
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemiconductor photocatalysts for hydrogen evolution: critical role of cocatalysts in enhancing performance
Bhagyashree Priyadarshini
Mishra
*,
Jasbir
Dahiya
and
Venkata
Krishnan
 *
*
School of Chemical Sciences and Advanced Materials Research Center, Indian Institute of Technology Mandi, Kamand, Mandi, 175075, Himachal Pradesh, India. E-mail: bhagyashree94dkl@gmail.com; vkn@iitmandi.ac.in
First published on 20th October 2025
Abstract
The conversion of abundant solar energy into green H2 fuel from photocatalytic water splitting has been extensively studied to address the growing energy demand. In this context, various semiconductors have been employed as photocatalysts; however, insufficient active sites for the redox reactions limit H2 evolution posing a significant challenge in semiconductor photocatalysis. Often, the use of a tiny amount of an additional constituent, known as a ‘cocatalyst’, can solve the issue by synergistically enhancing the performance of the semiconductor. Over the past few decades, noble metals and their derivatives have attracted huge research interest as cocatalysts; nevertheless, their limited availability and high cost significantly hinder large-scale applications. Hence, tremendous research attention has been devoted recently to design and develop inexpensive, earth-abundant, and highly stable materials which can be used as H2 evolution cocatalysts. This review article gives an outline of the recent advances in the high performance materials that have been used as efficient H2 evolution cocatalysts alongside photoactive semiconductors. Specifically, the H2 evolution cocatalysts summarized in this article are classified under metal nanoparticles, single atoms, bimetallic alloys, metal oxides, transition metal dichalcogenides, metal phosphides, metal carbides, metal borides and carbon based cocatalysts. Finally, the article discusses the pivotal challenges that restrict future advancements and provide potential research directions in the field of cocatalyst-driven photocatalytic H2 evolution using active semiconductors.
 Bhagyashree Priyadarshini Mishra | Dr Bhagyashree Priyadarshini Mishra received her MSc degree from Ravenshaw University, Odisha, India. She completed her PhD from Siksha ‘O’ Anusandhan University, Odisha, where she worked in the field of photocatalytic hydrogen evolution and H2O2 production. She joined the research group of Prof. Venkata Krishnan in 2024 as a postdoctoral researcher under the National Postdoctoral Fellowship (NPDF) scheme of Government of India. Her current research focuses on the development of effective photocatalysts for hydrogen evolution, nitrogen fixation and carbon dioxide reduction. |
 Jasbir Dahiya | Jasbir Dahiya received his BSc degree in Chemistry from M.D.U. University, Haryana, in 2022 and completed his MSc degree in Chemistry from the Indian Institute of Technology Mandi, India, in 2025. During his master's degree, he worked on the design of defect-rich transition metal oxide catalysts for photocatalytic hydrogen evolution and nitrogen fixation under the supervision of Prof. Venkata Krishnan. His research interests include heterogeneous catalysis, photocatalysis, and defect engineering strategies for sustainable energy conversion. |
 Venkata Krishnan | Prof. Venkata Krishnan obtained his PhD from the University of Stuttgart, Germany, in July 2006. Later, he worked as a postdoctoral researcher at the University of Pennsylvania, USA, from 2006 to 2010 and then as a research associate at the National Institute for Materials Science, Japan, from 2010 to 2012. He joined IIT Mandi in 2012 and is currently working as a Full Professor in the School of Chemical Sciences. His research interest is in the broad field of green chemistry and heterogeneous catalysis for energy and environmental applications. |
1. Introduction
Rapid industrialization has increased the demand for fossil fuels like coal, oil, and natural gas, which has put a heavy burden on the environment and led to a sharp increase in greenhouse gas emissions.1–3 A key tactic for solving the energy problem and environmental issues is switching to clean and renewable energy sources.3–5 Hydrogen (H2) is increasingly recognized as a clean and sustainable energy source, making it a strong contender to replace carbon-based fuels.6,7 There are several ways to produce H2, including water electrolysis, reforming natural gas, converting biomass, reforming methanol, and breaking down ammonia.8–12 Each of these methods has its own benefits and challenges, adding to the growing appeal of H2 as an environmentally friendly energy option. In this regard, solar energy is noteworthy because of its enormous availability and potential for a variety of uses. Overall splitting of water utilizing solar energy results in the evolution of both H2 and O2 in a stoichiometric ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and is considered an attractive aspect that contributes to energy revolution.13–16 However, focusing on selective H2 production from water splitting is one potential technique that not only provides a way to convert sunlight into clean chemical energy but also offers a practical solution to the current energy and environmental issues.17–19
:1 and is considered an attractive aspect that contributes to energy revolution.13–16 However, focusing on selective H2 production from water splitting is one potential technique that not only provides a way to convert sunlight into clean chemical energy but also offers a practical solution to the current energy and environmental issues.17–19
In this regard, after the revolutionary research by Fujishima and Honda in 1972 that water could be split into O2 and H2 using an electrochemical cell with a TiO2 electrode, illuminated by ultraviolet light, and coupled to a platinum electrode without the need for an external voltage, numerous UV active semiconducting materials have been utilized for photocatalysis.20,21 The photocatalytic process typically entails the absorption of light followed by the migration of photoexcited electron–hole pairs to surface active sites of the semiconductor, where they initiate reduction and/or oxidation reactions. The equilibrium of the thermodynamics and kinetics of the charge generation and separation process determines the overall efficiency of photocatalytic water splitting.22 Moreover, the efficiency of these materials is constrained by their narrow light absorption, mostly limited to the ultraviolet region, which accounts for just 5% of the solar spectrum, and their high rate of charge carrier recombination, creating a significant barrier to practical implementation. Thus, it is essential to enhance the absorption capabilities of the wide-bandgap materials by boosting light absorption, suppressing the recombination of photoinduced electron–hole pairs, and expanding their sensitive wavelength range.
Therefore, various strategies have been employed to obtain highly efficient semiconductor photocatalysts with increased potential for selective H2 evolution from aqueous solution, like bandgap engineering (by doping metals/non-metals), modulating size and morphology, coupling with various narrow-bandgap semiconductors, and incorporating cocatalysts that exhibit localized surface plasmon resonance (SPR) effects.23–27 Among these techniques, the combination of semiconductors with efficient cocatalysts has attracted a lot of interest nowadays. So far, a variety of cocatalysts have been used in photocatalytic H2 evolution reactions, with the majority of attention focused on noble metal-based cocatalysts. Elements such as Pt, Pd, Au, Ag, and Ru have been thoroughly studied and found to significantly improve the photocatalytic H2 evolution performance of the semiconductor.27 Furthermore, cocatalysts derived from noble metals, such as PtS and RhxP, have garnered a lot of interest because of their higher catalytic activity compared to their elemental counterparts.28,29 However, the scarcity and high cost of these precious metals prevent their widespread application. Because of this, increasing attention is being directed toward affordable cocatalysts made of earth-abundant elements that can act as electron sinks to enable charge migration and encourage hydrogen evolution. These cocatalysts include metal nanoparticles, single atoms, bimetallic alloy cocatalysts, metal oxide cocatalysts, transition metal dichalcogenide cocatalysts, metal phosphide cocatalysts, metal carbide cocatalysts, carbon based cocatalysts, and metal boride cocatalysts obtained from inexpensive and earth-abundant elements that can show their excellence in photocatalytic H2 evolution.
With the recent surge in advancements related to the development of cost-effective, stable, and efficient cocatalysts, it is an opportune moment to systematically evaluate and summarize photocatalytic systems where semiconductors are integrated with cocatalysts. Some impressive reports have summarized the synthesis of cocatalysts with their loading process over the support semiconductor, along with their photocatalytic applications in various fields like carbon dioxide reduction, nitrogen fixation, and pollutant degradation. Similarly, different cocatalyst loading over a particular support material has been summarized recently. Moreover, reviews summarizing the localized surface plasmon effect based cocatalysts have also been reported. However, a comprehensive assessment concerning the critical role of various cocatalysts over well-known support materials in photocatalytic H2 evolution is scanty. In this article, the contribution of cocatalysts in enhancing the photocatalytic H2 evolution from water splitting is discussed in detail. Furthermore, the cocatalysts have been classified as metal nanoparticles, single atoms, bimetallic alloys, metal oxides, transition metal dichalcogenides, metal phosphides, metal carbides, metal borides and carbon based cocatalysts and their modification with various widely used semiconductors is categorically discussed. Advantages as well as disadvantages of the cocatalysts have been discussed briefly. Finally, an assessment of the present state and future prospects of the field is given to stimulate creative ideas for developing cocatalyst-modified heterojunction systems in catalytic applications within this rapidly expanding area. This study thus concentrates on new developments in the use of cocatalysts in photocatalytic H2 evolution reactions. The various cocatalysts utilized for photocatalytic H2 evolution and covered in this article are shown in Scheme 1.
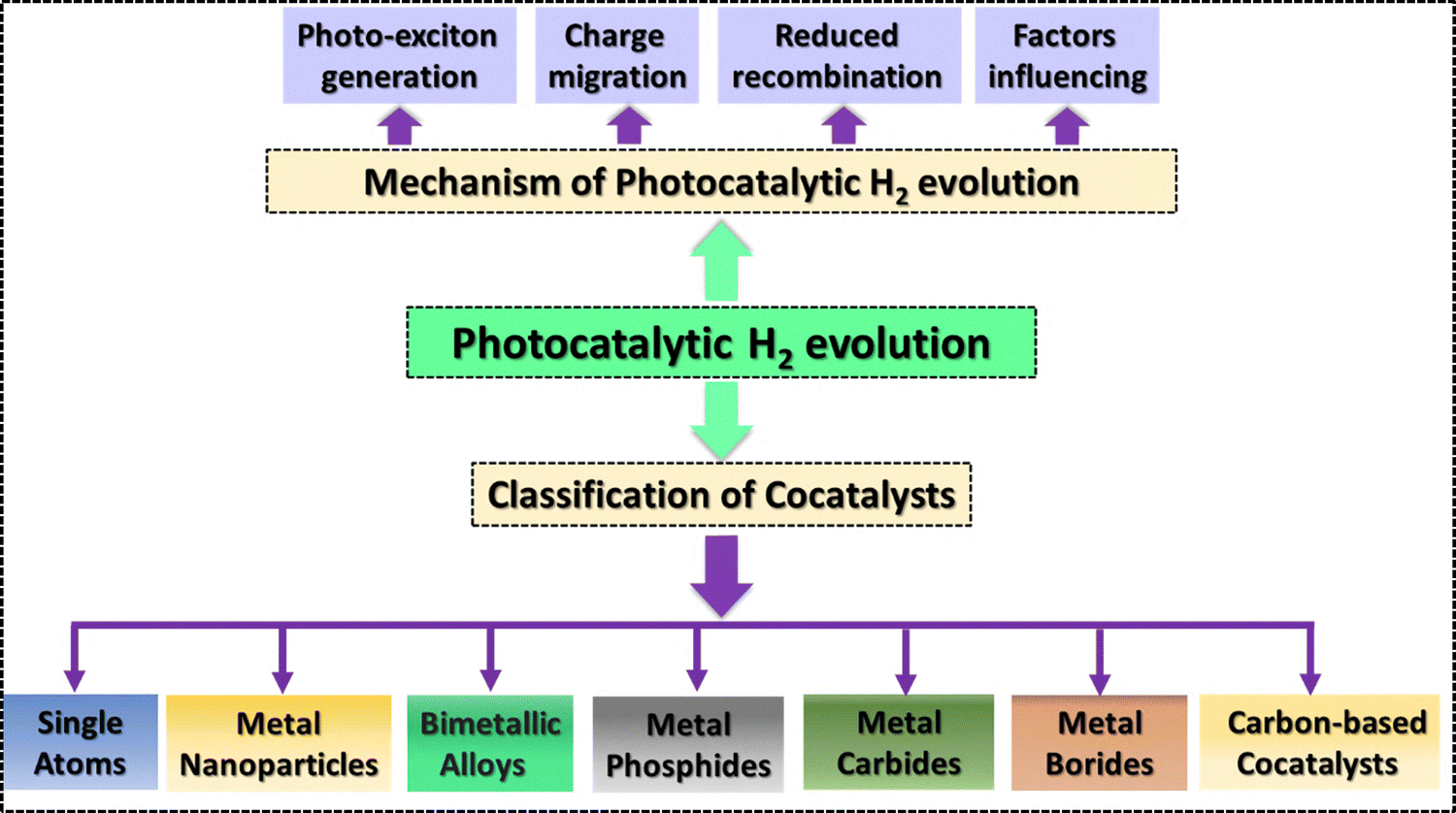 | ||
| Scheme 1 Schematic illustration of various aspects discussed in this article. | ||
2. Mechanistic insight into improved photocatalytic activity by cocatalysts
The basic principle of photocatalysis involves absorption of photons by the semiconducting material with an appropriate bandgap, resulting in the generation of photogenerated charge carriers (electrons and holes). These photoexcited charge carriers then migrate to the surface to participate in various redox reactions. The sequential steps after photo-exciton generation are represented in Scheme 2a and b. The photocatalytic hydrogen evolution process can be divided into three primary stages: electron generation, electron transport, and electron utilization. The initial stage is electron generation in which the photocatalyst, typically in powder form, is dispersed in an aqueous medium. Upon light irradiation, electrons in the valence band (VB) are excited to the conduction band (CB), leaving behind holes in the VB. For efficient photocatalysis, the material should exhibit strong light absorption over a broad wavelength range and a high quantum yield. Additionally, the CB edge must be more negative than the standard reduction potential of H+/H2 to ensure that the excited electrons possess sufficient reducing power. The subsequent step is electron transport. After excitation, the charge carriers migrate to the surface of the photocatalyst. However, some electrons and holes recombine within the bulk or on the surface, leading to energy loss as heat or light. This recombination significantly diminishes overall efficiency. To counteract this, effective spatial charge separation is essential. Achieving this separation under a suitable driving force remains one of the main challenges in photocatalytic systems. Scheme 2c represents the respective timescales for the photoinduced charge carriers. The final step is electron utilization. Photocatalytic water splitting using a semiconducting material denotes the stoichiometric splitting of water into H2 and O2 in the absence of sacrificial agents. To promote H2 evolution in aqueous medium, a half reaction of water splitting, electron donors or hole scavengers have been used to irreversibly oxidize the reducing agent instead of water. Basically, surface holes are effectively scavenged by electron donors to prevent recombination. The most commonly used hole scavengers include alcohols (e.g., methanol, ethanol, ethylene glycol, glycerol), organic acids (e.g., ascorbic, lactic, acetic), EDTA salts, sulfide species (Na2S–Na2SO3), and various reducing agents like glucose, triethanolamine, and triethylamine. Subsequently, electrons will be available over the surface of the photocatalyst. Hence, the retained electrons then drive the hydrogen evolution reaction, reducing protons to molecular hydrogen. For this step to proceed efficiently, the electron potential must be sufficiently negative, and the system must overcome kinetic overpotentials. It has also been proven that the Gibbs energy involved in hydrogen evolution accompanied with the oxidative decomposition of the sacrificial reagent is typically smaller than that of overall water splitting. This means that the energy stored in ‘hydrogen’ obtained using a sacrificial reagent is smaller compared to the case of hydrogen obtained from overall water splitting. Therefore, the ultimate goal is H2 production using water as a hydrogen and electron source. Moreover, the H2 evolution rate is influenced by several factors, including the number of available active sites, the specific reaction pathway, and mass transport limitations at the solid–liquid interface.4,30 Hence, to enhance H2 evolution from water splitting without using sacrificial agents, it is essential to utilize the photoinduced surface electrons and this is being explored by the scientific community.31 | ||
| Scheme 2 (a) Illustration of the photocatalytic hydrogen evolution mechanism involving a cocatalyst-loaded semiconductor, (b) depiction of the sequential processes like light absorption, charge carrier separation, migration, and recombination that occur within a cocatalyst-modified semiconductor during photocatalytic hydrogen production, (c) respective timescales in unmodified semiconductors and in metal–semiconductor heterostructures. Reproduced with permission.40 Copyright 2021, Wiley. | ||
As discussed before, photocatalytic water splitting causes both reduction of water to produce H2 and oxidation of water to produce O2, and back reaction between H2 and O2 to form water is possible as it is an uphill reaction. This can also affect the overall efficiency of photocatalytic H2 evolution. Hence, in order to increase the H2 evolution quantum efficiency, photocatalysts with surface properties that suppress back reactions are required. In this regard, cocatalysts are being loaded to accelerate the transfer of photogenerated electrons from the conduction band of the semiconductor to the surface of active cocatalysts, thereby achieving effective electron–hole separation. In addition, cocatalysts can reduce the activation energy or overpotential required for hydrogen evolution reactions occurring on semiconductor surfaces.1,32 Another important role of cocatalysts is mitigating photo-corrosion and enhancing the stability of semiconductor photocatalysts. This cocatalyst-induced mechanism for enhancing charge transfer can be categorized into two distinct components:
