DOI:
10.1039/D4CE00992D
(Paper)
CrystEngComm, 2025,
27, 46-54
Positional isomers of (E)-2-(anthracen-9-ylmethylene)-N-(aryl)hydrazinecarbothioamide, zinc complexes and polymorphic solvates†
Received
30th September 2024
, Accepted 20th November 2024
First published on 21st November 2024
Abstract
The synthesis, energy calculation and analysis of the weak interactions of two sets of positional isomers of (E)-2-(anthracen-9-ylmethylene)-N-(aryl)hydrazinecarbothioamide, one set with aryl = 3-OCH3- or 4-OCH3-phenyl and the other with aryl = 2,3 or 3,4-dichlorophenyl, were carried out. The nature of substituent/s and their positions in the rings rendered large differences in the angle between the anthracenyl and aryl planes at the distal ends. The respective bis-chelated zinc complexes of these positional isomers had a see-saw geometry. There was one dimethylformamide (DMF) molecule of crystallisation in the zinc complex of the bis-chelated 3-methoxyphenyl containing ligand, but a similar zinc complex with a 4-methoxyphenyl containing ligand had three DMF molecules per complex, due to the packing differences contributed through interplay of weak interactions. The zinc complex of the 2,3-dichloroaryl derived ligand had chloroform as a solvent crystallisation molecule and it showed two polymorphs of the chloroform solvated complex. One of the solvate polymorphs had intermolecular association with chloroform, it lost crystallinity very easily. Meanwhile, the other polymorphic solvate had intra-molecular hydrogen bonded chloroform molecules, crystals of this polymorph were stable enough to be characterized by several spectroscopic tools. The stable polymorph had S⋯π interactions forming a dimeric assembly. A similar zinc complex of the 3,4-dichloro-aryl group derived ligand had extensive stacking among the anthracenyl units in the lattice. It accommodated chloroform molecules of crystallization embedded in the lattice.
Introduction
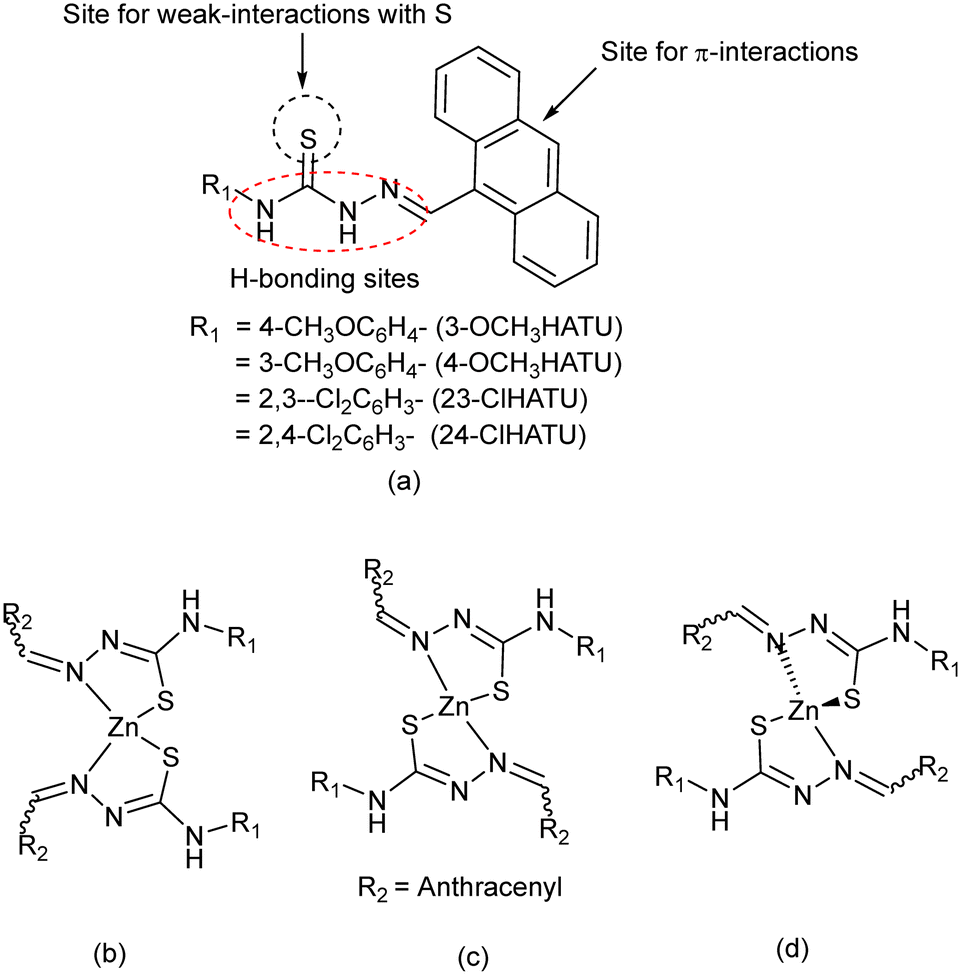
Thiourea-based compounds have generated interest from supramolecular chemistry1 and organo-catalysis,2 as medicinal compounds3 and in pesticides.4 Metal complexes of hydrazinethiocarbazides belong to such a class of complexes5 which show biological activities6 and serve as hosts for molecular recognitions.7 Positional isomers of thiocarbazides have also generated interest for solvation8 and have been functionalised to show interesting properties in different fields, such as emissive9 and energy materials,10 stabilisation of less-stable intermediates,11 signal transductions,12 and to generate different packing arrangements.13 The thiocarbazides have options to adopt the thione or thiol form,14 and also to adopt equilibrating geometries.15,16 The stabilisation of any such form is influenced by intramolecular interactions or solute–solvent interactions. We have chosen to study a series of compounds depicted in Fig. 1a which have close structural features of potent drug molecules.6 Each of these compounds has an anthracenyl group and an aryl-ring at two sides. The anthracenyl group being held by a single bond may adopt different orientations to modulate the stacking interactions of such semi-rigid molecules. Those may occur by involving π-stacking, C–H⋯π or X⋯π interactions (X = S or Cl).17,18 The sulphur atom may be involved in S⋯carbonyl interactions that occur at a separation distance between 3.2 and 3.5 Å and have 2–3 kcal per mole. They have great significance in biological systems and were observed in related imine systems too.19–21 Besides these, the presence of one or more substituent/s, such as –OMe or –Cl group/s on the aryl-ring, will change the electronic environments as well as the directional weak interactions. In these examples, the hydrogen bond acceptor –OMe substituents and chlorine atoms at different positions of a ring are expected to guide the directional properties. As such, the presence of chlorine atoms at different positions of a set of positional isomers would provide scope for a comparative study. In general, the weak interactions of the Cl atom vary between 0.5 and 3 kcal mol−1. These interactions are well known to guide self-assembled structures22,23 and polymorphs.24 Such studies contribute to enhancement of solubility25 and to improvement of synthetic methodology26 and medicinal aspects.27 These semi-flexible ligands also have extended conjugations over the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds extending to the CS bonds,28 which provide different geometries in metal complexes as listed in Fig. 1b–d by considering possibilities in zinc complexes. Similar ligands form four coordinate conformational isomeric complexes.29 Any geometrical change at the zinc centre would affect the weak interactions of the outer periphery of such complexes. In this work, we have synthesised and studied the structural aspects, energy and weak interactions in the positional isomers that are listed in Fig. 1a. Structural comparisons through DFT studies and Hirschfeld surface analyses on the ligands, bis-chelated zinc(II) complexes, and polymorphic solvate are detailed here.
N bonds extending to the CS bonds,28 which provide different geometries in metal complexes as listed in Fig. 1b–d by considering possibilities in zinc complexes. Similar ligands form four coordinate conformational isomeric complexes.29 Any geometrical change at the zinc centre would affect the weak interactions of the outer periphery of such complexes. In this work, we have synthesised and studied the structural aspects, energy and weak interactions in the positional isomers that are listed in Fig. 1a. Structural comparisons through DFT studies and Hirschfeld surface analyses on the ligands, bis-chelated zinc(II) complexes, and polymorphic solvate are detailed here.
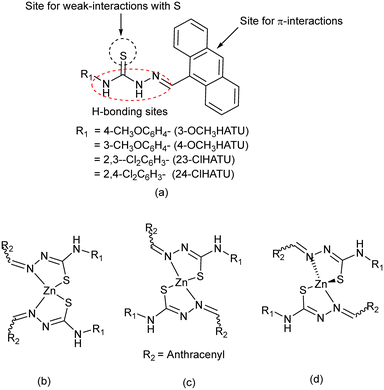 |
| | Fig. 1 (a) The ligands and (b–d) some possible zinc complexes with the ligands. | |
Experimental
Crystallographic study
Single-crystal X-ray diffraction data were collected for all the ligands and metal complexes studied here at 296 K with Mo Kα radiation (λ = 0.71073 Å) using a Bruker Nonius SMART APEX CCD diffractometer equipped with a graphite monochromator and an Apex CCD camera. Data reductions and cell refinement for the Bruker Nonius SMART APEX CCD diffractometer were performed using SAINT and XPREP software. Structures were solved by direct method and were refined by full-matrix least-squares on F2 using SHELXL-2014 software. All non-hydrogen atoms were refined in an anisotropic approximation against F2 of all reflections. Hydrogen atoms were placed at their geometric positions by riding and refined in the isotropic approximation. The crystal and refinement parameters are listed in Table S1.† Some of the hydrogen bond parameters of the ligands and complexes are listed in Table S2.†
General procedure for synthesis of the ligands
To a solution of (E)-(anthracen-9-ylmethylene)hydrazine (414 mg, 2 mmol) in diethyl ether (60 ml), respective bis-chlorine or mono-methoxy substituted phenyl isothiocyanate (2 mmol) was added and stirred for 10 h at 25 °C. In each case, this resulted in formation of a precipitate. The precipitate was filtered and washed with diethyl ether three times (3 × 10 mL). The residue in each case was dissolved in dimethylformamide, and the solutions were allowed to evaporate slowly in open air, which provided the crystalline product in each case.
Synthesis of [Zn{3-OCH3ATU)}2·DMF] and [Zn{4-OCH3ATU)}2·3DMF]
A solution of the respective ligand (1 mmol) and zinc(II) acetate dihydrate (219 mg, 1 mmol) in DMF solvent (20 mL) was stirred at room temperature for 24 h. A yellow precipitate was slowly formed at the bottom of the reaction flask. The precipitate obtained in each case was washed with methanol twice (15 mL × 2). The crude product that was insoluble in methanol was re-dissolved in DMF and the solution was filtered, and the filtrate was kept undisturbed for slow evaporation to obtain the crystals of the respective zinc(II) complex.
Synthesis of [Zn{23-ClATU)}2·CHCl3]-1 and [Zn{34-ClATU)}2·CHCl3]
The synthetic procedure for these two complexes was similar to that of the other zinc complexes described above, but the respective ligand namely 23- or 34-ClHATU was used in these cases. Each complex was crystallised by dissolving the crude complex in chloroform, followed by slow evaporation under ambient conditions. The spectroscopic details are provided in the ESI.†
Synthesis of [Zn{23-ClATU)}2·CHCl3]-2
A solution of 23-ClHATU (423 mg, 1 mmol) and zinc acetate dihydrate (219 mg, 1 mmol) in 20 mL of DMF was stirred at 100 °C for 24 h. This resulted in the formation of a yellow precipitate. The precipitate was washed with methanol to remove excess zinc salts. The crude product was dissolved in chloroform and filtered. The solution was then left undisturbed to undergo slow evaporation, which yielded crystals of [Zn{23-ClATU)}2·CHCl3]-2.
The spectroscopic details and yields are provided in the ESI.†
Results and discussion
Four hydrazinethiocarbazide ligands, (E)-2-(anthracen-9-ylmethylene)-N-(aryl)hydrazine carbothioamide with different aryl groups listed in Fig. 1a, were prepared by reacting (anthracen-9-ylmethylene)hydrazide with the corresponding aryl-thiocyanate as illustrated in Scheme 1. These were characterised by NMR and IR spectroscopy and by determining their crystal structures. The ligands were crystallised without a solvent of crystallisation with the exception of 3-OCH3HATU, which crystallised as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvate of dimethylformamide (DMF). The crystalline forms of two sets of positional isomers, one having aryl = 3- or 4- methoxy-phenyl and the other set with aryl = 2,3- or 2,4-dichloro-phenyl groups, had a syn–anti geometry across the thiourea part and had the projections of the N–H bond trans to each other as shown in Fig. 2. The syn–anti orientation is generally observed in the structures of N-functionalised thiourea and this was the case in the structure of the present ligands. As a consequence, they had the projections of the two N–H bonds trans to each other and the ligands had an E-geometry across the imine bond. The C16–S1 bond lengths in the respective structures varied between 1.664(5) Å and 1.679(2) Å, characteristic of the CS bond. The N1–N2 distances were 1.373(3) Å to 1.386(2) Å which confirmed the assigned sp2–sp2 hybridised nature of the NHN bonds in the compounds. Further to these, the N2–C16 bond lengths were in the range of 1.340(6) Å to 1.354(7) Å. These distances confirmed the presence of the CN bond as well as the CS bond (thiourea form) of the ligands. The torsion angles of the hydrazine-thiocarbazides were analysed by choosing three torsion angles listed in Table 1. The N1–N2–C16–S1 torsion in each case was similar but had some differences. Each pair of isomers had slight differences in the other two torsion angles listed in Table 1. As a consequence, the angle between the aromatic planes of the positional isomers had large differences. The angle between two aromatic planes in 3OCH3HATU and 4OCH3HATU was 34.91° and 86.34°, respectively, and in 23ClHATU and 34ClHATU, it was 4.15° and 40.33°, respectively. In the DMF solvate of 3-OCH3HATU, this DMF molecule was hydrogen bonded to the ligand by an N–H⋯O(carbonyl) hydrogen bond. The two methoxy derived compounds had difference in solvation, but the rest did not as the solvation did not cause a drastic change in the geometry of the semi-flexible ligand from the unsolvated positional isomer. Hence, it was the steric requirement of the individual ligand molecule to have tight packed structures that guided the geometries of the ligands observed in the crystalline state. In general, the C–H⋯halogen bonds contribute to stabilisation of conformations,30 and in the present cases, there were no such effects to mention.
:1 solvate of dimethylformamide (DMF). The crystalline forms of two sets of positional isomers, one having aryl = 3- or 4- methoxy-phenyl and the other set with aryl = 2,3- or 2,4-dichloro-phenyl groups, had a syn–anti geometry across the thiourea part and had the projections of the N–H bond trans to each other as shown in Fig. 2. The syn–anti orientation is generally observed in the structures of N-functionalised thiourea and this was the case in the structure of the present ligands. As a consequence, they had the projections of the two N–H bonds trans to each other and the ligands had an E-geometry across the imine bond. The C16–S1 bond lengths in the respective structures varied between 1.664(5) Å and 1.679(2) Å, characteristic of the CS bond. The N1–N2 distances were 1.373(3) Å to 1.386(2) Å which confirmed the assigned sp2–sp2 hybridised nature of the NHN bonds in the compounds. Further to these, the N2–C16 bond lengths were in the range of 1.340(6) Å to 1.354(7) Å. These distances confirmed the presence of the CN bond as well as the CS bond (thiourea form) of the ligands. The torsion angles of the hydrazine-thiocarbazides were analysed by choosing three torsion angles listed in Table 1. The N1–N2–C16–S1 torsion in each case was similar but had some differences. Each pair of isomers had slight differences in the other two torsion angles listed in Table 1. As a consequence, the angle between the aromatic planes of the positional isomers had large differences. The angle between two aromatic planes in 3OCH3HATU and 4OCH3HATU was 34.91° and 86.34°, respectively, and in 23ClHATU and 34ClHATU, it was 4.15° and 40.33°, respectively. In the DMF solvate of 3-OCH3HATU, this DMF molecule was hydrogen bonded to the ligand by an N–H⋯O(carbonyl) hydrogen bond. The two methoxy derived compounds had difference in solvation, but the rest did not as the solvation did not cause a drastic change in the geometry of the semi-flexible ligand from the unsolvated positional isomer. Hence, it was the steric requirement of the individual ligand molecule to have tight packed structures that guided the geometries of the ligands observed in the crystalline state. In general, the C–H⋯halogen bonds contribute to stabilisation of conformations,30 and in the present cases, there were no such effects to mention.
 |
| | Scheme 1 The synthesis of the ligands. | |
 |
| | Fig. 2 The crystal structure on the left and the angle between two aromatic planes of (a) 3-OCH3HATU (the DMF molecule is omitted for clarity), (b) 4-OCH3HATU, (c) 23-ClHATU and (d) 24ClHATU. | |
Table 1 Comparison of torsion angles among the positional isomers of arylHATU
| Torsion angle (°) |
N1–N2–C16–S1 |
N1–N2–C16–N3 |
C2–C1–C14–C15 |
| 3-OCH3HATU·DMF |
178.3(2) |
0.2(5) |
9.2(5) |
| 4-OCH3HATU |
174.91(15) |
4.6(3) |
3.2(3) |
| 23-ClHATU |
175.4(4) |
3.2(8) |
5.2(8) |
| 24-ClHATU |
177.4(4) |
2.5(6) |
4.4(8) |
Zinc complexes
Each ligand provided the respective bis-chelated zinc complex. All the complexes had close structural similarity with other thiocarbazide complexes in the literature.5 In each complex, the ligand was involved in N, S coordination using the thiolate form of the respective ligand. A close examination showed that each complex had different orientations of the anthracenyl plane of the ligand with respect to the same angle in the parent ligand. The structural aspects of each complex are discussed in the next section independently. The phase-purity of each complex was determined by recording powder X-ray diffraction (XRD). Those patterns matched with the simulated powder-XRD patterns from the crystallographic information file (Fig. S30†).
Positional isomeric complexes with methoxy-aryl derivatives
The bis-chelated complexes of the complexes of (E)-2-(anthracen-9-ylmethylene)-N-(aryl)hydrazine carbothioamide (aryl = 4-OCH3-phenyl or 3-OCH3-phenyl) were obtained as DMF solvates with different amounts of DMF. The complex with 3-OCH3ATU had a composition [Zn{3-OCH3ATU)}2·DMF]. Meanwhile, the complex of 4-OCH3ATU was a solvate of three DMF molecules, having a composition [Zn{4-OCH3ATU)}2·3DMF]. This showed that the amount of solvent depended on the ligand. In our earlier study, we found that the supramolecular effects of the ligands guided the compositions of the zinc complexes.31 In the present examples, the two complexes had different projections of –OCH3 groups making a difference in host systems to accommodate DMF guest molecules in different environments.
The [Zn{3-OCH3ATU)}2·DMF] complex had two similar Zn–S bond distances as well as two similar Zn–N bonds. The metal ligand bond distances and bond angles are listed in the Table 2. The [Zn{3-OCH3ATU)}2·DMF] complex had two dissimilar Zn–S bonds, as well as two Zn–N bonds. Accordingly, the former was more symmetric than the latter. The S1–C16 distances of the ligands of the two complexes were 1.756(3) Å and 1.754(4) Å. These distances were typical of a C–S bond, whereas, the N2–N1 bond distances were 1.374(4) Å and 1.378(4) Å, respectively. These were representative of an NN bond. These bond distances supported the corresponding thiolate form of the ligand in the complexes as assigned in the structures.
Table 2 Comparative bond distances and bond angles of [Zn{3-OCH3ATU)}2·DMF] and [Zn{4-OCH3ATU)}2·3DMF]
| Bond (Å) |
[Zn{3-OCH3ATU)}2·DMF] |
[Zn{4-OCH3ATU)}2·3DMF] |
| Zn1–N1 |
2.042(3) |
2.042(3) |
| Zn1–S1 |
2.261(1) |
2.260(12) |
| Zn1–N4 |
— |
2.049(3) |
| Zn1–S2 |
— |
2.251(12) |
| Bond-angle (°) |
[Zn{3-OCH3ATU)}2·DMF] |
[Zn{4-OCH3ATU)}2·3DMF] |
| N1–Zn1–N1 |
109.95(16) |
— |
| N1–Zn1–S1 |
111.60(8) |
86.06(8) |
| N1–Zn1–S1 |
85.92(8) |
— |
| S1–Zn1–S1 |
150.02(6) |
143.13(5) |
| N1–Zn1–N4 |
— |
119.56(12) |
| N1–Zn1–S2 |
— |
110.70(8) |
| N4–Zn1–S2 |
— |
85.84(9) |
| N4–Zn1–S1 |
— |
114.99(9) |
The angle between the two aromatic planes of a chelated ligand of the complex [Zn{3-OCH3ATU)}2·DMF] and the ligand 3-OCH3HATU was 78.88° and 86.14°, with a small difference of −7.26°. On the other hand, such an angle of [Zn{4-OCH3ATU)}2·3DMF] was 84.04°, this angle was 34.91° in the free ligand. Thus, there was a rotation of 49.13° of one of the planes or adjustment of the orientations of both the planes of each ligand upon coordination. This occurred due to the positional steric effect of the 3-methoxy group, as those groups projected away to maintain a trans-disposition across the two ligands. To do so, the plane of the chloroanthracenyl unit adjusted to an orientation to keep them apart. Meanwhile in the 4-OCH3 containing complex, the 4-OCH3 groups were located at the distal ends; hence, it was not required to exert a steric effect to settle them apart (Fig. 3).
 |
| | Fig. 3 The crystal structures of (a) [Zn{3-OCH3ATU)}2·DMF] and (b) [Zn{4-OCH3ATU)}2·3DMF], (for clarity solvent of crystallisation DMF molecules are omitted). (c) and (d) The respective angles between the aromatic planes of the same ligand. | |
Both the complexes had a τ4 value of 0.69 [τ4 = {360 − (α + β)}/141] (α and β are the two largest ligand metal–ligand bond angles). The τ4 is a measure to assign four coordinate geometries;32 a value falling in the range of 0.64–0.87 is assigned to a seesaw geometry (C2v point group). Accordingly, both the complexes had a see-saw geometry.
Positional isomeric complexes and polymorphism of dichloro-aryl derivatives
The independent reactions of 23-ClHATU with zinc(II) acetate dihydrate at room temperature followed by crystallisation from solution provided [Zn{23-ClATU)}2·CHCl3]-1; whereas, a similar reaction using 34-ClHATU provided [Zn{34-ClATU)}2·CHCl3]. The crystals of [Zn{23-ClATU)}2·CHCl3]-1 were very unstable; under open conditions, it lost its crystalline nature very fast and provided powder. With several attempts, we could obtain crystals that could be mounted for single-crystal diffraction. The structure was determined. However, the reaction of 23-ClHATU under refluxing conditions in DMF followed by recrystallisation from a solution in chloroform provided a packing polymorph [Zn{23-ClATU)}2·CHCl3]-2 that was stable. The crystal structures showed that each complex had one chloroform molecule of crystallisation. All these complexes had four coordinate chelated ligands forming a five-membered ring through the N2S2 mode of coordination. The metal–ligand bond-distances and the bond-angles of the complexes are listed in Table 3. As in the positional isomeric complexes with OCH3–aryl derived ligands, here also the C16–S1 bond tallied with a C–S bond, and the N1–N2 distances tallied with NN, establishing the chelate structure through the thiolate form of the respective ligand. The respective τ4 value of the two polymorphs was 0.71 and 0.74, respectively, whereas, the complex [Zn{34-ClATU)}2·CHCl3] had a τ4 value 0.71. Thus, each adopted a see-saw geometry.
Table 3 The metal–ligand bond distances and angles in the zinc complexes with dichloro-aryl functionalised ligands
| Bond |
Bond-length (Å) |
Bond-angle |
Angle (°) |
| [Zn{23-ClATU)}2·CHCl3]-1 |
| Zn1–N1 |
2.088(4) |
N1–Zn1–S2 |
113.96(13) |
| Zn1– S1 |
2.2685(18) |
N1–Zn1–S1 |
86.96(13) |
| Zn1–N4 |
2.048(4) |
N4–Zn1–S1 |
117.32(12) |
| Zn1–S2 |
2.284(2) |
S1–Zn1–S2 |
143.02(6) |
|
|
|
N4–Zn1–N1 |
106.47(16) |
|
|
|
N4–Zn1–S2 |
86.89(12) |
| [Zn{23-ClATU)}2·CHCl3]-2 |
| Zn1–N1 |
2.046(4) |
N1–Zn1–N1 |
108.1(2) |
| Zn1–S1 |
2.2573(12) |
N1–Zn1–S1 |
86.94(11) |
|
|
|
N1–Zn1–S1 |
86.93(11) |
|
|
|
N1–Zn1–S1 |
119.16(11) |
|
|
|
S1–Zn1–S1 |
136.66(7) |
| [Zn{34-ClATU)}2·CHCl3] |
| Zn1–N1 |
2.023(4) |
N1–Zn1–N1 |
122.6(2) |
| Zn1–S1 |
2.2630(13) |
N1–Zn1–S1 |
86.49(11) |
|
|
|
N1–Zn1–S1 |
86.49(10) |
|
|
|
S1–Zn1–S1 |
137.19(8) |
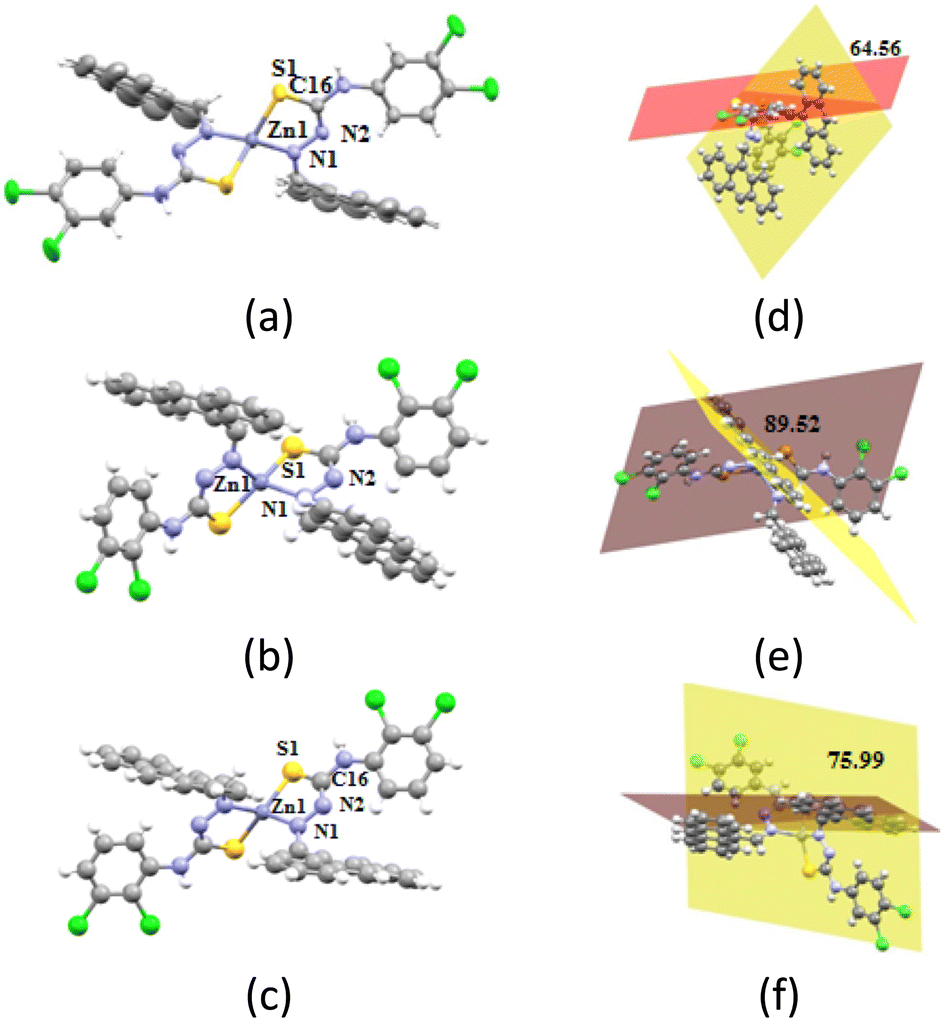
The angles between the planes containing the anthracenyl group and the plane containing the dichloro-aryl group of the ligands are shown in Fig. 4d–f. They showed extensive changes from the free ligands (refer to Fig. 2) while forming the complex. 34-ClHATU had such an angle of 40.33°, but in the zinc complex it was 78.88°, suggesting a twist of 36.55°. 23-ClHATU had a geometry that is very close to a planar structure, having an angle between the two distal aromatic planes of 4.15°. This angle in the two polymorphs was 89.52° and 75.99°, respectively. Hence, there was a difference of 13.35° between the planes of the anthracenyl unit and the plane containing the dichlorophenyl unit in the two polymorphs. The overlaid diagram showing the slight dissimilarity in the geometry is shown in Fig. 5. The difference in the orientations of the ligands provided the differences in the second coordination sphere leading to a packing difference between the two. In [Zn{23-ClATU)}2·CHCl3]-1, the self-assembly in the solid state was formed by different hydrogen bonded dimers formed by two S⋯π(N6) interactions (dS–π, 3.889 Å) at two sides of the two neighbouring molecules. Such interactions are not common, but are found in many disulphide-containing biological compounds.19
 |
| | Fig. 4 The crystal structures of (a) [Zn{34-ClATU)}2·CHCl3], (b) [Zn{23-ClATU)}2·CHCl3]-1, and (c) [Zn{23-ClATU)}2·CHCl3]-2 (solvent of crystallisation chloroform molecules are omitted for clarity in each case). (d)–(f) Figures showing the respective angle between the two aromatic planes of a ligand in the complexes. | |
 |
| | Fig. 5 Overlaid diagram of the inorganic part of the polymorphic solvate complex. | |
These dimers were held by chloroform weak hydrogen bonding of the C16–H···Cl6 hydrogen bond to one dimer and C45–H···π(centroid of C17C18) interaction with a centroid to centroid distance of separation of 3.531 Å (Fig. 6a). The self-assembly of the polymorph [Zn{23-ClATU)}2·CHCl3]-2 was composed of assemblies of discrete units through face to face parallel but staggered stacking among anthracenyl rings which was stabilised by supporting C15–H⋯π(C4) interactions to provide a one-dimensional chain-like structure. These chains were once again transformed into double chains through stacking between the dichloro-aryl groups with a centroid to centroid distance of 3.642 Å. The chloroform molecules in this case were held within the intra-molecular spaces of the ligands. The chloroform molecules were held through bifurcated C–H···π interactions (C11–H···Cl4 and C12–H···Cl4) with the anthracenyl ring and C23–H···π(C17) interactions as well as Cl···π(C18) interactions with the dichloro-aryl unit (Fig. 6b). On the other hand, the packing of [Zn{34-ClATU)}2·CHCl3] had a self-assembly of dimers with anthracenyl–anthracenyl stacking with a centroid to centroid distance of 4.037 Å. The chloroform molecules were held in such an assembly through insignificant interactions; alternatively, the chloroform molecules were trapped within the assembly. There are examples in the literature where stimuli9a and guest molecules33 affected the packing patterns of positional isomers. In the present case, chloroform played a key role in generating the crystalline product as in each case the loss of chloroform from the crystals resulted in the transformation to amorphous anhydrous forms (from naked eye observation and as confirmed by flat powder X-ray diffraction patterns). The chloroform solvent of crystallisation generally binds to the second sphere of a complex through different weak interactions.34,35
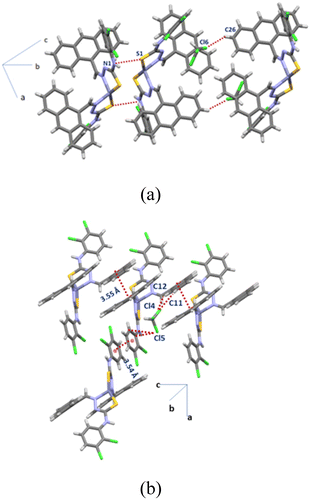 |
| | Fig. 6 The packing diagrams of (a) [Zn{23-ClATU)}2·CHCl3]-1 showing the intermolecular S–π interactions and (b) [Zn{23-ClATU)}2·CHCl3]-2. | |
Hirshfeld surface analysis and energy comparisons
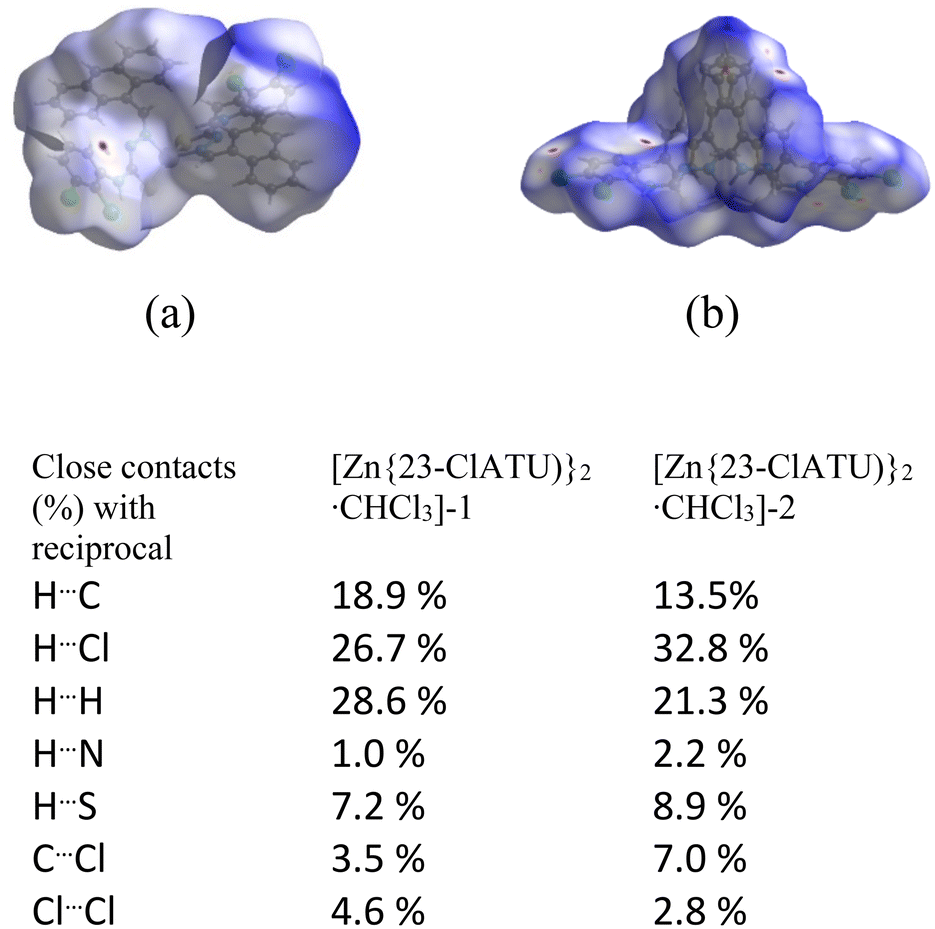
The Hirshfeld surface analyses36 of the positional isomers were carried out, and the percentages of different contacts from fingerprint plots of the ligands as well as complexes within 3.8 Å obtained are listed in tables (Fig. S31–S35,† Tables S3 and S4†). The solvated ligand 3-OCH3HATU·DMF had 4% higher C⋯H and 7.8% less C⋯C contacts than 4-OCH3HATU. Hence, there is a balance of different π-interactions in the two positional isomers. 3-OCH3HATU·DMF had 1.3% C⋯O interactions whereas 4-OCH3HATU had none of these interactions. This is attributed to the presence of the DMF molecule in the former case. These contacts were reflected in the fingerprint plots and the dnorm plots that are also listed in the ESI.† In the other positional isomeric pair, 23-ClHATU had 3.5% and 4% higher C⋯C and C⋯H contacts, respectively, than 34-ClHATU. The former Cl⋯all contacts were 12.9%, whereas, the latter had 30.1%, showing that the change in the position of chlorine changed the proximity of the chlorine atoms with respect to other interacting atoms in the vicinity of 3.8 Å. Among the ligands, only 23-ClHATU showed 0.3% C⋯S contacts, whereas the rest had none of such interactions. Thus, the involvement of the S atom in the weak interactions in the ligands was not significant. The Hirshfeld analysis and contacts of the complexes [Zn{3-OCH3ATU)}2·DMF], [Zn{4-OCH3ATU)}2·3DMF] and [Zn{34-ClATU)}2·CHCl3] are listed in Table S3.† The inorganic portions of the two solvate polymorphs were very similar as illustrated in the overlaid diagram; the surfaces and the contacts in the polymorphs [Zn{23-ClATU)}2·CHCl3]-1 or -2 are shown in Fig. 7 for comparison. Notable differences are seen in the C⋯Cl contacts and Cl⋯Cl contacts. Polymorph-1 had a dimer-like assembly, which suggested the presence of a S⋯π contact. It was reflected in the 8.3% S⋯C interaction and 0.3% S⋯N interaction in the surface analysis of Zn{23-ClATU)}2·CHCl3]-1. These contacts in the other polymorph were 0.5% and nil, respectively.
 |
| | Fig. 7 The Hirshfeld surfaces of (a) [Zn{23-ClATU)}2·CHCl3]-1 and (b) [Zn{23-ClATU)}2·CHCl3]-2. The percentages of contacts obtained from the fingerprint plots are listed below the diagram of the surfaces. | |
The energies of all the ligands and the zinc complexes at the singlet state were determined by DFT calculations at the B3LYP level using the LANL2DZ basis set. The LANL2DZ basis set is a useful basis set in DFT calculation for energy calculation of first row transition metal complexes.37
The pairs of positional isomers of the ligands were calculated without a solvent. Meanwhile, the methoxy-group containing zinc complexes had different amounts of solvents, hence the energy of the complexes without a solvent was calculated. 3-OCH3HATU had 0.15 kcal mol−1 less energy than 4-OCH3HATU, and the energy of the complex [Zn{3-OCH3ATU)}2] was 7.89 kcal mol−1 lower than that of [Zn{4-OCH3ATU)}2]. The positional isomer 34-ClHATU was less stable by 27.42 kcal mol−1 than 23-ClHATU (Table S5†). A similar trend in stability was observed in the energy associated with the complexes derived from them, [Zn{34-ClATU)}2·CHCl3] had 144.50 kcal mol−1 higher energy than [Zn{23-ClATU)}2·CHCl3] (Pcnn space group).
The energy difference between the two polymorphic solvates showed that the polymorph [Zn{23-ClATU)}2·CHCl3]-1 was stable by 0.15 kcal mol−1 compared to the other (Fig. 8). As the polymorph [Zn{23-ClATU)}2·CHCl3]-1 (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group) had a dimeric assembly with a S⋯C contact, we calculated the energy of the dimers of both the polymorphic complexes including two chloroform molecules. The dimers of the polymorphs also had the same trend in the order of energies but the energy gap was 0.45 kcal mol−1. Hence by dimer formation, there was a gain in energy, which is due to the contribution from S⋯π interactions. Though this energy was small, the intermolecular S–π interactions had a minor role in stabilisation (∼0.23 kcal mol−1), yet made a difference in stabilising the polymorph. A calculation without the chloroform showed that the energy difference had the same order but the gap was 123.17 kcal mol−1. Thus, there was a definite gain in each case but different in quantities due to chloroform solvation. This was indeed reflected in the different packing patterns. It may be noted that despite the small stability of the dimeric form from energy calculation, in reality it was less stable as solids. This was due to the propensity of the dimer to break down into monomers by loss of chloroform. The role of chloroform in packing of different compounds was studied through database analysis; it revealed different interactions depending on the structure of host molecules.34,35
space group) had a dimeric assembly with a S⋯C contact, we calculated the energy of the dimers of both the polymorphic complexes including two chloroform molecules. The dimers of the polymorphs also had the same trend in the order of energies but the energy gap was 0.45 kcal mol−1. Hence by dimer formation, there was a gain in energy, which is due to the contribution from S⋯π interactions. Though this energy was small, the intermolecular S–π interactions had a minor role in stabilisation (∼0.23 kcal mol−1), yet made a difference in stabilising the polymorph. A calculation without the chloroform showed that the energy difference had the same order but the gap was 123.17 kcal mol−1. Thus, there was a definite gain in each case but different in quantities due to chloroform solvation. This was indeed reflected in the different packing patterns. It may be noted that despite the small stability of the dimeric form from energy calculation, in reality it was less stable as solids. This was due to the propensity of the dimer to break down into monomers by loss of chloroform. The role of chloroform in packing of different compounds was studied through database analysis; it revealed different interactions depending on the structure of host molecules.34,35
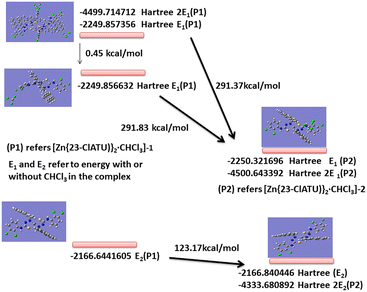 |
| | Fig. 8 The energy of complexes and polymorphs from DFT calculation at B3LYP level using the LANL2DZ basis set. | |
These results have shown that the relative stability of the positional isomers of the free ligands and the relative stability among the bis-chelated zinc complexes followed a similar hierarchy trend; however, in the complex the stability differences were not significant from the one between the positional isomeric free ligands. In an earlier study, it was observed that zinc mixed ligand complexes with different positional isomeric ligands of pyridine amides provided different compositions under the same reaction conditions. In that case, theoretical calculation suggested that out of various possibilities, it was not the thermodynamically stable isomers that were observed.31 Those reactions were guided by supramolecular interactions. Moreover, the chloroform getting into different intermolecular or intramolecular interactions in a distinct manner has interest, as such interactions influence exciplex emissions of ceratin metal complexes in solutions.38 The ligands used in the present study were suitable for chelation; hence, they formed stable complexes with uniform binding modes to the central zinc ion.
Conclusions
Distinguishable energies and weak interactions of the positional isomers arise from steric as well as electronic reasons. The type of solvent as well as the amounts of solvent in the assemblies of free ligands guided the assemblies. The orientations of aromatic groups across the hydrazine-carbazide units affected by the substituents provided planar to twisted geometries. It was possible to have higher amounts of solvent molecules per host, by changing the position of a methoxy-substituent on a host. The central coordination sphere of the zinc complexes guided the anchoring sites rather than the supramolecular features. The outer periphery of the complex by decorated ligands had different orientations of the aromatic units. This caused the packing difference in accommodating chloroform molecules at different positions, contributing to the stability of crystals and polymorphs. The S⋯π interactions contributed to dimer formation in one of the polymorphic solvates.
Data availability
The data supporting this article have been included as part of the ESI.† The CCDC no. of the ligands and complexes are 2346243, 2346245–2346248, 2346250, 2346253, 2346254 and 2387524. The spectroscopic details of the ligands and complexes, powder X-ray diffraction patterns, crystallographic table, Hirshfeld analysis, and theoretical energy data are available in the ESI.†
Author contributions
This work was carried by JN during doctoral study under the supervision of JBB, and both have equal contributions.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Thanks are due to the Department of Science and Technology India (project no. SR/FST/CS-II/2017/23C and project no. SR/FST/ETII-071/2016(G).
References
-
(a) U. Boas, A. J. Karlsson, B. F. de Waal and E. W. Meijer, J. Org. Chem., 2001, 66, 2136–2145 CrossRef CAS;
(b) X. Zhao, L. Wang, G. Zhou, S. Feng and L. Li, Stiff, Polymer, 2024, 290, 126596 CrossRef CAS;
(c) B. M. McVey, J. Rumble, C. E. V. Nathan, I. A. Gass, M. R. Sambrook and P. J. Cragg, Supramol. Chem., 2023, 34, 208–222 CrossRef CAS;
(d) H. Jiyun, F. Zanca, P. Lambe, M. Tsuji, S. Wijeweera, S. Todisco, P. Mastrorilli, W. Shirley, M. Benamara, P. Z. Moghadam and H. Beyzavi, ACS Appl. Mater. Interfaces, 2020, 12, 29212–29217 CrossRef PubMed;
(e) I. Garcia-Santos, A. Castineiras, G. Mahmoudi, M. G. Babashkina, E. Zangrando, R. M. Gomila, A. Frontera and D. A. Safin, CrystEngComm, 2022, 24, 368–378 RSC.
-
(a) O. Reis, S. Eymur, B. Reis and A. S. Demir, Chem. Commun., 2009, 1088–1090 RSC;
(b) Q. Li, M. Shi and T. C. W. Mak, Chin. Sci. Bull., 2001, 46, 1761–1763 CrossRef CAS.
-
(a) R. Ronchetti, G. Moroni, A. Carotti, A. Gioiello and E. Camaioni, RSC Med. Chem., 2021, 12, 1046–1064 RSC;
(b) J. E. Mendieta-Wejebe, M. C. Rosales-Hernandez, I. I. Padilla-Martínez, E. V. Garcia-Baez and A. Cruz, Int. J. Mol. Sci., 2023, 24, 9488 CrossRef CAS PubMed.
- J. Shang, Y. Zhang, N. Yang, L. Xiong, Q. Bian and B. Wang, Phosphorus, Sulfur Silicon Relat. Elem., 2023, 198, 659–672 CrossRef CAS.
-
(a) A. Tarai and J. B. Baruah, Dalton Trans., 2018, 47, 4921–4930 RSC;
(b) E. A. Hill, N. Zhao, A. S. Filatov and J. S. Anderson, Chem. Commun., 2020, 56, 7861 RSC.
-
(a) M. C. C. de Oliveira, C. M. R. SantAnna, J. C. Netto-Ferreira, A. Echevarria, D. Sousa-Pereira, O. A. Chaves and C. M. D. Reis, Bioorg. Chem., 2018, 81, 79–87 CrossRef PubMed;
(b) K. Chithra, D. Satheesh, K. Jayanthi, S. V. Kumar, V. Muthulakshmi, K. Kalaivani, R. Saravanan and P. Sellam, Chem. Data Collect., 2021, 32, 100652 CrossRef CAS;
(c) E. D. Dincel, Ç. Akdag, T. Kayra, E. D. Coşar, M. O. Aksoy, G. Akalın-Çiftçi and N. Ulusoy-Güzeldemirci, J. Mol. Struct., 2022, 1268, 133710 CrossRef CAS;
(d) S. Singh, P. K. Mandal, N. Singh, A. K. Misra, S. Singh, V. Chaturvedi, S. Sinha and A. K. Saxena, Bioorg. Med. Chem. Lett., 2010, 20, 2597–2600 CrossRef CAS PubMed.
-
(a) G. Luchini, D. M. H. Ascough, J. V. Alegre-Requena, V. Gouverneur and R. S. Paton, Tetrahedron, 2019, 75, 697–702 CrossRef CAS;
(b) A. Tarai and J. B. Baruah, ACS Omega, 2017, 2, 6991–7001 CrossRef CAS PubMed.
-
(a) N. Phukan and J. B. Baruah, CrystEngComm, 2016, 18, 3877–3890 RSC;
(b) N. Phukan and J. B. Baruah, Cryst. Growth Des., 2014, 14, 2640–2653 CrossRef CAS.
-
(a) G. Fan and D. Yan, Sci. Rep., 2014, 4, 4933 CrossRef CAS PubMed;
(b) P. Hao, H. Zhu, Y. Pang, J. Shen and Y. Fu, CrystEngComm, 2020, 22, 3371–3377 RSC;
(c) B. R. Jali and J. B. Baruah, Dyes Pigm., 2014, 110, 56–66 CrossRef CAS;
(d) Z. Yang, S. Zhu, Y. Liu, X. Niu, R. Chen, Y. Su, J. Yuan, W. Zhang, X. Chen and Z. An, ChemistrySelect, 2024, 9, e202302977 CrossRef CAS.
- Q. Sun, N. Ding, C. Zhao, J. Ji, S. Li and S. Pang, Chem. Eng. J., 2022, 427, 130912 CrossRef CAS.
- G.-P. Yong, C. Shen, Y. Feng, X.-R. Zhang and Y.-M. Zhao, CrystEngComm, 2015, 17, 6338–6345 RSC.
- N. Phukan, A. Goswami and J. B. Baruah, Inorg. Chim. Acta, 2015, 435, 239–243 CrossRef CAS.
-
(a) A. Verma, M. K. Tiwari, B. Show and S. Saha, ACS Omega, 2020, 5, 448–459 CrossRef;
(b) S. Verma, R. Brahma and J. B. Baruah, CrystEngComm, 2024, 26, 1976–1985 RSC.
- D. Delaere, G. Raspoet and M. T. Nguyen, J. Phys. Chem. A, 1999, 103, 171–177 CrossRef CAS.
- W. R. Carroll, C. Zhao, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Org. Lett., 2011, 13, 4320–4323 CrossRef CAS PubMed.
- W. R. Carroll, P. Pellechia and K. D. Shimizu, Org. Lett., 2008, 10, 3547–3550 CrossRef CAS PubMed.
- M. H. Sadr, Z. Khalilizadeh and E. R. T. Tiekink, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2007, 63, o4126 CrossRef CAS.
- N. Selvakumaran, R. Karvembu, S. W. Ng and E. R. T. Tiekink, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2011, 67, o602 CrossRef CAS PubMed.
- Z. Wang, T. Lu, F. Xie, T. Yang, Y. Xu, X. Li, M. Schnell and G. Feng, J. Phys. Chem. Lett., 2024, 15, 8917–8923 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- M. V. Vener, A. V. Shishkina, A. A. Rykounov and V. G. Tsirelson, J. Phys. Chem. A, 2013, 117, 8459–8467 CrossRef CAS.
- M. Capdevila-Cortada, J. Castelló and J. J. Novoa, CrystEngComm, 2014, 16, 8232–8242 RSC.
- A. Tarai and J. B. Baruah, CrystEngComm, 2019, 21, 1397–1406 RSC.
- F. Shakeel, M. A. Bhat and N. Haq, J. Chem. Eng. Data, 2014, 59, 2660–2664 CrossRef CAS.
- A. A. Aly, A. B. Brown, T. I. El-Emary, A. M. M. Ewas and M. Ramadan, Arkivoc, 2009,(i), 150–197 Search PubMed.
-
(a) J. Easmon, G. Purstinger, G. Heinisch, T. Roth, H. H. Fiebig, W. Holzer, W. Jager, M. Jenny and J. Hofmann, J. Med. Chem., 2001, 44, 2164–2171 CrossRef CAS;
(b) P. Jutten, W. Schumann, A. Hart, H. M. Dahse and U. Grafe, J. Med. Chem., 2007, 50, 3661–3666 CrossRef PubMed.
-
(a) V. B. Arion, Coord. Chem. Rev., 2019, 387, 348–397 CrossRef CAS;
(b) A. R. Aguirre, G. L. Parrilha, S. R. W. Louro, O. C. Alves, R. Diniz, F. Durval, W. Rocha and H. Beraldo, Polyhedron, 2022, 217, 115724 CrossRef CAS;
(c) C. Chieh, L. P. C. Lee and C. Chiu, Can. J. Chem., 1978, 56, 2526–2531 CrossRef CAS.
- J. Nath and J. B. Baruah, ACS Omega, 2023, 8, 42827–42839 CrossRef CAS PubMed.
- M. D. Prasanna and T. N. Guru Row, Cryst. Eng., 2000, 3, 135–154 CrossRef CAS.
- R. Brahma and J. B. Baruah, ACS Omega, 2020, 5, 3774–3785 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- J.-R. Wu, G. Wu, D. Li, D. Dai and Y.-W. Yang, Sci. Adv., 2022, 8, eabo2255 CrossRef CAS PubMed.
- F. H. Allen, P. A. Wood and P. T. A. Galek, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2013, 69, 379–388 CrossRef CAS PubMed.
- L. Brunel, F. Carré, S. G. Dutremez, C. Guérin, F. Dahan, O. Eisenstein and G. Sini, Organometallics, 2001, 20, 47–54 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- S. Chiodo, N. Russo and E. Sicilia, J. Chem. Phys., 2006, 125, 104107 CrossRef CAS PubMed.
- J. Zhang, A. Kundu, T. Elsaesser, P. Macchi, M. Kalter, G. Eickerling and W. Scherer, J. Phys. Chem. Lett., 2022, 13, 4447–4454 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic details of ligands and complexes, powder X-ray diffraction patterns, crystallographic table, Hirshfeld analyses, and theoretical energy data. The CCDC numbers of the ligands and complexes are 2346243, 2346245–2346248, 2346250, 2346253, 2346254 and 2387524. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce00992d |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Jubaraj B.
Baruah
and
Jubaraj B.
Baruah