
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the formation of coordination polymers based on dicarboxylic ligands derived from fluorene and their adsorption abilities†
Nizami
Israfilov
 a,
Nathalie
Kyritsakas
b,
Abdelaziz
Jouaiti
*a and
Sylvie
Ferlay
*a
a,
Nathalie
Kyritsakas
b,
Abdelaziz
Jouaiti
*a and
Sylvie
Ferlay
*a
aUniversité de Strasbourg, CNRS, CMC UMR 7140, F-67000 Strasbourg, France. E-mail: jouaiti@unistra.fr; ferlay@unistra.fr
bService de radiocristallographie de la Fédération de Chimie Le Bel – UAR 2042, Université de Strasbourg and, CNRS, F-67000 Strasbourg, France
First published on 29th January 2025
Abstract
Two new coordination polymers (compounds 1 and 2) based on the use of a V-shaped ligand L (9,9-bis(4-carboxyphenyl)fluorene) combined with Cu(NO3)2 or Zn(NO3)2 were synthesized and structurally characterized by single-crystal X-ray diffraction. These compounds add to the list of very few examples of reported CPs derived from ligands with a fluorene core. X-ray diffraction analysis revealed the structures of the compounds; 2D (1) and robust 3D (2) structures were reported, and their sorption and emission properties were analysed. Additionally, it was possible to isolate the following compounds as side products: compound 4, a 2D compound that is isomorphous to 1, and compound 3, a polymorph of 1.
Introduction
The fluorene moiety is well-known, especially for its photophysical properties, and finds applications in a wide variety of fields, such as materials1,2 in optical and 3D data storage,3 organic light-emitting devices4 and dye-sensitized solar cells.5 Active fluorene molecules are also found in biomedical research for photodynamic therapy6 and in sensing applications for ion detection.7Coordination polymers (CPs)8,9 are extended molecular assemblies built from metallic nodes and ligands and studied for their multiple properties and applications. The formation of metal-based functional materials involving fluorene derivatives has also been well documented.10 Starting from fluorene derivatives bearing carboxylic groups, there have been few luminescent CPs that have been described: one can cite their use for the recognition of Fe(III) ions,11 sensing of picric acid12 or the mechanoluminescence behaviour13 as an emitter.14 Fluorene moieties have also been appended with pyridyl derivatives for the formation of CPs, with their use as an anion sensor being thoroughly described.15
We are interested in the generation of porous coordination polymers (CPs) or MOFs (molecular organic frameworks),16,17 which are molecular extended architectures presenting different functionalities depending on their structures, with applications in fields such as catalysis, gas storage, luminescence, and sensing.17,18 The development of crystal engineering strategies19 has enabled the possibility to obtain and elucidate a multitude of crystalline structures. To achieve a certain degree of predictability in the obtained molecular structures, the ligands used must be structurally rigid.
We intend to provide a crystal engineering approach to illustrate the formation of new fluorene-based compounds, together with their sorption properties, which have not been commonly reported for such compounds. For this purpose, we intend to use a “short” V-shaped ditopic ligand, bearing carboxylate coordinating moieties. The design of H2L (9,9-bis(4-carboxyphenyl)fluorene) (Fig. 1) appears appropriate for this purpose. H2L has been known for the formation of fluorene-based polymers;20 however, it has never been reported for the formation of CPs. Due to the V shape of this ditopic ligand, the predictability of the formed coordination polymers appears much more accessible. We aim to investigate the influence of the flexibility of H2L and the nature of the used metallic salts, on the nature of the formed CPs.
 | ||
| Fig. 1 Fluorene-based ligand H2L used for the formation of CPs and flexibility on the tetrahedral central C atom. | ||
In this contribution, we report the structure of the obtained coordination polymers when H2L is combined with Cu(NO3)2 or Zn(NO3)2 salts in solvothermal conditions. We focused on the use of divalent metal ions presenting different coordination abilities, favouring the formation of metallic clusters, like the well-known paddle-wheel-shaped dimeric units. The structural investigations together with the properties (gas sorption and emission) of the corresponding CPs are described here below.
Results and discussion
Synthesis of the CPs
The synthesis of H2L was performed using an already reported method.20 The corresponding CPs were synthesized using solvothermal conditions in DMF, with drops of HCl, starting from Cu(NO3)2 or Zn(NO3)2 salts (see Experimental section). Crystals of four compounds, 1–4, suitable for single-crystal X-ray diffraction analysis, were analysed by XRD (see Experimental part). Compounds 3 and 4, present as side products using different synthetic conditions, are reported in the ESI† and the very low observed yields did not allow any further characterization.Compounds 1 and 2, which could be obtained in a reproducible way, with a high degree of purity and in high yields, are described here below, and their properties were studied.
For both the described compounds, the bond length values found in L are in accordance with previously reported analogue ligands bearing the fluorene moiety10 and are not discussed here.
Structural description for 1, a 2D compound
As revealed by the X-ray diffraction study on a single crystal, the compound displays the following formula (C27H16O4)Cu(C3H7NO)·nS (S being solvent molecules) (1). The coordination polymer crystallizes in the orthorhombic Cmca space group (see the crystallographic data in Table 1) and its asymmetric unit is composed of half of a ligand L2− located on a twofold axis, one Cu2+ cation, located on a mirror and half of a disordered DMF molecule, coordinated to Cu2+. The SQUEEZE instruction in PLATON21 was applied (which defined 430 electrons per cell). The residual electron density was assigned and estimated to have 43 molecules of the water solvent per cell.| CCDC | 2383979 | 2395186 |
|---|---|---|
| 1 | 2 | |
| Empirical formula | C30H23CuNO5, solvent | 2(C81H50O13Zn3.50), 6(C3H7NO), solvent |
| Formula weight | 541.03 | 3354.71 |
| Temperature | 120(2) | |
| Wavelength | 0.71073 Å | |
| Crystal system, space group | Orthorhombic, Cmca | Trigonal, R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
| Unit 2D Cocell dimensions | a = 23.4998(8) Å, α = 90 deg. | a = 25.5168(6) Å, α = 90 deg. |
| b = 18.6597(6) Å, β = 90 deg. | b = 25.5168(6) Å, β = 90 deg. | |
| c = 17.5655(6) Å, γ = 90 deg. | c = 20.8378(7) Å, γ = 120 deg. | |
| Volume | 7702.5(4) Å3 | 11749.9(7) Å3 |
| Z, calculated density | 8, 0.933 Mg m−3 | 3, 1.422 Mg m−3 |
| Absorption coefficient | 0.594 mm−1 | 1.132 mm−1 |
| F(000) | 2232 | 5178 |
| Crystal size | 0.150 × 0.120 × 0.100 mm | 0.140 × 0.130 × 0.120 mm |
| Theta range for data collection | 2.472 to 27.898 deg. | 2.161 to 27.887 deg. |
| Limiting indices | −30 ≤ h ≤ 26 | −33 ≤ h ≤ 33 |
| −24 ≤ k ≤ 23 | −33 ≤ k ≤ 33 | |
| −23 ≤ l ≤ 23 | −27 ≤ l ≤ 26 | |
| Reflections collected/unique | 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 079/4717 [R(int) = 0.0763] 079/4717 [R(int) = 0.0763] |
38206/6242 [R(int) = 0.0609] |
| Goodness-of-fit on F2 | 1.046 | 1.019 |
| Final R indices [I > 2sigma(I)] | R 1 = 0.0464, wR2 = 0.1285 | R 1 = 0.0544, wR2 = 0.1401 |
| R indices (all data) | R 1 = 0.0631, wR2 = 0.1400 | R 1 = 0.0767, wR2 = 0.1588 |
| Largest diff. peak and hole | 0.656 and −0.633 e Å3 | 0.862 and −0.741 e Å3 |
1 results from the bridging of the V-shaped ligand L2− by the paddle-wheel unit formed by the Cu2+ cations, acting as a square, thus the recognition pattern is a corrugated 2D grid formed in the xOz plane, and presents an M/L ratio of 1/1. The grids are made up of lozenges with side lengths of 14.652(2) Å and angles of 106.4° and 73.6° (see Fig. 2a and b). The metal centre displays a distorted pyramid square base coordination geometry (see bonds and angles in Table 2 and Fig. 2c), as frequently found in paddle-wheel derivatives, with a short Cu⋯Cu distance of 2.6302(7) Å. It is interesting to note, that paddle-wheel units have been frequently encountered in the formation of robust MOFs.22 The four equatorial positions of the metal centre are occupied by four oxygen atoms (coming from four different ligands) thus forming the basal plane of the metal, and the apical position is occupied by an oxygen atom of a disordered DMF molecule, as shown in Fig. 2c. As shown in Table 2, the Cu–O bond lengths are equal to 1.9619(15) and 1.9684(16) Å, whereas the apical Cu–ODMF distance is equal to 2.142(3).
 | ||
| Fig. 2 Portion of the X-ray structure for 1 showing the formation of the 2D neutral coordination network resulting from the bridging of the consecutive ligand L by Cu2+ cations: a) view of the corrugated plane along the b axis, b) view of the grid in the xOz plane c) details of the environment around Cu2+ cations. H atoms are not all presented for clarity. Disordered DMF molecules are represented. | ||
| 1 | 2 | |
|---|---|---|
| M–O | 1.9619(15) | 1.935(2) |
| 1.937(2) | ||
| 1.9684(16) | 1.9472(11) | |
| 2.019(2) | ||
| M–ODMF | 2.142(3) | — |
| M–M | 2.6302(7) | 2.707(6) |
| 2.838(15) | ||
| 2.998(4) | ||
| 3.000(14) |
In the CP, the α angle within the L2− ligand (Fig. 1) is equal to 102.19°.
In the crystal, the corrugated planes are stacked along the b axis (Fig. 2a and S1 in the ESI†), and the distance between the two layers is equal to ca. 9.3 Å. There is a CH–π contact between the coordinated DMF molecule and a phenyl ring of the L2− ligand in the crystal.
As already mentioned, in slightly different synthetic conditions (lowering the synthetic temperature from 85 °C to 80 °C, see the Experimental section), the presence of one polymorph 3 of formula (C27H16O4)Cu(OH2)·nS, presenting also a 2D structure with the same connectivity but a different stacking (interpenetrated 2D networks) was observed (see crystallographic Tables S1 and S2 and Fig. S3 in the ESI†). In addition, using slightly different conditions and Zn(NO3)2, it is also possible to observe a Zn isomorph of 1, compound 4 with the formula (C27H16O4)Zn(OH2)·nS (see crystallographic Tables S1 and S2 and Fig. S2 in the ESI†), where the molecule coordinated in the apical position of the metal is water (for 4) instead of DMF (for 1), and presenting the same packing.
Structural description for 2, a 3D compound
X-ray diffraction study on a single crystal evidenced a compound of the following formula (C27H16O4)6Zn7(OH)2·6DMF·nS or (C27H16O4)6Zn7O2·6DMF·2C·nS (S being solvent molecules and C is a monovalent cation) (2). The coordination polymer crystallizes in the trigonal R space group (see the crystallographic data in Table 1) and its asymmetric unit is composed of one ligand L2−, two disordered Zn2+ cations on special positions, one OH− (or O2−) anion and one DMF molecule (see Fig. S1 in the ESI†). It was not possible to precisely assign the nature of the anions within the Zn tetrahedron (OH− or O2−). The SQUEEZE instruction in PLATON21 was applied (which defined 300 electrons per cell). The residual electron density was assigned and can be estimated to be 30 molecules of the water solvent per cell, or water molecules and cations (probably derived from ammonium), located into the pores.
2 results from the bridging of V-shaped ligand L2− (see schematic representation Fig. 3a) by a Zn cluster composed of two fused Zn-tetrahedra, as shown in Fig. 3b, and inside of each tetrahedra can be found an anion (OH− or O2−), leading to the formation of [Zn7(μ4-OH)2]12+ (or [Zn7(μ4-O)2]10+) clusters (Secondary Building Units). The clusters are surrounded by 12 bridging ligands, coordinated through Zn–O bonds, and this results in a 3D coordination polymer, as schematically shown in Fig. 3c and d. The network adopts a typical 3D hexagonal structure. Other representations of this compound can be found in Fig. S4, in the ESI.†
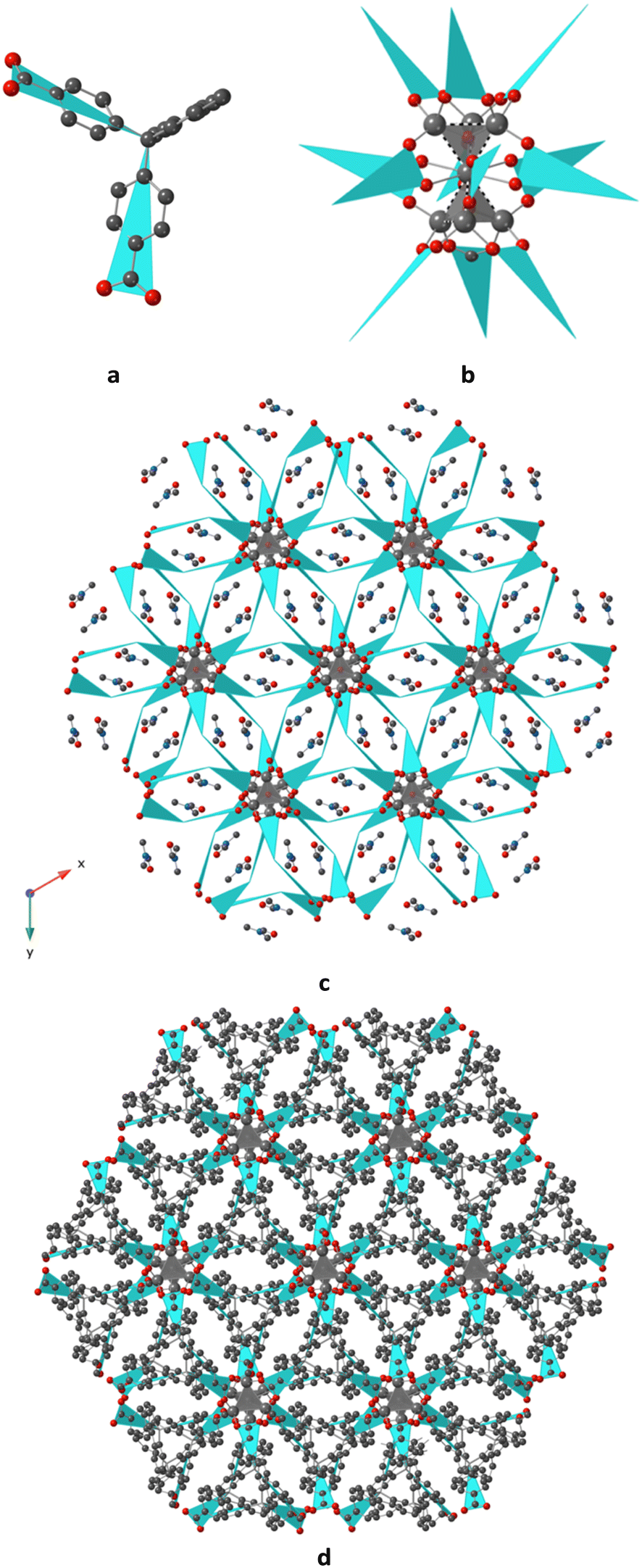 | ||
| Fig. 3 Portion of the X-ray structure for 3D 2 showing a) the schematic representation of L2−, adopting a V shape, in blue, b) the surrounding of the [Zn7(μ4-OH)2]12+ (or [Zn7(μ4-O)2]10+) units (only half of a schematic ligand is represented in blue colour, for the sake of clarity), by 12 L2− bridging ligands (the dashed lines represent the Zn–Zn interactions, c) the resulting schematic view of the 3D coordination polymer, with uncoordinated DMF molecules located in the free space, projection in the xOy plane and d) in the same plane, the resulting 3D structure where all atoms of the ligands are represented and the solvents are not represented. H atoms, the disordered Zn cations are not represented, for the sake of clarity. | ||
In the network, the Zn2+ cations display distorted tetrahedral O4 geometry (see bonds and angle in Table 2, and Fig. 2c), and the Zn atom fusing two tetrahedra adopts a deformed octahedral O6 geometry, with O atoms provided by the carboxylate moieties of L2− and from the OH− (or O2−) anions located inside the cavity of the tetrahedra, with Zn–O distances varying between 1.935(2) and 2.019(2) Å, as seen in Table 2. Short Zn⋯Zn distances, varying between 2.707(6) and 3.000(14) Å, are observed within the [Zn7(μ4-OH)2]12+ (or [Zn7(μ4-O)2]10+) clusters, as seen in Table 2.
In the CP, the α angle within the L2− ligand (Fig. 1) is equal to 101.29°.
The presence of [Zn7(μ4-O)2]10+ clusters in coordination compounds, analogous to what is reported for 2, and has been already reported in the literature: MOFs obtained from NBD (2-nitrobenzene-1,4-dicarboxylate) ligand,23,24 pyrazolate-dicarboxylate MOFs25 or -(4-carboxyphenyl)succinamate based MOFs.26 The presence of [Zn7(μ4-OH)n]m+ units has been scarcely reported in the literature.27
The behaviour of the corresponding microcrystalline powders of both compounds has been investigated, and XRPD measurements were performed (Fig. 4). The air-dried powdered 1 and 2 samples present the same crystalline phases, as derived from the simulated XRDs on the related single crystals.
 | ||
| Fig. 4 For 1 (a) and 2 (b), PXRD diagrams of the air-dried microcrystalline compounds and after BET measurements, compared with the simulated phases obtained from X-ray diffraction data. | ||
These measurements evidence that the compounds are crystalline, maintain their structures after being exposed to the air and that there is only one crystalline phase in the microcrystalline powder of 1 and 2.
Thermal behaviour of the formed CPs
The thermal behaviour of the corresponding microcrystalline powder of both compounds was investigated by TGA analysis (Fig. S5 in the ESI†).1 and 2 exhibit a two-step degradation profile. For 2, the first step, with an insignificant ∼3% weight loss up to 140 °C, is likely due to moisture and/or weakly bound DMF molecules. The second step involved a ∼20% weight loss up to 275 °C, corresponding to DMF molecules strongly trapped in the structure. Additionally, 2 shows a very high thermal stability, with degradation beginning around 420 °C.
For 1, despite several attempts, we were unable to obtain a well-defined degradation profile. The first step occurs up to 60 °C, followed by a second step between 60 °C and 230 °C, with the latter corresponding to a significant ∼40% weight loss. Degradation of 1 begins around 300 °C, which is lower than that observed for 2.
Gas sorption studies
After an activation phase (see Experimental section) on microcrystalline samples, N2(g) adsorption was performed for 1 and 2. For both compounds, a type I isotherm was observed (Fig. S6 in the ESI†), with a slight increase at the end of the isotherm, indicating interparticle condensation. The estimated SA is ca. 30 m2 g−1 for 1 and 60 m2 g−1 for 2, which are very low.CO2(g) adsorption was also performed (Fig. S6 in ESI†). 2 exhibits an adsorption capacity of 1.53 mmol g−1, with a single-point surface area of 168 m2 g−1. 1 demonstrates an even higher adsorption capacity of 2.85 mmol g−1 at 273 K and 1200 mbar, with a corresponding surface area of 310 m2 g−1.
After the adsorption measurements, a PXRD diagram of the microcrystalline compounds was recorded (Fig. 4). An amorphization process was observed for both compounds, Nevertheless, 1 and 2 reveal to maintain crystalline phases, with broadening of the observed peaks.
Emission properties of 2
The luminescence spectrum of 2 in the solid state was recorded at room temperature. As shown in Fig. 5, upon excitation (λexc = 280 nm), 2 emits violet light, presenting a large peak centered at 400 nm. The solid-state absorption spectrum of 2 is presented in Fig. S8 in the ESI.† | ||
| Fig. 5 For 2, solid-state emission spectra at room temperature with λexc = 280 nm. | ||
Compound 1 did not present any emission properties.
Discussion
The reproducible formation of the coordination polymers was found through the recognition of a V-shaped ligand (ligand H2L (9,9-bis(4-carboxyphenyl)fluorene)) and divalent metal, that are involved in clusters, and their formation is favoured using solvothermal conditions and acting as nodes in the formation of CPs. Grid-like compounds (1 (but also 3 and 4)) with an M/L ratio of 1/1 were formed with deformed lozenge of side 14.652(2) Å for 1 (or 13.890(2) Å for 3 and 14.798(2) Å for 4). The α angle around the tetrahedral carbon was found to be in the 101.8–103.9° range. The distortion from the tetrahedral environment of the C atom in H2L is low. For these compounds, solvent molecules (DMF, H2O) are present in the network and interact through H bonds with the planes for 1. During the activation process, the removal of the solvent molecules disorganizes the stacking of the planes, leading to the beginning of an amorphization process, as observed in PXRD. This partial amorphization is probably responsible for the very low N2 and CO2 uptake (corresponding SA are low). For the 3D 2 compound, the situation is different. The robust compound is built from the high nuclearity [Zn7(μ4OH)2]12+ (or [Zn7(μ4-O)2]10+) clusters: it is stable at high temperatures, and the DMF molecules are strongly trapped into the 3D network. After the activation process, the presence of only one crystalline phase was observed. Nevertheless, in this compound, the voids are very low, and again, the amount of N2 and CO2 uptake is low. This compound exhibits a strong violet luminescence.It is interesting to note that in all the structures involving H2L, the deformation around the tetrahedral central C atom (α angle) is not pronounced, and it acts, as expected, as a rigid V-shaped ligand.
Experimental
Synthetic procedures
All reagents were purchased from commercial sources and used without further purification. The synthesis of ligand L was performed using an already-reported method, without modifications.20In all the compounds, there is a large amount of solvent present inside the cavities, which is released when the crystals are exposed to the air, therefore, the elemental analysis data is not reproducible and are not shown here.
Crystallization conditions for CPs
Using the same conditions, but a temperature of 80 °C, it was possible to observe a few crystals of another crystalline phase. The crystals were isolated and revealed to be 3 (crystallographic Tables S1 and S2 and Fig. S3 in the ESI†).
For IR, see Fig. S10 in the ESI.†
Using the same conditions, but a temperature of 90 °C, it was possible to observe very few crystals of another crystalline phase. The crystals were isolated and revealed to be 4 (crystallographic Tables S1 and S2 and Fig. S2 in the ESI†).
Materials and methods
Crystal X-ray data collection. For all the compounds, the crystals were placed in oil, and a single crystal was selected, mounted on a glass fibre and placed in a low-temperature N2 stream.
X-ray analyses and structural resolutions for 1, 2 and 4 were performed at the Service de radiocristallographie, Fédération de Chimie “Le Bel”, UAR 2042, Strasbourg.
X-ray diffraction data collection was carried out on a Bruker PHOTON III DUO CPAD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using Mo-Kα radiation (λ = 0.71073 Å). The crystal-detector distance was 40 mm. The cell parameters were determined (APEX3 software)28 from reflections taken from one set of 180 frames at 1 s exposure. The structure was solved using the program SHELXT-2018.29 The refinement and all further calculations were carried out using SHELXL-2018.30 The H atoms were included in the calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using SADABS in APEX328; transmission factors: Tmin/Tmax = 0.6592/0.7456, for 1, Tmin/Tmax = 0.6942/0.7456 for 2 and Tmin/Tmax = 0.5694/0.7456 for 4.
For 3, X-ray diffraction data collection was carried out on a Bruker APEX II Kappa-CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using Mo-Kα radiation (λ = 0.71073 Å). The crystal-detector distance was 40 mm. The cell parameters were determined (APEX2 software)28 from reflections taken from three sets of 6 frames, each at 10 s exposure. The structure was solved using the program SHELXT-2018.29 The refinement and all further calculations were carried out using SHELXL-2019.30 The H atoms were included in the calculated positions and treated as riding atoms using SHELXL default parameters. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using SADABS in APEX2.28
PXRD. Powder diffraction studies (PXRD) on the air-dried powdered samples were performed using microcrystalline powders at room temperature (293(2) K), on a Bruker D8 diffractometer using a monochromatic Cu-Kα radiation at a scanning range between 2° and 40° at a scan step size of 2° min−1 and sample holder rotation at 15 rpm.
All measurements were performed with ultrahigh-purity gases. The specific surface areas were determined using the Brunauer–Emmett–Teller and Langmuir equations from the low-pressure sorption data on Micromeritics ASAP 2020: N2 at 77 K, and CO2 at 273 K.
Conclusions
The presented results showed the formation of four new coordination polymers based on a rigid V-shaped fluorene derivative ligand, bearing two coordinating carboxylic moieties. The reproducible formation of two 2D and 3D compounds, while using either divalent Cu or Zn cations, combined with ditopic dicarboxylic (9,9-bis(4-carboxyphenyl)fluorene) ligand L, under solvothermal conditions, attests for the recognition of metallic clusters (paddle-wheel or fused tetrahedra) with the carboxylate groups. Two compounds 1 (2D) and 2 (3D) of formula LCu(DMF)·nS and L6Zn7(OH)2·6DMF·nS or L6Zn7O2·6DMF·2C·nS (S being solvent molecules and C a monovalent cation) were thoroughly analysed from a structural point of view and they present stability when exposed in the air. When activated, they display very low porosity, but a subsequent CO2 uptake at low pressures. The scaffold of the compound appears to remain after the activation process, according to the PXRD data. Two other CPs, that were obtained as side products are reported from a structural point of view.This is a rare example of ordered molecular compounds, obtained from fluorene-based ligands. Furthermore, fluorene-based ligands are still under investigation together with their properties.
Data availability
CCDC 2383979 (1), 2395186 (2), 2379878 (3) and 2379879 (4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/datarequest/cif.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the University of Strasbourg and the CNRS (CNRS Chimie) are acknowledged.Notes and references
- R. Abbel, A. P. H. J. Schenning and E. W. Meijer, Fluorene-based materials their supramolecular properties, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4215–4233, DOI:10.1002/pola.23499.
- I. V. Kurdyukova and A. A. Ishchenko, Organic dyes based on fluorene and its derivatives, Russ. Chem. Rev., 2012, 81, 258–290, DOI:10.1070/RC2012v081n03ABEH004211.
- K. Ogawa, Two-photon absorbing molecules as potential materials for 3D optical memory, Appl. Sci., 2014, 4, 1–18, DOI:10.3390/app4010001.
- S.-Y. Ku, L.-C. Chi, W.-Y. Hung, S.-W. Yang, T.-C. Tsai, K.-T. Wong, Y.-H. Chen and C.-I. Wu, High-luminescence non-doped green OLEDs based on a 9,9-diarylfluorene-terminated 2,1,3-benzothiadiazole derivative, J. Mater. Chem., 2009, 19, 773–780, 10.1039/B814082.
- K. Lim, C. Kim, J. Song, T. Yu, W. Lim, K. Song, P. Wang, N. Zu and J. Ko, Enhancing the performance of organic dye-sensitized solar cells via a slight structure modification, J. Phys. Chem. C, 2011, 115, 22640–22646, DOI:10.1021/jp2070776.
- F. Bolze, S. Jenni, A. Sour and V. Heitz, Molecular photosensitisers for two-photon photodynamic therapy, Chem. Commun., 2017, 53, 12857–12877, 10.1039/C7CC06133A.
- D. M. Nguyen, A. Frazer, L. Rodriguez and K. D. Belfield, Selective fluorescence sensing of zinc and mercury ions with hydrophilic 1,2,3-triazolyl fluorene probes, Chem. Mater., 2010, 22, 3472–3481, DOI:10.1021/cm10059.
- B. F. Abrahams, B. F. Hoskins and R. Robson, A new type of infinite 3D polymeric network containing 4-connected, peripherally-linked metalloporphyrin building blocks, J. Am. Chem. Soc., 1991, 113, 3606–3607, DOI:10.1021/ja00009a065.
- J. R. Long and O. M. Yaghi, The pervasive chemistry of metal-organic frameworks, Chem. Soc. Rev., 2009, 38, 1213–2114, 10.1039/B903811F.
- W.-Y. Wong, Metallated molecular materials of fluorene derivatives and their analogues, Coord. Chem. Rev., 2005, 249, 971–997, DOI:10.1016/j.ccr.2004.10.007.
- Z.-H. Wang, X.-Z. Wang, X.-Y. Lai, Q. Hou, J.-X. Ma, J.-H. Li, K. Yue and Q.-F. Yang, Three new coordination polymers based on a fluorene derivative ligand for the highly luminescent sensitive detection of Fe3+, J. Mol. Struct., 2020, 1202, 127341, DOI:10.1016/j.molstruc.2019.127341.
- J. Cheng, Z. Liu, D. Wang, K. Chen, Y. You and X. Zhou, Fluorene-based Lanthanide Coordination Polymer: Structure, Luminescence and Sensing of Picric Acid, Appl. Organomet. Chem., 2019, 33, e5168, DOI:10.1002/aoc.5168.
- S. Jindal, G. Anjum, V. K. Maka and J. N. Moorthy, Mechanoluminescence and aggregation-enhanced emission (AEE) of an In-MOF based on a 9,9′- diphenyl-9H-fluorene tetraacid linker, Nanoscale, 2021, 13, 9668–9677, 10.1039/D1NR00898F.
- J. Robin, N. Audebrand, C. Poriel, J. Canivet, G. Calvez, T. Roisnel, V. Dorceta and P. Roussel, A series of chiral metal-organic frameworks based on fluorene di- and tetra-carboxylates: syntheses, crystal structures and luminescence properties, CrystEngComm, 2017, 19, 2042–2056, 10.1039/C7CE00108H.
- J.-P. Ma, Y. Yu and Y.-B. Dong, Fluorene-based Cu(II)-MOF: a visual colorimetric anion sensor and separator based on an anion-exchange approach, Chem. Commun., 2012, 48, 2946–2948, 10.1039/C2CC16800F.
- H. C. J. Zhou and S. Kitagawa, Metal-Organic Frameworks (MOFs), Chem. Soc. Rev., 2014, 43, 5415–5418, 10.1039/C4CS90059F.
- M. Dincă and J. R. Long, Chem. Rev., 2020, 120, 8037–8038 CrossRef PubMed , Porous framework chemistry special issue.
- C. Janiak and J. K. Vieth, MOFs, MILs and more: concepts, properties and applications for porous coordination networks (PCNs), New J. Chem., 2010, 34, 2366–2388, 10.1039/C0NJ00275E.
- G. R. Desiraju, Crystal Engineering: From Molecule to Crystal, J. Am. Chem. Soc., 2013, 135, 9952–9967, DOI:10.1021/ja403264c.
- (a) G.-S. Liou, H.-J. Yen, Y.-T. Su and H.-Y. Lin, Synthesis and Properties of Wholly Aromatic Polymers Bearing Cardo Fluorene Moieties, J. Polym. Sci., Part A: Polym. Chem., 2007, 4352–4357, DOI:10.1002/pola.22182; (b) J. Y. Han, J. Y. Lee, H.-J. Kim, M.-H. Kim, S. G. Han, J. H. Jang, E. A. Cho, S. J. Yoo and D. Henkensmeier, Synthesis and Characterization of Fluorene-Based Polybenzimidazole Copolymer for Gas Separation, J. Appl. Polym. Sci., 2014, 40521, DOI:10.1002/app.40521.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13, DOI:10.1107/S0021889802022112.
- D. Ongari, D. Tiana, S. J. Stoneburner, L. Gagliardi and D. Smit, Origin of the Strong Interaction between Polar Molecules and Copper(II) Paddle-Wheels in Metal Organic Frameworks, J. Phys. Chem. C, 2017, 121, 15135–15144, DOI:10.1021/acs.jpcc.7b02302.
- S. B. Choi, H. Furukawa, H. J. Nam, D. Y. Jung, Y. H. Jhon, A. Walton, D. Book, M. O'Keeffe, O. M. Yaghi and J. Kim, Reversible Interpenetration in a Metal-Organic Framework Triggered by Ligand Removal and Addition, Angew. Chem., Int. Ed., 2012, 51, 8791–8795, DOI:10.1002/anie.201202925.
- S. S. Iremonger, R. Vaidhyanathan, R. K. Mah and G. K. H. Shimizu, Zn7O2(RCOO)10 Clusters and Nitro Aromatic Linkers in a Porous Metal–Organic Framework, Inorg. Chem., 2013, 52, 4124–4126, DOI:10.1021/ic302127y.
- K.-J. Chen, C.-T. He, P.-Q. Liao, Y.-S. Wei, P.-X. Zhang, W. Xue, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, A flexible, porous, cluster-based Zn-pyrazolate-dicarboxylate framework showing selective adsorption properties, New J. Chem., 2014, 38, 2002–2007, 10.1039/C3NJ00918A.
- E. J. Carrington, R. Pétuya, R. K. Hylton, Y. Yan, D. Antypov, G. R. Darling, M. S. Dyer, N. G. Berry, A. P. Katsoulidis and M. J. Rosseinsky, The Anisotropic Responses of a Flexible Metal-Organic Framework Constructed from Asymmetric Flexible Linkers and Heptanuclear Zinc Carboxylate Secondary Building Units, Cryst. Growth Des., 2019, 19, 5604–5618, DOI:10.1021/acs.cgd.9b00558.
- Q. Ding, Y. Pan, Y. Luo, M. Zhou, Y. Guan, B. Li, M. Trivedi, A. Kumar and J. Liu, Photocatalytic and Ferric Ion Sensing Properties of a New Three-Dimensional Metal–Organic Framework Based on Cuboctahedral Secondary Building Units, ACS Omega, 2019, 4, 10775–10783, DOI:10.1021/acsomega.9b01008.
- M86-EXX229V1 APEX3 User Manual, Bruker AXS Inc., Madison, USA, 2016 Search PubMed.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
Footnote |
| † Electronic supplementary information (ESI) available: Asymmetric units for 1 and 2, stacking in 1 and other representations for 2. For 3 and 4, crystallographic table, bonds table and representations of the compounds. For 1 and 2, TGA traces and N2 and CO2 adsorption isotherms, IR spectra in the solid state, together with XRPD diagrams after sorption measurements. For 2, solid-state reflectance spectra at RT. CCDC 2379878, 2379879, 2383979 and 2395186. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01201a |
| This journal is © The Royal Society of Chemistry 2025 |