Firstly, the integration of noble metals or metallic cocatalysts with semiconductor photocatalysts can induce the formation of Schottky barriers at the interface, which generate an internal electric field that facilitates efficient charge separation. This process is driven by the variance in Fermi levels between the metal cocatalysts and the semiconductor, resulting in charge redistribution until Fermi level alignment is achieved. The ensuing band bending and Schottky barrier formation effectively suppress the recombination of photogenerated charge carriers by hindering the backflow of electrons. This promotes the participation of a greater number of electrons in reduction reactions such as hydrogen evolution. Furthermore, certain noble metals, including Au and Ag, exhibit localized surface plasmon resonance (LSPR), which enhances light absorption and contributes to increased charge carrier utilization. In addition, cocatalysts composed of precious metals (e.g., PtS2, RuP), transition-metal phosphides, sulfides, and carbides are also capable of forming Schottky junctions, thereby further improving interfacial charge separation and photocatalytic performance. Secondly, conductive non-metallic carbon-based materials such as graphene and carbon nanotubes have the ability to form Schottky junctions which can also improve the charge separation and enhance the photocatalytic hydrogen evolution.33,34
However, the efficiency of cocatalysts in photocatalytic hydrogen evolution reactions is influenced by several factors, including the method of deposition, loading amount, particle size, morphology, dispersion uniformity, and chemical composition.35 Consequently, there is typically an optimal set of parameters for cocatalyst application that leads to the highest photocatalytic performance. For instance, in the case of cocatalyst loading amount, insufficient deposition can lead to suboptimal separation of photogenerated charge carriers, thereby limiting H2 evolution efficiency. Conversely, excessive loading may hinder photocatalytic activity by blocking active surface sites, reducing light absorption, and causing non-uniform particle dispersion. Morphology of the cocatalysts employed also affects the activity of the overall semiconductor–cocatalyst system. In this regard, Park et al. studied how the morphology of Pt (round, cube, and rough shape) over CdSe nanorods affects photocatalytic H2 evolution. They found that the rough tip Pt cocatalyst showed the highest photocatalytic H2 evolution followed by round and cubic tips.36 In addition, the facets over which a cocatalyst is loaded are also crucial in determining the efficiency of photocatalytic processes. Different crystal facets of a semiconductor often preferentially accumulate either photogenerated electrons or holes due to intrinsic anisotropic charge transport. For instance, photogenerated electrons and holes preferentially migrate to distinct facets of BiVO4 – electrons accumulate on the {010} facets, while holes accumulate on the {110} facets. This spatial separation enables reduction reactions to occur on {010} facets and oxidation reactions to occur on {110} facets. Hence, when the cocatalysts are selectively deposited on their corresponding active facets, with reduction cocatalysts on {010} and oxidation cocatalysts on {110}, the charge carriers are effectively trapped and utilized, minimizing recombination losses and significantly enhancing photocatalytic and photoelectrochemical activity. In contrast, random or misaligned deposition of cocatalysts leads to inefficient charge separation, lower activity, and even suppressed performance.37 In another study, it was shown that by selectively loading Rh/Cr2O3 reduction cocatalysts on electron-rich facets and CoOOH oxidation cocatalysts on hole-rich facets, the hydrogen evolution reaction and oxygen evolution reaction can be enhanced separately.38 Moreover, it has been reported that local heat generated by light absorption by cocatalyst nanoparticles may positively affect the photocatalytic activity. When plasmonic or light-absorbing cocatalyst nanoparticles (such as Au, Ag, or certain transition metal oxides) interact with incident photons, they not only facilitate charge transfer but also convert part of the absorbed light into localized thermal energy. This nanoscale heating can enhance surface reaction kinetics by lowering the activation energy barriers of redox processes and improving the mobility of reactants and intermediates near the active sites. Furthermore, the photothermal effect can promote desorption of reaction products from the catalyst surface, preventing site blockage and sustaining activity. Therefore, cocatalysts with dual photonic and photothermal properties represent a promising strategy to synergistically boost photocatalytic efficiency.39
3. Classification of co-catalysts for enhanced photocatalytic H2 evolution
3.1. Single-atoms as co-catalysts
Recently, single-atom (SA) metals with an even smaller size than metal NPs have emerged as highly promising cocatalysts in photocatalysis due to their unsaturated coordination environments and exceptional catalytic activity, offering significant potential for applications such as hydrogen evolution. Notably, metal SA catalysts can exhibit high catalytic performance while significantly reducing material usage and overall cost. Unlike, metal nanoparticles where the atoms inside the nanoparticles cannot contribute to the reaction as the catalytic reactions occur on the surface, SAs, which lack inactive internal atoms, can utilize every metal atom as an active site, significantly enhancing the catalytic performance on a per-metal-atom basis. Hence, SACs decorated over semiconductors have been constructed for enhancement of photocatalytic H2 evolution.41,42 In this context, Fu et al. synthesized Cu SACs supported on the (101) surface of TiO2, in which the incorporation of Cu atoms in place of Ti vacancies was confirmed from the HAADF-STEM imaging, as shown in Fig. 1a.43 They explored that the Cu atoms in Cu-SAC/TiO2 change their coordination during photocatalytic hydrogen evolution. In addition, it is reported that the local Cu–O structure plays a vital role in boosting catalytic activity as shown in Fig. 1b and c. Simulations reveal that the Cu dz2 orbital efficiently captures photogenerated electrons and transfers them to water molecules, driving the H2 evolution reaction. Briefly, the generated H atom during the Volmer process adsorbed to a neighboring bridging O atom of the Cu SAC, which led to changes in the valence state as well as the color of Cu. It is concluded that the adsorption of the H atom is not responsible for the activation of Cu-SAC/TiO2; instead, it creates an inert state by adding an extra electron into the Cu dz2 orbital. This single atom decorated metal oxide exhibited higher stability towards the photocatalytic H2 evolution even when subjected to varying pressure and irradiation over a 20-h period, as shown in Fig. 1d.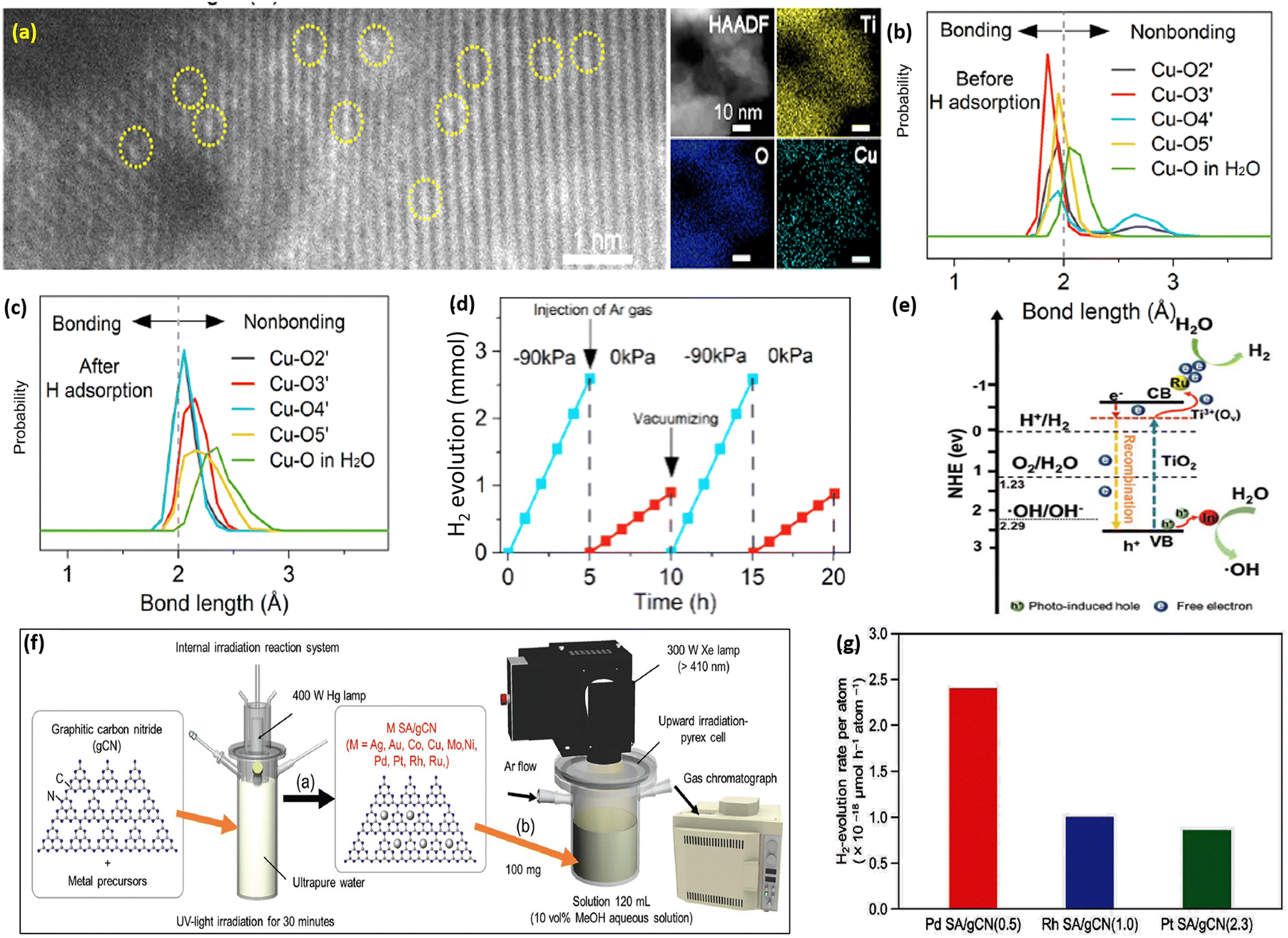 | ||
| Fig. 1 (a) High-resolution STEM and STEM-EDS mapping of Cu-SAC/TiO2. (b) and (c) Cu–O bond length distributions before and after hydrogen adsorption at 300 K. The observed elongation of Cu–O bond lengths after hydrogen adsorption indicates that the electron donated by the hydrogen atom enters Cu atom's d orbital, resulting in a repulsive interaction between the Cu atom and the H2O molecule. (d) Photocatalytic H2 evolution of Cu-SAC/TiO2 under varying dynamic pressure in an Ar-protected environment, with 0 kPa being the standard atmospheric pressure. Reproduced with permission.43 Copyright 2023, American Chemical Society. (e) The tentative mechanism for photocatalytic H2 production over Ru–In SA/TiO2. Reproduced with permission.44 Copyright 2022, Wiley. (f) (a) Synthesis of M SA-loaded g-CN and (b) photocatalytic H2 evolution for M SA/g-CN photocatalysts. (g) H2 evolution rate per metal atom for M SA/g-CN photocatalysts. Reproduced with permission.45 Copyright 2023, Wiley. | ||
Similarly, Peng et al. introduced an advanced photocatalyst, Ru/In dual-single atoms doped on TiO2 nanosheets (Ru–In SA/TiO2), designed to address the challenges of efficient hydrogen production through photocatalytic water splitting without sacrificial agents. This novel catalyst outperformed its counterparts such as Ru-doped, In-doped, and pristine TiO2 by 6, 18, and 53 times, respectively, with a remarkable hydrogen evolution rate of 174.1 μmol h−1. The dual-single atom doping approach increased the separation of photogenerated electrons and holes enhancing charge carrier mobility and reducing recombination by boosting Ti3+ sites and oxygen vacancies. Moreover, Ru facilitated electron transfer and In enhanced hole transfer, which inhibited electron–hole recombination and accounted for this efficiency (Fig. 1e). The catalyst provides an affordable and environmentally friendly way to produce hydrogen, which aligns with the goals of renewable energy. It also functions effectively without a sacrificial agent and shows remarkable stability.44
Furthermore, Akinaga and co-workers explored the importance of single-atom cocatalysts on graphitic carbon nitride (g-C3N4) for enhancing the photocatalytic H2 evolution reaction.45 They examined ten different transition metal elements (Ag, Au, Co, Cu, Mo, Ni, Pd, Pt, Rh, and Ru) supported on g-C3N4, by a photo-deposition method to achieve high weight loading (≈3 wt%) while preventing aggregation as shown in Fig. 1f. Experimental results demonstrated that Pd, Pt, and Rh single-atom catalysts supported on g-C3N4 exhibited superior H2 evolution activity, with Pd SA catalysts achieving the highest H2 evolution rate per active site owing to their favorable electronic configuration, which enhances proton adsorption and facilitates efficient charge transfer. Transient absorption spectroscopy, electrochemical studies, and X-ray absorption fine structure all verified that SA cocatalysts effectively promote charge separation and considerably lower the H2 evolution overpotential. It is noteworthy that g-C3N4 loaded with Pd SAs demonstrated an 8.6-fold increase in H2 evolution efficiency (Fig. 1g) per active site in comparison to Pd nanoparticles, demonstrating the benefits of single-atom dispersion in maximizing photocatalytic hydrogen generation. In a subsequent study, Ren and co-workers studied the effect of particle size of co-catalysts on photocatalytic H2 evolution.46 According to the study, the H2 evolution activity of the covalent organic layers supported on SiO2 nanoparticles was greatly increased when Pd nanoparticles were downsized to the atomic or cluster scale (Fig. 2a). An enhanced hydrogen evolution rate with an apparent quantum efficiency of 7.3% was observed with the Pd SAs/Cs cocatalyst. The enhancement in photocatalytic activity was attributed to the enhanced charge separation efficiency of COF layers rather than proton reduction.
 | ||
| Fig. 2 (a) Schematic illustration of the synthesis of Pd SAs/Cs on TP-TTA COF layers and the photoactive Pd0.033/TP TTA/SiO2/PAN film. Reproduced with permission.46 Copyright 2022, American Chemical Society. (b) Assembly of MOC-Q3 using ligand L-2 and Pd2+, crystal structure of MOC-Q3, EA-COF preparation using TP and EA, and structure of MOC-Q3/EA-COF. (c) Working mechanism of the MOC-Q3/EA-COF system. Reproduced with permission.47 Copyright 2024, American Chemical Society. (d) Schematic representation of the synthesis of the CoP-Vp@CZS photocatalyst. (e) FESEM images. (f) High magnification image of CoP-Vp@CZS. (g) Recyclability study of CoP-Vp@CZS in successive H2 evolution cycles. Reproduced with permission.48 Copyright 2023, Wiley. | ||
A unique Z-scheme single atom photosystem has been developed by Liang et al. which appears as a triangular prism shaped metal–organic cage (MOC-Q3) embedded with 3 catalytic Pd2+ sites and two photosensitive ligands, and the MOC-Q3 was loaded over a highly crystalline β-ketoenamine-linked COF (EA-COF) as shown in Fig. 2b.47 The optimized catalyst, MOC-Q3/EA-COF, was observed to achieve a higher H2 evolution with a TONPd of 118521 when ascorbic acid was used as a sacrificial agent. Furthermore, the H2 evolution rate for the Z-scheme system increased about 1.8 times under piezo-photocatalytic conditions. The enhanced photocatalytic performance was due to the broad light absorption, effective charge separation, and uniform distribution of the Pd SA active sites. The MOC-Q3/EA-COF Z-scheme photocatalyst also showed better photocurrent and less charge transfer resistance compared to the other counterparts. The photocatalytic mechanism was revealed by calculating the band edge potentials combining the outputs from Mott–Schottky measurements, valence band X-ray photoelectron spectroscopy, and UV-vis DRS as shown in Fig. 2c.
Zhang and co-workers reported a novel strategy to enhance photocatalytic hydrogen production by engineering single-atom phosphorus vacancies into the CoP cocatalyst (CoP-Vp) and the cocatalyst was coupled with Cd0.5Zn0.5S to form the CoP-Vp@Cd0.5Zn0.5S (CoP-Vp@CZS) heterojunction.48 The phosphorus defect enriched CoP hollow microspheres were synthesized from phosphorization of ZIF-67 followed by the liquid phase corrosion process as shown in Fig. 2d. The CoP obtained from phosphorization of the intermediate Co–OH had a hollow structure composed of a uniform assembly of vertically aligned nanoflakes (Fig. 2e and f). However, anchoring of CZS increased the thickness of the nanoflakes to 38 nm. In photocatalytic H2 evolution, the CoP-Vp@CZS catalyst achieved a remarkable hydrogen evolution rate of 2.052 mol h−1 per 30 mg of catalyst, which was 1.41 and 14.66 times greater than that of the pure CoP@CZS and ZCS samples, respectively, and a little decline in activity was noticed after 40 h of uninterrupted stability tests as shown in Fig. 2g. The enhanced H2 evolution was supported by the results obtained from photocurrent and time-resolved photoluminescence analysis. Hence, the incorporation of single atom CoP-Vp on to the CZS semiconductor successfully enhanced the photocatalytic H2 evolution efficiency. Similarly, Zhao et al. anchored cobalt single atoms over nitrogen-doped graphene (NG) to create an effective cocatalyst (Co-NG).49 They found that the photocatalytic H2 evolution increased to 1382 μmol h−1 when Co-NG was incorporated with CdS which is 3.42 times higher than that achieved with the NG-loaded CdS photocatalyst. This study also proved that SACs improve photocatalytic H2 evolution reaction. In another study, Zeng and coworkers fabricated Pt single atom incorporated carbon nitride with a significant content of 8.7 wt%. The photocatalyst exhibited a H2 evolution rate of 22650 μmol g−1 h−1 with 22.5% AQE (∼420 nm). They found that the location of Pt SAs plays a crucial role in promoting the photocatalytic performance.50 A series of Pd-loaded TiO2 photocatalysts were synthesized by Lv et al., suggesting that Pd size has a considerable impact on photocatalytic hydrogen evolution.51 Amongst these, the maximum activity was shown by Pd0.75/TiO2, which was 15, 1.5, and 1.6 times greater than that of Pdf/TiO2, Pd0.5/TiO2, and Pd1.0/TiO2, respectively. DFT studies showed that the combination of sub-nano cluster and single-atom Pd (PdSA) enhances the H* adsorption–desorption balance by optimising the d-band centre and bringing ΔGH* to zero. In keeping with the noted greater charge separation, PdSNC also encouraged electron transfer from PdSA. The design of sophisticated photocatalysts was guided by this work, which provides important insights into photocatalytic H2 evolution processes. Despite spectacular advantages of SACs in the field of photocatalytic H2 evolution, they suffer from several shortcomings, which limits their potential as co-catalytic materials. Firstly, stability of SACs remains a critical issue as isolated metal atoms tend to migrate and aggregate under reaction conditions, leading to nanoparticle formation and loss of catalytic activity. Secondly, the limited density of active sites restricts overall reaction rates, especially for large-scale hydrogen production, since loading beyond a certain threshold often causes aggregation. Thirdly, the coordination environment of single atoms is highly sensitive to the support surface; hence any structural change during operation (e.g., photo-corrosion or surface reconstruction) may drastically alter catalytic performance. Lastly, fabricating SACs with precise control over dispersion is a significant synthetic challenge.
3.2. Metal nanoparticles as co-catalysts
Metal nanoparticles significantly enhance photocatalytic efficiency due to their localized surface plasmon resonance (LSPR) effect, a current focus in energy research. Under resonant light excitation, free electrons in these nanocrystals oscillate collectively, generating intense electromagnetic fields and producing energetic “hot electrons”. The LSPR properties like strong light absorption and near-field enhancement are highly dependent on the nanoparticles’ shape, size, and composition. Pt nanoparticles, the highest performing H2 evolution cocatalysts, exhibit work function above 5.5 eV. Other metal nanoparticles show work function values in the range of 4.3–5.6 eV. Additionally, when plasmonic metals (e.g., Au, Ag, Cu) contact n-type semiconductors, Fermi level alignment facilitates hot electron injection into the semiconductor's conduction band, enabling kinetically demanding multielectron reactions by lowering activation barriers. Furthermore, metal nanoparticles are highly promising for H2 evolution due to their large surface-to-volume ratio, abundance of active surface atoms, and distinct electronic properties compared to their bulk forms.52,53In this regard, Zhang et al. effectively utilized the LSPR effect of Ag metal nanoparticles to utilize solar energy efficiently. They decorated the plasmonic Ag nanoparticles over a covalent organic framework by an in situ method (Fig. 3a). The photocatalyst is evidenced to achieve an outstanding H2 evolution rate of 801.4 μmol g−1 h−1 with 3% loading of Ag NPs, which is nearly 4 times more than the rate of the pristine TpPa-1-COF. The red shift in the absorption edge compared to that of TpPa-1-COF as obtained from UV-Vis DRS indicates that the deposition of Ag NPs expands the visible-light responsive nature of the catalyst. The absorption peak at around 465 nm as depicted in Fig. 3b was identified due to the LSPR effect of Ag NPs. The hot electron generation was confirmed by finite-difference time-domain (FDTD) simulation study which showed that an increase in the local interfacial electric field significantly facilitates easy passage of hot electrons at the surface. The mechanistic study revealed that the hot electrons generated by the plasma action of Ag NPs flow back to the CB of TpPa-1-COF where the reduction process is carried out to form H2 as the work function of n-type TpPa-1-COF is more than that of Ag NPs (Fig. 3c).54
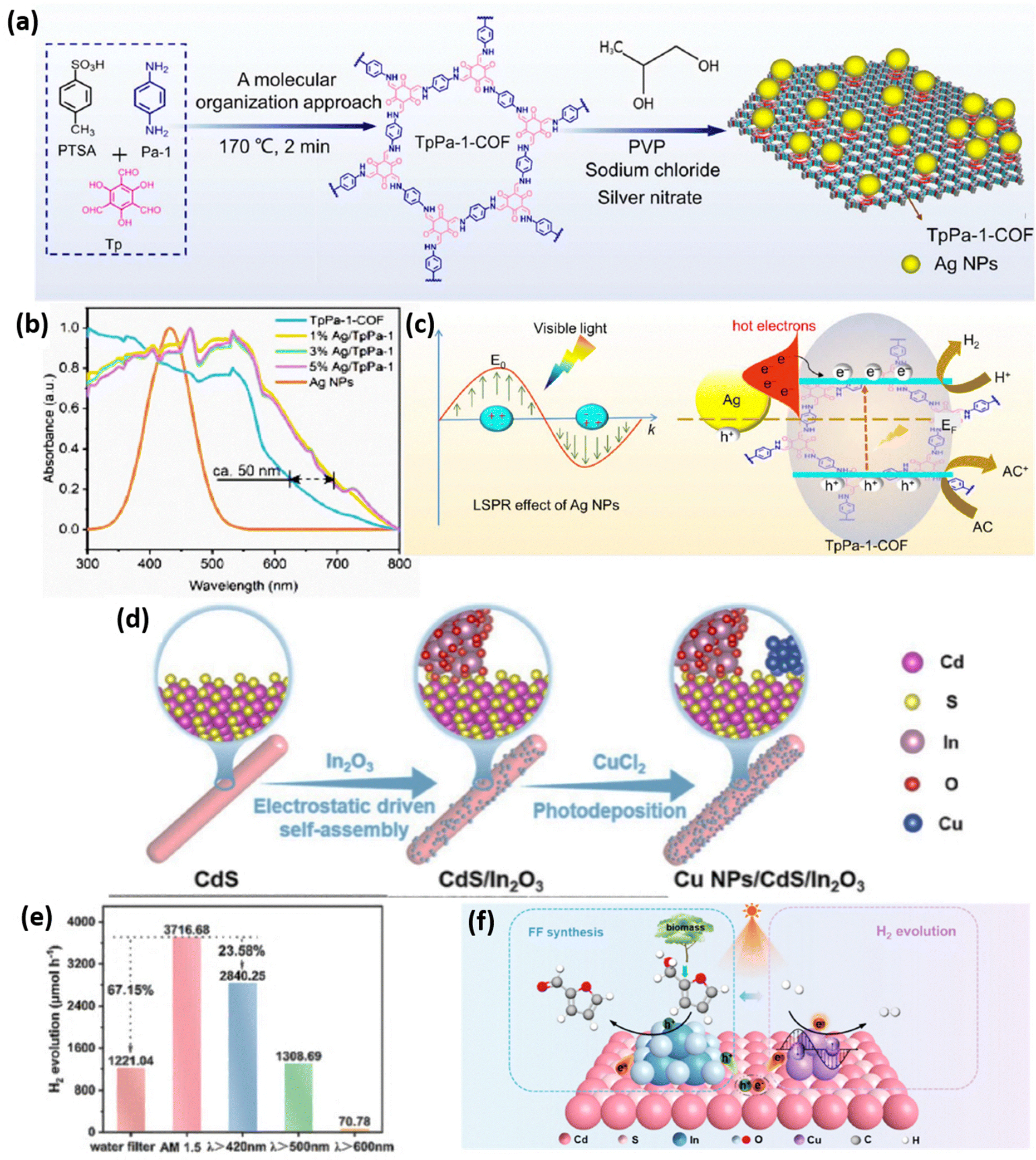 | ||
| Fig. 3 (a) Schematic depiction of the Ag/TpPa-1-COF synthesis. (b) UV-vis DRS spectra of the as-synthesized photocatalysts. (c) Suggested mechanism for the release of H2 by photocatalysis over Ag/TpPa-1. Reproduced with permission.54 Copyright 2024, Royal Society of Chemistry. (d) Schematic illustration of the synthesis of Cu NPs/CdS/In2O3. (e) Photocatalytic H2 evolution rate of Cu NPs/CdS/In2O3 under light irradiation at different wavelengths. (f) Mechanism over the photocatalyst Cu NPs/CdS/In2O3 for H2 evolution coupled with selective FFA oxidation to FF. Reproduced with permission.55 Copyright 2024, American Chemical Society. | ||
In another study, Liu and coworkers prepared a series of Cu/In2O3/CdS photocatalysts to intrinsically regulate the photogenerated charge carrier transfer for elevated photocatalytic H2 evolution (Fig. 3d). Metal nanoparticles like Au, Ag, etc. exhibit a LSPR effect that helps in broadening the light absorption range, refining charge separation, and facilitating the adsorption and dissociation of reactive molecules attributing to the generation of hot-electrons. Instead of using expensive NPs, herein, the cost-effective Cu NPs have been used to enhance the photocatalytic H2 evolution by facilitating the reduction of H+ to H2. The LMM spectra showed that the valence state of Cu in both binary and ternary composites is mainly Cu0 instead of Cu+. The study reported a better photocatalytic H2 evolution activity of CuNPs/In2O3/CdS (2840.25 μmol h−1) which exceeds the performance of pristine CdS based photocatalysts (Fig. 3e). The in situ XPS study verified the transfer of photoelectrons from CdS to Cu NPs as the binding energy of Cu 2p shifts negatively. The surface reaction thermodynamics confirmed the superior H+ adsorption ability of Cu and the electron transfer from Cu to H+. The increased concentration of high-energy hot electrons, which enhances H2 evolution, originated from two sources: one is the direct excitation due to the LSPR effect of Cu NPs and other is the transfer of photogenerated electrons from CdS to Cu NPs (Fig. 3f).55
Xu et al. synthesized a WS2@Cu hybrid by a two-step process in which Cu NPs are deposited over WS2 by a photochemical deposition method. The XPS profile from the Cu 2p region showed two peaks at 932.7 and 952.5 eV for Cu 2p3/2 and Cu 2p1/2 that are so close to Cu+ and Cu0 states, which was further confirmed by Cu LMM XAES analysis identifying the presence of Cu0 state. The WS2@Cu photocatalyst under irradiation with NIR light exhibited an average H2 evolution rate of around 178.6 μmol g−1 h−1, while bare Cu NPs showed a H2 evolution rate of 71.4 μmol g−1 h−1. Under NIR irradiation, WS2 exhibited no noticeable H2 evolution (Fig. 4a). This result suggested that NIR irradiation generated hot electrons that were injected to the CB of WS2 from plasmonic Cu over the Schottky barrier, facilitating the transfer of hot electrons to WS2 and suppressing the recombination rate in Cu NPs, thereby enhancing the photocatalytic activity (Fig. 4b). In addition, the H2 evolution activity further increases upon UV-vis light illumination as WS2 can also generate photoexcited electrons in the CB in addition to hot electrons and thus the increase in H2 evolution activity is due to the synergistic effect of two optical excitations.56
 | ||
| Fig. 4 (a) Photocatalytic H2 evolution on photocatalysts upon irradiation of >750 nm NIR light. (b) Proposed schematic representation of photoinduced charge migration in WS2@Cu for photocatalytic H2 evolution in the presence of sacrificial agent LA. Reproduced with permission.56 Copyright 2018, Wiley. (c) H2 evolution and associated field enhancement across diverse plasmonic photonic crystals shown, with the inset displaying electromagnetic field strengths for different plasmonic photocatalysts, in agreement with earlier SERS analyses. (d) The superior light absorption of Ag/SiO2 opal-270 for improved H2 evolution. Reproduced with permission.57 Copyright 2024, Wiley. (e) H2 evolution as a function of irradiation time under visible light (λ > 400 nm) for 0.5 wt% Cu/SrTiO3 and Au/SrTiO3. Reproduced with permission.58 Copyright 2019, Elsevier. (f) HRTEM image of Ag@SiO2@CdSAu. Reproduced with permission.59 Copyright 2020, American Chemical Society. (g) SEM image of Ag SHIN–Au@CdS nanoparticles. The inset illustrates a TEM image and the associated EDS mapping analysis of an individual Ag SHIN–Au@CdS nanoparticle. (h) Schematic illustration of the mechanism for enhancement in H2 evolution over Ag SHIN–Au@CdS dual antennas. Reproduced with permission.60 Copyright 2021, American Chemical Society. | ||
Mogan and co-workers unprecedently designed a plasmonic photonic crystal by taking a non-photo absorbing photonic counterpart to intensify the plasmonic field by enhancing the slow photon generation to promote light-to-chemical transformation. The deposited Ag NPs over SiO2 minimized the excessive competition between plasmonic metals on the photonic effect, mainly by mitigating the adverse effects of random plasmonic light scattering and optical screening. The plasmonic photonic catalyst achieved an outstanding H2 evolution rate outperforming other high-performance hybrid photocatalysts by up to 280-fold and surpassing bare Ag nanoparticles by >106-fold (Fig. 4c). This is attributed to the effective trapping and concentration of light within the plasmonic photonic crystals that enhance the photo absorption by the crystal and intensify the local plasmonic field on the Ag catalyst via LSPR (Fig. 4d). A surplus of hot carriers was produced as a result of non-radiative decay of the surface plasmons which promotes H2 evolution.57
To increase the lifetime of the hot electrons, various strategies like the use of heterojunctions, adjusting the reaction environment, and cocatalyst integration have been adopted, with metal particle size modulation being frequently overlooked. In this regard, Zhang et al. examined the effect of Cu NP size on the photocatalytic production of H2 by depositing Cu NPs ranging in size from 2.8 nm to 7.7 nm on the SrTiO3 support. The maximum detectable H2 generation activity of 76.3 μmol g−1 h−1 was noted at a 0.5 weight percent Cu loading and an average diameter of 3.9 nm, which is five times more than that of Au/SrTiO3 (Fig. 4e). Larger Cu particle sizes were responsible for the increment in photocatalytic H2 generation activity because they strengthen the SPR effect and lengthen the carrier lifespan. It is crucial to remember that Cu-based catalyst stability is often low, and further study is required to find solutions.58
Ren and co-workers developed a core–shell-satellite-type plasmonic nanocomposite photocatalyst, Ag@SiO2@CdS–Au, with varying core and shell thicknesses by a seeded growth method (Fig. 4f). They validated the significant impact of near-field improvement and PRET mechanisms in enhancing photocatalysis by comparing the reactivity towards photocatalytic H2 evolution. The inclusion of gold satellite nanoparticles helped to introduce hot electrons, expanding the range of light absorption for the photocatalyst and enhanced the electromagnetic field through Ag–Au coupling. The experimental findings demonstrated a significant improvement (approximately 200 times) in the efficiency of the photocatalytic hydrogen production process. The AQY spectra along with UV-vis spectra of the Ag@SiO2@CdS–Au core–shell-satellite nanocomposite and Ag@SiO2@CdS NPs were compared and it was observed that the Ag@SiO2@CdS–Au nanocomposite presented strong light absorption in the longer wavelength region (500-800 nm) due to the loading of Au satellite nanoparticles, outperforming Ag@SiO2@CdS NPs. The AQY plot of Ag@SiO2@CdS–Au displayed a similar trend but with much stronger light absorption in the short wavelength region (<500 nm) compared to Ag@SiO2@CdS.59
Another plasmon-mediated enhanced H2 evolution performance was reported by Yang et al. using a dual-plasmonic-antenna that efficiently generated energetic hot electrons and strong electromagnetic fields simultaneously as shown in Fig. 4g. Briefly, they fabricated the dual plasmonic catalyst by assembling Au@CdS with a pinhole on Ag@SiO2 and the H2 evolution rate by the photocatalyst was improved by almost 7000 times compared with CdS. The dual-plasmonic-antenna nanocomposites demonstrate superior photocatalytic activity for H2 evolution reaction under visible light irradiation. This is attributed to the efficient generation of a strong magnetic field and energetic hot electrons. The study shows that the hot electrons are produced in the Au cores of the Au@CdS satellites and then transferred to the conduction band of CdS, initiating the photocatalytic H2 evolution. Additionally, the strong electromagnetic field generated by the Ag SHINs promotes the production and migration of hot electrons, providing additional uplifting in the overall activity (Fig. 4h).60
3.3. Bimetallic alloys as co-catalysts
In recent years, bimetallic alloy nanoparticles (NPs) have increasingly drawn attention as cocatalysts for the photocatalytic generation of hydrogen from water. Studies have demonstrated that alloying a metal catalyst with a second metal can restrict the poisoning of the metal catalyst while enhancing its activity and selectivity.61 In accordance with the fundamental principles governing photocatalytic H2 production from H2O, bimetallic cocatalysts are anticipated to enhance the separation and migration of photogenerated excitons while providing adequate active sites for H2 evolution. Given the vast array of elements in the periodic table, countless bimetallic compositions are possible when selecting two metals at random. However, only a limited number of these compositions exhibit remarkable synergistic effects, leading to superior co-catalytic performance. As interest in bimetallic cocatalysts grows across various catalytic applications, significant advancements have been made in the development of bimetallic combinations, including bi-noble metals, noble–non-noble bimetals, and bi-non-noble metal alloy cocatalysts for the light-driven reduction of water to H2. Noble metals from the periodic table, including platinum (Pt), palladium (Pd), gold (Au), and silver (Ag), are frequently employed as highly effective cocatalysts in various reduction reactions due to their low Fermi levels. These metals trap photogenerated electrons and facilitate the generation of a Schottky barrier at the interface of the metal and semiconductor. This mechanism effectively inhibits the rapid recombination of photo-induced charge carriers, thereby significantly enhancing the overall performance of photocatalytic processes.62Zhang et al.63 synthesized Pt–Pd bimetal NPs by a dual-step chemical reduction method and Zn0.5Cd0.5S nanorods were synthesized utilizing the solvent thermal method. Additionally, platinum–palladium (Pt–Pd) was uniformly deposited over the exterior surface of Zn0.5Cd0.5S through an in situ synthesis process and used in H2 evolution from light-driven water-splitting. They found a significant improvement in the H2 evolution rate; under visible light illumination. The PtPd/Zn0.5Cd0.5S nanorods demonstrated an optimum rate of H2 production. Additionally, these nanorods achieved a notable apparent quantum yield (AQY) efficiency of up to 10.43% at a wavelength of 420 nm. The interface of PtPd NPs, characterized by high surface energy, functions as an active site for charge transfer. This characteristic facilitates improved photogenerated electron and hole separation, thereby enhancing the efficiency of the photocatalytic process and accelerating the overall rate of hydrogen evolution reaction (Fig. 5a and b). Furthermore, the synergistic effect of both the metals Pt and Pd within the alloy structure is evidenced by the superior photocatalytic activity of PtPd/Zn0.5Cd0.5S in comparison to Pt/Zn0.5Cd0.5S.
 | ||
| Fig. 5 (a) and (b) Mechanism of H2 evolution on the PtPd/ZnCdS surface. Reproduced with permission.63 Copyright 2021, Elsevier. (c) Illustration of the proposed mechanism for charge transfer and H2 evolution in the AuPd/TiO2 composite under UV-visible light. (d) and (e) ESR spectra of AuPd/TiO2 photocatalysts showing O2− detected in methanol and ˙OH detected in water. (f) SPC spectra of the AuPd/TiO2 photocatalyst with varying AuPd nanoparticle concentrations. Reproduced with permission.64 Copyright 2015, Royal Society of Chemistry. (g) Graphical illustration of electron flow induced by the Au LSPR effect in the Z-scheme system (g-C3N4/TiO2), culminating in their transfer to the catalytic active sites of palladium (Pd) within the Pd/Au@g-C3N4/TiO2 composite. (h) The mechanism of reaction involved in solar water splitting for the generation of hydrogen utilizing the Pd/Au@g-C3N4/TiO2 composite. Reproduced with permission.65 Copyright 2024, American Chemical Society. (i) The rate of hydrogen evolution was assessed for both pure g-C3N4 and g-C3N4 integrated with metal and metal alloy nanoparticles. Reproduced with permission.66 Copyright 2018, American Chemical Society. (j) Schematic representation of synthetic Ni–Pt/TiO2 photocatalysts. (k) and (l) Proposed photocatalytic mechanisms for monometallic and bimetallic modifications over TiO2. Reproduced with permission.67 Copyright 2023, American Chemical Society. | ||
Xin et al.64 synthesized AuPd bimetallic nanoalloy-decorated ultrathin two-dimensional titanium dioxide (TiO2) nanosheets via an in situ method for light-driven H2 evolution from water using methanol as the sacrificial agent. 0.3 wt% AuPd/TiO2 achieved an H2 evolution rate of 526 μmol h−1 g−1, surpassing the performance of pure TiO2 by a factor of 31. The AuPd alloy nanoparticles, with their lower Fermi level, served as efficient electron traps, facilitating the transfer of photogenerated electrons from the conduction band of TiO2 to the alloy surface (Fig. 5c). ESR and SPC analyses were employed to examine the roles of reactive species and photogenerated charge carriers. ESR results indicated the generation of superoxide (O2˙−) and hydroxyl radicals (˙OH) under UV-vis light irradiation (Fig. 5d and e). The SPC analysis revealed that the 0.3 wt% AuPd/TiO2 sample exhibited the highest signal intensity, indicating maximal charge carrier separation efficiency (Fig. 5f). However, excessive loading of AuPd nanoparticles reduced light absorption by TiO2, leading to diminished SPC signal intensity and lower photocatalytic performance. The findings underline the importance of optimizing bimetallic nanoparticle content to achieve maximum hydrogen production efficiency.
The quaternary system of Au–Pd NPs on TiO2/g-C3N4/TiO2 has also been studied for co-catalyzing photocatalytic H2 production from H2O, using triethanolamine as a sacrificial agent to stabilize the system and enhance hydrogen production. Rauf and coworkers65 successfully synthesized Pd/Au@g-C3N4/TiO2 through a two-step process comprising chemical reduction, hydrothermal treatment, and subsequent calcination. The hydrothermal treatment effectively enabled the in situ incorporation of Pd/Au cocatalysts, and the H2 evolution was found to be 11.23- and 6.97-fold higher than that of pristine TiO2 and g-C3N4, respectively. The integration of Pd–Au nanoparticles with the TiO2/g-C3N4 heterostructures significantly promoted electron transfer, attributed to the Z-scheme interaction between the components, as well as the localized surface plasmon resonance (LSPR) effect of Au and the Schottky barrier effect of Pd (Fig. 5g). This combination enhanced the photocatalytic hydrogen production by facilitating the separation of photogenerated electron–hole pairs and thus inhibiting recombination (Fig. 5h).
Bhunia et al.66 conducted an analysis examining the performance of platinum–gold (Pt–Au) bimetallic nanoparticles (3–5 nm) incorporated onto g-C3N4 at varying concentrations for the photocatalytic production of H2 from water, utilizing Na2SO3 and Na2S as sacrificial agents. The alloy nanoparticles with a Pt:Au atomic ratio of 1 demonstrated the highest photocatalytic activity when supported on g-C3N4, in addition to favorable results in recyclability assessments (Fig. 5i). The reported H2 evolution rate for bare g-C3N4 was 94 μmol g−1 h−1, while the PtAu-2/g-C3N4 configuration exhibited a markedly elevated rate of 1009 μmol g−1 h−1, reflecting an increase of nearly tenfold.
Considering the limited reserves and elevated costs associated with noble metals, significant efforts have been directed toward enhancing catalytic performance while minimizing the quantity of these metals utilized. Another notable strategy involves the alloying of two different categories of metals like noble metals with cost-effective and earth abundant non-noble transition metals, including Ni, Cu, and Ru. Notably, numerous noble–non-noble bimetal alloy NPs have demonstrated exceptional efficacy as co-catalysts, frequently surpassing the performance of pure noble metals in the photocatalytic evolution of H2 from water.
The study by Wang et al.67 investigated the photocatalytic H2 evolution using Ni–Pt bimetallic cocatalysts supported on TiO2via radiolytic reduction. The authors synthesized ultrasmall (1.9 nm) Ni–Pt NPs with precise dispersion on the TiO2 surface (Fig. 5j). Ni serves as an electron donor, thereby extending the lifetime of photogenerated charge carriers by providing additional electrons to the system. In contrast, Pt functions as an electron trap due to its elevated work function, effectively utilizing electrons for proton reduction. This electron transfer pathway is facilitated by the close contact between Ni–Pt NPs, which creates a multijunction structure on TiO2. In the context of monometallic systems, as illustrated in Fig. 5k, electrons migrate from TiO2 to the metal nanoparticles, where they are subsequently captured for proton reduction, with Pt exhibiting superior activity owing to its enhanced electron affinity. Conversely, in bimetallic systems, as depicted in Fig. 5l, a distinct pathway is established: photogenerated electrons are initially donated by Ni and are then efficiently captured by Pt at the Ni–Pt interface. This mechanism significantly enhances electron utilization and suppresses recombination. Furthermore, optimization of the Ni–Pt ratio has revealed that the Ni2–Pt1 configuration achieves the highest performance and an apparent quantum yield (AQY) of 77.5%, approximately 40 times greater than the performance of bare TiO2.
Pelicano et al.68 conducted a comprehensive investigation into the photocatalytic H2 evolution capabilities of ultrafine PtRu bimetallic alloy NPs supported on Al-doped SrTiO3. The study underscored the synergistic interactions between Pt and Ru atoms. Utilizing the application of a polyol method of synthesis, the researchers produced PtRu NPs with an average diameter of 1.0 ± 0.2 nm, which were uniformly deposited onto SrTiO3:Al via liquid-phase adsorption, followed by annealing in both air and hydrogen. From a mechanistic perspective, the inclusion of Ru within the Pt lattice is shown to reduce the work function of Pt, as demonstrated in Fig. 6a and b. This modification results in a decreased Schottky barrier height, thereby enhancing the transfer of photoexcited electrons from SrTiO3:Al to the cocatalyst. Data obtained from transient diffuse reflectance spectroscopy, as illustrated in Fig. 6c, confirm that the PtRu/SrTiO3:Al catalyst facilitates a more rapid decay of photoexcited electrons, achieving an apparent quantum yield of 65% at 365 nm, which exceeds the performance of monometallic Pt and Ru catalysts. Furthermore, the catalyst demonstrates exceptional stability, maintaining its activity for more than 20 hours without degradation. This work positions PtRu alloys as cost-effective alternatives to conventional noble-metal-only cocatalysts, highlighting their promising potential for scalable photocatalytic H2 production.
 | ||
| Fig. 6 (a) Air–H2–PtRu/SrTiO3:Al. (b) BF–STEM. (c) The time profile of the TDR signal measured at 0.48–0.67 eV following a 3.54 eV pump pulse for SrTiO3:Al, with and without the cocatalysts (pump fluence was 35 μJ per pulse, resulting in a photon density of 2.21 × 1018 photons cm−3, Cr2O3 was used as a cocatalyst to prevent oxidation). Reproduced with permission.68 Copyright 2022, John Wiley and Sons. (d) Mechanism of photocatalytic hydrogen production utilizing Cu2/Ag1@CdS catalysts. Reproduced with permission.69 Copyright 2024, American Chemical Society. (e) The synthesis of a 5 wt% CuNi (3:1)/h-BN/g-C3N4 nanocomposite. (f)–(i) Transmission electron microscopy (TEM) images of a 5 wt% CuNi (3:1)/h-BN/g-C3N4 nanocomposite at various magnifications. (j) The rates of hydrogen gas evolution were investigated for various materials, including g-C3N4, h-BN, a composite of 0.9% h-BN/g-C3N4, and a formulation of 5 wt% CuNi(3:1)/h-BN/g-C3N4, under conditions that included the presence of a scavenger. (k) Arrhenius plots serving as the method for defining the activation energy (Ea) of chemical reactions. Reproduced with permission.70 Copyright 2024, American Chemical Society. (l) Depiction of Cu–Ni/CdS hybrid fabrication. (m) and (n) Comparative analysis of the H2 generation rates for various compositions of Cu–Ni and a range of different metals. Reproduced with permission.71 Copyright 2024, Royal Society of Chemistry. | ||
Hussain et al.69 conducted a thorough investigation into the application of a Cu–Ag cocatalyst on CdS for the generation of hydrogen through sunlight-driven processes. The cocatalyst was synthesized through a hydrothermal method, during which Cu and Ag were chemically reduced and deposited onto the surface of CdS. This process resulted in the formulation Cu2%–Ag1%/CdS, which exhibited a notable hydrogen evolution rate with a quantum efficiency of 45.04% at 420 nm. Upon exposure to sunlight, CdS absorbs photons, thereby exciting electrons from the VB to the CB, generating holes in the VB. The surface plasmon resonance properties of Cu facilitate the generation of hot electrons that migrate to the CB of CdS, thereby enhancing the electron density at the surface of the semiconductor. Concurrently, Ag forms Schottky junctions at the CdS interface, functioning as electron sinks that efficiently capture photogenerated electrons and mitigate their recombination with holes (Fig. 6d). This research illustrated the significant potential of utilizing bimetallic cocatalysts in addressing the challenges associated with photocorrosion and increasing the reusability of the catalyst thereby enhancing the efficiency of hydrogen evolution under sunlight exposure.
To achieve cost-effective, competent, and durable cocatalysts for photocatalytic H2 evolution, considerable attention has been directed toward low-cost non-noble transition metals, including Ni, Cu, and, Ru. Nevertheless, the performance of individual non-noble transition metal cocatalysts remains limited, and a propensity for oxidation compromises their long-term stability. The strategic combination of these metals may facilitate the optimization of their properties, potentially resulting in a level of performance that exceeds that of noble metals. The CuNi alloy nanoparticles supported on h-BN/g-C3N4 were synthesized using a surfactant-free deposition–precipitation method (Fig. 6e), ensuring a uniform distribution of nanoparticles. Structural analysis via XPS and XANES confirmed the metallic state of the nanoparticles. At the same time, TEM revealed well-dispersed CuNi alloy nanoparticles with an average size of 90–100 nm anchored onto the h-BN/g-C3N4 nanosheets (Fig. 6f–i). The 5 wt% CuNi (3:1)/h-BN/g-C3N4 nanocomposite exhibited the highest photocatalytic hydrogen evolution rate of 3658.9 μmol g−1 h−1, outperforming pristine g-C3N4 and CuNi (3:1) nanoparticles by 4.31 and 1.82 times, respectively (Fig. 6j). This enhanced activity is attributed to the synergistic effects of Cu and Ni, with Cu enhancing visible light absorption through its localized surface plasmon resonance and Ni serving as an electron sink to suppress charge recombination. The optimization of CuNi composition and decoration on the h-BN/g-C3N4 matrix significantly improved the material's hydrophilicity, charge mobility, and photocatalytic efficiency. Additionally, the temperature-dependent study demonstrated that increasing the reaction temperature to 45 °C reduced the activation energy to 14.49 kJ mol−1 (Fig. 6k), further boosting hydrogen production. This study highlights the potential of CuNi/h-BN/g-C3N4 composites for sustainable and efficient solar-driven hydrogen evolution.70
Zeng et al.71 developed a ternary photocatalyst system that integrates plasmonic Cu–Ni bimetal nanoparticles with ultrathin CdS nanosheets through a solution-phase synthesis method (Fig. 6l). The optimized 8% Cu–Ni/CdS nanocomposite achieved an outstanding photocatalytic hydrogen generation under visible light, representing an improvement of 88 times compared to pure CdS, and it significantly outperformed its monometallic counterparts, as illustrated in Fig. 6m. Moreover, the composite exhibited a high apparent quantum yield (AQY) of 21.5% at 400 nm and 12.1% at 520 nm, indicating its efficiency across a wide range of the light spectrum. Fig. 6n further demonstrates that the performance of the ternary composite exceeded that of Cu/CdS and Ni/CdS. This enhancement can be attributed to the synergistic effects of Cu and Ni, where Cu improves visible light absorption through surface plasmon resonance (SPR), while Ni establishes Schottky junctions that effectively reduce charge recombination. This study highlights the potential of non-noble bimetallic cocatalysts in facilitating cost-effective solar energy conversion. However, structural stability of the bimetallic alloy cocatalysts is often compromised under photocatalytic operating conditions where selective leaching, phase segregation, or oxidation of one component can disrupt the alloyed state and reduce activity.
3.4. Metal phosphides as co-catalysts
Metal phosphides are promising materials for energy storage and conversion due to their distinctive properties, including exceptional electrical conductivity, catalytic activity, and chemical stability. Their application as photocatalysts and cocatalysts for hydrogen generation has garnered considerable attention in recent research. Metal phosphides based on elements such as Ni, Fe, Co, Cu, Mo, W, Ru, and Rh are particularly noteworthy. These materials typically exhibit metallic characteristics with work function values in the range of 4.5–5.1 eV, facilitating the effective capture and transfer of photoinduced electrons from semiconductor photocatalysts to the reaction sites. The metallic nature of these phosphides ensures low resistance to electron transport, which minimizes charge recombination within the photocatalyst and improves overall catalytic efficiency. Moreover, metal phosphides offer abundant active sites for hydrogen evolution, attributable to their surface composition and electronic structure, which promote proton reduction. However, establishment of a Schottky junction at the metal phosphide and semiconductor photocatalyst interface promotes band bending and prevents back flow of electrons which further enhances charge separation, thereby extending the lifetime of photogenerated electrons and holes.The development and study of non-precious metal co-catalysts are crucial for achieving efficient photocatalytic hydrogen production. Iron due to its abundance and low cost is a promising material for iron-based co-catalysts for photocatalytic hydrogen evolution. Zhao et al.72 have introduced a novel g-C3N4/FexP hybrid photocatalyst intended for visible-light-driven hydrogen evolution. This innovative photocatalyst integrates g-C3N4 with a noble-metal-free iron phosphide (FexP) cocatalyst. The synthesis of the hybrid occurs through a two-step process: initially, g-C3N4/FeOOH is prepared via hydrothermal methods, followed by phosphidation using sodium hypophosphite at 300 °C in an argon environment. The g-C3N4/FexP hybrid demonstrated remarkable photocatalytic performance, achieving a hydrogen evolution rate that is 277 fold greater than that of pristine g-C3N4, which can be comparable to systems loaded with platinum. This substantial enhancement is attributed to the synergistic dual-site mechanism present in FexP, where Fe(δ+) and P(δ−) atoms serve as centers for hydride and proton adsorption, respectively. This configuration facilitates efficient charge separation and promotes water reduction. As depicted in Fig. 7a and b, photogenerated electrons are transferred from g-C3N4 to FexP, thereby accelerating the hydrogen evolution process. Concurrently, the sacrificial agent (TEOA) functions to scavenge holes, reducing the likelihood of recombination. This robust interaction not only ensures high stability and efficiency but also presents a cost-effective and sustainable alternative to noble-metal-based photocatalysts for hydrogen production.
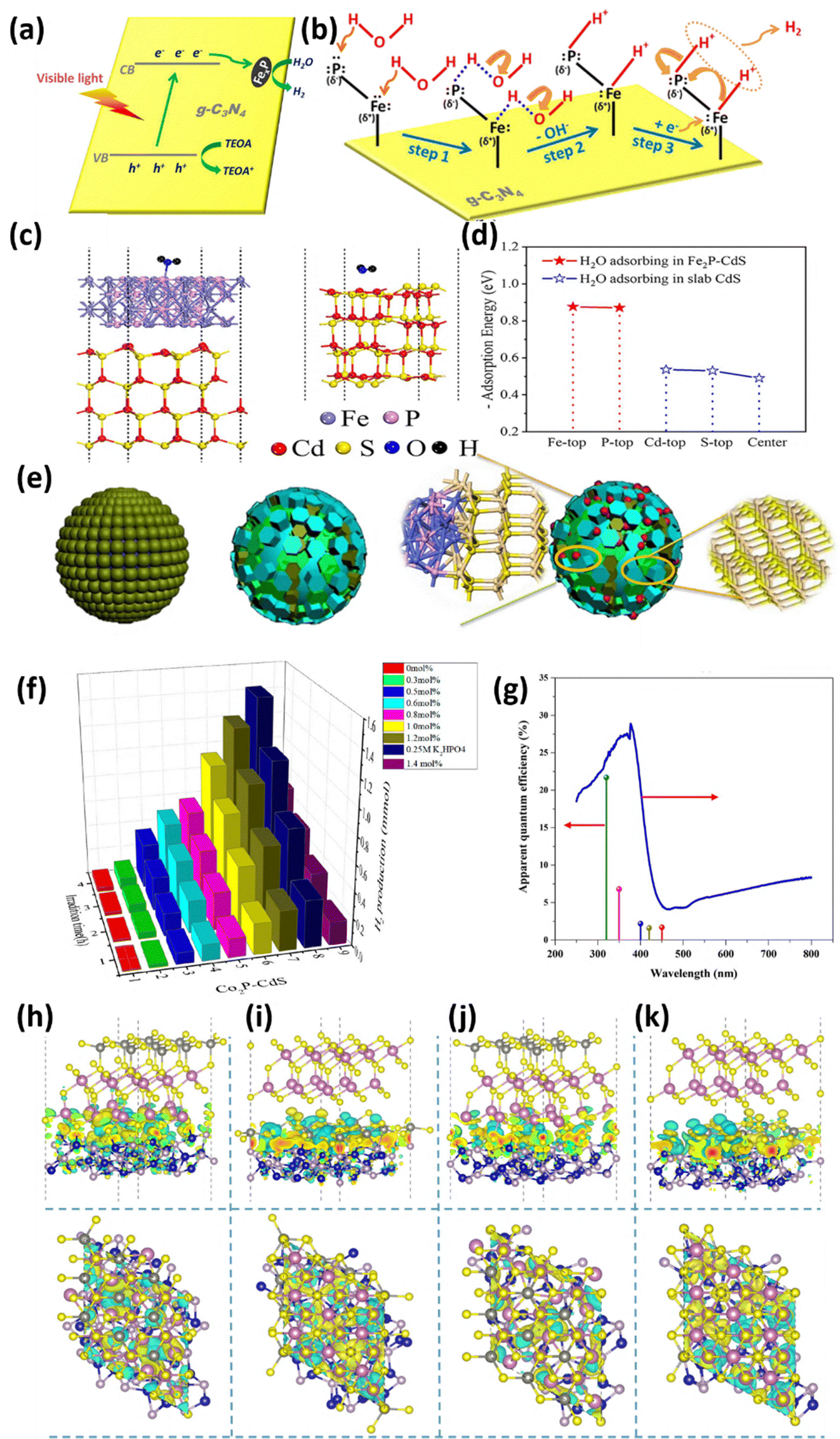 | ||
| Fig. 7 (a) Diagrammatic representation of the photocatalytic H2 evolution over g-C3N4/FexP-0.08 when exposed to visible light. (b) Schematic representation of charge transfer and reaction mechanism for the enhanced H2 evolution by the FexP cocatalyst on the g-C3N4 surface. Reproduced with permission.72 Copyright 2017, American Chemical Society. (c) H2O adsorbed structures on the Fe2P–CdS and slab CdS surfaces. (d) H2O adsorption energy at various Fe2P–CdS and slab CdS sites. Reproduced with permission.73 Copyright 2021, Elsevier. (e) Method for creating cauliflower-shaped CdS–Co2P sub-microspheres. (f) Under visible light, photocatalytic H2 evolution across H-Co2P–CdS composite samples with varying Co2P concentrations. Reproduced with permission.75 Copyright 2018, American Chemical Society. (g) Apparent quantum efficiency over 10% Co2P/ZIS NCGs. (h)–(k) Front and top images of the interface between (001) Co2P and (001) ZnIn2S4 showing four atomically matched interface topologies and electron density distributions. S, In, Zn, P, and Co atoms are represented by the yellow, purple, gray, brown, and blue balls, respectively. The accumulated and insufficient electron density is shown by the yellow and green area. Reproduced with permission.76 Copyright 2021, Elsevier. | ||
Liang et al.73 advanced the application of metal phosphides in the realm of photocatalysis by presenting a highly efficient 2D–1D Fe2P–CdS heterojunction photocatalyst. This innovative composite is designed to address the inherent challenges associated with CdS nanorods, notably rapid electron–hole recombination and insufficient stability. The Fe2P nanosheets, which are synthesized through a mild solvothermal method, exhibit an ultrathin structure (∼1.41 nm) characterized by an abundance of defect sites that facilitate exceptional catalytic activity. The integration of Fe2P nanosheets with CdS nanorods results in the formation of robust heterojunctions, which significantly augment charge transfer and separation efficiency. This composite has demonstrated a remarkable H2 evolution under visible light, surpassing the performance of bare CdS by a factor of 78.7. The superior catalytic performance of the Fe2P–CdS composite can be attributed to several synergistic mechanisms. Notably, a strong ionic interaction between Fe2P and CdS, as established by charge density difference calculations, promotes the migration of electrons from CdS to Fe2P, thereby mitigating recombination losses. Furthermore, as evidenced in Fig. 7c and d, the presence of Fe2P significantly enhances water adsorption, with calculated adsorption energy values ranging from −0.870 to −0.876 eV, which exceed those of pristine CdS (−0.490 to −0.536 eV). This enhanced water adsorption, in combination with a lower hydrogen reduction overpotential, serves to accelerate the hydrogen evolution. Employing experimental techniques such as time-resolved photoluminescence and electron spin resonance, it has been demonstrated that the 2D–1D heterostructure effectively suppresses charge recombination, resulting in the generation of abundant photo-reactive species. Additionally, the Fe2P–CdS hybrid exhibits notable stability over repeated cycles, underscoring its potential as a sustainable, noble-metal-free photocatalyst for H2 evolution.
The promising performance of iron phosphide-based systems, such as FeP/CdS and FeP/g-C3N4, has been further enhanced by the incorporation of nickel, as demonstrated by Zhu et al.74 This development led to the formation of NiFeP/g-C3N4, which exhibits superior photocatalytic activity for H2 evolution. The synthesis of this system was achieved through a controlled phosphidation process of NiFe(OH)2 supported on g-C3N4. The optimized NiFeP/g-C3N4 configuration achieved a remarkable H2 evolution under visible light, significantly outperforming single-metal phosphides. This enhancement is ascribed to the synergistic interactions between nickel and iron, which promote directional electron migration and lower the energy barriers for H2 evolution. Evaluations utilizing photocurrent and electrochemical impedance spectroscopy indicated improved charge separation and transfer efficiency. Furthermore, fluorescence lifetime measurements underscored effective electron localization at the NiFeP surface. The system displayed considerable stability, maintaining its activity over multiple cycles, and exhibited an apparent quantum yield of 4.98% at 420 nm. Mechanistic insights revealed the efficient redirection of photogenerated electrons towards the NiFeP sites, while holes on g-C3N4 facilitated water decomposition. These conclusions emphasize the significant potential of bimetallic phosphides as co-catalysts for sustainable H2 evolution.
The study by Li et al.75 unveils the remarkable potential of Co2P–CdS nanohybrids as a non-noble-metal based photocatalyst, setting a new benchmark for H2 evolution. The photocatalyst was synthesized via an innovative in situ hydrothermal method; Co2P nanoparticles were seamlessly integrated onto CdS sub-microspheres, forming a unique cauliflower-like structure (Fig. 7e) that enhances light absorption and provides abundant active sites for catalytic activity. This strong interfacial bonding facilitates efficient charge transfer while minimizing recombination losses, driving the optimized nanohybrid (1.2 mol% Co2P) to achieve a remarkable H2 evolution rate, outperforming Pt-loaded CdS by threefold and surpassing pure CdS by 41 times. The performance further soared with the addition of K2HPO4 as a sacrificial agent, as illustrated by the time-dependent hydrogen production profile (Fig. 7f), highlighting the catalyst's stability and prolonged efficiency under visible-light irradiation. This synergistic interplay of tailored structure and enhanced charge separation establishes Co2P–CdS as a trailblazer in sustainable H2 production, redefining the role of non-noble-metal materials in photocatalysis.
In another study, Zhang et al.76 developed a hierarchical photocatalyst by coating hollow cobalt Co2P nanocages with a thin layer of ZnIn2S4, offering a highly efficient and stable noble-metal-free solution for photocatalytic H2 evolution. The Co2P nanocages, derived from ZIF-67 precursors through a calcination and phosphiding process in a nitrogen atmosphere, feature a hollow structure that enhances the surface area and facilitates electron diffusion. The subsequent hydrothermal deposition of ZnIn2S4 forms a closely integrated heterojunction, significantly improving interfacial charge transfer and reducing electron–hole recombination. The interfacial charge migration and electronic structure of the (001) Co2P and (001) ZnIn2S4 heterojunction were investigated using DFT calculations (Fig. 7h–k). These simulations revealed that electrons accumulate on the Co2P surface, while ZnIn2S4 exhibits electron deficiency, indicating a robust electron transfer from ZnIn2S4 to Co2P at the heterostructure interface. This electron migration aligns with XPS findings, highlighting the robust interfacial interaction that facilitates the efficient separation of photoexcited charge carriers. The hollow nanocage structure of Co2P further enhances this effect by providing an abundant surface area for charge transfer, ultimately boosting photocatalytic activity. The photocatalyst achieved an exceptional hydrogen evolution rate under simulated sunlight, which was approximately 10 times higher than that of pristine ZnIn2S4, and surpassed the performance of 1% noble-metal-loaded ZnIn2S4. With an apparent quantum efficiency of 21.7% at 320 nm, the Co2P/ZnIn2S4 nanocages demonstrated superior activity and stability, maintaining performance over multiple cycles with minimal structural degradation (Fig. 7g). This work not only highlights the potential of transition metal phosphides like Co2P as effective noble-metal-free cocatalysts but also provides mechanistic insights into the interfacial electron dynamics, establishing a sustainable and cost-effective approach for hydrogen production.
Bimetallic systems such as cobalt phosphorus decorated with MCo2P (M = Ru, Ni) as a cocatalyst loaded on g-C3N4 can be a good alternative to Pt/g-C3N4 catalysts. In this regard, Meng et al.77 and Li et al.78 separately studied Ru-CoP/g-C3N4 and NiCoP/g-C3N4 based catalysts, respectively, contributing to significant advancements in designing cost-effective and highly efficient systems for photocatalytic H2 evolution. In the Ru-CoP/g-C3N4 system, a 2D/2D assembly structure was employed, integrating Ru-modulated CoP nanosheets with g-C3N4 nanosheets. This configuration achieved an outstanding H2 evolution rate under visible light irradiation with an apparent quantum efficiency of 3.49% (∼420 nm), nearer to the performance of Pt-based systems (Fig. 8a). The superior activity was attributed to the synergistic effects of Ru and CoP, which promoted efficient charge separation, accelerated surface water reduction kinetics, and created a strong Schottky interface effect. Similarly, the NiCoP/g-C3N4 heterostructure demonstrated remarkable photocatalytic efficiency, with a hydrogen evolution rate 2.27 times higher than that of Pt/g-C3N4 under comparable conditions. This system leveraged a mesoporous 3D/2D design, where NiCoP nanoclusters were anchored on g-C3N4 nanosheets. The presence of dual Co–N and Ni–N bonding states facilitated enhanced charge transfer and separation while maintaining structural stability over prolonged cyclic tests. The apparent quantum efficiency reached 4.8% at 420 nm, and the catalyst demonstrated exceptional durability, sustaining stable activity over 60 hours. Both systems underscore the potential of transition metal phosphides as noble-metal-free alternatives in photocatalysis, with tailored heterostructures and interfacial bonding playing crucial roles in optimizing performance. These studies provide valuable insights for developing advanced photocatalysts that combine high activity, stability, and scalability for sustainable hydrogen production.
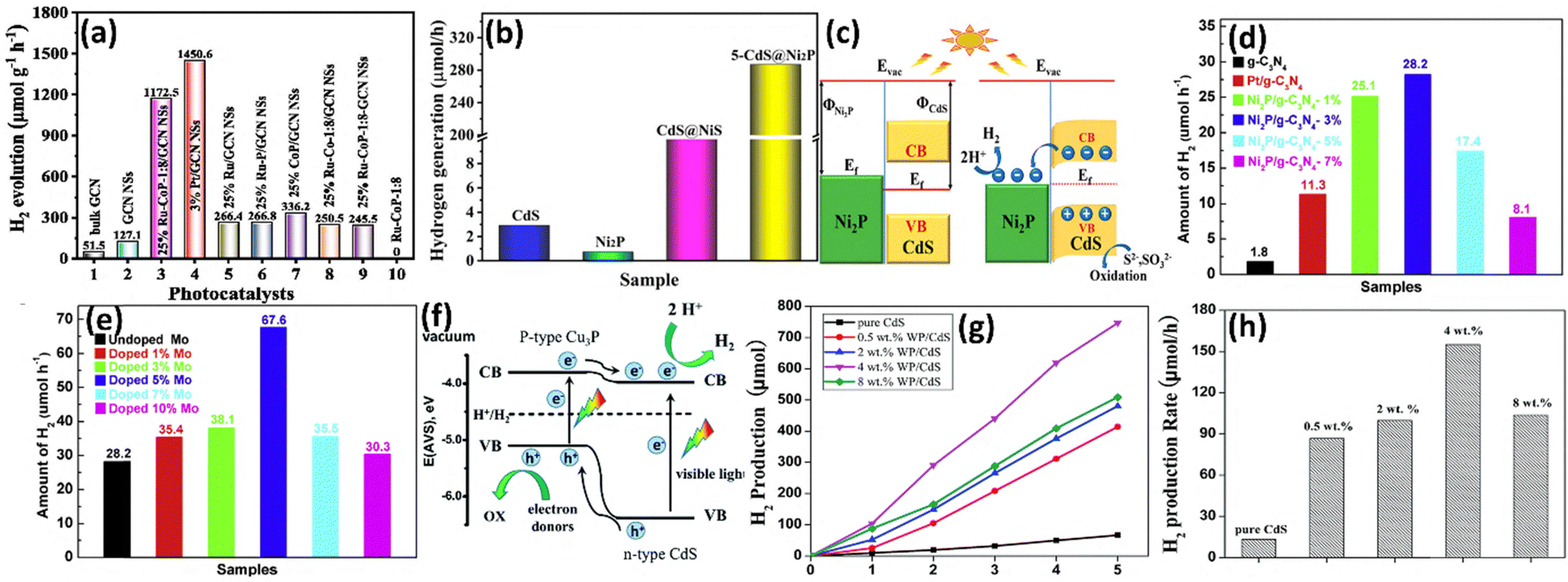 | ||
| Fig. 8 (a) Comparison of the typical rate of hydrogen evolution for the photocatalysts over 5 hours. Reproduced with permission.77 Copyright 2021, Elsevier. (b) The rate at which various catalysts produce hydrogen by photocatalysis. (c) Diagrammatic representation of CdS@Ni2P charge transport. Reproduced with permission.78 Copyright 2022, American Chemical Society. (d) H2 evolution rates over samples of Ni2P/g-C3N4 with varying Ni2P contents. (e) H2 evolution rates over the Ni2P/g-C3N4–3% sample with varing Mo doping. Reproduced with permission.80 Copyright 2020, Elsevier. (f) A potential reaction pathway for the Cu3P/CdS heterojunction-based photocatalytic evolution of H2. Reproduced with permission.82 Copyright 2015, Royal Society of Chemistry. (g) and (h) The rate of production of hydrogen (g) with time and (h) over WP/CdS photocatalysts at different WP loading concentrations (0.5 wt%, 2.0 wt%, 4.0 wt%, and 8.0 wt%). Reproduced with permission.84 Copyright 2017, Royal Society of Chemistry. | ||
Wang's group79 initially described Ni2P as a remarkably effective cocatalyst for visible light driven photocatalytic H2 production. It is evident that by employing Ni2P/CdS as a cocatalyst–semiconductor system, a substantial improvement in photocatalytic H2 evolution under visible light was achieved. Ni2P/CdS was synthesized by a hydrothermal method followed by an in situ photodeposition method. Optimized Ni2P/CdS exhibits an H2 evolution rate of 287 μmol h−1, surpassing that of bare CdS by over 98.3-fold (Fig. 8b). This remarkable improvement can be ascribed to the suitable Fermi level alignment (Fig. 8c). Additionally, Mo-doped Ni2P/g-C3N4 is an approach to increase the photocatalytic H2 evolution activity in visible light. Dong's group80 reported that Mo-Ni2P/g-C3N4 surpassed Pt/g-C3N4 and Ni2P/g-C3N4 by over 6- and 2.4-fold, respectively, with an evolution rate of 67.6 μmol h−1 at an optimized 5% Mo doping level (Fig. 8d and e). This enhancement is mainly attributed to the synergistic effect charge transfer to the MoNi2P nanodots from the nanosheet of g-C3N4 and hole scavenging by TEOA from the valence band. Molybdenum-based materials show a high photocatalytic H2 evolution rate. Especially, the metallic nature of MoP enables electron capture and effectively enhances the migration of photo-induced charges. The study by Yue et al.81 reported a first-time MoP cocatalyst synthesized by temperature-programmed reduction and doped on CdS for high photocatalytic H2 activity, with a quantum yield of ∼5.8% and an evolution rate of 163.2 μmol h−1 mg−1, surpassing that of pure and calcined CdS by over 9- and 20-fold, respectively.
Copper phosphide (Cu3P), a p-type semiconductor, has been validated as an effective co-catalyst for numerous photocatalytic reactions such as CO2 reduction and H2 production when integrated with semiconductors like CdS, g-C3N4, TiO2, etc. A study by Sun et al.82 first discovered Cu3P as an effective co-catalyst for H2 evolution when loaded onto CdS by a solvothermal method. The combination of Cu3P and CdS forms a p–n junction, with Cu3P acting as a p-type junction and CdS as an n-type junction (Fig. 8f). Under visible light, a H2 evolution rate of ∼200 μmol h−1 mg−1 with a quantum yield of ∼25% was achieved under optimized conditions. Further Zhou et al.83 conducted research to demonstrate the remarkable photocatalytic H2 production performance of g-C3N4 loaded with Cu3P and Cu97P3 synthesized by an in situ grinding and phosphorylation method. The photocatalytic H2 evolution rate of Cu3P/g-C3N4 and Cu97P3/g-C3N4 was reported to be 343 μmol h−1 g−1 and 162.9 μmol h−1 g−1, respectively.
Based on the similarities between Rh and Co and between W and Mo as they present in the same group, some W and Rh phosphide-based co-catalysts were developed for photocatalytic H2 evolution. In a 2017 study, Zhang et al.84 synthesized WP NPs via temperature programming reduction and doped them on semiconductor CdS. Following optimization, the 4% doped WP co-catalyst on CdS exhibited a high photocatalytic H2 evolution rate of 155.2 μmol h−1, surpassing that of pristine CdS by over 11.6-fold and being 1/3 times the rate of Pt/CdS (Fig. 8g and h). Chen et al.85 synthesized single-site-like Rh-phosphide doped on g-C3N4via a hydrothermal method followed by phosphating. As indicated by DFT calculations of reaction coordination of RhP reduces the Gibbs free energy of g-C3N4, resulting in a 33-fold increase in the H2 evolution rate compared to that of bare g-C3N4.
Despite their strong initial activity for photocatalytic H2 evolution, metal phosphide co-catalysts suffer from some drawbacks, which limits their practical use. Under light irradiation in aqueous media, they readily undergo surface oxidation or reconstruction, forming metal oxides or oxyhydroxides and leaching phosphorus, which degrades the true active sites and diminishes long-term performance. Many phosphides also suffer from photocorrosion and poor durability in strongly acidic or alkaline medium. In addition, precise control of stoichiometry and nanostructure is difficult in the case of metal phosphides, which makes them non-uniform, and hence large-scale fabrication of these cocatalysts is challenging. Moreover, phosphorus loss during synthesis or operation raises environmental concerns.
3.5. Metal carbides as co-catalysts
Metal carbides have emerged as a fascinating category of cocatalysts effectively used for photocatalytic hydrogen evolution due to their exceptional properties like unique electronic structures, excellent electrical conductivity, and robust chemical stability. These metal carbides exhibit work function values in the range of 4.4–5.0 eV. These properties enable efficient electron transfer to their surfaces, improved separation of photogenerated carriers, and enhanced catalytic activity when coupled with semiconductor photocatalysts.86,87 In this regard, He et al.88 synthesized Ni3C via the pyrolysis of nickel acetylacetone at 250 °C in oleyl amine, and then ground it with g-C3N4 followed by calcination for 1 h at 200 °C to prepare Ni3C/g-C3N4 (Fig. 9a). After optimization, the 15 wt% Ni3C loaded g-C3N4 showed a H2 evolution rate of 15.18 μmol h−1, which is approximately 116.7 times higher than that of bare g-C3N4, with a quantum yield of 0.40% (at 420 nm) which is comparable with that of Pt/g-C3N4 (Fig. 9b and c). Furthermore, Tao's group89 used MAPbI3 as the semiconductor and Ni3C was loaded as a cocatalyst. This approach reported 2362 μmol g−1 h−1 H2 evolution rate, surpassing the rate of bare MAPbI3 by 55 times (Fig. 9d). The mechanistic pathway of both the catalysts Ni3C/g-C3N4 and Ni3C/MAPbI3 is depicted in Fig. 9e and f. In another study, the effect of the Mo2C cocatalyst was studied by Liu et al.90 and it was found that Mo2C is an outstanding cocatalyst with g-C3N4. The optimized catalyst 7 wt% 2D/2D Mo2C/g-C3N4 exhibited a H2 production rate of 675.27 μmol g−1 h−1. They compared its activity with 0.5 wt% Pt/g-C3N4 and 7 wt% B/2D Mo2C/g-C3N4 and found 3.5- and 5.1-fold higher H2 evolution rates, respectively. The work suggested the creation of a 2D/2D heterostructure that increases the interfacial contact between the co-catalyst and the semiconductor providing more active sites for electrons to migrate efficiently to the catalyst surface and prevents the recombination of charge carriers (Fig. 9g). | ||
| Fig. 9 (a) Diagrammatic representation of the Ni3C/g-C3N4 photocatalyst production process. (b) The photocatalytic H2 evolution time courses are as follows: (a) g-C3N4, (b) CNi5, (c) CNi15, (d) CNi10, (e) CNi20, and (f) 0.5 weight percent Pt/g-C3N4. (c) The mean H2 evolution rates across g-C3N4, 0.5 weight percent Pt/g-C3N4, and Ni3C/g-C3N4 composites. Reproduced with permission.88 Copyright 2018, Royal Society of Chemistry. (d) Performance of photocatalytic H2 evolution in the presence of visible light. Reproduced with permission.89 Copyright 2019, American Chemical Society. (e) Diagrammatic representation of the Ni3C/g-C3N4 composite photocatalysts' photo-induced charge separation process. Reproduced with permission.88 Copyright 2018, Royal Society of Chemistry. (f) Band diagram for the HI-splitting photocatalytic process of Ni3C/MAPbI3. Reproduced with permission.89 Copyright 2019, American Chemical Society. (g) Schematic representation of electron transfer processes within the 2D/2D Mo2C/g-C3N4 nanojunction photocatalyst. Reproduced with permission.90 Copyright 2022, American Chemical Society. (h) The rate of evolution of photocatalytic H2 using g-C3N4, PMC/g-C3N4, MC-Li/g-C3N4, and Pt/g-C3N4, (i) Synthesis process of Mo2C nanodots. Reproduced with permission.91 Copyright 2020, Elsevier. (j) Schematic representation of the rGO–Mo2C/TiO2 production process. (k) H2 evolution rate of TiO2, TiO2/rGO (0.5 wt%), and TiO2/rGO–Mo2C (0.5 wt%). (l) and (m) Photocatalytic H2 evolution mechanism of rGO–Mo2C/TiO2. Reproduced with permission.92 Copyright 2021, American Chemical Society. (n) UV-vis absorption spectra, inset: photograph of different weight percent doped catalyst. (o) H2 evolution rate bar graph at different weight percentages. Reproduced with permission.93 Copyright 2019, American Chemical Society. (p)–(r) SEM image of WC-S (sheet), WC-F (flower), and WC-B (bulk). (s) Mechanism of H2 evolution with Pd-WC-F/CdS. Reproduced with permission.94 Copyright 2021, Elsevier. (t) Comparison of photocatalytic H2 evolution rate between different heterointerfaces. (u) Schematic representation of band gap difference between cubic and hexagonal CdS. Reproduced with permission.96 Copyright 2017, Elsevier. | ||
The effect of lithium (Li) incorporation in ultrasmall Mo2C nanodots was studied by Yang et al.91 with respect to their efficiency as a cocatalyst for photocatalytic H2 evolution. It was observed that the H2 evolution rate was 12.23 times higher than that achieved with pristine MoC loaded on CdS. In addition, the photocatalytic performance of Mo2C/CdS was 5.81 times higher than that of Pt/CdS in lactic acid aqueous solution. They also compared the photocatalytic activity with Pt/g-C3N4 and the results are shown in Fig. 9h. Lithium incorporation assisted synthesis of ultrasmall Mo2C nanodots is presented in Fig. 9i. Another study on Mo2C was carried out by Liu and coworkers,90,92 who used the calcination method followed by sonication to synthesize rGO–Mo2C/TiO2 (Fig. 9j). This ternary heterostructure successfully achieved higher photocatalytic activity compared to TiO2 and rGO/TiO2 as shown in Fig. 9k. The high photocatalytic activity is ascribed to the synergistic effect arising from rGO nanosheets functioning as electron mediators to enhance photoelectron transfer and Mo2C nanoparticles acting as catalytic sites that boost the interfacial hydrogen evolution reaction. In brief, under UV irradiation, electrons from the valence band of TiO2 are excited to the conduction band, and move via rGO sheets, which act as a fast electron promoter, to the Mo2C NPs where the H+ ions get absorbed; the whole mechanism is illustrated in Fig. 9l and m. Another non-noble transition metal compound, tungsten (W) carbide, was studied by Tong and co-workers.93 They synthesized the tungsten carbide (WC1−x) cocatalyst by a calcination process to obtain nanocrystals of size 1.98 ± 0.29 nm followed by their incorporation on g-C3N4 at different weight percentages. As shown in Fig. 9n, UV-Vis absorption spectra showed successful incorporation and a change in color according to the weight percentage, with the color shifting from light to dark with increasing WC1−x weight percentage. The photocatalytic H2 evolution activity of g-C3N4 was significantly improved by loading the WC1−x cocatalyst, and the optimized system, WC1−xCN5, exhibited the highest H2 evolution among all weight percentages as shown in Fig. 9o, which can be attributed to the multifunctional role of WC1−x, namely, significantly enhancing charge separation and transportation while offering abundant active sites for catalytic reactions.
The effect of different morphologies of WC cocatalysts in photocatalytic H2 evolution was studied by Dang et al.,94 as shown in Fig. 9p–r. As confirmed from the SEM images, three types of morphologies were designed such as WC-S (sheet), WC-F (flower), and WC-B (bulk), amongst which the 3D flower-like WC exhibited the highest co-catalytic property. Among all three, WC-F/CdS shows the highest H2 evolution activity of 24502 μmol h−1 g−1. The most efficient noble metal cocatalyst Pd doped on WC and combined over CdS was compared with the WC-F cocatalyst and it was found that the Pd-doped composite showed a slightly higher activity (1.16 times), as WC-F has a high specific capacitance (12.07 F g−1) which helps to trap electrons from the conductance band of CdS. These trapped electrons are consumed for H+ reduction to H2 and some unconsumed electrons are transferred to Pd and help in further H2 evolution as shown in Fig. 9s. Furthermore, Tian et al.95 studied vanadium carbide (VC) integrated on CdS and found that the 15 wt% VC/CdS catalyst showed the highest H2 photocatalytic activity with a quantum yield of 8.7% (420 nm) which is 12 times higher than that of pure CdS. This approach gives better results than noble metal Ag doped on CdS and almost close results to Pt/CdS. Different wt% VC was integrated over CdS by a sol–gel method.
Research by Peng et al.96 demonstrated how the lattice compatibility between a semiconductor and its cocatalyst can significantly influence the rate of H2 evolution. In their study, two different catalysts were synthesized using a hydrothermal method by varying the temperature to form distinct heterointerfaces: one hexagonal-SiC and hexagonal-CdS, and second cubic-CdS and hexagonal-SiC. The lower band gap difference between hexagonal-CdS and hexagonal-SiC facilitates more efficient charge transfer, resulting in a four times increase in the H2 evolution rate compared to cubic-CdS and hexagonal-SiC (Fig. 9t and u). Overall, this strategy offers a promising route for designing highly efficient photocatalysts through lattice-matched interface engineering.
The drawbacks of metal carbide cocatalysts during the photocatalytic H2 evolution reaction mainly include their susceptibility to photo-corrosion. In addition, particle aggregation and surface carbon deposition decrease the catalytic activity. Moreover, precise control of stoichiometry, phase purity, and nanoscale morphology is difficult and energy-intensive, making large-scale, low-cost synthesis less practical. These issues like poor long-term durability, interfacial resistance, and scalability constraints remain key hurdles to the reliable use of metal carbide cocatalysts in photocatalytic H2 evolution.
3.6. Metal borides as cocatalysts
Noble metals have played a starring role in the dazzling performance of photocatalytic hydrogen evolution reactions, driving innovation with their exceptional efficiency. Yet their exorbitant price and fleeting abundance make them the rare gems of the catalyst world, challenging their widespread adoption in our pursuit of a sustainable future. Platinum, palladium, gold, and silver are noble metals that can enhance the photolytic activity of semiconducting materials. Metal borides with work function values in the range of 4.4–4.6 eV can exhibit similar H2 evolution performance. Hence, metal borides can be used as a substitute for these metals to improve the photolytic H2 evolution performance. A study reported97 the use of nickel boride (NiB), which showed enhanced hydrogen evolution reaction performance, with a hydrogen production rate of 464.4 μmol g−1 h−1 and an impressive quantum yield of 10.92% when exposed to 365 nm light. Nickel boride nanoparticles, when used as cocatalysts with the C3N4 photocatalyst and when 7.5 wt% of NiB was combined with C3N4, led to optimal H2 evolution (Fig. 10a). The enhancement of H2 evolution reaction is attributed to the presence of strong B(δ−)–Ni(δ+)–N(δ−) bonds between NiB and C3N4. The Mott–Schottky test showed that the conduction band potential of C3N4/NiB7.5 is lower at −1.35 V (Fig. 10b), which means electrons can move from C3N4 to NiB. The photoinduced electrons from C3N4 quickly transfer to NiB through the B(δ−)–Ni(δ+)–N(δ−) bonds. NiB then helps these electrons to reach the active sites for H2 evolution, while photoinduced holes in C3N4 are consumed by TEOA molecules. This research work showed that metal boride cocatalysts are a good replacement for noble metal cocatalysts. Hence, nickel boride can enhance H2 evolution. Another report98 utilizing cobalt demonstrated its potential as an alternative co-catalyst to noble metals. Cobalt boride (CoB) was loaded onto the semiconductor g-C3N4 nanosheets. The optimized CoB–gC3N4 composite showcased a significantly higher hydrogen generation rate, approximately 60 times greater than that of the bare gC3N4 nanosheets. Importantly, it also exhibited commendable stability. To produce gCN nanosheets (NS), melamine and NH4Cl were heated together at 550 °C for 4 hours, resulting in exfoliated gCN NS that were then used to load CoB co-catalysts. Initially, 100 mg of gCN NS was mixed with 100 mL of deionized water and sonicated. CoCl2·6H2O was added to this mixture and stirred for 1 hour to allow Co2+ ions to adsorb onto the gCN NS. Subsequently, NaBH4 was added to reduce the Co2+ ions, forming CoB nanoparticles (NPs) (Fig. 10c). The solution turned dark, indicating successful reduction. After 1 hour, the mixture was centrifuged, washed with deionized water and 2-propanol, and dried under vacuum. Different amounts of Co2+ (1%, 3%, 5%, 7%, 9%, and 11% by weight) were used to create samples with varying CoB loadings, named 1-CoB-gCN, 3-CoB-gCN, 5-CoB-gCN, 7-CoB-gCN, 9-CoB-gCN, and 11-CoB-gCN. At 9 wt%, the hydrogen evolution reaction is maximum, and beyond 11 wt%, the co-catalyst covers the surface of the gCN nanosheet which causes shielding and affects the absorption of photons; beyond 5 wt% loading there is increment in the H2 evolution. The plausible mechanism of H2 evolution is shown in Fig. 10d. A report by Kurenkova et al.99 showed metal borides to be effective co-catalysts for oxide semiconductors. This research shows that tungsten boride (WB) increases the hydrogen evolution reaction. Loading the WB co-catalyst on TiO2 semiconductors increases the rate of H2 evolution by 23 times in comparison to pure TiO2. The H2 evolution reaction rate increases as the concentration of the co-catalyst increases from 0 wt% to 5 wt% and the maximum activity recorded is 81 μmol h−1 gcat−1 (Fig. 10e). When TiO2 is exposed to light, electrons move from the valence band to the conduction band, creating electron–hole pairs. In pure TiO2, these charge carriers recombine rapidly, resulting in a short lifespan for the electrons and holes. By adding WB5−x particles to TiO2, these particles capture some of the electrons from the TiO2 conduction band (Fig. 10f). This transfer of electrons to the WB5−x particles helps to separate the electron–hole pairs more effectively. As the amount of WB5−x particles increases, more contact sites between TiO2 and WB5−x are created, enhancing the efficiency by decreasing the gap between the conducting band and the valence band. Boride creates a Fermi level. Tungsten boride (WB) increases the lifetime of photogenerated charge carriers and photocatalytic activity. | ||
| Fig. 10 (a) H2 evolution rate of NiB/C3N4 at different wt%. (b) Mechanism of H2 evolution with g-C3N4/NiB7.5. Reproduced with permission.97 Copyright 2018, American Chemical Society. (c) Schematic representation of the synthesis and formation of the CoB-g-C3N4 composite. (d) Schematic illustration of the photocatalytic H2 generation mechanism of CoB-g-C3N4 composites. Reproduced with permission.98 Copyright 2024, Elsevier. (e) H2 evolution rate at different wt% of WB5−x–WB2TiO2. (f) Mechanism of electron transfer and H2 evolution on WB5−x/TiO2. Reproduced with permission.99 Copyright 2024, Elsevier. (g) and (h) H2 evolution rate at different wt% of CdS@NiB and with pure CdS and CdS@Pt. (i) Schematic representation of the plausible mechanism of CdS@NiB H2 evolution. Reproduced with permission.100 Copyright 2022, American Chemical Society. (j) Recyclability H2 evolution rate of 15 wt% NiB/ZnCdS over five cycles. Reproduced with permission.101 Copyright 2022, Elsevier. (k) and (l) Mechanism of H2 evolution on NixPB–rGO/CdS. (m) Schematic representation of electron transport on NixB and NixPB. Reproduced with permission.102 Copyright 2022, Elsevier. (n) Synthesis process of layered boron. (o) Synthesis process of exfoliated Pt doped MgB2. Reproduced with permission.103 Copyright 2021, American Chemical Society. (p) H2 evolution mechanism over Nix–B/ZnIn2S4. (q) Diagrammatical representation of the synthesis scheme of Nix–B/ZnIn2S4. Reproduced with permission.104 Copyright 2022, American Chemical Society. | ||
A drawback of loading co-catalysts on semiconductors is the shielding of the active site of the semiconductor (TiO2). CdS@NiB composites (0.005, 0.01, 0.015, 0.02, 0.03) were prepared by Lv et al.100 by the water bath method; the harsh conditions of hydrothermal synthesis reduce CdS activity, while a one-step water bath method preserves the active sites of CdS. The rate of H2 evolution was reported as 28112 μmol h−1 g−1 with an improvement of 5.7 times for CdS@Pt and 11 times for pure CdS (Fig. 10g and h). The enhanced performance of H2 evolution supported by ‘density functional theory’ calculations reveals that incorporating NiB into CdS results in a strong electron coupling effect that enhances electron participation in H2 production. The d-band center of Ni atoms in the CdS@NiB composite shifts from −1.96 eV to −1.84 eV upon adsorption of water. This shift promotes the movement of electrons across the Fermi level, improving the electron transfer from NiB → CdS → H2O. This interaction results in a significant increase in the bond length of H2O from 0.975 Å to 0.986 Å and an increase in the H–O–H bond angle from 104.274° to 105.365°. This facilitates decomposition into hydrogen and hydroxyl ions. Also, the incorporation of NiB narrows the band gap of CdS from 2.12 eV to 1.91 eV. The charge density redistribution in CdS@NiB-H2O leads to substantial charge accumulation around CdS and H2O. Water molecules preferentially adsorb on the Cd atoms of CdS. The electron-rich sites on CdS facilitate the transfer of hydrogen atoms (H*) and hydroxyl ions (OH−), resulting in the formation of adsorbed hydrogen intermediates (H2O + e− → H* + OH−). These intermediates then combine to form H2 (2H* + 2e− → H2), leading to the release of a substantial amount of H2 from the CdS@NiB composite (Fig. 10i). Despite the efficient performance of CdS in photocatalytic hydrogen production, its practical application is hindered due to its photoinduced corrosive nature. Song et al.101 reported the synthesis of ZnCdS by a hydrothermal method. Since the rapid recombination of photogenerated electrons and holes limits the photocatalytic efficiency of ZnCdS, the cocatalyst NiB at different wt% was doped into it by calcination method at 773 K. The highest hydrogen evolution was reported at 15 wt%, being 17 times higher than that achieved with pure ZnCdS with a rate of 8137.1 μmol h−1 g−1; a further increase in wt% led a decrease in H2 evolution. The recycling stability test over five cycles showed that hydrogen production remained high and relatively stable throughout all cycles (Fig. 10j).
Furthermore, Long et al.102 reported another approach to improve the H2 evolution, by increasing the efficiency of the co-catalyst. Long et al. incorporated P in between NixB, loaded it on reduced graphene oxide, and combined it with the CdS NixPB–rGO/CdS photocatalyst. This improvement is attributed to the decreased electron density of the Ni atoms in NixB, enhancing the hydrogen-adsorption capacity at the Ni active sites and thereby boosting the hydrogen-evolution process by 1.8 times compared to NixB–rGO/CdS and by 9.9 times compared to rGO/CdS. Upon exposure to visible light, the NixPB–rGO/CdS photocatalyst generates electron–hole pairs within the CdS component (Fig. 10k and l). The electrons and holes are then separated and migrate to the Ni active sites on the NixPB cocatalysts. Hydrogen is produced through this process, with the rGO nanosheets facilitating the efficient transfer of electrons to the Ni sites (Fig. 10m). The NixPB–rGO/CdS photocatalysts were synthesized using a simple chemical reduction method at room temperature. This process involved mixing cadmium sulfide with graphene oxide using sonication, followed by the in situ formation of amorphous NixPB. The mass ratio of the NixPB cocatalyst to CdS was set at 5 wt%. The molar ratios of phosphorus (P) to boron (B) were varied at 0.2, 0.5, 1, 2, and 5.
Besides, a novel material MgB4 MXene has been developed to drive the H2 evolution as well. Xiao's group103 found that doping of Pt on nanosheets (Pt–MgB4) results in an enhanced ability to absorb and utilize solar energy due to electron trapping by Pt and the abundant terminal metal sites on their surface and exceptional metallic-layer conductivity. MgB4 MXene-like nanosheets are achieved by exfoliation and selective etching. The MgB2 powder is first dispersed and exfoliated using ultrasound and then subjected to selective etching with HCl. HCl is used to remove Mg atoms (Fig. 10n and o). The recyclability test over four cycles showed no decreases in Pt deposit, and H2 evolution in the 1st cycle was 1250 μmol and 1175 μmol in the 4th cycle. Without platinum (Pt), the MgB4 MXene-like nanosheets generate about 120 μmol of hydrogen after 4 hours, which is roughly 10% of the amount produced by the Pt-deposited MgB4 nanosheets. The photocatalytic activity of these Pt-deposited MgB4 nanosheets is comparable to that of Pt-deposited P25 under ultraviolet light. Additionally, MgB4 MXene-like nanosheets with Pt exhibit superior performance under visible light because their smaller bandgap allows for better solar energy absorption and utilization. The valence band maximum shifts upward in Pt-MgB4 due to the influence of Pt atoms, which introduces surface defects or oxygen vacancies. This shift enhances visible light absorption and improves photocatalytic hydrogen production. Additionally, UV-Vis DRS results indicate that Pt–MgB4 has a characteristic bandgap, but absorption in the infrared region suggests an even smaller bandgap. The top of the valence band (VB) is approximately 0.34 eV for MgB4 and around 1.52 eV for Pt-deposited MgB4.
Water splitting is the most common way for H2 evolution; Li et al.104 reported organic transformation for H2 by using a Nix–B cocatalyst doped on 2D ultrathin ZnIn2S4 nanosheets. The rate of H2 production reported with a quantum yield of 24% is 3.1 times higher than that of the traditional H2 photocatalytic half-reaction. When ZnIn2S4 absorbs light energy, electrons are promoted from the valence band to the conduction band, generating electron–hole pairs. Aromatic alcohols absorbed on the surface capture holes and the holes oxidize C–H bonds to form radicals and protons, and these radicals are converted into aldehydes with the concomitant release of protons. The electrons in the conduction band are transferred to the Nix–B cocatalyst. Protons are reduced by the cocatalyst into H2 gas. The nanosheet facilitates quick separation of electron–hole pairs and cocatalyst reduction (Fig. 10p). The nanosheets are prepared by a facile reflux method and the ZnIn2S4/Nix–B heterostructure with different compositions (x = 0.3, 0.5, 1, 1.5%) is prepared by an in situ growth method as shown in Fig. 10q. The ZnIn2S4 nanosheet has a negative zeta potential leading to the adsorption of nickel ions due to electrostatic attraction. Sodium borohydride reduces nickel ions into nickel–boron compounds like Nix–B. For comparison, ZnIn2S4/Pt, ZnIn2S4/NiS, and ZnIn2S4/Ni(OH)2 were prepared utilizing photodeposition, reflux and coprecipitation method, respectively. The photoactivity performance test shows that the performance of ZnIn2S4/Nix–B 0.5% is higher than that of ZnIn2S4/NiS, ZnIn2S4/Ni(OH)2 and ZnIn2S4/Pt. High-temperature synthesis to control stoichiometry and phase variations with very small stoichiometric differences hinders their practical application as a cocatalyst.
3.7. Carbon-based cocatalysts
Carbon-based materials, including carbon nanotubes (CNTs), activated carbon, and graphene, are highly abundant on Earth. Carbon-based cocatalysts possess photocatalytic properties similar to those of noble metals, enhanced by features such as extended light absorption that provides a localized photothermal effect and a large surface area. They show a work function of 4.5–4.7 eV which is favorable for electron uptake from the CB of the semiconductor for the H2 evolution reaction. Additionally, carbon materials are inert and thermally stable. They serve as nucleation sites for semiconductor nanoparticle growth due to their defect-rich surfaces that facilitate nucleation. Moreover, they increase the number of active sites for photocatalytic activity. Typically, reactions occur at the interface of the semiconductor material and the cocatalyst, but with carbon materials, reactions also take place on the surface of the carbon surrounding the interface. This section discusses four carbon-based cocatalysts: carbon nanotubes (CNTs), carbon quantum dots, graphene, and fullerene (C60).In this regard, Shen et al.105 reported a semiconductor-free Cu3P-supported CNT system and the Cu3P-CNT was synthesized via a sol–gel method followed by thermal reduction. They reported a quantum yield of 10.23% with Cu3P-CNT-EY (Eosin Y). This evolution rate was 2.5 times more than that of bare Cu3P and higher than that of some other reported systems like Pt/RGO/TPPH, [ZnTm-PyP]4+-MoS2/RGO and ZnTCPP-MoS2/TiO2. The photocatalytic reaction setup used 50 mg of catalyst Cu3P-CNT and 18 mg of EY in a 15% volume solution of TEOA. Different wt% of CNTs was loaded on Cu3P, and the activity increased until 3 wt% and also showed the highest H2 evolution rate as represented in Fig. 11a and b. Fig. 11a shows a time study of H2 evolution; from the starting hours Cu3P-3%CNT shows the highest yield. A plausible mechanism of this system is shown in Fig. 11c, in which first in the presence of light, the EY dye electron got excited to a singlet state, underwent intersystem crossing and then was transferred to the triplet state. Then in the presence of TEOA, it got reduced to EY−. The non-covalent interaction between CNT and EY dye facilitated the electron transfer from EY− to Cu3P. This further transfers electrons for H2O reduction to H2.
 | ||
| Fig. 11 (a) H2 evolution study by time. (b) H2 evolution study of different systems ((a) Cu3P, (b) Cu3P-0.5% CNT, (c) Cu3P-1% CNT, (d) Cu3P-3% CNT, (e) Cu3P-5% CNT, (f) Cu3P-10% CNT). (c) Mechanism of H2 evolution via Cu3P-CNT. Reproduced with permission.105 Copyright 2018, American Chemical Society. (d) Synthesis scheme of CNT/Au/g-C3N4. (e) Mechanism of H2 evolution via CNT/Au/g-C3N4. Reproduced with permission.106 Copyright 2022, John Wiley and Sons. (f)–(h) SEM images of bulk MoS2 and ball milled MoS2-C60-BM3. (i) The left side shows the mechanism of H2 evolution via the MoS2-C60 hybrid and the right side shows the ball milling process. Reproduced with permission.107 Copyright 2018, Elsevier. (j) and (k) H2 generation rate study via UV-Vis and visible light of pure TiO2 and different wt% loaded CQDs. (l) Plausible mechanism of photocatalytic H2 evolution via CQDs/TiO2. Reproduced with permission.108 Copyright 2015, American Chemical Society. (m) Schematic representation of the synthesis scheme of CMCN-11/8%SCQDs. (n) Mechanism of photocatalytic H2 evolution of CMCN-11/8%SCQDs. Reproduced with permission.109 Copyright 2024, Elsevier. (o) Schematic representation of the synthesis scheme of rGO–SO3H/TiO2. (p) Formation scheme of rGO–SO3H. (q) Digital pictures of (a) rGO, (b) rGO–SO3H, and (c) rGO–SO3H/TiO2. (r) Mechanism of photocatalytic H2 evolution via rGO–SO3H/TiO2. Reproduced with permission.110 Copyright 2022, Elsevier. | ||
In another study, Xia et al.106 reported that CNTs can enhance the H2 evolution rate of the noble metal incorporated catalyst, Au/g-C3N4. Fig. 11d shows the synthesis scheme of the CNT/Au/g-C3N4 catalyst. First, g-C3N4 was synthesized via hydrolysis followed by annealing and then Au nanoparticles were incorporated on g-C3N4 sheets via a deposition–precipitation method. Thereafter, the CNT was grown on Au/g-C3N4via a chemical vapor deposition method. The use of CNTs for the modification of the catalyst helps as an electron highway as shown in Fig. 11e and as electrons jump from the valence band to the conductance band when g-C3N4 absorbs light, these electrons diffuse on Au via CNTs and the gathered electrons reduce water to H2 gas with an evolution rate of 950 μmol g−1 h−1 which is 3-fold higher than that of the Au/g-C3N4 binary counterpart.
Another approach to increase photocatalytic activity via carbon based co-catalysts is using C60. Guan et al.107 reported a C60/MoS2 photocatalyst which showed an enhanced H2 evolution in the presence of EY. By using the ball milling technique, C60 was deposited on the edge of MoS2. The ball milling technique helped to convert bulk MoS2 to small-size sheets as represented in SEM images (Fig. 11f–h), which show that before ball milling the size of MoS2 was 10 micrometers and after ball milling, the size was reduced to 1 micrometer. This phenomenon is also diagrammatically shown in Fig. 11i, which shows that in bulk form MoS2 its edges were captured by C60 while after ball-milling MoS2 breaks into thin sheets; the mechanism of the photocatalytic H2 evolution of the MoS2/C60-EY catalyst is also shown in Fig. 11i. It also followed the same pathway as discussed in the above cases; the EY dye first transfers electrons to MoS2 and then to C60 which is present on the edges, which further helps in H2 evolution from H2O. The authors tried many morphologies for the analysis: bulk MoS2 + EY showed a zero H2 evolution rate and the ball milled MoS2 showed H2 evolution, with MoS2-(2.8 wt%) C60-BM + EY showing the highest H2 evolution of all tried morphologies. The ball-milled MoS2-(2.8 wt%) C60 exhibited a higher H2 evolution rate than the physically blended MoS2/C60. Lian et al.108 reported C60/CdS/TiO2 which was synthesized via evaporation-induced self-assembly (EISA). Different wt% of C60 (0.25–1.5 wt%) was integrated out of which 0.50 wt% showed the highest H2 evolution rate of 6.03 μmol h−1, with a quantum yield of 2% at 420 nm; however, without C60 integration, H2 evolution was quite low, i.e., 0.71 μmol h−1. Hence, C60 acted as a transit station when electrons get excited from the valence band to the conduction band in the presence of visible light from both TiO2 and CdS; C60 acted as an electron acceptor from both CdS and TiO2 and used these trapped electrons for the H2 evolution.
Another type of carbon-based co-catalyst is carbon quantum dots (CQDs). Yu et al.108 synthesized CQDs/TiO2via a hydrothermal method which showed a 4-fold higher H2 evolution rate compared to that of bare TiO2. Different wt% of CQDs (i.e., 0.5 to 2.5 wt%) was integrated on TiO2, and out of these 1.5 wt% integration showed the highest photocatalytic activity. However, a further increase in integration wt% led to a decrease in the H2 evolution rate due to a decrease in active sites as doping increased. Photocatalytic experiments were carried out under UV-Vis and visible light as shown in Fig. 11j and k. Also, UV-vis doping with 1.5 wt% and visible light doping with 2 wt% showed the highest photocatalytic activity. In this approach, CQDs act as photosensitizers and also help to slowdown the charge recombination rate by accepting electrons from the TiO2 conduction band (Fig. 11l). Xu et al.109 synthesized sulfur carbon quantum dots (SCQDs) and integrated them on CoMoO4/g-C3N4 (CMCN). The detailed synthesis scheme is shown in Fig. 11m, in which different wt% of SCQDs was loaded on the 1:1 mass ratio of CM/CN, and the CMCN-11/8%SCQDs exhibited the highest H2 evolution rate of 4916.63 μmol g−1 h−1 at 9 pH with a quantum yield of 1.78%. This yield was possible due to the fast electron transfer provided by the EY dye while absorbing light which then transferred to SCQDs that slowed the recombination rate. CM/CN also absorbs light and forms an n-type semiconductor and this is converted into an S-type heterojunction via bending of Fermi levels. This alignment creates an internal electric field which helps move electrons in the right direction. Hence, electrons from SCQDs and CN transfer to CM and these electrons react with H2O molecules to produce H2 gas (Fig. 10n).
The last type of carbon-based cocatalyst is graphene. Wang et al.110 reported TiO2 doped with edge sulfonated graphene (rGO–SO3H). The rGO–SO3H catalyst was prepared by reduction of graphene in the presence of NaBH4, followed by acid sulfonation at the graphene edges via a diazotization reaction This process involved the formation of a diazonium salt and subsequent sonication to modify TiO2, resulting in the formation of rGO–SO3H/TiO2 (Fig. 10o–q). Under UV light, electrons are excited to the conduction band of TiO2 and then transferred to graphene and then to the –SO3H group on benzene which forms the –SO3− ion which first absorbs H+ and then reduces it by donating electrons. As shown in the last step of Fig. 10r, H2 gas is released through the combination of two hydrogen atoms leaving the –SO3− ion available to repeat the reaction. A H2 evolution rate of 197.1 μmol h−1 g−1 was reported.
Regardless of the abundant surface area for photocatalytic H2 evolution provided by the carbon based cocatalysts, they show limited intrinsic catalytic activity, often requiring additional modifications like heteroatom doping, hybridization with metals, defect creation, and modulated dimensions to achieve competitive activity.
4. Summary and future perspectives
In summary, the photocatalytic H2 evolution efficiency of different semiconductors may be enhanced by the use of cocatalysts, which is promising and extremely effective and has gathered significant attention in recent decades. The practical use of noble-metal-based cocatalysts in large-scale energy applications is hampered by their high cost and restricted availability, despite their exceptional performance. Thus, significant research efforts have gone into creating effective cocatalysts devoid of noble metals. Different non-noble metal based cocatalysts have drawn a lot of interest among the alternatives because of their great charge carrier-capturing capacity, low cost, wide availability, and outstanding stability, which makes them appealing options for photocatalytic applications. This article provides an in-depth overview of the key developments in widespread and new generation cocatalysts for boosting the H2 evolution performance by photocatalytic water splitting, including the effect of cocatalysts in enhancing the photocatalytic H2 evolution reaction, their classification as metal nanoparticles, single atom cocatalysts, bimetallic alloy cocatalysts, metal oxide cocatalysts, transition metal dichalcogenide cocatalysts, metal phosphide cocatalysts, metal carbide cocatalysts, carbon based cocatalysts, and metal boride cocatalysts, and their modification over various well-known photocatalysts.Despite tremendous advancements in the development of stable and effective cocatalysts and in the understanding of their underlying mechanisms, considerable challenges remain in substantially enhancing the efficiency and ensuring long-term stability of photocatalytic water splitting systems using these cocatalysts. The following problems need to be addressed.
(i) Since the cocatalysts are essential for attaining effective photocatalytic H2 evolution with semiconductors, their performance needs to be greatly improved in order to further improve the efficiency and durability of the cocatalysts. Continuous research efforts are therefore essential in identifying and designing high-performance cocatalysts. Among the strategies to be emphasized, one is improving the intrinsic properties of existing cocatalysts, such as enhancing their charge-trapping ability, increasing the number of surface active sites, and reducing the overpotentials of surface reactions. These goals can be addressed through appropriate modification attempts. Another one is discovering innovative forms of cocatalysts that can offer superior charge extraction and can optimize adsorption and dissociation of water molecules.
(ii) Though many efforts have been devoted to lab-scale H2 evolution using co-catalyst–semiconductor systems, their scalability for industrial-scale H2 production remains a formidable challenge. The practical implementation of these catalytic systems at the scale required for significant H2 production needs an understanding of materials, engineering, and economic bottlenecks. However, the uniform and reproducible deposition of the cocatalyst over a large area of the semiconductor is necessary; state-of-the-art techniques like photodeposition, electrodeposition, and atomic layer deposition (ALD) can be adopted to control particle size and distribution. In addition, the design and architecture of multicomponent systems like type-II, Z-scheme, and S-scheme heterojunctions can be fabricated in which selective deposition of cocatalysts over the surface of the collective component will lead to improved photocatalytic H2 production. In this regard, a large-scale prototype of a photocatalytic water-splitting device has indeed been demonstrated by Nishiyama et al., who reported safe, continuous hydrogen production on a 100 m2 outdoor photocatalytic panel reactor array, operating for several months under natural sunlight and achieving a solar-to-hydrogen efficiency of up to 0.76% while autonomously recovering hydrogen using a commercial polyimide membrane.111 Although this represents the first field-scale implementation of photocatalytic overall water splitting, the technology remains at the research stage. For a commercial scale, efficiencies must reach 5–10%, photocatalyst lifetime and visible-light activity must improve, and cost-effective reactor and gas-separation designs are still needed before commercialization. Ongoing global efforts are addressing these challenges to translate such demonstrations into economically viable industrial systems.
(iii) Comprehensive research is necessary to fully understand the photocatalytic process pertaining to the migration pathway of photogenerated charge carriers and the photocatalytic reaction active sites. There is currently no concrete proof of the migratory route and real active locations of photogenerated electrons and holes. Additionally, the literature provides conflicting pathways for interfacial charge transfer. For instance, the photocatalytic H2-production activity may be increased by intimate contact or spatial charge separation by reduction cocatalysts. These problems should be carefully examined since they are important for the choice of components and the design of the photocatalytic system.
(iv) Much of the existing knowledge is based on conjectural interpretations, and the fundamental processes underpinning the enhanced performance of the photocatalytic hydrogen evolution reaction are still controversial. Therefore, in-depth mechanistic studies are crucial. By allowing for real-time monitoring of charge transfer at the cocatalyst/semiconductor interface and surface catalytic processes, operando techniques like in situ X-ray photoelectron spectroscopy (XPS), in situ transmission electron microscopy (TEM), and in situ extended X-ray absorption fine structure (EXAFS) provide insightful information. Furthermore, theoretical computations are essential for directing the choice of the best cocatalysts and clarifying the processes behind the noted increase in photocatalytic H2 evolution activity after cocatalyst deposition.
Conflicts of interest
The authors declare no conflict of interest.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
The authors are thankful to IIT Mandi for the facilities provided. We are thankful to Dr Ashish Kumar, Sardar Patel University Mandi, Himachal Pradesh, India, for his insightful inputs while compiling this article. BPM acknowledges the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India, for the National Postdoctoral Fellowship (NPDF). VK acknowledges the DST-SERB project (CRG/2022/003559) for financial support.References
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- D. Sharma, P. Choudhary, S. Kumar and V. Krishnan, Small, 2023, 19, 2207053 CrossRef CAS PubMed.
- A. Kumar and V. Krishnan, Adv. Funct. Mater., 2021, 31, 2009807 CrossRef CAS.
- B. P. Mishra and K. Parida, J. Mater. Chem. A, 2021, 9, 10039–10080 RSC.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- X. Yi, T. Lu, Y. Li, Q. Ai and R. Hao, Renewable Sustainable Energy Rev., 2025, 210, 115147 CrossRef CAS.
- J. Yang, X. Zheng, S. S. A. Shah, C. Wang, X. Li, Z. Yan and L. Peng, Carbon Energy, 2025, e695 CrossRef CAS.
- H. Karibe, S. Sair, A. Faik and H. Ait Ousaleh, Int. J. Hydrogen Energy, 2025, 133, 200–213 CrossRef CAS.
- M. A. Bashir, J. Tuo, J. Weidman, Y. Soong, M. L. Gray, F. Shi and P. Wang, Energy Adv., 2025, 4, 330–363 RSC.
- H. Wang, E. Harkou, A. Constantinou, S. M. Al-Salemc, G. Manos and J. Tang, Chem. Soc. Rev., 2025, 54, 2188–2207 RSC.
- P. Saini, D. Jampaiah, S. Periasamy, A. P. Kulkarni and S. K. Bhargava, Chem. Commun., 2025, 61, 6027–6054 RSC.
- G. Zou, Q. Wang, G. Ye, Z. Pan, S. Wang, M. Anpo and G. Zhang, Adv. Funct. Mater., 2025, 35, 2420899 CrossRef CAS.
- H. Zhuzhang, X. Liang, J. Li, S. Xue, Y. Lin, B. Sa, S. Wang, G. Zhang, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2025, 64, e202421861 CrossRef CAS PubMed.
- Q. Wang, D. Zheng, Z. Pan, W. Xing, S. Wang, Y. Hou, M. Anpo and G. Zhang, Adv. Funct. Mater., 2025, 2501889 CrossRef CAS.
- Y. Zou, S. Li, D. Zheng, J. Feng, S. Wang, Y. Hou and G. Zhang, Sci. China Chem., 2024, 67, 2215–2223 CrossRef CAS.
- J. Yang, S. Tian, Z. Song, Y. Hao and M. Lu, Coord. Chem. Rev., 2025, 523, 216257 CrossRef CAS.
- S. Raza, I. Sadiq, S. Shaheen, M. Saniya and T. Ahmad, Sustainability Sci. Technol., 2025, 2, 012002 CrossRef CAS.
- S. A. Ali, S. Majumdar, P. K. Chowdhury, S. M. Alshehri and T. Ahmad, ACS Appl. Energy Mater., 2024, 7, 7325–7337 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Kumar, A. Kumar and V. Krishnan, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS.
- A. Kumar, P. Choudhary, A. Kumar, P. H. C. Camargo and V. Krishnan, Small, 2022, 18, 2101638 CrossRef CAS PubMed.
- M. A. Khan, S. Mutahir, I. Shaheen, Y. Qunhui, M. Bououdina and M. Humayun, Coord. Chem. Rev., 2025, 522, 216227 CrossRef CAS.
- B. Guan, J. Chen, Z. Li, Z. Zhuang, Y. Chen, Z. Ma, J. Guo, C. Zhu, X. Hu, S. Zhao, H. Dang, L. Chen, K. Shu, Z. Guo, K. Shi, Y. Li, C. Yi, J. Hu and Z. Huang, Energy Fuels, 2024, 38, 806–853 CrossRef CAS.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- H. Zhao, L. Jian, M. Gong, M. Jing, H. Li, Q. Mao, T. Lu, Y. Guo, R. Ji and W. Chi, Small Struct., 2022, 3, 2100229 CrossRef CAS.
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 RSC.
- G. Zhao and X. Xu, Nanoscale, 2021, 13, 10649–10667 RSC.
- Z. Lin, L. Li, L. Yu, W. Li and G. Yang, J. Mater. Chem. A, 2017, 5, 5235–5259 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- C. Bie, L. Wang and J. Yu, Chem, 2022, 8, 1567–1574 CAS.
- S. A. Ali, I. Sadiq and T. Ahmad, J. Mol. Chem., 2025, 5, 1204 CrossRef CAS.
- A. K. Ghosh, U. Saha, S. Biswas, Z. A. Alothman, M. A. Islam and M. Dolai, Ind. Eng. Chem. Res., 2022, 61, 175–186 CrossRef CAS.
- J. Xu, Q. Ji, X. Yan, C. Wang and L. Wang, Appl. Catal., B, 2020, 268, 118739 CrossRef CAS.
- B. Park, W.-W. Park, J. Y. Choi, W. Choi, Y. M. Sung, S. Sul, O.-H. Kwon and H. Song, Chem. Sci., 2023, 14, 7553–7558 RSC.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Y. Kageshima, H. Inuzuka, H. Kumagai, B. Ohtani, K. Teshima and H. Nishikiori, J. Phys. Chem. C, 2023, 127, 18327–18339 CrossRef CAS.
- L. Yuan, Z. Geng, J. Xu, F. Guo and C. Han, Adv. Funct. Mater., 2021, 31, 2101103 CrossRef CAS.
- H. Liu, W. Xiong, C. Zhou, C. Lai, L. Li, G. Wang, X. Huo, G. Zeng and M. Cheng, Coord. Chem. Rev., 2025, 529, 216468 CrossRef CAS.
- S. M. Wu and P. Schmuki, Adv. Mater., 2025, 37, 2414889 CrossRef CAS PubMed.
- Y. Fu, K. Lu, A. Hu, J. Huang, L. Guo, J. Zhou, J. Zhao, O. V. Prezhdo and M. Liu, J. Am. Chem. Soc., 2023, 145, 28166–28175 CrossRef CAS PubMed.
- H. Peng, T. Yang, H. Lin, Y. Xu, Z. Wang, Q. Zhang, S. Liu, H. Geng, L. Gu, C. Wang, X. Fan, W. Chen and X. Huang, Adv. Energy Mater., 2022, 12, 2201688 CrossRef CAS.
- Y. Akinaga, T. Kawawaki, H. Kameko, Y. Yamazaki, K. Yamazaki, Y. Nakayasu, K. Kato, Y. Tanaka, A. T. Hanindriyo, M. Takagi, T. Shimazaki, M. Tachikawa, A. Yamakata and Y. Negishi, Adv. Funct. Mater., 2023, 33, 2303321 CrossRef CAS.
- X. Ren, C. Li, J. Liu, H. Li, L. Bing, S. Bai, G. Xue, Y. Shen and Q. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 6885–6893 CrossRef CAS PubMed.
- Z.-Z. Liang, X.-A. Li, Q.-Z. Chen, X.-L. Wang, P.-Y. Su, J.-F. Huang, Y. Zhou, L.-M. Xiao and J.-M. Liu, ACS Catal., 2024, 14, 10447–10461 CrossRef CAS.
- J. Zhang, L. Li, M. Du, Y. Cui, Y. Li, W. Yan, H. Huang, X. A. Li and X. Zhu, Small, 2023, 19, 2300402 CrossRef CAS.
- Q. Zhao, W. Yao, C. Huang, Q. Wu and Q. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 42734–42741 CrossRef CAS.
- Z. Zeng, Y. Su, X. Quan, W. Choi, G. Zhang, N. Liu, B. Kim, S. Chen, H. Yu and S. Zhang, Nano Energy, 2020, 69, 104409 CrossRef CAS.
- T. Lv, B. Xiao, F. Xia, M. Chen, J. Zhao, Y. Ma, J. Wu, J. Zhang, Y. Zhang and Q. Liu, Chem. Eng. J., 2022, 450, 137873 CrossRef CAS.
- T. Kawawaki, Y. Mori, K. Wakamatsu, S. Ozaki, M. Kawachi, S. Hossain and Y. Negishi, J. Mater. Chem. A, 2020, 8, 16081–16113 RSC.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- L. Zhang, X. Lu, J. Sun, C. Wang and P. Dong, J. Mater. Chem. A, 2024, 12, 5392–5405 RSC.
- B. Liu, Y. Li, Y. Guo, Y. Tang, C. Wang, Y. Sun, X. Tan, Z. Hu and T. Yu, ACS Nano, 2024, 18, 17939–17949 CrossRef CAS PubMed.
- X. Xu, F. Luo, W. Tang, J. Hu, H. Zeng and Y. Zhou, Adv. Funct. Mater., 2018, 28, 1804055 CrossRef.
- T. Raja Mogan, J. Zhang, L. S. Ng, S. K. Boong, C. Chong, J.-K. Lee, H. Li and H. K. Lee, Angew. Chem., Int. Ed., 2024, 63, e202401277 CrossRef CAS PubMed.
- X. Zhang, A. Fu, X. Chen, L. Liu, L. Ren, L. Tong and J. Ye, Catal. Today, 2019, 335, 166–172 CrossRef CAS.
- H. Ren, J.-L. Yang, W.-M. Yang, H.-L. Zhong, J.-S. Lin, P. M. Radjenovic, L. Sun, H. Zhang, J. Xu, Z.-Q. Tian and J.-F. Li, ACS Mater. Lett., 2021, 3, 69–76 CrossRef CAS.
- J.-L. Yang, Y.-L. He, H. Ren, H.-L. Zhong, J.-S. Lin, W.-M. Yang, M.-D. Li, Z.-L. Yang, H. Zhang, Z.-Q. Tian and J.-F. Li, ACS Catal., 2021, 11, 5047–5053 CrossRef CAS.
- B. T. Sneed, A. P. Young, D. Jalalpoor, M. C. Golden, S. Mao, Y. Jiang, Y. Wang and C. K. Tsung, ACS Nano, 2014, 8, 7239–7250 CrossRef CAS PubMed.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, H. Xue, J. Gao, Y. Peng, W. Song and L. Ge, Chin. J. Catal., 2021, 42, 1677–1688 CrossRef CAS.
- Y. Xin, L. Wu, L. Ge, C. Han, Y. Li and S. Fang, J. Mater. Chem. A, 2015, 3, 8659–8666 RSC.
- K. U. Sahar, K. Rafiq, M. Z. Abid, U. U. Rehman, R. H. Althomali, A. Rauf and E. Hussain, Energy Fuels, 2024, 38, 17995–18009 CrossRef CAS.
- K. Bhunia, M. Chandra, S. Khilari and D. Pradhan, ACS Appl. Mater. Interfaces, 2019, 11, 478–488 CrossRef CAS PubMed.
- C. Wang, D. Dragoe, C. Colbeau-Justin, P. Haghi-Ashtiani, M. N. Ghazzal and H. Remita, ACS Appl. Mater. Interfaces, 2023, 15, 42637–42647 CrossRef CAS.
- C. M. Pelicano, M. Saruyama, R. Takahata, R. Sato, Y. Kitahama, H. Matsuzaki, T. Yamada, T. Hisatomi, K. Domen and T. Teranishi, Adv. Funct. Mater., 2022, 32, 2202987 CrossRef CAS.
- E. Hussain, A. Ishaq, M. Z. Abid, M. Z. Waleed, A. Rauf, R. Jin and K. Rafiq, ACS Appl. Energy Mater., 2024, 7, 1914–1926 CrossRef CAS.
- V. Rout, B. Maji, H. V. Annadata, R. R. Maharana, D. K. Panda, J. Samantaray, U. K. Goutam, K. Samanta, M. Mishra and P. Dash, ACS Appl. Mater. Interfaces, 2024, 16, 69333–69358 CrossRef CAS PubMed.
- Q. Zeng, Y. Bao, S. Ning, Q. Yu, Y. Wei and D. Zeng, J. Mater. Chem. A, 2024, 12, 17286–17294 RSC.
- H. Zhao, J. Wang, Y. Dong and P. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 8053–8060 CrossRef CAS.
- Z. Liang, C. Yang, J. Lu and X. Dong, Appl. Surf. Sci., 2021, 566, 150732 CrossRef CAS.
- Q. Zhu, B. Qiu, H. Duan, Y. Gong, Z. Qin, B. Shen, M. Xing and J. Zhang, Appl. Catal., B, 2019, 259, 118078 CrossRef CAS.
- S. Li, L. Wang, S. Liu, B. Xu, N. Xiao, Y. Gao, W. Song, L. Ge and J. Liu, ACS Sustainable Chem. Eng., 2018, 6, 9940–9950 CrossRef CAS.
- Q. Zhang, X. Wang, J. Zhang, L. Li, H. Gu and W.-L. Dai, J. Colloid Interface Sci., 2021, 590, 632–640 CrossRef CAS PubMed.
- S. Meng, P. An, L. Chen, S. Sun, Z. Xie, M. Chen and D. Jiang, J. Colloid Interface Sci., 2021, 585, 108–117 CrossRef CAS PubMed.
- C. Li, H. Wu, Y. Du, S. Xi, H. Dong, S. Wang and Y. Wang, ACS Sustainable Chem. Eng., 2020, 8, 12934–12943 CrossRef CAS.
- K. Li, X. Chen, J. Zhao, H. She, J. Huang, L. Wang and Q. Wang, ACS Appl. Energy Mater., 2022, 5, 10207–10215 CrossRef CAS.
- C. Li, H. Wu, S. Hong, Y. Wang, N. Song, Z. Han and H. Dong, Int. J. Hydrogen Energy, 2020, 45, 22556–22566 CrossRef CAS.
- Q. Yue, Y. Wan, Z. Sun, X. Wu, Y. Yuan and P. Du, J. Mater. Chem. A, 2015, 3, 16941–16947 RSC.
- Z. Sun, Q. Yue, J. Li, J. Xu, H. Zheng and P. Du, J. Mater. Chem. A, 2015, 3, 10243–10247 RSC.
- H. Zhou, R. Chen, C. Han, P. Wang, Z. Tong, B. Tan, Y. Huang and Z. Liu, J. Colloid Interface Sci., 2022, 610, 126–135 CrossRef CAS PubMed.
- J. Zhang, W. Yao, C. Huang, P. Shi and Q. Xu, J. Mater. Chem. A, 2017, 5, 12513–12519 RSC.
- Z. Chen, Y. Bu, L. Wang, X. Wang and J.-P. Ao, Appl. Catal., B, 2020, 274, 119117 CrossRef CAS.
- K. He, E. Campbell, Z. Huang, R. Shen, Q. Li, S. Zhang, Y. L. Zhong, P. Zhang and X. Li, Small Struct., 2022, 3, 2200104 CrossRef CAS.
- N. S. Powar, C. B. Hiragond, D. Bae and S.-I. In, J. CO2 Utiliz., 2022, 55, 101814 CrossRef CAS.
- K. He, J. Xie, Z.-Q. Liu, N. Li, X. Chen, J. Hu and X. Li, J. Mater. Chem. A, 2018, 6, 13110–13122 RSC.
- Z. Zhao, J. Wu, Y.-Z. Zheng, N. Li, X. Li and X. Tao, ACS Catal., 2019, 9, 8144–8152 CrossRef CAS.
- W. Liu, D. Zhang, R. Wang, Z. Zhang and S. Qiu, ACS Appl. Mater. Interfaces, 2022, 14, 31782–31791 CrossRef CAS PubMed.
- F. Yang, D. Liu, Y. Li, L. Cheng and J. Ye, Chem. Eng. J., 2020, 399, 125794 CrossRef CAS.
- J. Liu, P. Wang, J. Fan, H. Yu and J. Yu, ACS Sustainable Chem. Eng., 2021, 9, 3828–3837 CrossRef CAS.
- R. Tong, Z. Sun, X. Wang, S. Wang and H. Pan, J. Phys. Chem. C, 2019, 123, 26136–26144 CrossRef CAS.
- Y. Dang, L. Feng, W. Hu, W. Wang, Q. Zhang and B. Ma, Int. J. Hydrogen Energy, 2021, 46, 39251–39261 CrossRef CAS.
- L. Tian, S. Min and F. Wang, Appl. Catal., B, 2019, 259, 118029 CrossRef CAS.
- Y. Peng, G. Han, D. Wang, K. Wang, Z. Guo, J. Yang and W. Yuan, Int. J. Hydrogen Energy, 2017, 42, 14409–14417 CrossRef CAS.
- Q. Zhu, B. Qiu, M. Du, M. Xing and J. Zhang, Ind. Eng. Chem. Res., 2018, 57, 8125–8130 CrossRef CAS.
- S. Gupta, C. Prapaitrakool, B. R. Bhagat, C.-L. Yeh, A. Dashora, A. Sirisuk, N. Patel, N. Daneu, A. Kocjan, M. Spreitzer, J. C. S. Wu and M. M. Kržmanc, Int. J. Hydrogen Energy, 2024, 60, 1288–1298 CrossRef CAS.
- A. Yu Kurenkova, A. D. Radina, V. S. Baidyshev, P. V. Povalyaev, E. E. Aidakov, E. Yu. Gerasimov, D. D. Mishchenko, A. V. Zhurenok, A. Y. Pak, E. A. Kozlova and A. G. Kvashnin, Appl. Surf. Sci., 2024, 661, 160095 CrossRef CAS.
- Z. Lv, Y. Wang, Y. Liu, J. Wang, G. Qin, Z. Guo and C. Zhang, J. Phys. Chem. C, 2022, 126, 9041–9050 CrossRef CAS.
- L. Song, S. Zhang, D. Liu, S. Sun and J. Wei, Int. J. Hydrogen Energy, 2020, 45, 8234–8242 CrossRef CAS.
- H. Long, P. Wang, X. Wang, F. Chen and H. Yu, Appl. Surf. Sci., 2022, 604, 154457 CrossRef CAS.
- L. Xiao, X. Li, J. Zhang and Z. He, ACS Appl. Nano Mater., 2021, 4, 12779–12787 CrossRef CAS.
- X. Li, S. Lu, J. Yi, L. Shen, Z. Chen, H. Xue, Q. Qian and M.-Q. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 25297–25307 CrossRef CAS PubMed.
- R. Shen, J. Xie, Y. Ding, S.-Y. Liu, A. Adamski, X. Chen and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 3243–3250 CrossRef CAS.
- Z. Xia, C. Chen, X. Qi, Q. Xu, H. Tang and G. Liu, Adv. Sustainable Syst., 2023, 7, 2200134 CrossRef CAS.
- J. Guan, J. Wu, D. Jiang, X. Zhu, R. Guan, X. Lei, P. Du, H. Zeng and S. Yang, Int. J. Hydrogen Energy, 2018, 43, 8698–8706 CrossRef CAS.
- Z. Lian, P. Xu, W. Wang, D. Zhang, S. Xiao, X. Li and G. Li, ACS Appl. Mater. Interfaces, 2015, 7, 4533–4540 CrossRef CAS PubMed.
- S. Xu, M. Li, Y. Wang and Z. Jin, Int. J. Hydrogen Energy, 2024, 51, 16–30 CrossRef CAS.
- P. Wang, P. Deng and Y. Cao, Int. J. Hydrogen Energy, 2022, 47, 1006–1015 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |