
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterplay of hydrogen bonding and π-stacking interactions in the solid-state architecture of pranoprofen: insights from X-ray crystallography and computational analyses
Rafel
Prohens
 *a,
Rafael
Barbas
b,
Guadalupe
Abrego
c and
Antonio
Frontera
*d
*a,
Rafael
Barbas
b,
Guadalupe
Abrego
c and
Antonio
Frontera
*d
aLaboratory of Organic Chemistry, Faculty of Pharmacy and Food Sciences, University of Barcelona, Avda. Joan XXIII, 08028 Barcelona, Spain. E-mail: rafel_prohens@ub.edu
bUnitat de Polimorfisme i Calorimetria, Centres Científics i Tecnològics, Universitat de Barcelona, Baldiri Reixac 10, 08028 Barcelona, Spain
cDepartment of Chemical and Instrumental Analysis, Faculty of Chemistry and Pharmacy, University of El Salvador, Ciudad Universitaria, 3026 San Salvador, El Salvador
dDepartament de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma, Spain. E-mail: toni.frontera@uib.es
First published on 12th February 2025
Abstract
We report the crystal structure of the anhydrous form of pranoprofen, a valuable non-steroidal anti-inflammatory drug, solved by direct space methodologies from synchrotron X-ray powder diffraction data. Through a detailed joint experimental and computational study we have explored the complex interplay of aromatic and hydrogen-bonding interactions present in the molecular arrangement of pranoprofen in the solid state. Cooperativity and reinforcement of π-stacking and hydrogen bonding interactions govern the singular crystal packing of pranoprofen, suggesting potential binding mechanisms with biological targets.
Introduction
Pranoprofen (α-methyl-5H-[1]-benzopyrano [2,3-b]-pyridine-7-acetic acid), is a non-steroidal anti-inflammatory drug which can be used as a safe and effective alternative anti-inflammatory treatment following strabismus and cataract surgery.1–3 This drug has the beneficial effect of reducing the ocular signs and symptoms of dry eye and decreasing the inflammatory markers of conjunctival epithelial cells.4 Its efficacy is equivalent to moderate-potency corticosteroids, but it has a better safety profile and it is considered for the treatment of chronic conjunctivitis of presumed nonbacterial origin.5 It also can be used for the treatment of the acute and long term management of osteoarthritis and rheumatoid arthritis. Although this drug has shown high anti-inflammatory and analgesic efficiency and a minimal risk of side effects on gastrointestinal tract after oral administration6 the pharmaceutical use of pranoprofen is limited due to its inadequate biopharmaceutical profile, with a short plasmatic half-life, low water solubility and is unstable in aqueous solution, particularly when exposed to light.7,8To improve these limitations, pranoprofen has been formulated in poly (lactic/glycolic) acid nanoparticles and pranoprofen-loaded in nanostructured lipid carriers as a means of exploring novel formulations to improve the biopharmaceutical profile of PF for topical administration.9–13 As a cyclooxygenase (COX) inhibitor, pranoprofen works by suppressing the synthesis of prostaglandins, key mediators of inflammation and pain. Recent studies have also explored its potential in combination therapies and its role in managing oxidative stress and inflammation-related complications, further expanding its scope of use in modern medicine.
In this manuscript, we present the solid-state X-ray structure solved by direct space methodologies from X-ray synchrotron powder diffraction data of pranoprofen (Scheme 1) and analyzed its crystal packing, providing valuable insights into the predominant noncovalent interactions that shape its solid-state architecture. These findings can contribute to understanding the potential binding mechanisms of this drug with biological systems, such as the active sites of cyclooxygenase enzymes. Specifically, the H-bonding and π-stacking interactions identified in the solid state have been thoroughly examined using DFT calculations and various computational tools, including MEP, NCIplot, and QTAIM, with a particular focus on their cooperative effects.
 | ||
| Scheme 1 Structure of pranoprofen. | ||
Materials and methods
Materials
Pranoprofen® used in this study was kindly supplied by Alcon Cusi (Barcelona, Spain).X-Ray Powder Diffraction analysis (XRPD)
X-ray powder diffraction patterns of pranoprofen were obtained at 298 K using synchrotron radiation at ALBA's beam line BL04-MSPD14 using Mythen detector15 and a wavelength of 0.61939 Å. Capillary of 0.7 mm was used in rotation during data collection. Data acquisition time of 10 min per pattern was used, and the final treated data are the addition of ten acquisitions over the angular range of 0.5–43.6° (2θ).Computational details
The best solution from the direct space methodology conducted with the FOX software of anhydrous pranoprofen was subjected to geometry optimization by DFT16,17 periodic calculations performed within the generalized gradient approximation (GGA),18 as provided by the module CASTEP19 in Materials Studio software,20 using a basis set cutoff energy of 520 eV, ultrasoft pseudopotentials,21 PBE functional, semi-empirical dispersion corrections (Grimme),22 fixed unit cell and periodic boundary conditions. The validation of the structure23,24 was conducted through a second calculation starting from the optimized structure obtained in the previous step by using the same DFT parameters but setting free the unit cell parameters. Atomic displacement RMSD values were calculated to assess the reliability of the optimization.The X-ray geometry was used to analyze the energetic features of anhydrous pranoprofen at the PBE0-D3/def2-TZVP level of theory.25–27 The positions of the H-atoms were optimized while keeping the heavy atoms fixed. Energetic calculations and wavefunction generation were performed using the Gaussian-16 program.28 The QTAIM29 and NCIplot30 analyses were carried out using the AIMAll program31 at the same computational level. Hydrogen bond energies were estimated using the values of the potential energy density and the methodology described in the literature.32 Dimerization energies were calculated as the difference between the energy of the dimer and the sum of the monomer energies. Similarly, tetramers were computed as dimers. All interaction energies were corrected for basis set superposition error (BSSE) using the counterpoise method introduced by Boys and Bernardi.33
Results and discussion
Structure determination from space direct methods
The crystal structure determination of Pranoprofen was carried out by means of direct-space methodologies34 and synchrotron high-resolution X-ray powder diffraction data obtained in the MSPD beam line in Alba (Fig. 1). | ||
| Fig. 1 Rietveld plot and comparative of asymmetric unit molecules after the Rietveld refinement (red) and after the validation procedure (blue) with DFT geometry optimization. The plot shows the experimental powder XRD profile (red marks), the calculated powder XRD profile (black solid line), the difference profile (blue, lower line) and Bragg positions (blue lines). Agreement factors: Rwp = 11.8%, Rexp = 0.34%; χ2 = 120. | ||
The powder diffractogram was indexed out to a monoclinic cell of about 1225 Å3 by means of Dicvol04,35,36 (figures of merit: M = 99, F = 520) and validated with a Le Bail fit of the data using FullProf37 (goodness of fit: Rwp: 7.20, Rexp: 0.35; χ2 = 417). The space group was deduced as P21/c from the systematic absences and confirmed with the SGAid program of the DAJUST38 software. According to a typical estimated density value 1.4 Mg m−3 the asymmetric unit was assumed to contain one molecule of pranoprofen (Z = 4). The structure solution was carried out by direct space methodologies starting from a molecular model optimized by DFT with SPARTAN39 by means of the program FOX40 with the parallel tempering algorithm. The used background (estimated from a set of experimentally read points and interpolated) and the resulting cell, zero error and shape parameters of the Le Bail fit were used in the structure solution procedure with FOX. Several trials of 20 million runs were performed. The best solution (based on the Rwp value) was refined by the Rietveld method using FullProf,37 in combination with DFT calculations in order to improve through geometry optimization the planarity of the aromatic rings and to locate the hydrogen atomic coordinates. Fig. 2 depicts the final Rietveld plot and the overlapped molecules of the asymmetric unit after the Rietveld refinement and the DFT validation procedure. A summary of crystal data with relevant refinement parameters is given in Table 1. Finally, the atomic coordinates (together with the cell parameters) were subjected to optimization by DFT calculations, aiming to validate the crystal structure (RMSD = 0.1027).41 The hydrogen bond geometric data are summarized in Table 2.
 | ||
| Fig. 2 (a) Numbering of atoms involved in hydrogen-bonding interactions. (b) Molecular packing, highlighting hydrogen bonds (blue arrows) and π-stacking interactions (orange arrows). Cell axes are also shown. | ||
| T (K) | 298 |
| System | Monoclinic |
| Space group | P21/c |
| a (Å), b (Å), c (Å) | 11.46400(16), 10.37828(14), 10.88732(18) |
| α (°), β (°), γ (°) | 90, 108.9328(10), 90 |
| Volume (Å3) | 1225.26(3) |
| Z | 4 |
| R (%) | 11.8 |
| Donor | Hydrogen | Acceptor | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) |
|---|---|---|---|---|---|---|
| O31 | H32 | N6 | 1.02 | 1.59 | 2.59 | 165 |
| C24 | H28 | O30 | 1.10 | 2.59 | 3.49 | 138 |
Structural description of pranoprofen crystal structure
Anhydrous pranoprofen crystallizes in the monoclinic P21/c space group and the crystal structure has one independent molecule in the asymmetric unit (Z = 4). In the structure the pranoprofen molecule establishes a strong intermolecular hydrogen bond between the carboxylic acid and the pyridine moieties. In addition, antiparallel orientated molecules are stacked through the establishment of aromatic interactions. Overall, the pranoprofen molecules are packed in a “zig-zag” pattern established by both hydrogen bond and aromatic interactions as depicted by blue and orange arrows in Fig. 2.We have analyzed the most relevant intermolecular interactions in the structure by means of the Hirshfeld surface calculation42 and the associated fingerprint plot43,44 by using the Crystal Explorer software.45
Fig. 3 highlights the intermolecular hydrogen bonds on the Hirshfeld surface as red areas, together with the reciprocal H⋯N and N⋯H contacts as sharp spikes on the fingerprint plots. Weaker interactions like H⋯O; H⋯H; H⋯C have been pointed out in the figure.
 | ||
| Fig. 3 (a) Hirshfeld surface mapped on the pranoprofen molecule with dnorm. Relevant contacts are pointed out. (b) Fingerprint plots computed from Hirshfeld surface showing the percentage contribution of the individual types of interaction to the total Hirshfeld surface area. Main close contacts are highlighted: H⋯N (blue), H⋯O (red), H⋯C (green), H⋯H (purple). | ||
DFT calculations
Fig. 4 provides a partial view of the solid-state structure of pranoprofen (P), highlighting the chromenopyridine scaffold of two P molecules engaged in antiparallel π-stacking interactions. In this arrangement, the pyridine ring of one molecule stacks over the phenyl ring of the adjacent molecule, and vice versa. Additionally, each molecule forms two very short OH⋯N hydrogen bonds: one acting as a donor via the propionic acid arm and the other as an acceptor via the pyridine nitrogen atom. We propose that the latter interaction decreases the π-basicity of the pyridine ring, likely mitigating electrostatic repulsion between the π-electrons of the aromatic rings and thereby reinforcing the π-stacking interaction. This hypothesis is analyzed in detail below.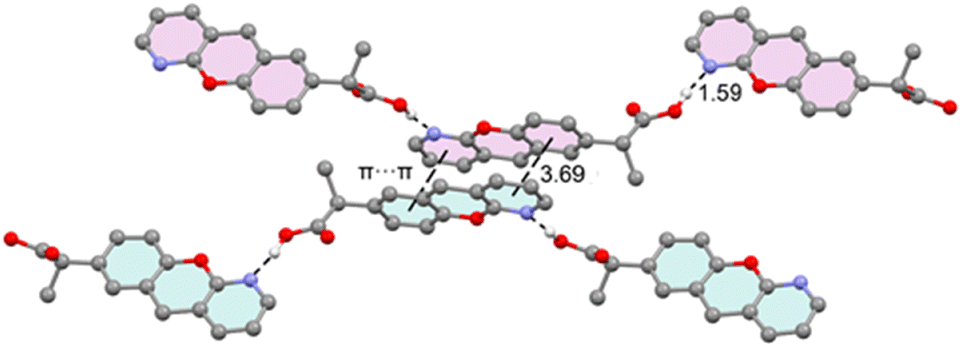 | ||
| Fig. 4 X-ray solid state structure of P, with indication of the π-stacking, H-bonding interactions. Distances in Å. H-atoms omitted for clarity apart from the carboxylic proton. | ||
We initiated our study by calculating the MEP (molecular electrostatic potential) surfaces of pranoprofen (P) and its hydrogen-bonded dimer (P2HB) to explore the electrophilic and nucleophilic regions of the molecule in the monomeric state and how these change upon dimer formation (Fig. 5). For the monomer, the MEP maximum is located at the carboxylic proton (+51.5 kcal mol−1), and the minimum is at the nitrogen atom of the pyridine ring (−39.5 kcal mol−1). This aligns with the very short POH⋯N H-bond observed in the X-ray structure of P (see Fig. 3). Notably, the MEP values are also substantial and negative at the oxygen atom of the chromene unit and the carboxylic oxygen atom (−31.4 and −33.9 kcal mol−1, respectively). The MEP over the aromatic rings is modest and negative for both the phenyl and pyridine rings.
 | ||
| Fig. 5 MEP surface analysis of P (a) and P2HB (b). Selected surface energies are given in kcal mol−1. | ||
The MEP surface of the P2HB dimer reveals significant changes in the molecule acting as the hydrogen bond acceptor. The MEP maximum increases to +54 kcal mol−1, indicating an enhanced ability of the carboxylic group to establish hydrogen bonds. More importantly, the MEP over the pyridine ring transitions from negative (−6.9 kcal mol−1) in the monomer to positive in the dimer (1.8 kcal mol−1), providing clear evidence that the electronic properties of the chromenopyridine system are influenced by hydrogen bonding. Additionally, the MEP value at the N-atom of the pyridine ring in the monomer acting as a hydrogen bond donor becomes more negative (−42.0 kcal mol−1), reflecting a strengthened H-bond acceptor capability.
This MEP analysis demonstrates that hydrogen bonding not only alters the polarity of the pyridine ring upon dimer formation but also enhances the dimer's overall ability to act as both a hydrogen bond donor and acceptor. Consequently, the infinite 1D hydrogen bond chains observed in the solid-state structure of P exhibit favorable cooperativity.
Fig. 6 presents the QTAIM/NCIplot analysis for the hydrogen-bonded and π-stacked dimers of P, designated as P2HB and P2π, respectively. The QTAIM analysis of the P2HB dimer indicates that the monomers are connected through two bond critical points (BCPs) and corresponding bond paths. One of these represents the short OH⋯N hydrogen bond, while the other corresponds to an ancillary CH⋯O interaction. The reduced density gradient (RDG) analysis reveals an isosurface coinciding with the BCP of the ancillary CH⋯O contact. Notably, the absence of an RDG isosurface for the OH⋯N hydrogen bond suggests a partial covalent character, supported by the high charge density at the BCP (0.0811 a.u.) and the negative value of the total energy density. The interaction energy for the P2HB dimer is calculated to be −10.6 kcal mol−1, with the ancillary CH⋯O interaction contributing only 0.8 kcal mol−1.
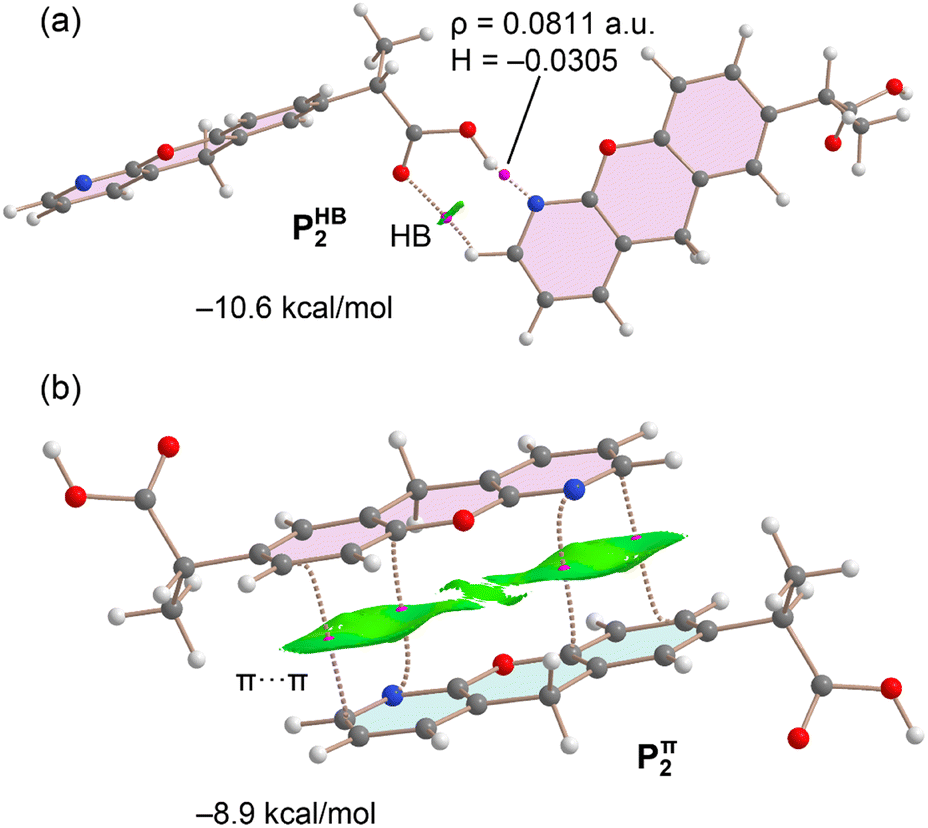 | ||
| Fig. 6 Overlapped QTAIM and NCIplot analyses of the P2HB (a) and P2π, (b) dimers. The dimerization energies are indicated. Bond critical points as fuchsia spheres and bond paths as dashed lines. Ring and cage critical points were omitted. For the NCIplot analysis, the following setting were used, S = 0.45, ρ cut-off = 0.04 a.u., color scale −0.035 ≤ sign(λ2)ρ ≤ −0.035 a.u. Only intermolecular interactions are represented. | ||
The QTAIM/NCIplot analysis of the P2π dimer (Fig. 6b) highlights an extended green RDG isosurface, characterizing the π-stacking interaction. Intriguingly, this isosurface also partially extends over the non-aromatic ring of the chromenopyridine moiety. The π-stacking interaction is further defined by four BCPs and bond paths linking the phenyl and pyridine rings of the stacked molecules. The calculated interaction energy for the π-stacking is significant at −8.9 kcal mol−1, underscoring the critical role of this interaction in stabilizing the solid-state architecture of P.
Fig. 7 illustrates the combined QTAIM/NCIplot analysis of two different tetramers, both featuring the π-stacked dimer as a common core. These tetramers, labeled as P4A and P4B, differ in their hydrogen bonding roles: in P4A, the dimeric core (P2π) acts as a hydrogen bond donor via the carboxylic groups, whereas in P4B, it serves as a hydrogen bond acceptor. In the tetramer P4A, no additional bond critical points or bond paths connect the monomers beyond those already observed in the isolated dimers P2HB and P2π. The interaction energy, calculated as the combination of two P2HB dimers, is −8.5 kcal mol−1—nearly identical to that of the isolated P2π dimer (see Fig. 4). This indicates that the monomers' participation in hydrogen bonding as donors does not influence the strength of the π-stacking interaction. In sharp contrast, the interaction energy for the P4B tetramer, also computed as the combination of two P2HB dimers, is significantly higher at −19.6 kcal mol−1, more than double the dimerization energy of P2π. QTAIM/NCIplot analysis reveals the formation of two symmetrically equivalent CH⋯O interactions between the propyl group of one monomer and the carboxylate group of the adjacent monomer, marked as yellow circles in Fig. 7b. These interactions contribute an energy of −2.8 kcal mol−1 (−1.4 kcal mol−1 per interaction). Consequently, the interaction energy attributed to the π-stacking within the tetramer increases to −16.8 kcal mol−1, nearly double that of the isolated π-stacking interaction.
 | ||
| Fig. 7 Overlapped QTAIM and NCIplot analyses of the P4A (a) and P4B, (b) dimers. The dimerization energies are indicated (the P2HB dimer is considered as monomer for the calculation). Bond critical points as fuchsia spheres and bond paths as dashed lines. Ring and cage critical points were omitted. For the NCIplot analysis, the following setting were used, S = 0.45, ρ cut-off = 0.04 a.u., color scale −0.035 ≤ sign(λ2)ρ ≤ −0.035 a.u. Only intermolecular interactions are represented. | ||
This analysis highlights that the involvement of the pyridine N-atoms in hydrogen bonding interactions significantly enhances the π-stacking interaction, as anticipated by the MEP surface analysis. This interplay between hydrogen bonding and π-stacking could influence pranoprofen's behavior as an inhibitor in biological systems, where such interactions are prevalent in enzyme active sites. The mutual reinforcement of hydrogen bonding and π-stacking interactions could be leveraged to design more effective inhibitors or biomimetic materials, optimizing binding affinities and stability in target environments.
Conclusions
The solid-state X-ray structure of pranoprofen reveals a complex interplay of π-stacking and hydrogen-bonding interactions that govern its molecular arrangement and stability. Computational analyses, including MEP, QTAIM, and NCIplot, provide a detailed understanding of the electronic and structural contributions to these interactions. The π-stacking interaction, supported by antiparallel aromatic ring alignment and mitigated electrostatic repulsion, plays a critical role in stabilizing the crystal structure. Moreover, the cooperative effects of hydrogen bonding, as evidenced by the enhanced polarity of key molecular regions upon dimer and tetramer formation, further reinforce the π-stacking interactions. These findings underline the importance of noncovalent interactions in shaping the solid-state architecture and offer insights into pranoprofen's potential binding mechanisms with biological targets, such as cyclooxygenase enzymes.46 The observed mutual reinforcement of π-stacking and hydrogen bonding interactions provides a valuable framework for designing biomimetic materials and improving drug efficacy through optimized molecular interactions.Data availability
Crystallographic data for anhydrous pranoprofen has been deposited at the CCDC under CCDC number 2410183 and can be obtained from https://www.ccdc.cam.ac.uk.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Research Project of MICIU/AEI of Spain (projects PID2020-115637GB-I00, PID2023-148453NB-I00 and PID2023-146632OB-I00 FEDER funds). We thank Prof. Ana Calpena (University of Barcelona) for providing a sample of pranoprofen for our study.Notes and references
- I. Akyol-Salman, D. Lece-Sertoz and O. Baykal, J. Ocul. Pharmacol. Ther., 2007, 23, 280–283 CrossRef CAS PubMed.
- M. Sawa, K. Masuda and M. Nakashima, Japanese Journal of Cataract and Refractive Surgery, 1999, 13, 193–200 Search PubMed.
- A. Z. McColgin and J. S. Heier, Curr. Opin. Ophthalmol., 2000, 11, 3–6 CrossRef CAS PubMed.
- X. Liu, S. Wang, A. A. Kao and Q. Long, Cornea, 2012, 31, 1235–1239 CrossRef PubMed.
- R. Notivol, M. Martinez and M. M. Bergamini, Am. J. Ophthalmol., 1994, 117, 651–656 CrossRef CAS PubMed.
- G. Kajal, C. Subrata and N. Arunabha, Pharm. Sin., 2011, 2, 152–168 Search PubMed.
- M. Narashino, H. Ichikawa, S. Narita and A. S. Yotsukaido, United States Pat., US005225206A, 1993 Search PubMed.
- K. Kobe, H. Otsu, Y. Kobe and Y. Kakogawa,United States Pat., US005856345A, 1999 Search PubMed.
- G. Abrego, H. L. Alvarado, M. A. Egea, E. Gonzalez-Mira, A. C. Calpena and M. L. Garcia, J. Pharm. Sci., 2014, 103, 3153–3164 Search PubMed.
- C. Cañadas, H. L. Alvarado, A. C. Calpena, A. M. Silva, E. B. Souto, M. L. García and G. Abrego, Int. J. Pharm., 2016, 511, 719–727 CrossRef PubMed.
- C. Cañadas, G. Abrego, H. L. Alvarado, A. C. Calpena and A. Boix-Montañes, J. Pharm. Biomed. Anal., 2018, 160, 109–118 CrossRef PubMed.
- M. Rincón, A. C. Calpena, M. J. Fabrega, M. L. Garduño-Ramírez, M. Espina, M. J. Rodríguez-Lagunas, M. L. García and G. Abrego, Nanomaterials, 2018, 8, 1022–1050 CrossRef PubMed.
- M. Rincón, A. C. Calpena, B. Clares, M. Espina, M. L. Garduño-Ramírez, M. J. Rodríguez-Lagunas and G. Abrego, Nanomedicine, 2018, 13, 2397–2413 CrossRef PubMed.
- F. Fauth, R. Boer, F. Gil-Ortiz, C. Popescu, O. Vallcorba, I. Peral, D. Fullà, J. Benach and J. Juanhuix, Eur. Phys. J. Plus, 2015, 130, 160–173 CrossRef.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360–S370 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev. A, 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev. A, 1965, 140, A1133 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- Materials Studio, version 7.0, Accelrys Software, Inc., San Diego, CA, USA, 2013 Search PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. van de Streek and M. A. Neumann, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2014, 70, 1020–1032 CrossRef CAS PubMed.
- J. van de Streek and M. A. Neumann, Acta Crystallogr., Sect. B:Struct. Sci., 2010, 66, 544–558 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019, https://aim.tkgristmill.com Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Ghosh, S. Islam, S. Pramanik and S. Kumar Seth, J. Mol. Struct., 2022, 1268, 133697 CrossRef CAS.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 1991, 24, 987–993 CrossRef CAS.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- J. Rodriguez–Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- O. Vallcorba, J. Rius, C. Frontera, I. Peral and C. Miravitlles, J. Appl. Crystallogr., 2012, 45, 844–848 CrossRef CAS.
- Spartan’10, Wavefunction Inc., Irvin, CA, USA, 2014 Search PubMed.
- V. Favre-Nicolin and R. Cerný, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- J. van de Streek and M. A. Neumann, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 1020–1032 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman and J. J. Mckinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. Mckinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017, https://hirshfeldsurface.net Search PubMed.
- S. Xu, D. J. Hermanson, S. Banerjee, K. Ghebreselasie, G. M. Clayton, R. M. Garavito and L. J. Marnett, J. Biol. Chem., 2014, 289, 6799–6808 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |