
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ru-catalyzed deoxydehydration (DODH) with hydrazine
Siyi
Luo†
,
Chen-Chen
Li†
 ,
Zihang
Qiu†
,
Ruofei
Cheng
and
Chao-Jun
Li
*
,
Zihang
Qiu†
,
Ruofei
Cheng
and
Chao-Jun
Li
*
Department of Chemistry, and FRQNT Centre for Green Chemistry and Catalysis, McGill University, Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 15th September 2025
Abstract
A novel method for the deoxydehydration (DODH) of diols to alkenes has been developed by using hydrazine as a reductant via HOME (hydrazone as an organometallic equivalent) chemistry catalyzed by Ru-PNP complexes. This process avoids harsh reaction conditions, minimizes waste, and can be applied to a variety of diol compounds.
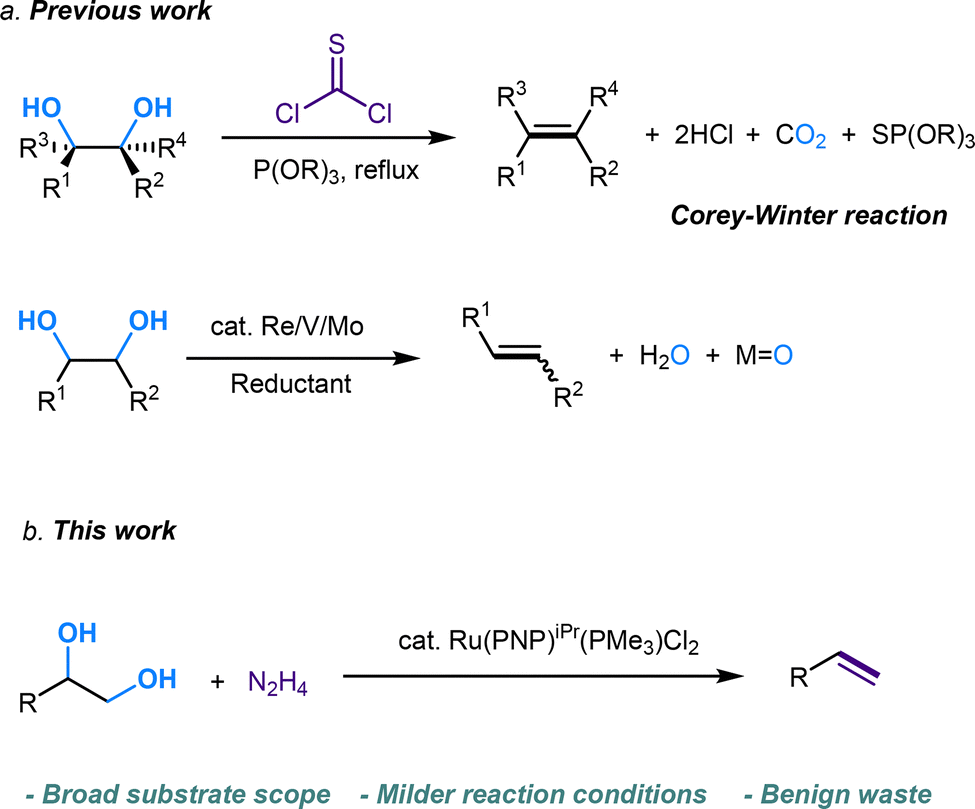
Diol and polyol structures are prevalent in natural compounds and biomolecules.1 As renewable feedstocks, biomass-derived diols and polyols have become attractive alternatives to fossil fuels in the production of value-added chemicals. Dehydration, hydrogenation, and hydrogenolysis have received great attention for the conversion of polyols.2–4 On the other hand, olefin serves as a fundamental building block in organic synthesis.5,6 However, methods for directly converting diols into alkenes are very limited. Thus, the selective and efficient conversion of diols into olefins via a deoxydehydration (DODH) process holds the potential to facilitate a wide range of transformations in synthetic chemistry. Compared to traditional, well-developed strategies starting from alkene or carbonyl compounds,7–10 the olefination derived from abundantly available alcohols via DODH offers a cost-effective pathway to achieve C
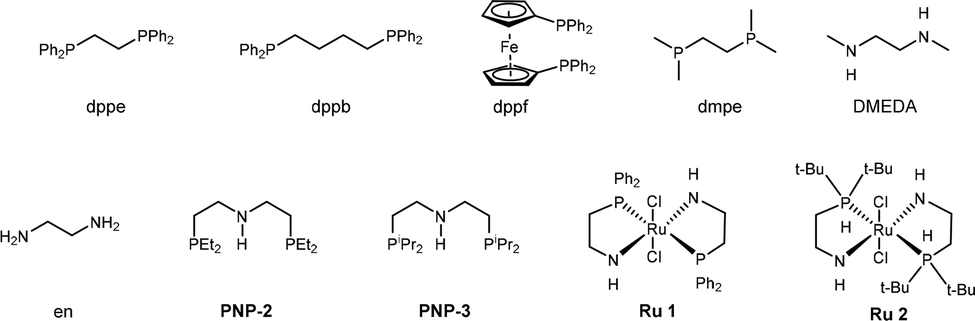
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C linkage from an economic perspective. Currently, DODH methods are quite limited, and existing techniques require toxic and hazardous chemicals (phosphite or phosphine) and harsh reaction conditions.11–14 Deoxydehydration (DODH) of diols toward alkenes with the extrusion of one oxygen atom and one equivalent of water molecule was exploited first by Corey et al., in a reaction known as the Corey–Winter reaction.15 However, the well-established Corey–Winter reaction usually involves prolonged heating at elevated temperatures and the use of phosphorylated reductive reagents (Scheme 1a).16 In addition to the Corey–Winter reaction, the DODH of vicinal diols and polyols is typically performed using Re,17 V,18 or Mo19 in combination with phosphine, secondary alcohols including 3-octanol, or H2 (for certain solid catalysts) as reductants.17–19 This process often necessitates rigorous reaction conditions. Furthermore, they exhibit inefficacy in facilitating the DODH of substrates with complex structures (Scheme 1a).13 According to HOME-chemistry (hydrazones as organometallic equivalents) and “borrowing hydrogen” strategies,20–22 our group has developed a HOME-chemistry-based olefination strategy from two carbonyl compounds, which experienced an E1cb process.23 Likewise, in 2018, Milstein's lab revealed a manganese-catalyzed dehydrogenative coupling of alcohols with hydrazine/hydrazone accompanied by a hydrogen transfer process to produce alkenes,24 following the example of the Wittig-type and Julia-type olefination of alcohols.25,26 Besides manganese, ruthenium enabled alcohols as carbonyl surrogates.27 Moreover, both ruthenium and titanium demonstrated high activity in the conversion of alcohols to alkenes and alkanes as well.28 Inspired by these works, we proposed that Ru could be a suitable catalyst for DODH with the assistance of hydrazine due to its versatile applications in coupling processes that involve alcohols. By leveraging the hydrogen transfer process, we developed a Ru-catalyzed DODH reaction enabled by hydrazine. Our approach offers the advantage of being operable under mild conditions with minimal environmental impact. It requires low temperatures and eliminates the need for gas, resulting in only N2, H2, and water as byproducts (Scheme 1b). Aromatic-substituted diols and aliphatic diols exhibit good performance within our method. Mechanistically, our approach involves a Ru-catalyzed dehydrogenation, followed by hydrazone formation and an intramolecular E1cb elimination. We started by investigating the reaction conditions of aliphatic diols and hydrazine. A set of ruthenium(II) catalysts was tested first in view of their high activity and good functional group tolerance in hydrazone chemistry involving alcohols as carbonyl surrogates in deoxygenation and Grignard-type reactions.27,29,30 We utilized 1,2-dodecanediol as our model substrate, attributed to its simple structure and high boiling point. It should be noted that no desired product was detected in the reaction system catalyzed by Ru(PPh3)3Cl2 with K3PO4 as the base at 100 °C (Table 1, entry 1). The combination of a ruthenium catalyst and an electron-rich phosphine ligand typically resulted in better performance in HOME chemistry.31 Therefore, after the replacement of Ru(PPh3)3Cl2 with more electron-rich Ru(PMe3)4Cl2 or more stable Ru(dppb)PPh3Cl2 as a catalyst, 9% and 7% yields were observed, respectively (Table 1, entries 2 and 3). Then, different bidentate ligands were tested in the presence of [Ru(p-cymene)Cl2]2 (Table 1, entries 4, 5 and 6). Compared with dppe and DMEDA, the catalyst with more electron-rich and less sterically hindered dmpe showed better activity (Table 1, entry 5). In contrast, ruthenium catalysts with other bidentate ligands proved ineffective in this reaction system (Table 1, entries 7–10). Since the dehydrogenative coupling of alcohols and hydrazine has shown high efficiency with transition-metal pincer catalysts,24,32,33 we investigated the efficiency of Ru, Mn and Ir-pincer catalysts (Table 1, entries 11–15).34 The yield was tremendously attenuated with Ru(PNP-2)Cl2 or Ru(PNP-3)Cl2 (Table 1, entries 11 and 12). It was reported that the ruthenium-pincer complex with a monodentate phosphine ligand significantly depressed some potential side reactions, including hydrazone dimerization.35 Thus, the highest yield was obtained by treating aliphatic diols with Ru(PNP-3)(PMe3)Cl2 (Table 1, entry 13). Organic bases such as DBU failed to furnish the final product (Table 1, entries 16). In contrast, K3PO4 (Table 1, entry 13) was the most effective base (Table 1, entries 17 and 18). Temperature has a significant influence on productivity as well. The yield demonstrated an increasing trend against temperature from 60 to 100 °C (Table 1, entries 13, 19 and 20) but decreased above 100 °C (Table 1, entry 21), probably due to the competing Wolff–Kishner reduction. Notably, when the catalyst loading of Ru(PNP-3)(PMe3)Cl2 was increased to 10 mol%, an even higher yield was detected at 80 °C (Table 1, entry 23) versus 100 °C (Table 1, entry 22). For aromatic diols, an alkyl side product was observed due to the hydrogen transfer ability of the Ru catalyst. Thus, further investigation into the reaction conditions was necessary, and 1-phenyl-1,2-ethanediol was chosen as the model substrate. It should be noted that an 88% yield was detected at 70 °C when we shortened the reaction time to 16 h, decreased the catalyst loading to 5 mol% and the amount of base to 25 mol%, and added 5 μL DMSO as an additive (Table 2, entry 7). When the reaction was charged with the identical catalyst loading and base quantity used for aliphatic diols, the resulting yield was diminished in comparison (Table 2, entries 1 and 2). In addition, a decreased yield was noted when the reaction was carried out at temperatures either above or below 70 °C. (Table 2, entries 5 and 8). With other conditions unchanged, a control experiment was performed without DMSO, leading to a diminished yield (Table 2, entry 9).30 We then applied the optimized reaction conditions to various compounds containing diols, giving rise to moderate to high yields in most cases. In the scope of aliphatic diols, aside from 1,2-dodecanediol affording a satisfactory 70% isolated yield (Table 3, 2a), a phenyl ring (Table 3, 2b), morpholine (Table 3, 2c and 2j), methoxy group (Table 3, 2d), alkene (Table 3, 2g) and carbazole (Table 3, 2i) were well tolerated. In contrast, lower reactivities were observed on substrates with substituted ethoxy benzenes (Table 3, 2e and 2f). Their products, 2e and 2f, were mixed with unreacted starting materials. Furthermore, meglumine exhibited a low reactivity, likely attributed to challenges in solubility and the incompatibility of amine functionalities (Table 3, 2h). It is worth noting that complex mixtures were formed in the presence of an additional terminal hydroxy group or aldehyde functional group, including glyceraldehyde. This could be explained by the possible intermolecular olefination side reaction and other competing reactions. Furthermore, fructose is not suitable for the reaction, indicating that condensation of N2H4 did not occur on the internal carbonyl group, which serves as evidence for the mechanism that dehydrogenation likely takes place at the terminal OH group. As for aromatic diols, apart from the 88% yield detected in the conversion of 1-phenyl-1,2-ethanediol (Table 3, 4a), the reaction is applicable to naphthalene (Table 3, 4d) and halo-substituted aromatic rings (Table 3, 4b and 4c). Substrates bearing sterically hindered polycyclic aromatic hydrocarbons such as fluorene (Table 3, 4e) and pyrene (Table 3, 4f) delivered 40% and 30% yields, respectively. Besides, the reactivity was dramatically attenuated in the presence of the methylenedioxy group (Table 3, 4h) compared to the alkoxy group (Table 3, 4g). This likely occurs due to the conflicting electronic effect of meta and para-oxygen on the aromatic ring, slowing down the E1cB elimination process. An alternative explanation could be that oxygen may coordinate with ruthenium, thereby hindering the reaction pathway. In contrast, the substrate with the para-electron-donating benzyloxy and tert-butoxy groups exhibited high reactivity due to their weaker electronic effect and higher steric hindrance (Table 3, 4i and 4j). Other common functional groups were well tolerated (Table 3, 4m and 4n), with the exception of heterocycles (Table 3, 4k and 4l). Furthermore, the compound 1-(3-nitrophenyl)ethane-1,2-diol underwent a conversion to vinylaniline alongside a reduction process (Table 3, 4o). Aromatic diols that featured an ester group (methyl 4-vinylbenzoate), a cyano group (4-vinylbenzonitrile), or an additional simple alkyl group at the 2-position (prop-1-en-2-ylbenzene) were found to be ineffective (see the SI). The reactions involving certain internal diols derived from their respective alkenes also proceeded effectively (Table 3, 4p and 4q). The 1-phenylpropane-1,2-diol-derived product 4p was obtained in an 84% yield with a 93
C linkage from an economic perspective. Currently, DODH methods are quite limited, and existing techniques require toxic and hazardous chemicals (phosphite or phosphine) and harsh reaction conditions.11–14 Deoxydehydration (DODH) of diols toward alkenes with the extrusion of one oxygen atom and one equivalent of water molecule was exploited first by Corey et al., in a reaction known as the Corey–Winter reaction.15 However, the well-established Corey–Winter reaction usually involves prolonged heating at elevated temperatures and the use of phosphorylated reductive reagents (Scheme 1a).16 In addition to the Corey–Winter reaction, the DODH of vicinal diols and polyols is typically performed using Re,17 V,18 or Mo19 in combination with phosphine, secondary alcohols including 3-octanol, or H2 (for certain solid catalysts) as reductants.17–19 This process often necessitates rigorous reaction conditions. Furthermore, they exhibit inefficacy in facilitating the DODH of substrates with complex structures (Scheme 1a).13 According to HOME-chemistry (hydrazones as organometallic equivalents) and “borrowing hydrogen” strategies,20–22 our group has developed a HOME-chemistry-based olefination strategy from two carbonyl compounds, which experienced an E1cb process.23 Likewise, in 2018, Milstein's lab revealed a manganese-catalyzed dehydrogenative coupling of alcohols with hydrazine/hydrazone accompanied by a hydrogen transfer process to produce alkenes,24 following the example of the Wittig-type and Julia-type olefination of alcohols.25,26 Besides manganese, ruthenium enabled alcohols as carbonyl surrogates.27 Moreover, both ruthenium and titanium demonstrated high activity in the conversion of alcohols to alkenes and alkanes as well.28 Inspired by these works, we proposed that Ru could be a suitable catalyst for DODH with the assistance of hydrazine due to its versatile applications in coupling processes that involve alcohols. By leveraging the hydrogen transfer process, we developed a Ru-catalyzed DODH reaction enabled by hydrazine. Our approach offers the advantage of being operable under mild conditions with minimal environmental impact. It requires low temperatures and eliminates the need for gas, resulting in only N2, H2, and water as byproducts (Scheme 1b). Aromatic-substituted diols and aliphatic diols exhibit good performance within our method. Mechanistically, our approach involves a Ru-catalyzed dehydrogenation, followed by hydrazone formation and an intramolecular E1cb elimination. We started by investigating the reaction conditions of aliphatic diols and hydrazine. A set of ruthenium(II) catalysts was tested first in view of their high activity and good functional group tolerance in hydrazone chemistry involving alcohols as carbonyl surrogates in deoxygenation and Grignard-type reactions.27,29,30 We utilized 1,2-dodecanediol as our model substrate, attributed to its simple structure and high boiling point. It should be noted that no desired product was detected in the reaction system catalyzed by Ru(PPh3)3Cl2 with K3PO4 as the base at 100 °C (Table 1, entry 1). The combination of a ruthenium catalyst and an electron-rich phosphine ligand typically resulted in better performance in HOME chemistry.31 Therefore, after the replacement of Ru(PPh3)3Cl2 with more electron-rich Ru(PMe3)4Cl2 or more stable Ru(dppb)PPh3Cl2 as a catalyst, 9% and 7% yields were observed, respectively (Table 1, entries 2 and 3). Then, different bidentate ligands were tested in the presence of [Ru(p-cymene)Cl2]2 (Table 1, entries 4, 5 and 6). Compared with dppe and DMEDA, the catalyst with more electron-rich and less sterically hindered dmpe showed better activity (Table 1, entry 5). In contrast, ruthenium catalysts with other bidentate ligands proved ineffective in this reaction system (Table 1, entries 7–10). Since the dehydrogenative coupling of alcohols and hydrazine has shown high efficiency with transition-metal pincer catalysts,24,32,33 we investigated the efficiency of Ru, Mn and Ir-pincer catalysts (Table 1, entries 11–15).34 The yield was tremendously attenuated with Ru(PNP-2)Cl2 or Ru(PNP-3)Cl2 (Table 1, entries 11 and 12). It was reported that the ruthenium-pincer complex with a monodentate phosphine ligand significantly depressed some potential side reactions, including hydrazone dimerization.35 Thus, the highest yield was obtained by treating aliphatic diols with Ru(PNP-3)(PMe3)Cl2 (Table 1, entry 13). Organic bases such as DBU failed to furnish the final product (Table 1, entries 16). In contrast, K3PO4 (Table 1, entry 13) was the most effective base (Table 1, entries 17 and 18). Temperature has a significant influence on productivity as well. The yield demonstrated an increasing trend against temperature from 60 to 100 °C (Table 1, entries 13, 19 and 20) but decreased above 100 °C (Table 1, entry 21), probably due to the competing Wolff–Kishner reduction. Notably, when the catalyst loading of Ru(PNP-3)(PMe3)Cl2 was increased to 10 mol%, an even higher yield was detected at 80 °C (Table 1, entry 23) versus 100 °C (Table 1, entry 22). For aromatic diols, an alkyl side product was observed due to the hydrogen transfer ability of the Ru catalyst. Thus, further investigation into the reaction conditions was necessary, and 1-phenyl-1,2-ethanediol was chosen as the model substrate. It should be noted that an 88% yield was detected at 70 °C when we shortened the reaction time to 16 h, decreased the catalyst loading to 5 mol% and the amount of base to 25 mol%, and added 5 μL DMSO as an additive (Table 2, entry 7). When the reaction was charged with the identical catalyst loading and base quantity used for aliphatic diols, the resulting yield was diminished in comparison (Table 2, entries 1 and 2). In addition, a decreased yield was noted when the reaction was carried out at temperatures either above or below 70 °C. (Table 2, entries 5 and 8). With other conditions unchanged, a control experiment was performed without DMSO, leading to a diminished yield (Table 2, entry 9).30 We then applied the optimized reaction conditions to various compounds containing diols, giving rise to moderate to high yields in most cases. In the scope of aliphatic diols, aside from 1,2-dodecanediol affording a satisfactory 70% isolated yield (Table 3, 2a), a phenyl ring (Table 3, 2b), morpholine (Table 3, 2c and 2j), methoxy group (Table 3, 2d), alkene (Table 3, 2g) and carbazole (Table 3, 2i) were well tolerated. In contrast, lower reactivities were observed on substrates with substituted ethoxy benzenes (Table 3, 2e and 2f). Their products, 2e and 2f, were mixed with unreacted starting materials. Furthermore, meglumine exhibited a low reactivity, likely attributed to challenges in solubility and the incompatibility of amine functionalities (Table 3, 2h). It is worth noting that complex mixtures were formed in the presence of an additional terminal hydroxy group or aldehyde functional group, including glyceraldehyde. This could be explained by the possible intermolecular olefination side reaction and other competing reactions. Furthermore, fructose is not suitable for the reaction, indicating that condensation of N2H4 did not occur on the internal carbonyl group, which serves as evidence for the mechanism that dehydrogenation likely takes place at the terminal OH group. As for aromatic diols, apart from the 88% yield detected in the conversion of 1-phenyl-1,2-ethanediol (Table 3, 4a), the reaction is applicable to naphthalene (Table 3, 4d) and halo-substituted aromatic rings (Table 3, 4b and 4c). Substrates bearing sterically hindered polycyclic aromatic hydrocarbons such as fluorene (Table 3, 4e) and pyrene (Table 3, 4f) delivered 40% and 30% yields, respectively. Besides, the reactivity was dramatically attenuated in the presence of the methylenedioxy group (Table 3, 4h) compared to the alkoxy group (Table 3, 4g). This likely occurs due to the conflicting electronic effect of meta and para-oxygen on the aromatic ring, slowing down the E1cB elimination process. An alternative explanation could be that oxygen may coordinate with ruthenium, thereby hindering the reaction pathway. In contrast, the substrate with the para-electron-donating benzyloxy and tert-butoxy groups exhibited high reactivity due to their weaker electronic effect and higher steric hindrance (Table 3, 4i and 4j). Other common functional groups were well tolerated (Table 3, 4m and 4n), with the exception of heterocycles (Table 3, 4k and 4l). Furthermore, the compound 1-(3-nitrophenyl)ethane-1,2-diol underwent a conversion to vinylaniline alongside a reduction process (Table 3, 4o). Aromatic diols that featured an ester group (methyl 4-vinylbenzoate), a cyano group (4-vinylbenzonitrile), or an additional simple alkyl group at the 2-position (prop-1-en-2-ylbenzene) were found to be ineffective (see the SI). The reactions involving certain internal diols derived from their respective alkenes also proceeded effectively (Table 3, 4p and 4q). The 1-phenylpropane-1,2-diol-derived product 4p was obtained in an 84% yield with a 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 E
:7 E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :Z ratio; however, 4q, due to its steric hindrance, gave only a 29% yield with poor stereoselectivity.
:Z ratio; however, 4q, due to its steric hindrance, gave only a 29% yield with poor stereoselectivity.
 | ||
| Scheme 1 Methods for the direct conversion of diols into olefins. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Catalyst loading (mol%) | Base | T (°C) | 2a (%) |
| Reaction conditions: 1a (40.5 mg, 0.2 mmol), Ru(PNP-3)(PMe3)Cl2 (11.1 mg, 0.02 mmol, 10 mol%), K3PO4 (31.8 mg, 0.15 mmol, 75 mol%), 1 M N2H4 solution in THF (0.5 mL, 0.5 mmol, 2.5 equiv.), 24 h at 80 °C, under N2. | ||||||
| 1 | Ru(PPh3)3Cl2 | — | 5 | K3PO4 | 100 | 0 |
| 2 | Ru(PMe3)4Cl2 | — | 5 | K3PO4 | 100 | 9 |
| 3 | Ru(dppb)PPh3Cl2 | — | 5 | K3PO4 | 100 | 7 |
| 4 | [Ru(p-cymene)Cl2]2 | dppe | 5 | K3PO4 | 100 | 0 |
| 5 | [Ru(p-cymene)Cl2]2 | dmpe | 5 | K3PO4 | 100 | 36 |
| 6 | [Ru(p-cymene)Cl2]2 | DMEDA | 5 | K3PO4 | 100 | 0 |
| 7 | Ru1 | — | 5 | K3PO4 | 100 | 7 |
| 8 | Ru2 | — | 5 | K3PO4 | 100 | 0 |
| 9 | Ru2 | dmpe | 5 | K3PO4 | 100 | 20 |
| 10 | Ru(dppf)(en)Cl2 | — | 5 | K3PO4 | 100 | 7 |
| 11 | Ru(PNP-2)Cl2 | — | 5 | K3PO4 | 100 | 11 |
| 12 | Ru(PNP-3)Cl2 | — | 5 | K3PO4 | 100 | 18 |
| 13 | Ru(PNP-3)(PMe3)Cl2 | — | 5 | K3PO4 | 100 | 64 |
| 14 | Mn(CO)2(PNP-3)Br | — | 5 | K3PO4 | 100 | 28 |
| 15 | Ir(PNP-3)H2Cl | — | 5 | K3PO4 | 100 | 6 |
| 16 | Ru(PNP-3)(PMe3)Cl2 | — | 5 | DBU | 100 | 0 |
| 17 | Ru(PNP-3)(PMe3)Cl2 | — | 5 | K2PO3 | 100 | 49 |
| 18 | Ru(PNP-3)(PMe3)Cl2 | — | 5 | KOtBu | 100 | 20 |
| 19 | Ru(PNP-3)(PMe3)Cl2 | — | 5 | K3PO4 | 60 | 42 |
| 20 | Ru(PNP-3)(PMe3)Cl2 | — | 5 | K3PO4 | 80 | 61 |
| 21 | Ru(PNP-3)(PMe3)Cl2 | — | 5 | K3PO4 | 120 | 51 |
| 22 | Ru(PNP-3)(PMe3)Cl2 | — | 10 | K3PO4 | 100 | 58 |
| 23 | Ru(PNP-3)(PMe3)Cl2 | — | 10 | K3PO4 | 80 | 71 |
|
||||||
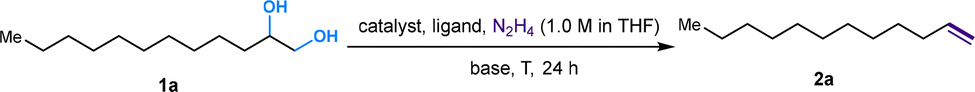
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Catalyst loading (mol%) | K3PO3 (mol%) | T (°C) | DMSO (μL) | 4a (%) |
| Reaction conditions: 3a (27.6 mg, 0.2 mmol), Ru(PNP-3)(PMe3)Cl2 (5.54 mg, 0.01 mmol, 5 mol%), K3PO4 (10.6 mg, 0.05 mmol, 25 mol%), 1 M N2H4 solution in THF (0.5 mL, 0.5 mmol, 2.5 equiv.), DMSO (5 μL, 0.07 mmol, 35 mol%), 16 h at 70 °C, under N2. | ||||||
| 1 | Ru(PNP-3)(PMe3)Cl2 | 10 | 75 | 80 | 5 | 34 |
| 2 | Ru(PNP-3)(PMe3)Cl2 | 5 | 75 | 80 | 5 | 51 |
| 3 | Ru(PNP-3)(PMe3)Cl2 | 5 | 100 | 80 | 5 | 43 |
| 4 | Ru(PNP-3)(PMe3)Cl2 | 5 | 50 | 80 | 5 | 53 |
| 5 | Ru(PNP-3)(PMe3)Cl2 | 5 | 25 | 80 | 5 | 58 |
| 6 | Ru(PNP-3)(PMe3)Cl2 | 5 | 10 | 80 | 5 | 40 |
| 7 | Ru(PNP-3)(PMe3)Cl2 | 5 | 25 | 70 | 5 | 88 |
| 8 | Ru(PNP-3)(PMe3)Cl2 | 5 | 25 | 50 | 5 | 23 |
| 9 | Ru(PNP-3)(PMe3)Cl2 | 5 | 25 | 70 | — | 68 |
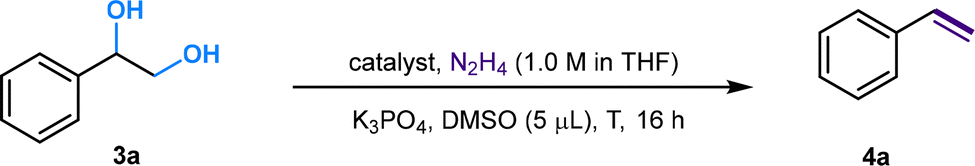
| a 1H NMR yields were reported using mesitylene as an internal standard. b The reaction was conducted at 100 °C. c The reaction was conducted at 120 °C. Reaction conditions A for aliphatic diols: diol (0.2 mmol), Ru(PNP-3)(PMe3)Cl2 (11.1 mg, 0.02 mmol, 10 mol%), K3PO4 (31.8 mg, 0.15 mmol, 75 mol%), 1 M N2H4 solution in THF (0.5 mL, 0.5 mmol, 2.5 equiv.), 24 h at 80 °C, under an N2 atmosphere. Yields of isolated products were reported unless otherwise noted. Reaction conditions B for aromatic diols: diol (0.2 mmol), Ru(PNP-3)(PMe3)Cl2 (5.54 mg, 0.01 mmol, 5.0 mol%), K3PO4 (10.6 mg, 0.05 mmol, 25 mol%), 1 M N2H4 solution in THF (0.5 mL, 0.5 mmol, 2.5 equiv.), DMSO (5 μL, 0.07 mmol, 35 mol%), 16 h at 70 °C, under an N2 atmosphere. Yields of isolated products were reported unless otherwise noted. |
|---|

|
In conclusion, we have disclosed for the first time a DODH process of terminal diols toward alkenes with hydrazine by using a ruthenium catalyst. Both aromatic and aliphatic diols were efficiently transformed, resulting in the release of benign water, hydrogen gas, and nitrogen gas as by-products. Compared to traditional DODH methodologies, this protocol exhibits high selectivity of terminal diols under mild reaction conditions. From a sustainability perspective, this DODH method sets up a promising avenue for the selective conversion of biomass-based feedstocks into high-value olefin products. Therefore, further studies of the mechanism and synthetic utilities of this DODH strategy are ongoing.
We are grateful to the Canada Research Chair Foundation (to C.-J. L.), the CFI, FRQNT Centre for Green Chemistry and Catalysis, NSERC, Killam Research Fellowship of the Canadian Council for the Arts, and McGill University for financial support. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. Supplementary information: General information, experimental procedures, unsuccessful substrates, spectroscopic data, and copies of 1H and 13C NMR spectra of the products. See DOI: https://doi.org/10.1039/d5cc04813c.Notes and references
- K. Yamatsugu and M. Kanai, Chem. Rev., 2023, 123, 6793–6838 CrossRef PubMed
.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Renewable Sustainable Energy Rev., 2020, 127, 109852 CrossRef CAS
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC
- M. Schuster and S. Blechert, Angew. Chem., Int. Ed. Engl., 1997, 36, 2036–2056 CrossRef
- Z. Wu, M. Hu, J. Li, W. Wu and H. Jiang, Org. Biomol. Chem., 2021, 19, 3036–3054 RSC
- A. Maercker, Org. React., 2004, 14, 270–490 Search PubMed
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS
- L. F. Van Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195–200 RSC
- P. R. Blakemore, J. Chem. Soc., Perkin Trans. 1, 2002, 2563–2585 RSC
-
C. Boucher-Jacobs and K. M. Nicholas, Deoxydehydration of polyols, Selective catalysis for renewable feedstocks and chemicals, 2014, pp. 163–184 Search PubMed
- A. R. Petersen and P. Fristrup, Chem. – Eur. J., 2017, 23, 10235–10243 CrossRef PubMed
- L. J. Donnelly, S. P. Thomas and J. B. Love, Chem. – Asian J., 2019, 14, 3782–3790 CrossRef
- N. N. Tshibalonza and J.-C. M. Monbaliu, Green Chem., 2020, 22, 4801–4848 RSC
- E. Block, Org. React., 1984, 30, 457–566 CrossRef
- E. E. López-López, J. A. Pérez-Bautista, F. Sartillo-Piscil and B. A. Frontana-Uribe, Beilstein J. Org. Chem., 2018, 14, 547–552 Search PubMed
-
(a) L. Sandbrink, E. Klindtworth, H.-U. Islam, A. M. Beale and R. Palkovits, ACS Catal., 2016, 6, 677–680 CrossRef
- A. R. Petersen, L. B. Nielsen, J. R. Dethlefsen and P. Fristrup, ChemCatChem, 2018, 10, 769–778 CrossRef
- J. R. Dethlefsen, D. Lupp, B. C. Oh and P. Fristrup, ChemSusChem, 2014, 7, 425–428 CrossRef PubMed
- C.-J. Li, Pure Appl. Chem., 2023, 95, 465–474 CrossRef
- X.-J. Dai, C.-C. Li and C.-J. Li, Chem. Soc. Rev., 2021, 50, 10733–10742 RSC
- C.-J. Li, J. Huang, X.-J. Dai, H. Wang, N. Chen, W. Wei, H. Zeng, J. Tang, C. Li and D. Zhu, Synlett, 2019, 1508–1524 Search PubMed
- W. Wei, X.-J. Dai, H. Wang, C. Li, X. Yang and C.-J. Li, Chem. Sci., 2017, 8, 8193–8197 RSC
- U. K. Das, S. Chakraborty, Y. Diskin-Posner and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 13444–13448 CrossRef
- D. Srimani, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 11092–11095 CrossRef
- E. Khaskin and D. Milstein, Chem. Commun., 2015, 51, 9002–9005 RSC
- C.-C. Li, H. Wang, M. M. Sim, Z. Qiu, Z.-P. Chen, R. Z. Khaliullin and C.-J. Li, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed
-
(a) B. Sardar, N. Biswas and D. Srimani, Organometallics, 2023, 42, 55–61 CrossRef
- C.-C. Li, J. Kan, Z. Qiu, J. Li, L. Lv and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 4544–4549 CrossRef PubMed
- X.-J. Dai and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5433–5440 CrossRef PubMed
- X.-J. Dai, H. Wang and C.-J. Li, Angew. Chem., Int. Ed., 2017, 56, 6302–6306 CrossRef
- U. K. Das, Y. Ben-David, Y. Diskin-Posner and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 2179–2182 CrossRef CAS PubMed
- J. O. Bauer, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2016, 6, 8415–8419 CrossRef CAS
- J.-L. Huang, X.-J. Dai and C.-J. Li, Eur. J. Org. Chem., 2013, 6496–6500 CrossRef CAS
- S. Luo, H. D. M. Pham, C.-C. Li, Z. Qiu, R. Cheng, R. Z. Khaliullin and C.-J. Li, Org. Lett., 2024, 26, 3004–3009 CrossRef CAS PubMed
Footnote |
| † Siyi Luo, Chen-Chen Li, and Zihang Qiu contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |