOFs incorporating tetracarboxylate linkers is largely influenced by the coordination modes of calcium centres and linker conformation as well as the synthesis conditions. Table 1 summarises previously reported Ca-MOFs with tetracarboxylate ligands, showing variations in SBU types and dimensionality. The molecular structures of the corresponding linkers are shown in Fig. S10.†| Formula | SBU type | Dimensionality | Ref. |
|---|---|---|---|
| [Ca2(MDIP)(H2O)4]·CH3OH·4H2O | Extended chain | 2D | 35 |
| [Ca2(H2SBF-TC)(DMF)2]·2DMF | [Ca4O24] tetramer | 3D | 36 |
| [Ca3(HATPTC)2(DMF)5] | [Ca3O18] trimer | 2D | 37 |
| Ca(H2PZTC)(H2O)3 | Isolated [CaO7N2] | 1D | 38 |
| [Ca2(BIPA-TC)(DMF)4]·2DMF | Extended chain | 3D | 27 |
| [Ca2(EDDA)(H2O)2(DMA)]·3H2O | [Ca2O12] dimer | 3D | 39 |
| Ca2(EDDA)(H2O)(DMF) | [Ca4O26] tetramer | 3D | 26 |
| [Ca2(DBBD)(H2O)2(DMF)]·2DMF·H2O | [Ca2O12] dimer | 3D | 39 |
| Ca2(QPDC) | Extended chain | 3D | 40 |
| Ca(H2TCPB) | Isolated [CaO6] | 3D | 10 |
| Ca3(HTCPP)2 | Extended chain | 3D | 41 |
| [Ca(H4BDCPO)(DMA)2]·2DMA | Isolated [CaO6] | 2D | 42 |
| Ca(H2NTTB)(DMA)2 | Isolated [CaO6] | 3D | This work |
| Ca5(H2NTTB)(NTTB)2(H2O)8 | Extended chain | 3D | This work |
| Ca5(H2NTTB)(NTTB)2(H2O)5 | Disrupted chain | 3D | This work |
Most Ca-MOFs with tetracarboxylate linkers feature polynuclear clusters or extended chain structures, where oxygen atoms are frequently shared between calcium cations rather than forming isolated SBUs. Examples include tetrameric Ca4O24 SBUs in [Ca2(H2SBF-TC)(DMF)2]·2DMF,36 trimeric Ca3O18 units in Ca3(HATPTC)2(DMF)5,37 and infinite calcium chains in Ca2(QPDC)40 and Ca3(HTCPP)2.41 Binuclear Ca2O12 motifs are also seen in Ca2(DBBD)(H2O)2(DMF) and Ca2(EDDA)(H2O)2(DMA).39 In these structures, bridging or coordination water to calcium cations are often observed, likely due to the high hydration energy of the Ca2+.43 Notably, isolated [CaO6] units are rare and have only been achieved under specific solvent conditions. To the best of our knowledge, mononuclear [CaO6] is only observed in Ca(H2TCPB) when ethanol is used as the sole solvent, without water. Additionally, in [Ca(H4BDCPO)(DMA)2]·2DMA, the [CaO6] unit is stabilised by coordination with two DMA molecules, a coordination mode that is also observed in compound 1.
Ca MOFs incorporating linkers with phenylcarboxylate groups exhibit significant conformational flexibility, where variations in the dihedral angles of the adjacent phenylcarboxylate planes influence metal coordination and overall assembly. Replacing H4TCPB10 (benzene core) with H4TCPP41 (pyrazine core) results in distinct frameworks: Ca(H2TCPB) (dihedral angle φ = 52.41°) features a rigid benzene core, whereas Ca3(HTCPP)2 (φ1 = 55.51°, φ2 = 63.93°), displays non-planar phenylcarboxylate configurations.
Solvent selection critically influences calcium coordination and framework assembly.24,44,45 In this study, we studied the formation of three distinct Ca-MOFs from the same tetracarboxylate linker, demonstrating how solvent-dependent variations govern metal–ligand interactions and structural outcomes. Compound 1, synthesised in a DMA/water mixture, features calcium centres capped by DMA molecules, preventing aggregation into polynuclear clusters. This results in a framework characterised by discrete [CaO6] units and improved thermal stability evidenced by TGA. Amide solvent is frequently observed coordinating to metal nodes in MOFs, through its oxygen atom.46,47 This coordination restricts ligand flexibility, reducing its dihedral angle to 52.31° from 64.97° in the free ligand. It is notable that the hydrolysis48,49 of DMA under solvothermal conditions may also contribute to framework modulation (crystal size and phase control) through in situ formation of acetic acid50 and dimethylamine.51
In compound 2, the ethanol/water system facilitates the formation of infinite calcium chains, interconnected via bridging ligands and water molecules with intramolecular hydrogen bonding, resulting in a highly connected framework. Ethanol's lower polarity and weaker coordination strength favour extended interactions between calcium ions and carboxylate ligands. The NTTB4− ligand adopts larger dihedral angles (55.19–57.45°), reflecting enhanced conformational flexibility. In compound 3, the introduction of 1 M HCl (pH ∼ 2), alters ligand protonation, leading to a distinctive arrangement where four calcium centres form a partially continuous chain bridged to an isolated Ca cation. This structure exhibits the broadest range of dihedral angles (51.53–60.73°), highlighting increased ligand adaptability under acidic conditions.
Conclusions
This study illustrates the importance of solvent selection on the structural assembly of calcium-based MOFs with a tetratopic carboxylate ligand. By varying the solvent system and reaction conditions, three distinct frameworks are synthesised, whose metal centres range from isolated calcium nodes to continuous and partially disrupted chains that lead to various possible modes of connection of carboxylate ligands. These structural variations show diverse binding modes and geometry of the carboxylate ligands, demonstrating the critical role of solvent composition, polarity, and pH in controlling the coordination of calcium ions and the overall framework topology. These results will contribute towards design principles for future discovery of MOF structures with complex ligands. Mapping the relationships between structural chemistry and synthesis conditions provides reference data for computational models and machine learning approaches, which might allow prediction of synthesis conditions for frameworks with desired characteristics.Experimental methods
Synthesis
Synthesis of 4,4′,4′′,4′′′-(naphthalene-2,3,6,7-tetrayl)tetrabenzoic acid (H4NTTB) was performed in the same way with similar yield as described in the literature.28 Monocrystals suitable for X-ray diffraction were obtained by diluting 10 mg of H4NTTB in 0.5 mL of DMSO and then adding 0.3 mL of tetrahydrofuran (THF). After a few weeks, colourless crystals of the DMSO solvate of the ligand were obtained.Characterisation
The crystal structures of all compounds were determined by single crystal X-ray diffraction.For H4NTTB(DMSO)2, single-crystal X-ray intensity data collection was carried out at room temperature with a four-circle kappa-axis Bruker D8 Venture diffractometer equipped with Mo wavelength X-ray microsource and photon III C14 detector.
Due to the small size of the crystals of compound 1, the X-ray data acquisition was performed at SOLEIL synchrotron by using the PROXIMA 2A beamline equipped with a micro-focused beam.
For compounds 2 and 3 a crystal was placed on a Rigaku Oxford Diffraction Synergy-S diffractometer equipped with a dual radiation source and a hybrid pixel array detector. Data collection was conducted at a temperature of 100(2) K. The structure was solved using intrinsic phasing with the ShelXT52 structure solution program and refined using the ShelXL refinement package, employing least-squares minimisation within the Olex2 (ref. 53) software environment.
During crystal structure analysis the SQUEEZE process54 was used to eliminate the contribution of disordered solvent to the calculated structure factors for H4NTTB(DMSO)2 and compounds 1 and 3. For compound 3 we were only able to locate hydrogen atoms on bound water O3B which are hydrogen bonded to neighbouring ligand carboxylic oxygens. The remaining metal bound water molecules are likely hydrogen-bonded to the disordered waters in the voids that have not been located and so the correct position of their hydrogens cannot be deduced, so these were left as oxygen atoms in the final refined crystal structure.
Powder XRD measurements were performed at room temperature using a 3rd generation Malvern Panalytical Empyrean diffractometer equipped with multicore (iCore/dCore) optics and a Pixcel3D detector operating in 1-D receiving slit mode. The radiation source was a Cu tube emitting Cu Kα1/2 rays with an average wavelength of 1.5418 Å. Powder XRD data were analysed by fitting the diffraction profiles using the GSAS software suite,55 using Pawley method to refine peak shape parameter and lattice parameters.
TGA was conducted using a Mettler-Toledo TGA/DSC1 instrument under an air atmosphere with a heating rate of 5 °C per minute.
Data availability
CCDC 2421320–2421323 contain the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BWZ thanks the China Scholarship Council for award of a PhD scholarship. Some of the equipment used in this research was provided by the University of Warwick's Research Technology Platforms.References
- C. S. Diercks, M. J. Kalmutzki, N. J. Diercks and O. M. Yaghi, ACS Cent. Sci., 2018, 4, 1457–1464 CrossRef CAS PubMed.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38–39 CrossRef CAS PubMed.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- Q. H. Yang, Q. Xu and H. L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- L. Y. Li, L. D. Guo, F. Zheng, Z. G. Zhang, Q. W. Yang, Y. W. Yang, Q. L. Ren and Z. B. Bao, ACS Appl. Mater. Interfaces, 2020, 12, 17147–17154 CrossRef CAS PubMed.
- S. K. Xian, Y. H. Lin, H. Wang and J. Li, Small, 2021, 17, 2005165 CrossRef CAS.
- R. K. Alavijeh, K. Akhbari and J. White, Cryst. Growth Des., 2019, 19, 7290–7297 CrossRef CAS.
- X. Y. Chen, A. M. Plonka, D. Banerjee, R. Krishna, H. T. Schaef, S. Ghose, P. K. Thallapally and J. B. Parise, J. Am. Chem. Soc., 2015, 137, 7007–7010 CrossRef CAS.
- P. George, R. K. Das and P. Chowdhury, Microporous Mesoporous Mater., 2019, 281, 161–171 CrossRef CAS.
- B. Przybył, J. Zoń and J. Janczak, J. Mol. Struct., 2013, 1048, 172–178 CrossRef.
- Y. Sun, Y. Sun, H. Zheng, H. Wang, Y. Han, Y. Yang and L. Wang, CrystEngComm, 2016, 18, 8664–8671 RSC.
- E. Gavilan and N. Audebrand, Polyhedron, 2007, 26, 5533–5543 CrossRef CAS.
- R. B. Nielsen, P. Norby, K. O. Kongshaug and H. Fjellvåg, Dalton Trans., 2012, 41, 12082–12089 RSC.
- S.-i. Noro, J. Mizutani, Y. Hijikata, R. Matsuda, H. Sato, S. Kitagawa, K. Sugimoto, Y. Inubushi, K. Kubo and T. Nakamura, Nat. Commun., 2015, 6, 5851 CrossRef CAS PubMed.
- R.-B. Lin, L. Li, H.-L. Zhou, H. Wu, C. He, S. Li, R. Krishna, J. Li, W. Zhou and B. Chen, Nat. Mater., 2018, 17, 1128–1133 CAS.
- X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CAS.
- X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CAS.
- M. Lammert, H. Reinsch, C. A. Murray, M. T. Wharmby, H. Terraschke and N. Stock, Dalton Trans., 2016, 45, 18822–18826 CAS.
- T. Islamoglu, D. Ray, P. Li, M. B. Majewski, I. Akpinar, X. Zhang, C. J. Cramer, L. Gagliardi and O. K. Farha, Inorg. Chem., 2018, 57, 13246–13251 CAS.
- J. Su, S. Yuan, T. Wang, C. T. Lollar, J.-L. Zuo, J. Zhang and H.-C. Zhou, Chem. Sci., 2020, 11, 1918–1925 RSC.
- J. Lyu, X. Zhang, K.-i. Otake, X. Wang, P. Li, Z. Li, Z. Chen, Y. Zhang, M. C. Wasson, Y. Yang, P. Bai, X. Guo, T. Islamoglu and O. K. Farha, Chem. Sci., 2019, 10, 1186–1192 CAS.
- Y. Chen, X. Zhang, M. R. Mian, F. A. Son, K. Zhang, R. Cao, Z. Chen, S.-J. Lee, K. B. Idrees, T. A. Goetjen, J. Lyu, P. Li, Q. Xia, Z. Li, J. T. Hupp, T. Islamoglu, A. Napolitano, G. W. Peterson and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 21428–21438 CAS.
- A. M. Plonka, X. Chen, H. Wang, R. Krishna, X. Dong, D. Banerjee, W. R. Woerner, Y. Han, J. Li and J. B. Parise, Chem. Mater., 2016, 28, 1636–1646 CAS.
- S. R. Miller, E. Alvarez, L. Fradcourt, T. Devic, S. Wuttke, P. S. Wheatley, N. Steunou, C. Bonhomme, C. Gervais, D. Laurencin, R. E. Morris, A. Vimont, M. Daturi, P. Horcajada and C. Serre, Chem. Commun., 2013, 49, 7773–7775 CAS.
- L. Han, L. Qin, L. Xu, Y. Zhou, J. Sun and X. Zou, Chem. Commun., 2013, 49, 406–408 CAS.
- H. Kubo, S. Konishi, R. Oketani, T. Hayashi and I. Hisaki, Chem. – Eur. J., 2024, 30, e202401645 CAS.
- M. Ichikawa, J. Cryst. Mol. Struct., 1979, 9, 87–105 CAS.
- K. S. P. Karunadasa, C. H. Manoratne, H. M. T. G. A. Pitawala and R. M. G. Rajapakse, J. Phys. Chem. Solids, 2019, 134, 21–28 CAS.
- R. K. Vakiti, B. D. Garabato, N. P. Schieber, M. J. Rucks, Y. Cao, C. Webb, J. B. Maddox, A. Celestian, W. P. Pan and B. B. Yan, Cryst. Growth Des., 2012, 12, 3937–3943 CrossRef CAS.
- J. M. Andrić, G. V. Janjić, D. B. Ninković and S. D. Zarić, Phys. Chem. Chem. Phys., 2012, 14, 10896–10898 RSC.
- Z. L. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- P. Ghosh, Y. J. Colón and R. Q. Snurr, Chem. Commun., 2014, 50, 11329–11331 RSC.
- X. Duan, J. Lin, Y. Li, C. Zhu and Q. Meng, CrystEngComm, 2008, 10, 207–216 RSC.
- X. Fang, L. Wang, X. He, J. Xu and Z. Duan, Inorg. Chem., 2018, 57, 1689–1692 CrossRef CAS.
- W.-M. Liao, J.-H. Zhang, S.-Y. Yin, H. Lin, X. Zhang, J. Wang, H.-P. Wang, K. Wu, Z. Wang, Y.-N. Fan, M. Pan and C.-Y. Su, Nat. Commun., 2018, 9, 2401 CrossRef PubMed.
- S. T. Golafale, C. W. Ingram, A. A. Holder, W.-Y. Chen and Z. J. Zhang, Inorg. Chim. Acta, 2017, 467, 163–168 CrossRef CAS.
- Y. Wang, W. Fan, X. Wang, D. Liu, Z. Huang, F. Dai and J. Gao, Polyhedron, 2018, 155, 261–267 CrossRef CAS.
- X. Li, Y. Lin, L. Yu, J. Zou and H. Wang, Inorg. Chem., 2022, 61, 13229–13233 CAS.
- Y. Lin, J. Zhang, H. Pandey, X. Dong, Q. Gong, H. Wang, L. Yu, K. Zhou, W. Yu, X. Huang, T. Thonhauser, Y. Han and J. Li, J. Mater. Chem. A, 2021, 9, 26202–26207 CAS.
- A. Margariti, S. Rapti, A. D. Katsenis, T. Friščić, Y. Georgiou, M. J. Manos and G. S. Papaefstathiou, Inorg. Chem. Front., 2017, 4, 773–781 CAS.
- S. E. Rodriguez-Cruz, R. A. Jockusch and E. R. Williams, J. Am. Chem. Soc., 1998, 120, 5842–5843 CAS.
- X. Zhou, P. Liu, W.-H. Huang, M. Kang, Y.-Y. Wang and Q.-Z. Shi, CrystEngComm, 2013, 15, 8125–8132 CAS.
- D. Frahm, F. Hoffmann and M. Fröba, Cryst. Growth Des., 2014, 14, 1719–1725 CAS.
- Y. Fu, Y. Yao, A. C. Forse, J. Li, K. Mochizuki, J. R. Long, J. A. Reimer, G. De Paëpe and X. Kong, Nat. Commun., 2023, 14, 2386 CAS.
- W.-Q. Zhang, W.-Y. Zhang, R.-D. Wang, C.-Y. Ren, Q.-Q. Li, Y.-P. Fan, B. Liu, P. Liu and Y.-Y. Wang, Cryst. Growth Des., 2017, 17, 517–526 CAS.
- M. M. Heravi, M. Ghavidel and L. Mohammadkhani, RSC Adv., 2018, 8, 27832–27862 CAS.
- S. M. A. H. Siddiki, M. N. Rashed, A. S. Touchy, M. A. R. Jamil, Y. Jing, T. Toyao, Z. Maeno and K.-i. Shimizu, Catal. Sci. Technol., 2021, 11, 1949–1960 CAS.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 CAS.
- N. Zhao, L. Yang, B. Xie, J. Han, Q. Pan, X. Li, M. Liu, Y. Wang, X. Wang and G. Zhu, Chem. Commun., 2018, 54, 11264–11267 CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CAS.
- A. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2421320–2421323. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00145e |
| This journal is © The Royal Society of Chemistry 2025 |