
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating trends in synthesis and structural periodicity in a series of tetravalent actinide–oxo hexamers†
Thomas L.
McCusker
 a,
Nicole A.
Vanagas
a,
Jennifer E. S.
Szymanowski
b,
Robert G.
Surbella
III
c,
Jeffery A.
Bertke
a,
Ana
Arteaga
*c and
Karah E.
Knope
*a
a,
Nicole A.
Vanagas
a,
Jennifer E. S.
Szymanowski
b,
Robert G.
Surbella
III
c,
Jeffery A.
Bertke
a,
Ana
Arteaga
*c and
Karah E.
Knope
*a
aDepartment of Chemistry, Georgetown University, 37th and O Streets NW, Washington, D.C., 20057, USA. E-mail: tlm102@georgetown.edu; nv137@georgetown.edu; jb2667@georgetown.edu; kek44@georgetown.edu
bDepartment of Civil & Environmental Engineering & Earth Sciences, University of Notre Dame, 301 Stinson-Remick, Notre Dame, Indiana 46556, USA. E-mail: JSzymanowski@nd.edu
cPacific Northwest National Laboratory, 902 Battelle Boulevard, Richland, WA 99354, USA. E-mail: robert.surbella@pnnl.gov; ana.arteaga@pnnl.gov
First published on 13th December 2024
Abstract
Metal ion hydrolysis and condensation reactions are critical to describing the chemical behavior of the tetravalent actinides (An) due to their high charge density. This recognition has fueled synthetic efforts targeting polynuclear actinide–oxo clusters. Oligomers ranging from trimers to octatriacontamers have been reported, with the hexameric unit, which typically exhibits a [An6(OH)4O4]12+ core, representing the most pervasive cluster. Hexamers decorated by a range of ligands, including carboxylates, sulfates, and chlorides, have been described. Previous reports have demonstrated the formation of hexamers for Th, U, Np, and Pu both in solution and the solid state, yet little work has focused on the synthesis and properties of structurally analogous clusters across the early An series using the same complexing ligand. Here, a series of benzoate (Bz) decorated actinide–oxo/hydroxo hexamers of the same general formula [An6O4(OH)4(Bz)12(H2O)n], where An = Th, U, Np, Pu and n = 6 for Th and 4 for U–Pu is reported. The title compounds were characterized by X-ray diffraction, UV-vis–NIR absorbance, Raman, and infrared spectroscopy. Notably isolation of these phases and elucidation of the parameters that underpin their formation provides insight into the ways differences in metal ion charge density manifest across the early tetravalent actinides, both in their synthetic and structural chemistry.
Introduction
The actinides have been the subject of research for decades, namely due to their application in nuclear energy and security; however, their radioactivity and limited abundance have caused our understanding of actinide (An) chemical behavior to lag behind that of the rest of the periodic table. Nonetheless, it has long been recognized that understanding the solution and solid-state speciation of the actinides is critically important to areas ranging from thermodynamics to waste management and environmental transport.1The speciation of actinide elements is dependent on several factors, but is principally governed by oxidation state.2,3 The early actinides, Th–Pu, adopt a common +4 oxidation state and the high charge density of the An4+ ions plays an integral role in describing the chemical behavior of the actinides in aqueous systems.4 Specifically, tetravalent actinides tend to undergo hydrolysis in the presence of water.2,5 The propensity of the An4+ ions to hydrolyze increases with increasing charge density, from Th4+ to Pu4+, as a result of radial contraction brought on by poor shielding of the f-electrons.6 Actinide–hydroxide products are formed as a result of hydrolysis, and these species undergo subsequent condensation reactions to yield oligomers,2 either through oxolation or olation, which are characterized by the formation of oxo or hydroxo bridges, respectively. It is broadly recognized that these species are important to describing fundamental and applied aspects of actinide behavior1,7–11 and have thus motivated investigations into the synthesis, characterization, and reactivity of actinide–oxo clusters.2,12–14
Tetravalent actinide–oxo clusters with nuclearities ranging from 3–38 have been described; the most prevalent structural unit is the hexanuclear cluster, with a [An6O4(OH)4]12+ core.12–44 Synthetic parameters such as pH, temperature, solvent, and ligands influence the assembly of An4+–oxo clusters.27,29,30,45,46 Inorganic ligands, such as sulfate, nitrate, and chlorides, are effective in directing the formation of An4+–oxo clusters. Moreover, hexamers capped by simple organic ligands with relevance to environmental systems, including glycine and formate, have also been reported.21,43,47,48 While there is a growing catalog of An oligomers, few reports have focused on the synthesis and characterization of a series of clusters with the same capping ligand.42 Such a series can be leveraged to understand trends in syntheses, which are reflective of the differences in metal ion acidity, and structural periodicity.
Here a family of tetravalent An4+–oxo clusters (An = Th, U, Np, Pu) stabilized by benzoate capping ligands is described. The structures were determined by single crystal X-ray diffraction (SCXRD), and the vibrational properties were examined using infrared (IR) and Raman spectroscopy. Further, comparison of the ultraviolet-visible-near infrared (UV-vis–NIR) absorption spectra collected for the reaction solutions and solid-state allow identification of the clusters in solution. Differences in synthetic conditions and structural systematics across the series are observed and reflect the contraction in ionic radii, increasing charge density, and resulting hydrolysis and condensation behavior from Th–Pu.
Results and discussion
Synthesis
Reactions of AnCl4 salts or An4+/HCl(aq) solutions with benzoic acid (Bz) yielded four phases; Th6, [Th6O4(OH)4(Bz)12(H2O)6], U6, [U6O4(OH)4(Bz)12(H2O)4]·2(C2H6O)·2.5(H2O), Np6, [Np6O4(OH)4(Bz)12(H2O)4]·x(C2H6O)·y(H2O), and Pu6, [Pu6O4(OH)4(Bz)12(H2O)4]·m(C2H6O)·n(H2O). These polynuclear species result from actinide hydrolysis and condensation reactions, and are important structural units for describing An behavior in aqueous systems.3,16,18,49,50 Isolation of a series of structurally analogous hexamers, as reported here, and consideration of the variations in syntheses underscore differences in Brønsted acidity of metals ions. Moreover, spectroscopic investigations further inform the signatures that can be used for the detection and monitoring of stability of An4+–oxo clusters.The increase in metal ion charge density that occurs from Th4+ to Pu4+ is reflected in the propensity of these metal ions to hydrolyze and is evidenced in thermodynamic data. For example, the formation constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K) for the first hydrolysis product, M(OH)3+, for Th(OH)3+, U(OH)3+, Np(OH)3+, and Pu(OH)3+ are −2.500 ± 0.500, −0.540 ± 0.060, 0.550 ± 0.200, and 0.600 ± 0.200.51,52 Tetravalent Th, with an ionic radius of 1.08 Å, is the largest of the An4+ and least likely to hydrolyze. The decreased susceptibility of Th to hydrolyze as compared to the other An4+ ions is evidenced in this work by observation of a non-hydrolyzed Th chain, [Th(Bz)4]n, which was previously reported by Falaise et al.53 Notably, efforts to prepare the title compound, Th6, by dissolution of ThCl4 in H2O/EtOH (ethanol) were unsuccessful irrespective of pH or metal to ligand ratio. The synthesis of the previously reported UiO-66 metal organic framework (MOF), a well reported MOF with a hexameric node of the general formula [M6O4(OH)4(C8H6O4)12], needed to be employed to target Th6.54 This allowed us to leverage water (which comes from the hydrated thorium chloride salt) as a reactant rather than a solvent, providing greater control over hydrolysis.
K) for the first hydrolysis product, M(OH)3+, for Th(OH)3+, U(OH)3+, Np(OH)3+, and Pu(OH)3+ are −2.500 ± 0.500, −0.540 ± 0.060, 0.550 ± 0.200, and 0.600 ± 0.200.51,52 Tetravalent Th, with an ionic radius of 1.08 Å, is the largest of the An4+ and least likely to hydrolyze. The decreased susceptibility of Th to hydrolyze as compared to the other An4+ ions is evidenced in this work by observation of a non-hydrolyzed Th chain, [Th(Bz)4]n, which was previously reported by Falaise et al.53 Notably, efforts to prepare the title compound, Th6, by dissolution of ThCl4 in H2O/EtOH (ethanol) were unsuccessful irrespective of pH or metal to ligand ratio. The synthesis of the previously reported UiO-66 metal organic framework (MOF), a well reported MOF with a hexameric node of the general formula [M6O4(OH)4(C8H6O4)12], needed to be employed to target Th6.54 This allowed us to leverage water (which comes from the hydrated thorium chloride salt) as a reactant rather than a solvent, providing greater control over hydrolysis.
By comparison, U6, Np6, and Pu6 were synthesized from aqueous H2O/EtOH solutions via slow evaporation (Table 1). For uranium, the second largest ion, slight differences in hydrolysis and condensation behavior are captured in the synthetic conditions such as pH and metal to ligand ratio. A much higher pH is required to isolate U6 than Pu6 and interestingly Np6 forms at a higher pH than U6; however, six times as much benzoic acid is required for both Np6 and Pu6 to limit hydrolysis and direct the assembly of the hexamer. It should be mentioned that for Pu6, this increase in the amount of benzoic acid could be necessary not only to thwart metal ion hydrolysis and condensation but also to offset the lower percentage of benzoic acid that is deprotonated at the lower pH of the Pu6-solution. For reference, the pKa of benzoic acid is 4.19. Ultimately, this study shows that while thorium and uranium are often used as surrogates for Np and Pu, under similar synthetic conditions they often demonstrate their own unique chemical behavior.
| Phase | Solvent | Temperature | M:L ratio |
pH |
|---|---|---|---|---|
| Th-chain | H2O/EtOH | Room Temp. (immediate precipitation) | 1:2–1:12 |
N/A |
| Th 6 | DMF | 130 °C | 1:3.7 |
N/A |
| U 6 | H2O/EtOH | Room Temp. (slow evap.) | 1:2 |
1.13 |
| Np 6 | H2O/EtOH | Room Temp. (slow evap.) | 1:12 |
2.28 |
| Pu 6 | H2O/EtOH | Room Temp. (slow evap.) | 1:12 |
0.64–1.01 |
Structure descriptions
All members of the An6 series consist of [An6O4(OH)4(Bz)12(H2O)n] (where An = Th, U, Np, Pu and n = 6 for Th and 4 for U, Np, Pu). A general description of these clusters is provided here, and further details in the ESI.† In each of the clusters, six An4+ metal centers are bridged through eight μ3-oxygens to form a hexamer of composition, [An6O4(OH)4]12+. Twelve benzoate ligands decorate the cluster core, and a variable number of waters are bound directly to the actinide atoms.Despite the similar core, there are several key differences between Th6 and the rest of the members of the An6 series. The Th6 compound consists of one unique Th coordination environment. The Th metal centers are 9-coordinate and adopt a monocapped square antiprism coordination geometry. Each Th is bound to one water molecule, four oxygens from four benzoates, and four μ3-O/OH groups (Fig. 1). Conversely, the rest of the members of An6 exhibit metal centers with two unique coordination environments (Fig. 2). Like the Th counterpart, one of the metal centers is 9-coordinate and adopts a distorted monocapped square antiprismatic coordination geometry. The An is bound to a water molecule, four oxygen atoms from three benzoate molecules, two of which are monodentate bridging and one that is bidentate, and four μ3-O/OH groups. The second metal is 8-coordinate and adopts a square antiprismatic coordination geometry with the metal bound to four μ3-O/OH and four oxygen atoms from four monodentate bridging benzoates.
 | ||
| Fig. 1 Illustration of (a) Th6 cluster and (b) the local coordination environment about the Th metal centers. Thorium and carbon are shown in blue and black respectively. Oxygen atoms from water and the benzoate ligands are shown in red, and μ3-oxygens in maroon. Hydrogen atoms and disorder have been omitted for clarity. | ||
 | ||
| Fig. 2 Illustration of (a) U6/Np6/Pu6 cluster, (b) the local coordination environment about the 9-coordinate metal centers, and (c) the local coordination environment about the 8-coordinate metal centers. Uranium/neptunium/plutonium and carbon are shown in green and black respectively. Oxygen, water, and benzoate oxygens are shown in red, and μ3-oxygens are shown in maroon. Hydrogen atoms and disorder have been omitted for clarity. | ||
Other structural differences are consistent with larger trends in periodicity in the actinide series. Most notably is the decrease in metal–oxygen bond lengths for An–μ3-O/OH, An–Owater, and An–Obenzoate from Th6 to Pu6, (Table 2). The decrease in bond length from Th6 to Pu6 is consistent with the decrease in ionic radii from Th to Pu that arises from the actinide contraction. Another notable difference is reflected in the coordination environment from Th6 to Pu6. The compound Th6 is entirely built from 9-coordinate metal centers while U6–Pu6 exhibit both 8- and 9-coordinate metal centers.
| Compound | An coordination number | An–μ3O (Å) | An–μ3OH (Å) | An–Obenzoate (Å) | An–Owater (Å) | An–An distance (Å) |
|---|---|---|---|---|---|---|
| a Only a preliminary refinement of Np6 was obtained, therefore average An–Obenzoate and An–Owater distances are not provided. | ||||||
| Th 6 | 9 | 2.31(1) | 2.51(1) | 2.49(2) | 2.70(1) | 3.944(3) |
| U 6 | 8/9 | 2.25(4) | 2.462(37) | 2.458(10) | 2.46(1) | 3.84(5) |
| Np 6 | — | 2.22(12) | 2.41(15) | N/A | N/A | 3.81(5) |
| Pu 6 | 8/9 | 2.23(3) | 2.44(4) | 2.45(1) | 2.41(3) | 3.80(5) |
Solution and solid-state UV-vis–NIR absorbance spectra
UV-vis–NIR absorption spectroscopy is a powerful technique for differentiating mononuclear and polynuclear An species.23,24,42,45,46 As such, the solution and solid-state absorption spectra for U6, Np6, Pu6 were collected and compared to the An4+/HCl stock solution. For U6 and Np6 the solution from which crystals precipitated was also analyzed.The UCl4 salt dissolved in 1 M HCl displays f–f transitions characteristic of U4+ (Fig. 3), with peaks observed at 495 nm, 549 nm, 648 nm, and 672 nm.55 Notably, the spectrum of U4+ in 1 M HCl is consistent with that reported for U–H2O–Cl species in aqueous solutions.45 Upon the addition of benzoic acid in ethanol (U6-solution), the peak centered at 663 nm increases in intensity relative to the peak centered at 648 nm. These bands are attributed to three transitions (3H4 → 3P0, 3H4 → 1G4, and 3H4 → 1D2), and previous work has shown the relationship between the higher wavelength (i.e., 663 nm) and lower wavelength (648 nm) is diagnostic for the hexanuclear U4+–oxo/hydroxo clusters in solution. The increased intensity of the 663 nm peak relative to the peak centered at 648 nm is consistent with the hexameric unit.44,45 Interestingly, comparison of the U6-crystals to solution shows notable differences, particularly between 600 nm and 700 nm; these differences have been observed in previously reported uranium hexamers and are attributed to differences in ligand binding and lattice solvent.46
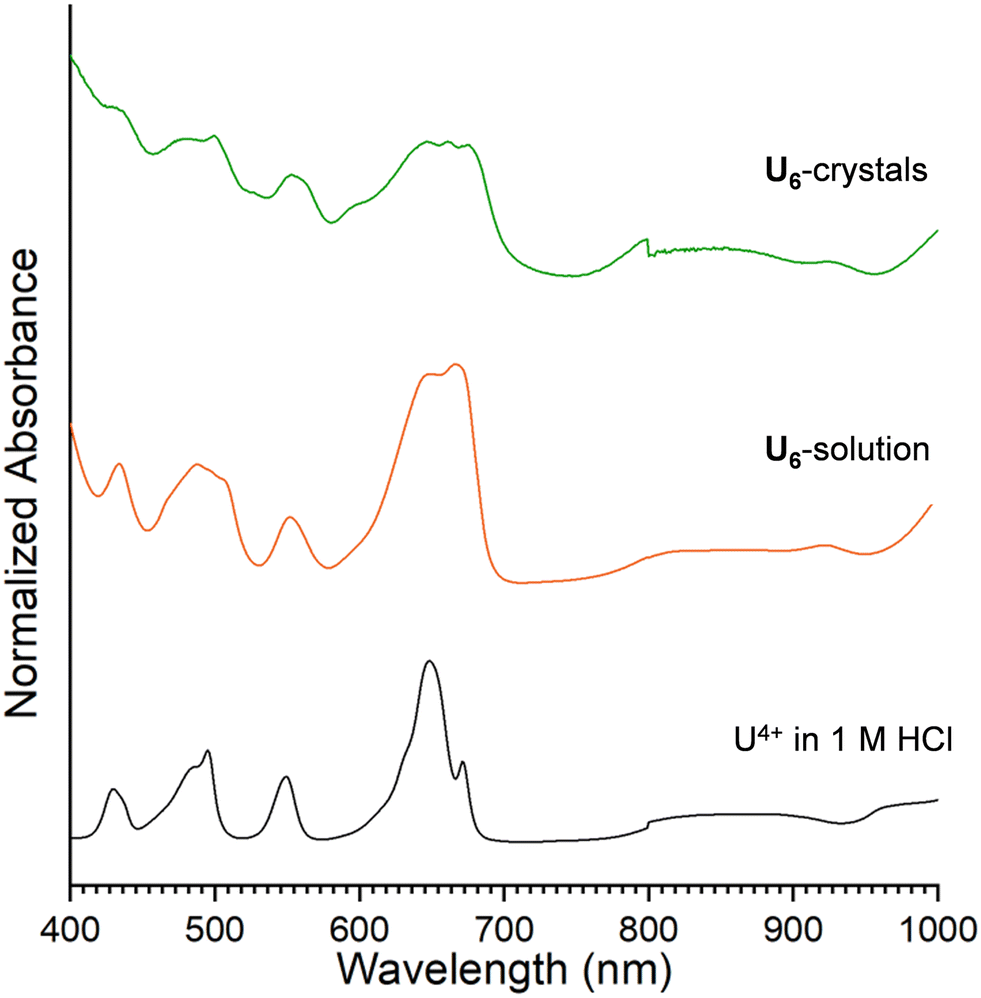 | ||
| Fig. 3 UV-vis–NIR absorption spectra of UCl4 in 1 M HCl (black), U6 reaction solution (orange), and U6 crystals (green). | ||
The UV-vis–NIR absorption data of Np4+ in 1 M HCl, Np6-solution, and Np6-crystals are characteristic of Np4+ transitions with peaks at 590 nm, 730 nm, 820 nm, and 960 nm (Fig. 4).56 Comparison of the Np4+ in 1 M HCl spectrum to that collected for the Np6-solution shows a significant shift in the band centered at 730 nm to 740 nm. Such a red shift was also observed by Takao et al. and may be attributed to hexamer formation.23 Also consistent with previous work is the growth in intensity of peaks at 604 nm and 899 nm in the Np6-crystals spectrum relative to the Np solutions.23 While the development of spectroscopic handles for neptunium-based clusters remains underdeveloped compared to uranium, this work further suggests spectral features may be diagnostic of neptunium oligomer formation and stability.
 | ||
| Fig. 4 UV-vis–NIR absorption spectra of Np4+ in 1 M HCl (brown), Np6-solution (dark red), and Np6-crystals (red). | ||
The UV-vis–NIR absorption spectra of Pu4+ in 1 M HCl and Pu6-crystals are shown in Fig. 5. The Pu4+ stock solution spectrum exhibits peaks at 470 nm, 660 nm, 800 nm, and 1080 nm, all characteristic of Pu4+ in HCl.57 Unfortunately, attempts to collect a UV-vis–NIR absorption spectrum of the Pu6-solution were precluded by the low concentration of the plutonium. Nonetheless, comparison of the UV-vis–NIR absorption spectra of Pu4+ in 1 M HCl and Pu6-crystals suggests a shift in the peak centered at ∼470 nm to lower wavelengths (i.e., 457 nm) (Fig. 5). The observed shift is diagnostic of Pu hexamers, as reported by Chupin et al. and Tamain et al., where a blueshift in the 475 nm peak is typically observed in acidic plutonium solutions to 457–458 nm, which both sources claim is indicative of plutonium complexation.24,42 Such observations further suggest that absorption spectroscopy is a powerful handle for monitoring oligomer formation and stability.
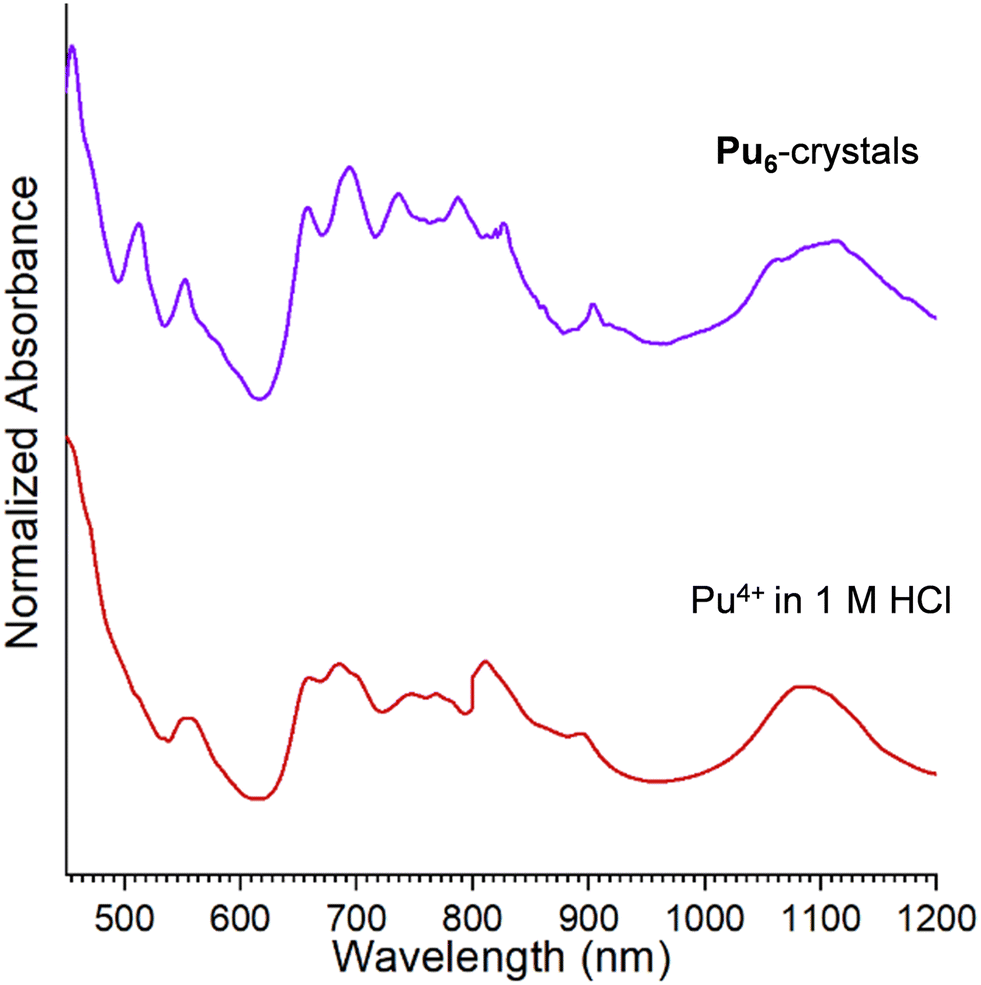 | ||
| Fig. 5 UV-vis–NIR absorption spectra of Pu4+ in 1 M HCl (red) and Pu6-crystals (purple). | ||
Vibrational spectroscopy
The vibrational spectra of Th6, U6, Np6, and Pu6 were collected to further examine how the actinide contraction manifests in spectroscopic properties. The IR spectra of the compounds are shown in Fig. 6 and a complete listing of the observed bands is provided in the ESI.† As expected, the spectra are largely dominated by vibrational modes of the benzoate ligands. The medium intensity peak centered at 1390 cm−1 is the result of a C–C aromatic stretch, the split bands between 1530–1690 cm−1 are a result of a conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, and the broad peak above 3000 cm−1 is indicative of aromatic C–H stretches.58 Notably, these stretches are all present within the spectrum of Np6, further suggesting that benzoates do in fact decorate the cluster. Also, the sharp peak above 3600 cm−1 is attributed to the μ3-OH stretch.59 This peak is evident for Th6, however is less intense for Pu6 and Np6 and not present for U6. This is likely the result of the overall lower IR intensity for U6 relative to the other phases. Somewhat surprisingly, no peaks within the IR appear to trend with differences in bond lengths across the series. This is likely due to the fact that An–O are likely low-frequency vibrations outside the range of the collected spectra.28
O stretch, and the broad peak above 3000 cm−1 is indicative of aromatic C–H stretches.58 Notably, these stretches are all present within the spectrum of Np6, further suggesting that benzoates do in fact decorate the cluster. Also, the sharp peak above 3600 cm−1 is attributed to the μ3-OH stretch.59 This peak is evident for Th6, however is less intense for Pu6 and Np6 and not present for U6. This is likely the result of the overall lower IR intensity for U6 relative to the other phases. Somewhat surprisingly, no peaks within the IR appear to trend with differences in bond lengths across the series. This is likely due to the fact that An–O are likely low-frequency vibrations outside the range of the collected spectra.28
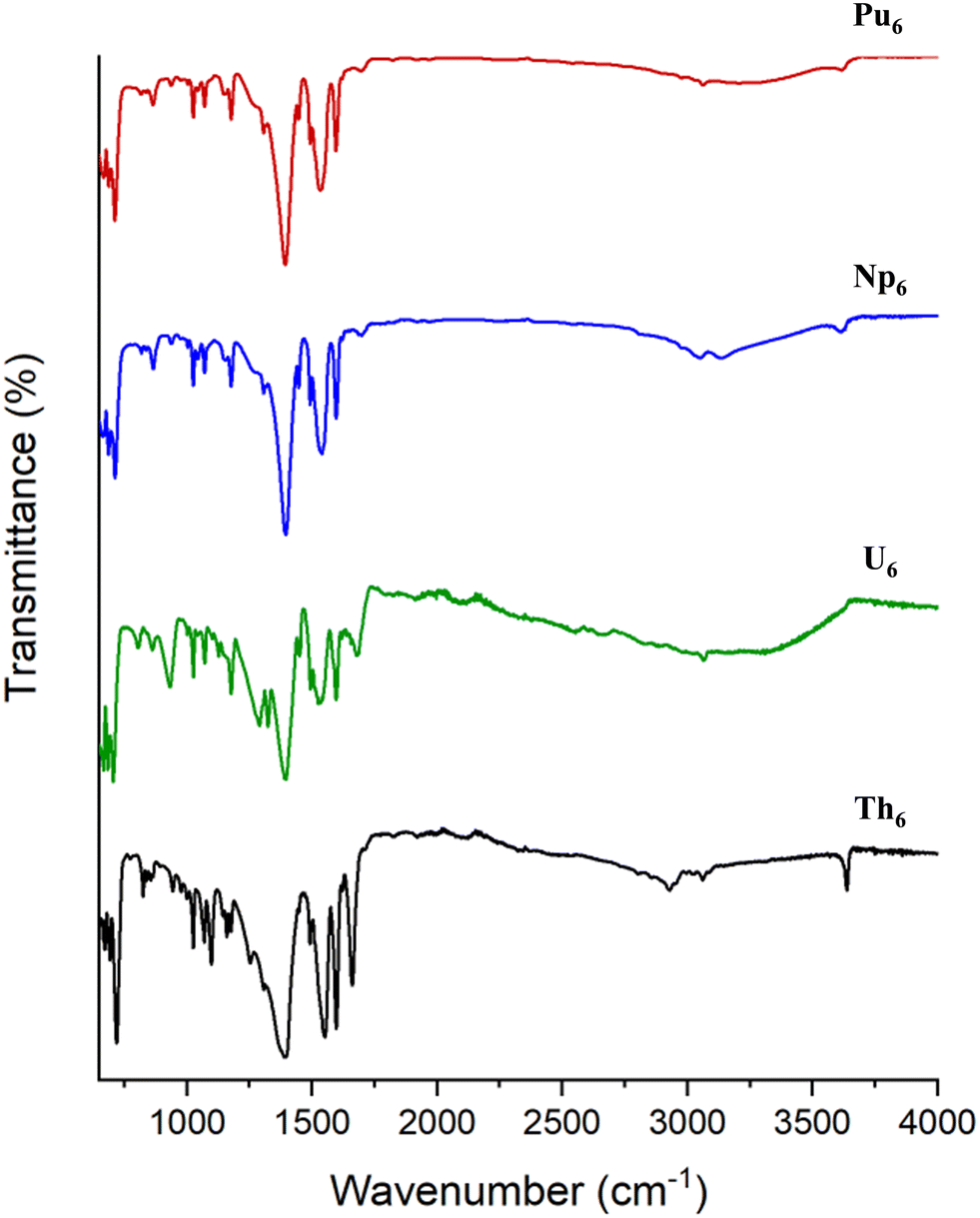 | ||
| Fig. 6 Infrared spectra of Th6 (black), U6 (green), Np6 (blue), and Pu6 (red). | ||
The Raman spectra of each of the clusters agree with each other and exhibit bands consistent with the decorating benzoate ligand (Fig. S9†). Specifically, peaks observed at ∼835 cm−1 (COO– in plane scissoring), 1003 cm−1 (aromatic ring stretch), and 1604 cm−1 (aromatic ring stretch) are all attributed to benzoate.60 As for the expected An–O stretches, based on the mass of the actinide ion relative to the rest of the atoms in the cluster, they would most likely appear within the low frequency region (≤500 cm−1).59 Unfortunately, like the IR spectra, stretches in this portion of the spectra are low resolution and therefore, their assignment cannot be made with certainty.
Conclusions
A series of four An4+–oxo hexamers (An = Th, U, Np, Pu) is reported. While such clusters have been previously described, this work examines the formation of an analogous series of hexamers across the early actinides, Th–Pu, such that the effects of increasing charge density, and hence metal ion acidity, on synthesis could be examined. The decreased Brønsted acidity of Th relative to the later actinides, U–Pu, is evidenced in this work by observation of a non-hydrolyzed Th chain, [Th(Bz)4]n and the inability to translate the synthetic conditions used to isolate U6, Np6, and Pu6. Further, structural characterization of the phases through single crystal X-ray diffraction show that U and Pu adopt the same cluster core, while Th differs in the number and connectivity of the decorating ligands; these differences reflect trends in periodicity that arise from the actinide contraction. Additionally, investigations into the UV-vis–NIR absorption behavior of these compounds and the solutions from which they crystallize lend insight into spectroscopic handles that can be used for the identification of these species in both in solution and the solid state.Experimental methods
Synthetic procedure
CAUTION: thorium-232 (t1/2 = 1.42 × 1010y), uranium-238 (t1/2 = 4.5 × 109y), neptunium-237 (t1/2 = 2.1 × 106y), and plutonium-239 (t1/2 = 2.4 × 104y) are hazardous α and γ emitting radionuclides. Bulk transuranium samples pose a serious health risk and must be studied in a facility designed and designated for the handling of highly radioactive and toxic heavy metals. Bulk material manipulations were conducted in a negative pressure radiological glovebox, while subsamples were handled within radiological fume hoods.Thorium chloride (ThCl4) (International Bioanalytical Industries, Inc.), uranyl oxide (UO3) (International Bioanalytical Industries, Inc.), hexachloropropene (Sigma Aldrich), benzoic acid (BA, Fisher Scientific), hydroxylamine hydrochloride (Fisher Scientific), and ethanol (EtOH, Fisher Scientific) were all used as received from the commercial suppliers. Dimethylformamide (DMF, Fisher Scientific) was dried overnight on molecular sieves (Fisher Scientific) prior to use. Uranium chloride (UCl4) was prepared from UO3 in hexachloropropene following the approach designed by Patel et al.61 Preparation of the Np4+ and Pu4+ stock solutions is described below. Nanopure water (≤0.05 μS; Millipore USA) was used in all reactions.
The compound Th6, [Th6O4(OH)4(C7H5O2)12(H2O)6], was prepared using an approach adapted from that described for Th–UiO-66, [Th6O4(OH)4(C8H6O4)12].54 ThCl4 (0.010 g, 0.0267 mmol) and 300 μL of a 0.328 M solution of benzoic acid in DMF (0.012 g, 0.0984 mmol) were added to a 23 mL Teflon cup. The Teflon cup was placed in a 23-mL Parr acid digestion vessel and heated statically at 130 °C for 24 hours. The vessel was removed from the oven and allowed to cool to room temperature prior to opening. Slow evaporation of the resulting colorless solution under ambient conditions in a 4 dram shell vial over 24 hours yielded colorless block-like crystals. Yield: 44% based on Th4+.
The compound U6, [U6O4(OH)4(C7H5O2)12(H2O)4]·2(C2H6O)·2.5(H2O), was synthesized at room temperature in a nitrogen filled glovebox. The latter was used to limit oxidation of uranium. The starting material, UCl4 (0.075 g, 0.197 mmol), was dissolved in H2O (1 mL) in a 4 dram shell vial. In a separate vial, BA (0.048 g, 0.393 mmol) was dissolved in EtOH (1 mL). The organic and aqueous solutions were combined, and the resulting solution was left to slowly evaporate for 12 hours. The initial pH of the solution was 1.13. Green crystals precipitated over 24 hours. Yield: 48% based on U4+.
The compound Np6, [Np6O4(OH)4(C7H5O2)12(H2O)4]·x(C2H6O)·y(H2O) was synthesized using a similar approach as U6. A Np4+ solution was prepared via chemical reduction of a Np5+/HCl solution (0.702 mL, 30 mM solution, 0.0211 mmol of 237Np) with hydroxylamine hydrochloride (21 mg, 0.757 mmol). The oxidation state of Np was confirmed via UV-vis absorption spectroscopy (Fig. 4). Following the reduction to Np4+, the solution was evaporated to near dryness and then reconstituted with 0.500 mL of water. Benzoic acid (32 mg, 0.262 mmol) was dissolved in EtOH (0.5 mL), and the resulting solution was added to the Np4+ stock solution; the pH was 0.12. The reaction mixture was diluted with EtOH (1.250 mL); the pH of the resulting solution was 2.28. The solution was left to evaporate and pale pink crystals precipitated over 24 hours. The crystals diffracted poorly; however, both the unit cell and a preliminary refinement of Np6 are consistent with a benzoate decorated hexamer. Powder X-ray diffraction (PXRD) further confirmed that Np6 was representative of the bulk precipitate (Fig. S8†).
The compound Pu6, [Pu6O4(OH)4(C7H5O2)12(H2O)4]·m(C2H6O)·n(H2O), was synthesized following the same general synthetic approach as that described for U6 and Np6. A Pu4+ stock solution was prepared by heating 239Pu (36 mg) in 6 M HCl to near dryness. This resulting residue was then redissolved in 2.4 mL of 0.5 M HCl (total concentration: 18.67 mg mL−1). A solution of BA (9.09 mg, 0.0744 mmol) in 800 μL of a 1:1 H2O/EtOH mixture was added to a Pu4+/HCl stock solution (80.0 μL, 1.5 mg, 0.00628 mmol of 239Pu). The initial pH was 0.6. The pH of the solution was adjusted to 1.0 using concentrated (50% wt/wt) NaOH. The solution was then slowly evaporated over 72 hours, after which red crystals and clear acicular needle-like crystals consistent with recrystallized BA were observed.
Powder X-ray diffraction
Powder X-ray diffraction data were collected for Th6, U6, and Np6 using a Rigaku Ultima IV diffractometer (Cu K = 1.524 Å, 2 = 3–40°). The Np6 compound was placed on a zero-background plate within a Bruker sample holder and contained using a Kapton® film.62 Calculated diffraction patterns were generated using CIFs of Th6, U6, and Np6 and were compared to the experimental patterns. Agreement between the calculated and experimental patterns suggest that the crystals used for structure determination were representative of the bulk reaction product.Vibrational spectroscopy
Raman spectra of Th6 and U6 were collected on single crystals using an HORIBA LabRAM HR Evolution Raman spectrometer equipped with a 532 nm laser. Spectra were collected at room temperature from 100–3200 cm−1. Raman spectra of Np6 and Pu6 were collected on single crystals using a Renishaw inVia™ Raman spectrophotometer equipped with a 532 nm and 785 nm laser, respectively. Data were collected at room temperature from 100–3200 cm−1. Infrared spectra for Th6 and U6 were collected using an Agilent Cary 630 FTIR. Single crystals were handpicked for measurement and spectra were collected from 650–4000 cm−1. Infrared spectra for Np6 and Pu6 were collected using a Bruker Lumos FTIR spectrophotometer equipped with an attenuated total reflection accessory. Spectra were collected from 600–4000 cm−1 and data were processed using the OPUS V.7.2 software.Optical UV-vis–NIR absorption spectroscopy
The solid-state UV-vis–NIR optical absorption spectrum was collected for U6 on an Agilent Cary 5000 UV-vis–NIR spectrophotometer. The sample was prepared by grinding individual crystals into dry KBr (Fisher Scientific) and placing the mixture into a solid-state sample holder. Spectra were collected from 200–1000 nm. The UV-vis–NIR absorption data for Np6 and Pu6 were collected on single crystals using a Craic solid-state UV-vis–NIR microscope spectrophotometer due to the limited yield. Crystals were transferred to a glass slide that was covered with a glass coverslip and sealed with epoxy. Spectra were collected from 200–1000 nm.Solution-state data for U6 and other uranium-based solutions were collected on an Agilent Cary 5000 UV-vis–NIR spectrophotometer. Samples were placed in a quartz cuvette and spectra were collected from 200–1000 nm. Solution-state data for Np6, Pu6, as well as neptunium and plutonium-based solutions, were collected using a Mikropack DH-2000-BALL deuterium and halogen light source coupled to an Ocean Optics Flame detector. Scattered light was collected with a fiber-optic cable. Data were then processed using OceanView spectroscopy software (V.2.0.8.).
Structure determination
Single crystals of Th6, and U6 were selected from the bulk reaction product, mounted on MiTeGen micromounts in mineral oil, and placed on a Bruker D8 QUEST diffractometer equipped with a Photon 100 detector and a Mo IμS source (K = 0.71073 Å). Crystals of Np6 and Pu6 were mounted and super glued to MiTeGen micromounts, then covered with a heat-shrinkable sheath that was glued to the mount for secondary containment.62 The single-crystal diffraction data were collected on a Bruker D8 Venture diffractometer equipped with a CMOS detector and a Mo IS source (K = 0.71073 Å). For all samples, data were reduced using SAINT and absorption corrections were applied using SADABS, both within the APEX III software.63–65 The structure solutions were all performed using intrinsic phasing method and structure refinements were conducted using the ShelXle software.66,67 It is important to note for Np6, only a preliminary data collection and refinement was possible due to poor crystal quality and hence limited diffraction. As such, only unit cell parameters are provided. Further details of the refinement of Th6, U6, and Pu6 are provided in the ESI.†Data availability
Data supporting the results presented in this manuscript including refinement details, thermal ellipsoid plots, powder X-ray diffraction patterns, and Raman spectra are provided as ESI.† Accession codes CCDC 2387949–2387951 contain the ESI† crystallographic data for this paper.Author contributions
Please use https://credit.niso.org/, we have been asked by reviewers to use this method to acknowledge the contribution of each of the authors.All authors have approved the final version of this manuscript. All authors contributed equally to this work. CRediT author statement. TLM: conceptualization, methodology, validation, formal analysis, investigation, writing, visualization. NAV: conceptualization, methodology, investigation. RGS: methodology, supervision. JESS: investigation. JAB: formal analysis. AA: methodology, formal analysis, investigation, resources, writing, supervision. KEK: conceptualization, methodology, formal analysis, resources, writing, and supervision.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Synthesis and characterization of the thorium and uranium compounds were performed at Georgetown University, under the support of the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Early Career Research Program under Award DE-SC0019190. Both TLM and KEK were supported by this award. The neptunium and plutonium work was conducted at Pacific Northwest National Laboratory by TLM, RGS, and AA. TLM was supported under award DE-SC0019190. The funding mechanism for work performed by TLM and AA at PNNL was the Laboratory Directed Research and Development program at Pacific Northwest National Laboratory, a multiprogram national laboratory operated by Battelle for the Department of Energy. AA is grateful for support from the Linus Pauling Distinguished Postdoctoral Fellowship. RGS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences, Heavy Element Chemistry program, under Award FWP 73200. The authors thank Ashley Hastings for providing insight into synthetic techniques which led to the successful isolation of the Th compound and Lulio Sanz for assistance in collecting solid-state UV-vis absorption data of U6.References
- G. Choppin, J. Radioanal. Nucl. Chem., 2007, 273, 695–703 CrossRef.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944–994 CrossRef.
- T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69–109 CrossRef.
- K. E. Knope and L. Soderholm, Inorg. Chem., 2013, 52, 6770–6772 CrossRef PubMed.
- V. Neck and J. I. Kim, Radiochim. Acta, 2001, 89, 1–16 CrossRef.
- W. Küchle, M. Dolg and H. Stoll, J. Phys. Chem. A, 1997, 101, 7128–7133 CrossRef.
- A. P. Novikov, S. N. Kalmykov, S. Utsunomiya, R. C. Ewing, F. Horreard, A. Merkulov, S. B. Clark, V. V. Tkachev and B. F. Myasoedov, Science, 2006, 314, 638–641 CrossRef.
- A. B. Kersting, Inorg. Chem., 2013, 52, 3533–3546 CrossRef PubMed.
- K. Maher, J. R. Bargar and G. E. Brown Jr, Inorg. Chem., 2013, 52, 3510–3532 CrossRef.
- L. S. Natrajan, A. N. Swinburne, M. B. Andrews, S. Randall and S. L. Heath, Coord. Chem. Rev., 2014, 266–267, 171–193 CrossRef.
- G. L. Johnson and L. M. Toth, Plutonium (IV) and thorium (IV) hydrous polymer chemistry. [Conversion of hydrolye-bridged polymer links to oxygen-bridged linkages], Oak Ridge National Lab. (ORNL), Oak Ridge, TN (United States), 1978 Search PubMed.
- I. Colliard, J. C. Brown and M. Nyman, Inorg. Chem., 2022, 62, 1891–1900 CrossRef PubMed.
- R. Faizova, F. Fadaei-Tirani, R. Bernier-Latmani and M. Mazzanti, Am. Ethnol., 2020, 132, 6822–6825 Search PubMed.
- D. Zhou, Y. Yang, Z. Weng, J. Wang, Y. Yan, L. Cheng, Y. Fan, L. Chen, H. Zhang and L. Chen, Inorg. Chem., 2024, 63, 14278–14283 CrossRef.
- G. E. Sigmon and A. E. Hixon, Chem. – Eur. J., 2019, 25, 2463–2466 CrossRef PubMed.
- N. P. Martin, C. Volkringer, P. Roussel, J. März, C. Hennig, T. Loiseau and A. Ikeda-Ohno, Chem. Commun., 2018, 54, 10060–10063 RSC.
- N. P. Martin, C. Volkringer, N. Henry, X. Trivelli, G. Stoclet, A. Ikeda-Ohno and T. Loiseau, Chem. Sci., 2018, 9, 5021–5032 RSC.
- C. Falaise, C. Volkringer, J.-F. Vigier, A. Beaurain, P. Roussel, P. Rabu and T. Loiseau, J. Am. Chem. Soc., 2013, 135, 15678–15681 CrossRef CAS PubMed.
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298–302 CrossRef.
- D. Ray, J. Xie, J. White, G. E. Sigmon, L. Gagliardi and A. E. Hixon, Chem. – Eur. J., 2020, 26, 8115–8120 CrossRef PubMed.
- R. E. Wilson, S. Skanthakumar, G. Sigmon, P. C. Burns and L. Soderholm, Inorg. Chem., 2007, 46, 2368–2372 CrossRef PubMed.
- C. Tamain, T. Dumas, D. Guillaumont, C. Hennig and P. Guilbaud, Eur. J. Inorg. Chem., 2016, 2016, 3536–3540 CrossRef.
- K. Takao, S. Takao, A. C. Scheinost, G. Bernhard and C. Hennig, Inorg. Chem., 2012, 51, 1336–1344 CrossRef PubMed.
- G. Chupin, C. Tamain, T. Dumas, P. L. Solari, P. Moisy and D. Guillaumont, Inorg. Chem., 2022, 61, 4806–4817 CrossRef PubMed.
- I. Colliard, G. Morrison, H.-C. zur Loye and M. Nyman, J. Am. Chem. Soc., 2020, 142, 9039–9047 CrossRef PubMed.
- S. T. Tsantis, D. I. Tzimopoulos, M. Holynska and S. P. Perlepes, Int. J. Mol. Sci., 2020, 21, 555 CrossRef PubMed.
- Y.-J. Hu, K. E. Knope, S. Skanthakumar and L. Soderholm, Eur. J. Inorg. Chem., 2013, 2013, 4159–4163 CrossRef.
- K. E. Knope, M. Vasiliu, D. A. Dixon and L. Soderholm, Inorg. Chem., 2012, 51, 4239–4249 CrossRef.
- C. Falaise, K. Kozma and M. Nyman, Chem. – Eur. J., 2018, 24, 14226–14232 CrossRef PubMed.
- L. Chatelain, R. Faizova, F. Fadaei-Tirani, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2019, 58, 3021–3026 CrossRef PubMed.
- Z.-J. Li, S. Guo, H. Lu, Y. Xu, Z. Yue, L. Weng, X. Guo, J. Lin and J.-Q. Wang, Inorg. Chem. Front., 2020, 7, 260–269 RSC.
- P. Woidy and F. Kraus, Z. Anorg. Allg. Chem., 2014, 640, 1547–1550 CrossRef.
- C. Falaise, C. Volkringer, C. Hennig and T. Loiseau, Chem. – Eur. J., 2015, 21, 16654–16664 CrossRef.
- Z. Yue, X. Guo, M.-L. Feng, Y.-J. Lin, Y. Ju, X. Lin, Z.-H. Zhang, X. Guo, J. Lin and Y.-Y. Huang, Inorg. Chem., 2020, 59, 2348–2357 CrossRef.
- S. T. Tsantis, A. Lagou-Rekka, K. F. Konidaris, C. P. Raptopoulou, V. Bekiari, V. Psycharis and S. P. Perlepes, Dalton Trans., 2019, 48, 15668–15678 RSC.
- N. E. Travia, B. L. Scott and J. L. Kiplinger, Chem. – Eur. J., 2014, 20, 16846–16852 CrossRef.
- K. E. Knope, S. Skanthakumar and L. Soderholm, Inorg. Chem., 2015, 54, 10192–10196 CrossRef PubMed.
- J. Lin, M. Qie, L. Zhang, X. Wang, Y. Lin, W. Liu, H. Bao and J. Wang, Inorg. Chem., 2017, 56, 14198–14205 CrossRef PubMed.
- X.-Y. Qian, T.-H. Zhou and J.-G. Mao, Dalton Trans., 2015, 44, 13573–13580 RSC.
- D. K. Unruh, J. de Groot, M. Fairley, A. Libo, S. Miller and T. Z. Forbes, Inorg. Chem., 2015, 54, 1395–1404 CrossRef.
- S. Fichter, T. Radoske and A. Ikeda-Ohno, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2021, 77, 847–852 CrossRef.
- C. Tamain, T. Dumas, C. Hennig and P. Guilbaud, Chem. – Eur. J., 2017, 23, 6864–6875 CrossRef.
- L. M. Mokry, N. S. Dean and C. J. Carrano, Angew. Chem., Int. Ed. Engl., 1996, 35, 1497–1498 CrossRef.
- N. A. Vanagas, J. N. Wacker, C. L. Rom, E. N. Glass, I. Colliard, Y. Qiao, J. A. Bertke, E. Van Keuren, E. J. Schelter, M. Nyman and K. E. Knope, Inorg. Chem., 2018, 57, 7259–7269 CrossRef.
- C. Falaise, H. A. Neal and M. Nyman, Inorg. Chem., 2017, 56, 6591–6598 CrossRef PubMed.
- N. A. Vanagas, R. F. Higgins, J. N. Wacker, D. R. C. Asuigui, E. Warzecha, S. A. Kozimor, S. L. Stoll, E. J. Schelter, J. A. Bertke and K. E. Knope, Chem. – Eur. J., 2020, 26, 5872–5886 CrossRef PubMed.
- I. Colliard, C. Falaise and M. Nyman, Inorg. Chem., 2020, 59, 17049–17057 CrossRef PubMed.
- X. Kong, X. Liao, Z. Huang, L. Mei, H. Wang, K. Hu and W. Shi, Chin. Chem. Lett., 2024, 35, 109642 CrossRef.
- C. Falaise, C. Volkringer, J.-F. Vigier, N. Henry, A. Beaurain and T. Loiseau, Chem. – Eur. J., 2013, 19, 5324–5331 CrossRef.
- N. P. Martin, J. März, H. Feuchter, S. Duval, P. Roussel, N. Henry, A. Ikeda-Ohno, T. Loiseau and C. Volkringer, Chem. Commun., 2018, 54, 6979–6982 RSC.
- M. Rand, J. Fuger, I. Grenthe, V. Neck and D. Rai, Chemical Thermodynamics of Thorium, Chemical Thermodynamics, OECD Publishing, Paris, 2008 Search PubMed.
- I. Grenthe, X. Gaona, L. Rao, A. Plyasunov, W. Runde, B. Grambow, R. Konings, A. Smith, E. Moore and M.-E. Ragoussi, Second update on the chemical thermodynamics of uranium, neptunium, plutonium, americium and technetium, Chemical thermodynamics, Organisation for Economic Co-Operation and Development, 2020, vol. 14 Search PubMed.
- C. Falaise, C. Volkringer and T. Loiseau, Inorg. Chem. Commun., 2014, 39, 26–30 CrossRef.
- C. Falaise, J.-S. Charles, C. Volkringer and T. Loiseau, Inorg. Chem., 2015, 54, 2235–2242 CrossRef PubMed.
- D. Cohen and W. T. Carnall, J. Phys. Chem., 1960, 64, 1933–1936 CrossRef.
- J. C. Hindman, L. B. Magnusson and T. J. LaChapelle, J. Am. Chem. Soc., 1949, 71, 687–693 CrossRef.
- M. H. Lee, Y. J. Park and W. H. Kim, J. Radioanal. Nucl. Chem., 2007, 273, 375–382 CrossRef.
- M. Kumar, R. Bala, V. S. Gondil, S. K. Pandey, S. Chhibber, D. V. S. Jain, R. K. Sharma and N. Wangoo, J. Mater. Sci., 2017, 52, 8568–8575 CrossRef.
- A. M. Hastings, D. Ray, W. Jeong, L. Gagliardi, O. K. Farha and A. E. Hixon, J. Am. Chem. Soc., 2020, 142, 9363–9371 CrossRef PubMed.
- W. Xi and A. J. Haes, J. Chem. Phys., 2020, 153, 184707 CrossRef PubMed.
- D. Patel, A. J. Wooles, E. Hashem, H. Omorodion, R. J. Baker and S. T. Liddle, New J. Chem., 2015, 39, 7559–7562 RSC.
- A. Arteaga, A. D. Nicholas, L. C. Ducati, J. Autschbach and R. G. Surbella III, Inorg. Chem., 2023, 62, 4814–4822 CrossRef PubMed.
- APEX2, version 2010.7, Bruker AXS Inc., Madison, WI, 2010 Search PubMed.
- SAINT, Bruker AXS Inc., Madison, WI, 2007 Search PubMed.
- SADABS, Bruker AXS Inc., Madison, WI, 2008 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic refinement details, thermal ellipsoid plots, powder X-ray diffraction patterns, and Raman spectra. Accession codes CCDC 2387949–2387951. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01042f |
| This journal is © The Royal Society of Chemistry 2025 |