
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in the understanding of molecular beryllium element bonding
Magnus R.
Buchner

Anorganische Chemie, Nachwuchsgruppe Hauptgruppenmetallchemie, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35032 Marburg, Germany. E-mail: magnus.buchner@chemie.uni-marburg.de; Fax: +49 (0)6421 2825669; Tel: +49 (0)6421 2825668
First published on 30th June 2025
Abstract
Molecular beryllium chemistry has made huge advances in the last ten years. Especially, low valent beryllium compounds and beryllium–element multiple bonds have emerged as highly investigated subjects. To understand these compounds and their reactivity, a detailed understanding of beryllium element bonding is necessary. Therefore, Be–C, Be–N, Be–P, Be–O, Be–S, Be–Se, Be–halide, Be–Be, Be–Al and Be–Mg bonds have been studied in detail in this review.
 Magnus R. Buchner | Magnus R. Buchner studied chemistry at the Technische Universität München where he received his PhD in 2011 under the supervision of Klaus Ruhland. After postdoctoral stays in the groups of Florian Kraus (Munich), Robin Perutz (York) and Sjörd Harder (Erlangen) and a stint at the patent department of the Fraunhofer-Gesellschaft, he started his independent research at the Philipps-Universität Marburg in 2015, funded by the Deutsche Forschungs Gemeinschaft and since 2019 within the Emmy Noether program. His research interests lie in the coordination, organometallic and bioinorganic chemistry of hard (pseudo) main group metals, with a focus on beryllium. |
1 Introduction
Since the turn of the century, s-block chemistry, especially alkaline earth metal chemistry, has seen a huge resurgence and demonstrated that reactivities far beyond the known applications of lithium-organic compounds and Grignard reagents are possible.1–13 However, one element was left aside. Due to the notorious “toxicity” of beryllium and its compounds, as well as the limited commercial availability of suitable precursors, very little research had been performed on its chemistry.14 However, with more research into beryllium associated health hazards, a more concise picture on the underlying biological mechanisms liable for beryllium associated diseases emerged.15,16 Based on this knowledge, a re-evaluation and update of the health and safety measurements necessary for safe handling of beryllium and its compounds were possible,17 which allowed for a renaissance in chemical beryllium research roughly ten years ago. This led to detailed studies on N-donor beryllium complexes by the Schulz group,18–21 while our group focused on the systematic investigation of beryllium chemistry with biologically relevant O-donor ligands and related systems.22–31 This enabled the isolation and structural authentication of the first homoleptic beryllium carboxylate (1PhFig. 1).24 Additionally, it can be shown that the speciation in water and anhydrous ammonia is similar and the latter leads to unprecedented adamantane shaped tetranuclear complex-cation 2.32–34 In the search for beryllium precursors with innocent leaving groups, we, additionally, performed some basic investigations into beryllium-organic compounds with anionic C-donor ligands in the hope of using these as Brønsted basic starting materials.35,36 Similar compounds and their applications were also studied by the Schulz group,37–39 while the Gilliard group concentrated on beryllium complexes with dative C-donor ligands.40–43 | ||
| Fig. 1 O- and N-donor ligand based multinuclear beryllium complexes.24,32 | ||
In parallel to the above mentioned research, a new field of beryllium chemistry evolved in the wake of the seminal Mg(I) dimer introduced by the Jones group,1 which evolved as a quasi-universal reducing agent.3 Therefore, numerous efforts were made to isolate low valent beryllium compounds. This led to cyclic alkyl amino carbene (CAAC) stabilised compounds with beryllium in the formal oxidation states 044 and I (3 and 4, Fig. 2).45,46 However, the interpretation of the oxidation state in these compounds is heavily discussed,47–49 which is also partially due to the observation that CAAC stabilised low valent Be compounds only show CAAC centred reactivity.50
 | ||
| Fig. 2 Low valent beryllium compounds (n = 0 and 1; R = Me and Et; E = Al, Ga, and In).44–46,51–55 | ||
An alternative approach to the realisation of low valent beryllium compounds is the formation of beryllium–element bonds with other main group metals with comparable electronegativity. In the case of aluminium as the bonding partner, this was first achieved by the Jones group (5Fig. 2),51 while a more generalised solution could be realised by us in collaboration with the Aldridge group through employment of the 4,5-bis(2,6-di-iso-propylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene ligand (NON). With this NON ligand at a group 13 element, beryllium element bonds with aluminium, gallium and indium are accessible (6, Fig. 2).52,54 The aluminium derivative of 6 features a non-nuclear attractor (NNA) on the Be–Al bond according to quantum theory of atoms in molecules (QT-AIM), which was the first time this was observed for a heterobimetallic bond.52 Accordingly, 6 acts as a reducing agent and readily inserts carbodiimide into the Be–Al bond (7, Scheme 1). The fact that beryllium is bound to the carbon atom of the former carbodiimide suggests that the beryllium atom in 6 acts as a nucleophile and was therefore the first example of actual low valent behaviour in beryllium.52
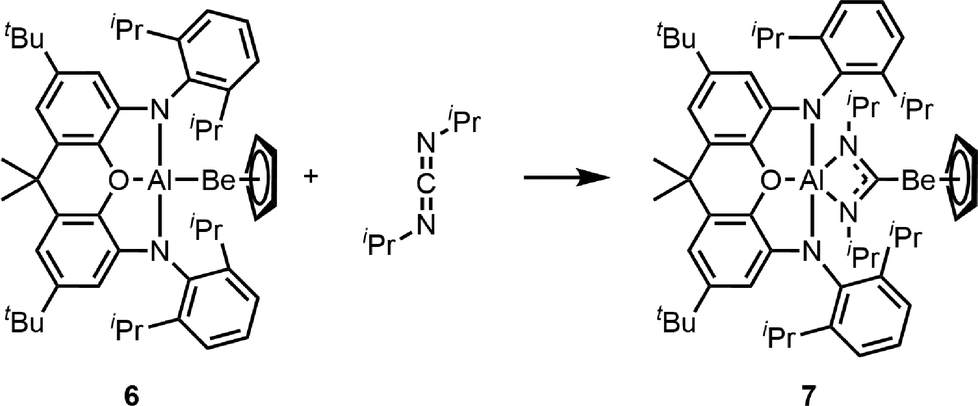 | ||
| Scheme 1 Nucleophilic attack of the beryllium atom at the electrophilic site of a carbodiimide.52 | ||
A breakthrough was achieved by Boronski, Aldridge and co-workers when they isolated diberyllocene (8, Fig. 2)53 and a derivative with one pentamethylcyclopentadienyl ligand,56 which are only the second known homoleptic dimetallocenes besides the seminal dizincocenes reported by Carmona.57,58 Interestingly, despite its homoleptic Be–Be bond, 8 only acts as a reducing agent when heteroleptic Be–metal bonds are formed.53,59,60 Additionally, two examples of ligand exchange under retention of the Be–Be bond have been described so far.56 In collaboration with the Harder group, we were able to synthesise the first compound with a Be–Mg bond (9Fig. 2).559 features a charge distribution, which is to date the closest to the formal oxidation state of Be(0). Furthermore, 9 is surprisingly stable towards heat and bulky molecules, while it readily reduces rod shaped molecules like organic azides. This results in the reductive coupling of two azide units to a hexazenediido ligand (Scheme 2).55 A similar reductive coupling of azides was previously observed with the Jones Mg(I)-dimer.61 However, the reaction of 9 with adamantyl azide does not stop with the hexazenediide formation but also one additional azide molecule is inserted into the Be–Cp* bond during the formation of 10.55 This insertion into a Be–Cp* bond is only the second example of the activation of a beryllium–cyclopentadienyl bond. A related reaction was observed when sterically demanding beryllocenes reacted with an organic isocyanide, as depicted in Scheme 3.62,63 This lack of knowledge on the reactivity of this fundamental bond is surprising considering the fact that beryllocenes are the organo-beryllium compounds for which, by far, the best understanding of bonding exists.37,64–66
 | ||
| Scheme 2 Reductive azide coupling at the Be–Mg bond and consecutive azide insertion into a Be–Cp* bond (Ad = adamantyl).55 | ||
 | ||
| Scheme 3 Insertion of an isocyanide carbon atom into a beryllium–cyclopentadienyl bond (R = H and Me).62,63 | ||
Besides low valent beryllium chemistry, beryllium–element multiple bonds have also emerged as a novel research topic in the last five years (Fig. 3). This ranges from double dative Be![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in 13 and 1467,68 to delocalised π-systems in 15 and 16.69 While evidence for partial Be
C bonds in 13 and 1467,68 to delocalised π-systems in 15 and 16.69 While evidence for partial Be![[partial double bond, top dashed]](https://www.rsc.org/images/entities/char_e12f.gif) N double bonds has been known for decades,70,71 this observation has been generalised with compounds like 1772 and 18.73 Based on these studies, the first Be
N double bonds has been known for decades,70,71 this observation has been generalised with compounds like 1772 and 18.73 Based on these studies, the first Be![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C and BeO triple bonds were only recently realised in 19 and 20.74
C and BeO triple bonds were only recently realised in 19 and 20.74
 | ||
| Fig. 3 Complexes with beryllium–element multiple bonds (R = Ph, Et, nBu, and iBu).67–69,72–74 | ||
Considering the huge advances that were made in beryllium chemistry in recent years, my group has moved more into the investigation of principle beryllium element bonding to understand the underlying processes stabilising low valent beryllium species as well as beryllium–element bonds with the hope of rationalising the observed reactivity. In this article, our advances in the understanding of Be–C, Be–N, Be–P, Be–O, Be–S, Be–Se and Be–halide bonds are summarised in the context of the findings by other research groups during this time period.
2 C-donor ligands
While attempting to prepare the dinuclear beryllium chloride analogue to dilithiated hexacarbodiphosphorane 21 as a precursor for a compound with a Be–Be bond, we serendipitously obtained di-ortho-beryllated hexaphenylcarbodiphosphorane (22) according to Scheme 4. 22 features the first isolated BeC double bond, and according to energy decomposition analysis in combination with the natural orbital for chemical valence (EDA-NOCV), the ligand interacts with the beryllium atom through one electron-sharing bond and three dative bonds.67 The rigid geometry at the beryllium atom, which is enforced through di-ortho-beryllation, facilitates an efficient overlap between the second lone pair at the carbone carbon atom and the empty p-orbital of beryllium, which is not the case in hexaphenylcarbodiphosphorane adducts to beryllium halides, which show perpendicular orientation of these two orbitals.75
 | ||
| Scheme 4 Synthesis of di-ortho-beryllated hexaphenylcarbodiphosphorane.67 | ||
However, 22 shows extremely poor solubility in solvents which do not decompose the compound.67 This low solubility is a property of all carbodiphosphorane adducts of beryllium, the origin of which is still unclear.75,76 This low solubility meant that the reactivity of 22 could not be investigated.67 These solubility issues could be mitigated by moving to mono-ortho-beryllated systems 13 (Fig. 3). These complexes can easily be prepared through the reaction of homoleptic organo-beryllium compounds with hexaphenylcarbodiphosphorane as exemplified through the reaction with diphenyl beryllium (23), yielding phenyl derivative 13Ph (Scheme 5). No di-ortho-beryllation occurs via this route and experiments with a less sterically demanding but more electron donating carbodiphosphorane confirm that the driving force for ortho-beryllation is the steric pressure in the system. Accordingly, with the latter carbodiphosphorane, no ortho-beryllation occurs and adduct 14 is formed (Fig. 3). Similar to 22, 13 and 14 also feature a double dative bond from the carbone carbon atom to the beryllium atom. Despite their improved solubility, no reactivity of the BeC bond with typical substrates could be observed, which is most likely due to strong steric shielding of this bond.68 Efforts are ongoing to reduce the steric demand of the ligands in these systems to make the BeC bond more accessible.
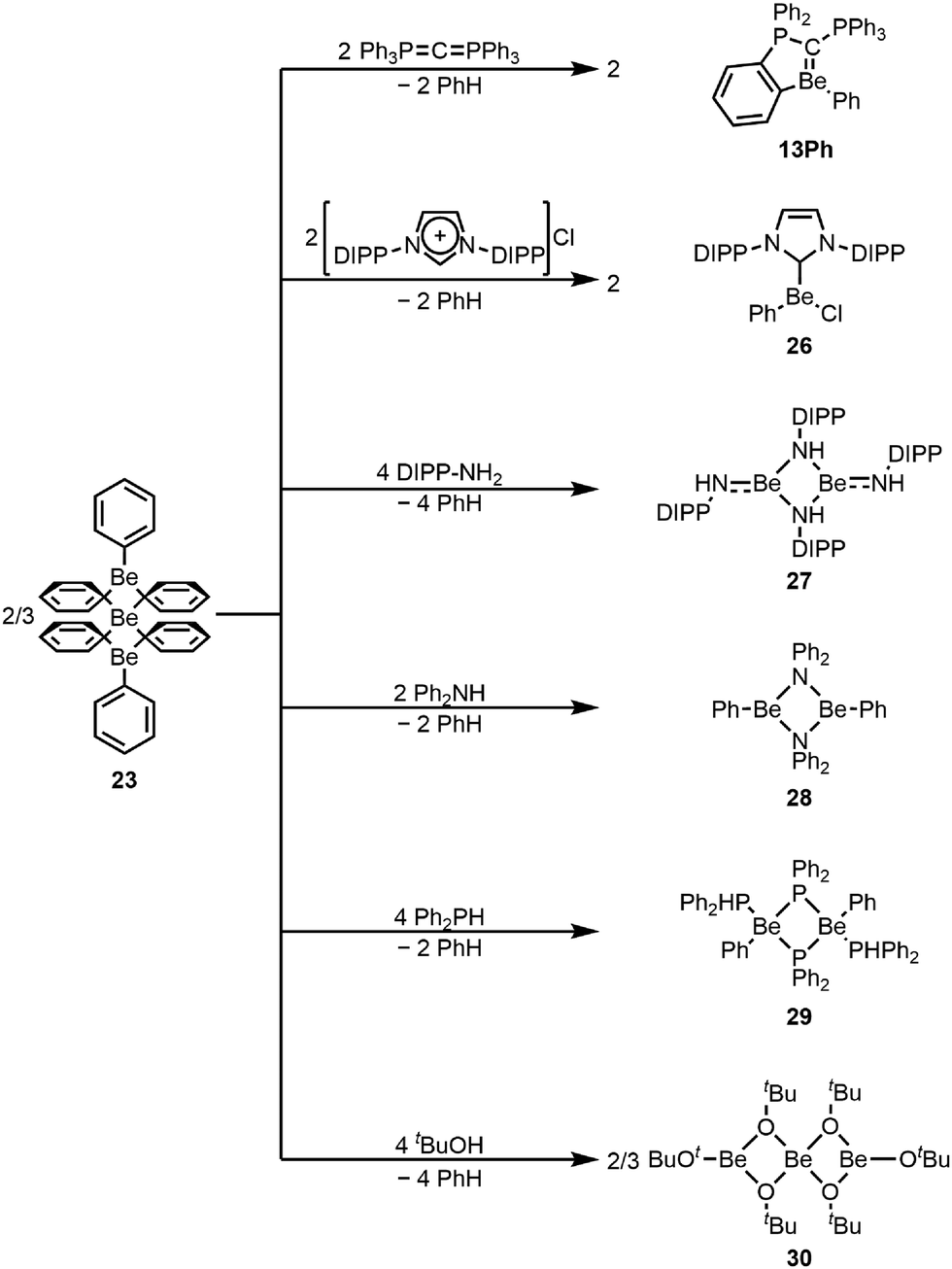 | ||
| Scheme 5 Versatile use of diphenylberyllium as a starting material.68,73,77 | ||
For comparison to the carbodiphosphorane adducts 13, 14 and 22 the bonding in N-heterocyclic carbene (NHC) adducts to diphenyl beryllium and beryllium dibromide was also investigated. This revealed that in these NHC systems, a double dative bond is also present. However, the bonding is completely different, since in these cases, one dative bond is formed from the carbene's lone pair to the beryllium atom, while the other dative bond originates from the BePh2 or BeBr2 fragment donating to the empty p-orbital of the carbene carbon atom.68,78 This interaction is the reason for the distinctive perpendicular orientation of the BePh2 or BeX2 (X = Cl, Br, and I) and the NHC plane found in all tri-coordinated NHC beryllium complexes.79–82 Hyperconjugation of the π-systems of the phenyl rings into the empty p-orbital at the beryllium atom results in the formation of partial BeC double bonds in Lewis base adducts of diphenyl beryllium (15, Fig. 3). However, if strong donor ligands like N-heterocyclic immines (NHIs) are used, these prevent partial BeC double bonds through the formation of a partial BeN double bond (16Fig. 3).69 The hyperconjugation and the resulting delocalisation of the π-system are also probably the reason why tri-coordinated Lewis base adducts to diphenyl beryllium (24, Scheme 6) are more preferentially formed than the tetra-coordinated analogue (25, Scheme 6).83,84
 | ||
| Scheme 6 Reactions yielding heteroleptic organo-beryllium halides (X = Cl, Br, and I; X′ = Cl and Br; L = THF and NEt3).85 | ||
Homoleptic diphenyl beryllium 23 acts as a versatile Brønsted base and can be used to directly prepare a wide range of beryllium complexes (Scheme 5). Through deprotonation of C–H acidic imidazolium salts, Grignard analogue beryllium complex 26 is accessible, while reaction with amines results in the formation of homo- (27) and heteroleptic (28) beryllium amides, depending on the sterics of the employed amines.73,77 Also phosphines can be deprotonated to give beryllium phosphide 29, while alcoholysis results in homoleptic beryllium tert-butoxide 30, all of which could be structurally authenticated.77
Until recently Grignard analogue beryllium complexes were rare.86,87 However, we could show that heteroleptic organo-beryllium halides (31Scheme 6) are actually the preferred species. This is evident from equilibria lying almost exclusively on the heteroleptic side if tri-coordinated Lewis base adducts of the beryllium halides (32) and 24 are mixed. The same observation was also made if either tetra-coordinated Lewis base adducts to the beryllium halides (33) and diphenyl beryllium or the beryllium halides and complexes 25 are allowed to react. Also simple mixing of beryllium halides, diphenyl beryllium and the desired donor ligand results in clean formation of Grignard analogues 31.85 Similar results were also obtained in parallel by the Jones group.88 Depending on the steric bulk of the donor ligand L and the properties of the halido ligand, either mononuclear compounds 31 or dinuclear complexes 34 are formed (Scheme 6).85 Similar observations have already been made in NEt3 adducts of the beryllium halides.31 At present, detailed studies on the bonding in dinuclear heteroleptic organo-beryllium halides are conducted to understand the mechanism of their formation from homoleptic precursors as well as the analogy to homoleptic organo-magnesium compounds, which can only be synthesised through an equilibrium shift via salt elimination.
In beryllium chemistry, beryllium–element bond lengths determined in the solid state have extensively been used to analyse the bond strengths and bond order. However, often compounds with different coordination numbers at the beryllium center have been used. To investigate, whether solid state parameters are an appropriate tool to draw these conclusions we prepared a set of 1-tris(pyrazolyl)borate (Tp) organo-beryllium complexes with anionic and neutral C-donor ligands, as depicted in Fig. 4.89
 | ||
| Fig. 4 Neutral and cationic 1-tris(pyrazolyl)borate organo-beryllium complexes.89 | ||
Extensive solution and solid state analyses in combination with quantum chemistry revealed that all Be–C bonds are almost identical in nature. No discrimination into dative, covalent or ionic bonds could be made regardless whether the C-donor was a carbene or an alkyl or aryl. Furthermore, it was found that the structural parameters obtained from X-ray diffraction analysis were similar and that sterics plays the decisive role.89 Therefore, solid state parameters can only be used with caution when deductions on bonding or oxidation states are drawn. This is only possible if systems with a similar steric demand and with the same coordination number at the beryllium center are compared. Also a quantum chemical analysis is crucial to understand the bonding mode.
In summary, while the number of well characterised organo-beryllium compounds has increased significantly, there is still a lack of systematic investigations into their reactivity and ligand exchange processes. At present, we are investigating the reactivity of beryllocenes as well as the formation and application of beryllium Grignard compounds.
3 N-donor ligands
To study the differences between Be–C, Be–N, Be–O and Be–S bonding, we investigated the coordination behaviour of ambiphilic pseudo-halides at the rigid [TpBe]+ fragment,90 which we also used to study Be–C bonds (see the above section).89 While cyanate and thiocyanate preferentially bind via the N-donor site, cyanide readily forms the cyanide and isocyanide beryllium complexes in a one-to-one ratio. Quantum chemical analysis of the bonding showed that N-coordination leads to strong polarisation of the pseudo-halido ligands, which result in charge accumulation at the nitrogen atoms. This partial negative charge is a strong stabilising factor for the highly polarised, mainly ionic Be–pseudo-halide bonds. However, in the case of cyanide, this polarisation has a significantly weaker influence on the bond towards metal atoms in general. The σ-donation is more effective if the C-donor site is coordinated, which is the reason why most metals form cyanide complexes. Only if strong orbital interactions, like with beryllium, are possible, N-coordination is preferred and then polarisation of the CN π-system additionally stabilises these isocyanide complexes.90
To gain insights into the electronic and steric effect onto Be–N bonds, a set of mono-, di- and trinuclear beryllium amide complexes have been synthesised (e.g.27 and 28, Scheme 5 as well as 37, Scheme 7). In these compounds, the terminal N-donor ligands are mainly bound through a 2-electron-2-centre σ-bond, while the μ2-N bridging interactions consist of 2-electron-3-centre σ-bonds. The electron deficiency at the tri-coordinated beryllium atoms is compensated through donation from the lone pairs of the terminal nitrogen atoms, which leads to partial BeN double bonds. In contrast to this, the lone pairs at the bridging nitrogen atoms remain mainly N-located. Further electron delocalisation is present if the molecular geometry allows orbital overlap with an aromatic π-system or if imines are employed. In all explored compounds, the charge distribution between beryllium and nitrogen atoms is almost identical and seems to have no influence on the structure of the compounds. The decisive factor for the observed molecular structures is the steric demand of the ligands, as illustrated by the space filling models in Scheme 7.73
 | ||
| Scheme 7 Conversion of organo-beryllium amide 28 into heteroleptic beryllium amide 37 (top) and respective space filling models of the compounds (bottom).73 | ||
While we investigated the beryllium species present in liquid ammonia,32–34 we realised that little is known about the solution behaviour of simple beryllium compounds in common organic N-donor solvents like acetonitrile, ethylenediamine or pyridine. Investigation of the solution behaviour of beryllium chloride, bromide, iodide and triflate in acetonitrile showed that in contrast to N,N-dimethyl formamide,29 no halide dissociation occurs and, therefore, no ionic species are formed. Only in the case of beryllium triflate, some indications for triflate dissociation were observed. Despite the lack of charged species in solution, halide exchange was observed as evident from the formation of heteroleptic berylliumhalide adducts of acetonitrile according to eqn (1)–(3), which predominantly lie on the right side.91
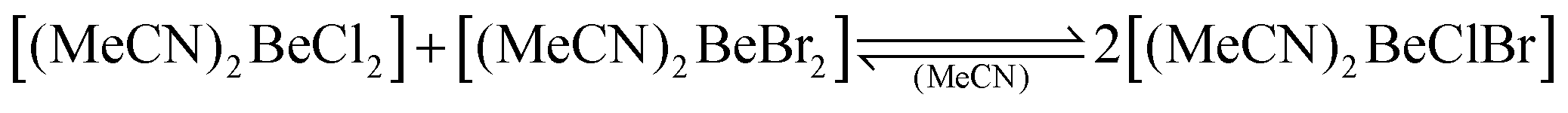 | (1) |
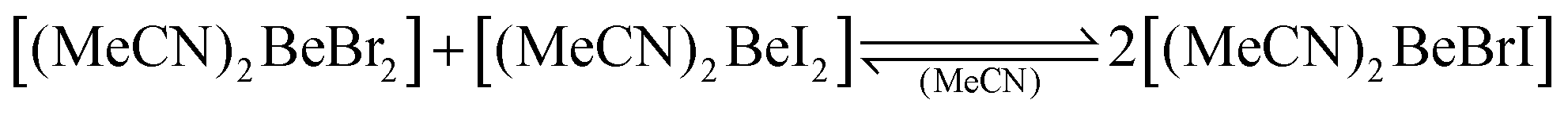 | (2) |
 | (3) |
These observations indicate that no large differences between the anionic ligands, like in the heteroleptic organo-beryllium halides (Scheme 6),85 are necessary but rather small electronegativity and steric differences between anionic ligands facilitate the preferred formation of heteroleptic beryllium complexes.91
In contrast to acetonitrile, ethylenediamine readily replaces chloride, bromide and iodide from the first coordination sphere of beryllium during the formation of beryllium dications. The same reactivity is also observed for the pseudo-halides N3−, CN− and SCN−, while ethylenediamine is not able to displace fluorido ligands.92 This reactivity is closely related to liquid ammonia,33,93 even though the solubility in ethylenediamine is significantly lower.92 Similar solubility issues were also encountered when quinolino[7,8-h]quinoline complexes of beryllium were investigated in acetonitrile and pyridine.94
Besides the above-mentioned studies, N-donor ligands have been mainly employed as auxiliary ligands, like Tp19–21,89,90,95 or 1,3-diketiminates,96,97 to enforce a rigid coordination environment. Recently, a sterically demanding amido ligand has also been used to stabilise an anionic dinuclear hydrido beryllate with three μ2-bridging hydrogen atoms.98 Accordingly, still very little is known about the reactivity of Be–N bonds. Though, due to the high Be–N bond strength, this bond should be hard to break, let alone respective compounds utilised in the activation of other molecules. However, this assumption needs to be tested experimentally.
4 P-donor ligands
Phosphine complexes of beryllium halides have been shown to be a feasible precursor for the synthesis of beryllium-organic compounds36 and are also capable of activating C–halide bonds,99 while phosphine adducts to beryllium hydrides have been shown to activate CO and CO2.100 However, little was known about the underlying ligand exchange processes, the influence of the steric demand of the phosphines or the stability of the Be–P bonds. Therefore, we conducted an extensive study on ligand exchange processes in beryllium phosphine complexes. While readily two small PMe3 ligands can coordinate with beryllium during the formation of tetra-coordinated beryllium complexes 38 (Scheme 8), the coordination sphere of beryllium can only accomodate one larger PCy3 (39, Scheme 9).101 The threshold cone angle up to which two phosphines can be coordinated is between 136° and 145°.102 The energy required for dissociation of a PMe3 ligand in 38 rises from the chloride via the bromide to the iodide derivatives, since the respective Be–halide bonds become weaker and, therefore, the resulting electron deficiency at the beryllium atom needs to be compensated through stronger Be–P bonds. This increase in Be–P bond strength can be directly observed in solution via the 1JPBe coupling constants in the 9Be and 31P NMR spectra. In the case of the iodo-derivative, actually phosphine dissociation and iodide dissociation are equal in energy, which is the reason why in the iodo system cationic species are also non-negligible. Therefore, their reactivity differs from those of the other halido complexes.101 While PMe3 dissociation followed by solvent coordination plays a role in C–Cl bond activation processes by [(PMe3)2BeCl2],99,101 this process is irrelevant for phosphine exchange, when an excess of phosphine is present.101 This ligand exchange process proceeds via an interchange mechanism and involves penta-coordinated intermediate 40 (Scheme 8).101 | ||
| Scheme 8 Ligand exchange at trimethylphoshine complexes of the beryllium halides via an interchange mechanism (X = Cl, Br, and I).101 | ||
 | ||
| Scheme 9 Dissociation processes at multinuclear beryllium phosphine complexes (X = Cl, Br, and I and R = Me and Cy).101 | ||
In the case of dinuclear complexes 39 two significantly different dissociation processes take place. For PMe3, homolytic cleavage to tri-coordinated intermediate 41 is energetically favoured, whereas for PCy3, phosphine loss resulting in the formation of asymmetric dinuclear complex 42 is less energy demanding (Scheme 9).101 The formation of 42 with its highly Lewis acidic tri-coordinated beryllium atom, which is not stabilised by a neutral donor ligand, is the explanation for the significantly higher reactivity of the PCy3 beryllium halide complexes.99,101
Due to the observation that 1JPBe NMR coupling constants are a direct measure for the Be–P bond strength,99,101 we developed a spectroscopic probe for the assessment of the base strength of various anionic ligands. Considering that the 1JPBe NMR coupling constant strongly depends on the Be–P distance, we employed a highly rigid monoanionic tridentate phosphaphenylborato ligand to facilitate a constant coordination environment at the beryllium atom. The reaction of the respective lithium salt 43 with a variety of homo- and heteroleptic beryllium (pseudo)-halides, triflate and beryllium-organic compounds gave the desired beryllium complexes 44, as depicted in Scheme 10.103,104 In these compounds the 1JPBe NMR coupling constant is a direct measure of the electron density of the beryllium atom and correlates with the base strength of the coordinated anionic ligand L. Furthermore, it was proven that the Be–P distances obtained from X-ray diffraction experiments are not suitable parameters for the assessment of the ligating properties of L.103
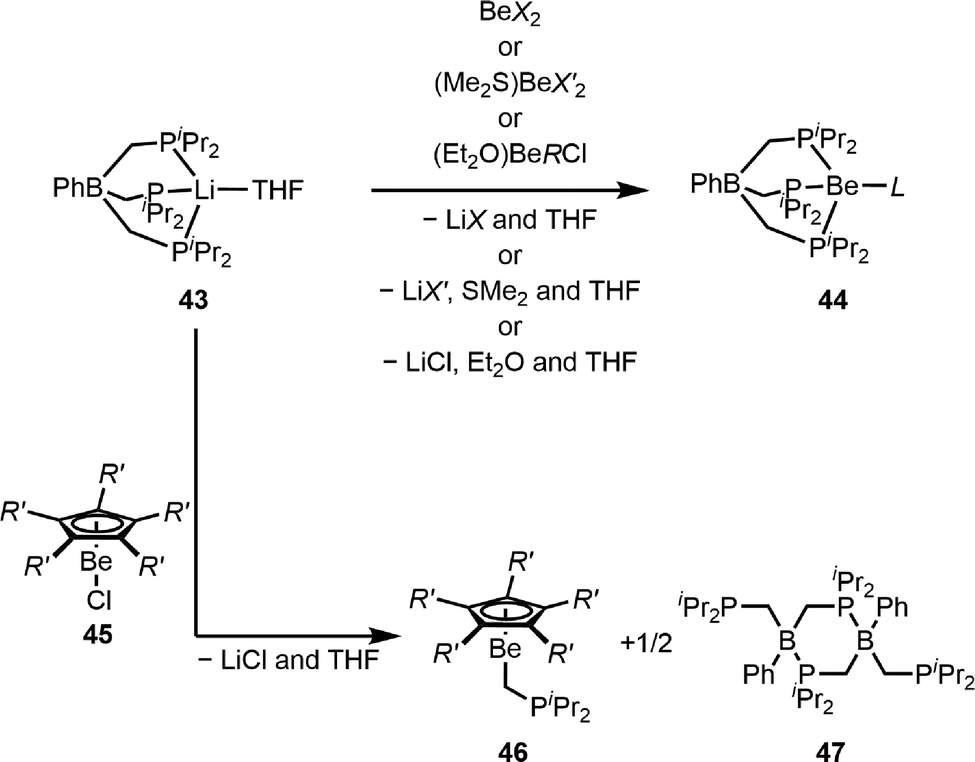 | ||
| Scheme 10 Synthesis of phosphorus-based beryllium scorpionate complexes and transmetallation from boron to beryllium (X = Cl, Br, I, and OTf; X′ = CN, N3, NCO, and NCS; R = Ph and nBu; L = Cl, Br, I, OTf, CN, N3, NCO, NCS, Ph, and nBu; and R′ = H and Me).103,104 | ||
Attempts to synthesise derivatives of 44 with cyclopentadienyl ligands through the reaction of 43 with half-sandwich beryllocenes (45) failed. Instead, the unprecedented transmetalation of a [CH2P(iPr)2]− group from boron onto beryllium was observed, which gave half-sandwich compounds 46 and neutral phosphaborane 47 (Scheme 10). This reaction shows that cyclopentadienyl as a six electron donor exhibits superior ligating properties compared to phosphaphenylborane. The formation of 46 is the first transmetalation reaction from a more to a less electronegative element, and similar reactivity can also be induced through steric overcrowding at the beryllium atom, withdrawal of electron density from the beryllium atom or weakening of the beryllium–scorpionate bond through oxidation of the phosphorus atoms.104
The Be–P bond strength seems to be in a sweet spot so that not only compounds can still be conveniently isolated but also bond cleavage is easily induced to facilitate further reactivity. At present, we investigate the properties of the Be–P bonds in detail and apply P-donor complexes of beryllium in Lewis acid–base reactions.105
5 O-donor ligands
Ethers, especially diethyl ether and tetrahydrofuran (THF), are among the most ubiquitous organic solvents. However, these solvents are rarely used in beryllium chemistry, except for the synthesis of the Et2O adducts of BeCl2, BeBr2 and BeI2 (48Fig. 5) from elemental beryllium.106 Even though these etherates are applied for further synthesis88 and their structures are well known,86,106,107 little was known about the actual species in etheral solutions. In Et2O, the diadducts 48 are present exclusively and no dissociation of halides occurs, which is analogous to the solution behaviour of the beryllium halides in acetonitrile.91,108 μ2-Halido bridged dinuclear complexes 49 can be prepared through the reaction of one equivalent Et2O with the respective beryllium halide in aromatic solvents and the solid state structure of the chlorido derivative has been determined.109 However, these dinuclear species play no role in diethyl ether solutions. Generally the etherates 48 do not dissolve well in diethyl ether, which is presumably the reason why this solvent is not commonly used.108 Similar diadducts are formed in THF (50), and the chlorido and iodo derivatives are also only sparingly soluble in the solvent; interestingly, the bromido compound dissolves well. However, in the case of BeI2, cationic species 51 and 52 are also presumably present in solution. These two complex cations are also observable in solutions of the THF diadduct of BeI2 in benzene, together with μ2-O bridged dinuclear 53, which results from ring opening of THF.108 Based on these results, ethers are non-ideal solvents for beryllium chemistry, since due to the high oxophilicity of beryllium, the formed ethrates show low solubility in ethers, while in the case of THF activation of the solvent also occurs. Nevertheless, diethyl ether complexes 48 are well soluble in aromatic solvents and the Be–O bond strength seems to be weak enough to remove the ether if desired.88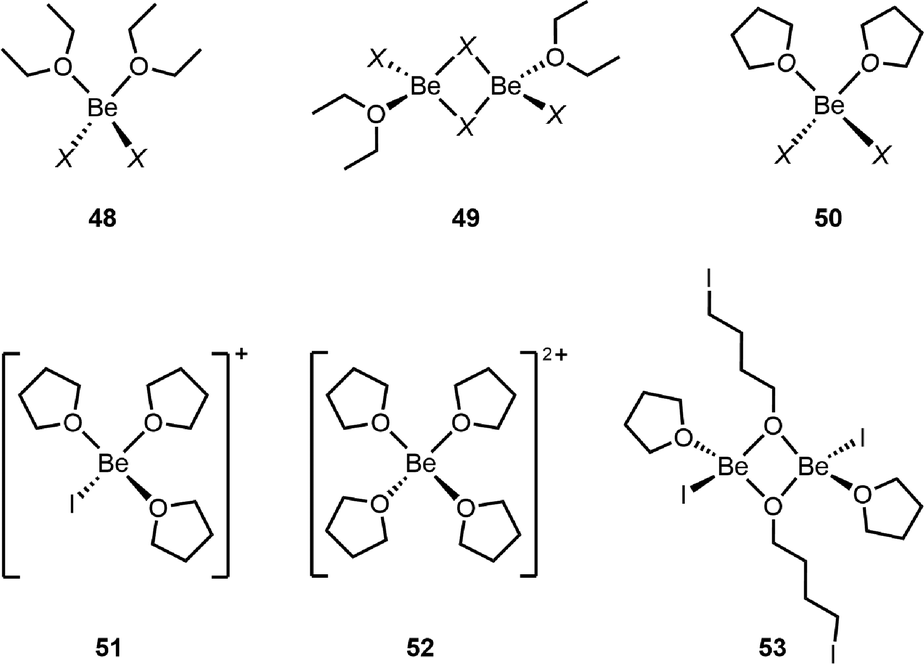 | ||
| Fig. 5 Structures of beryllium etherates (X = Cl, Br, and I).108 | ||
Ring opening also occurs if cyclic silaethers react with beryllium halides or diphenyl beryllium. Bromide, iodide and phenyl are selectively transferred onto the silicon atoms of hexamethylcyclotrisiloxane (D3) from BeBr2, BeI2 and 23, respectively (Scheme 11). In the case of BeCl2, no selective reaction occurs. The obtained trinuclear silanolates 54 are the first examples of α-bromo and α-iodo silanolates. However, only in the case of dimethylphenyl silanolate, hydroylsis gives free silanol PhMe2SiOH, while free α-bromo and α-iodo silanols are not stable and react to a mixture of cyclic methylsiloxanes (Dn).110 The stabilisation of α-halido silanolates, in which the parent free silanols are not stable, is in analogy to the stabilisation of α-iodo alcoholates through beryllium coordination.30
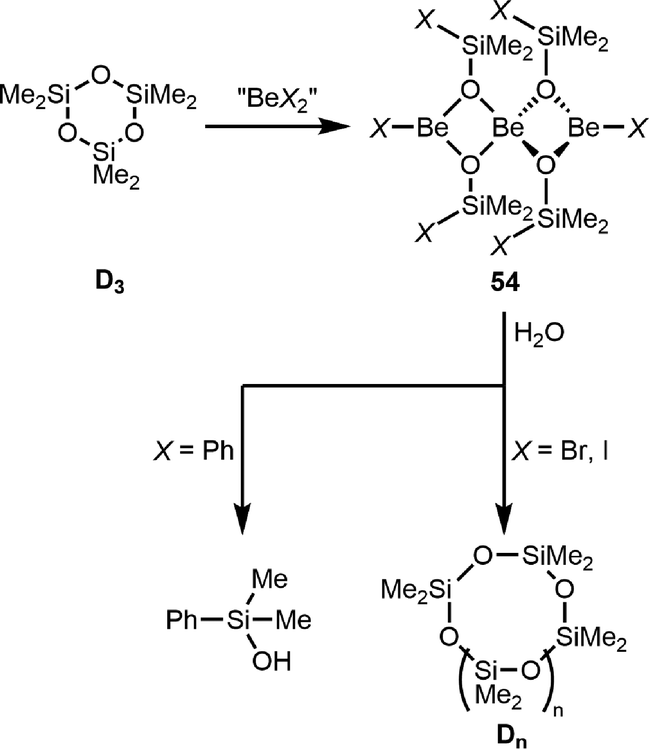 | ||
| Scheme 11 Beryllium mediated transfer of chloro-, bromo- and phenyl-groups onto silicon and their subsequent hydrolysis (X = Ph, Br, and I).110 | ||
In the hope of obtaining a beryllium precursor with a weakly coordinating and inert anionic ligand, we expanded our research into beryllium halide and pseudo-halide complexes onto triflate (OTf−).111 While Be(OTf)2 can be readily prepared, triflate does not act as a weakly coordinating ligand in beryllium chemistry. While triflates show weaker coordination to beryllium atoms than chlorides and bromides, they exhibit better coordination properties than iodides. This is due to the high oxophilicity of beryllium and the good hard–hard match according to the hard and soft acids and bases (HSAB) concept. In contrast to this, the soft iodo ligand only forms weak bonds with beryllium atoms. According to this observation, the structural chemistry of beryllium triflates (Fig. 6) is closely related to that of BeCl2 and BeBr2 complexes. In the diethyl ether, complex 55 is formed, which is similar to the halides 48,108,111 while removal of the solvent results in the formation of one-dimensional polymer 56, which is comparable to aldehyde adducts of BeCl2.30,111 With bulky ligands like NEt3 triflate bridged dinuclear complexes 57, similar to the respective BeCl2 and BeBr2 complexes, are formed.31,111 Furthermore, auto-ionisation in the presence of 12-crown-4 to 58 occurs, which has also been observed for diphenyl beryllium and BeCl2.22,35,111 Eventually, SMe2 activation and formation of hexatriflatodiberyllate 59 have also been encountered, in analogy to beryllium halides.111,112
 | ||
| Fig. 6 Examples for the highly variable structures of neutral and cationic beryllium triflates as well as triflato beryllates.111 | ||
Based on our findings concerning the interaction of Be2+ ions with biologically relevant functional groups,24 we concentrated on the interactions with carboxy functions. If beryllium chloride reacts with one equivalent of a carboxylic acid, hexanuclear beryllium chloro carboxylates 60 are formed (Scheme 12). 60 react with further carboxylic acid to give tetranuclear 61, which is the precursor compound class to the well known beryllium oxo-carboxylates 62113 and rare homoleptic beryllium carboxylates 1. This finding enabled a first rationalisation on how these two compound classes are formed.114 Considering the assumption that 62 are the actual species liable for beryllium associated diseases,115 we conducted an in-depth study on the electronic and steric influence of the carboxy ligands on the Be4O core of 62. This study revealed that only the steric demand of the carboxylic acid has an impact on the spectroscopic properties of 62, while atomic distances are not affected by the carboxylic acids.113
 | ||
| Scheme 12 Formation of beryllium chloro carboxylates and subsequent formation of homoleptic beryllium carboxylates as well as oxo-carboxylates (R = Ph, o-Tol, and Mes).114 | ||
To evaluate whether the peptide backbone of proteins is a likely coordination site for Be2+ ions, beryllium carboxamide complexes were investigated.116 The Be–O bonds in these compounds seem to be stronger than in related beryllium complexes with esters25 or ketones.116 However, an evaluation of the relative binding strength of carboxamides versus carboxy and hydroxy functions was not possible in organic solvents due to solubility issues. The low solubility of the beryllium carboxamide compounds is caused by the formation of extended and very strong hydrogen bond networks.116 To overcome these issues, reactions in inorganic, water-related solvents are ongoing.
The high oxophilicity of beryllium is also highlighted by the fact that μ2-O is preferred over μ2-Cl bridging not only for anionic O-donor ligands but also for neutral phosphine oxides. This is highlighted by the reaction of dinuclear phosphine adduct 63 with p-cresol to give 64 and with Me3PO to give 65, as depicted in Scheme 13.117 This oxophilicity can reach an extent, which prevents the isolation of beryllium compounds due to the formation of beryllium oxide under ligand decomposition.118
 | ||
| Scheme 13 Formation of μ2-oxygen bridged dinuclear beryllium p-cresolate 64 and trimethylphosphine oxide complex 65.117 | ||
Unsurprisingly, O-donors form extremely strong bonds with beryllium atoms. This often results in low solubilities or reactivity. This limits the use of O-donor based solvent due to the competition between solvent and ligand coordination. While O-donor complexes are likely less relevant for the synthesis of low valent beryllium compounds, they are highly relevant to understand the physiological processes liable for beryllium metabolisation. Therefore, efforts are ongoing to better understand these compounds and their speciation in solution.
6 Further donor ligands
To evaluate the different Be–chalcogen bond strengths, 44Cl was exposed to elemental oxygen, sulphur, selenium and tellurium (Scheme 14). With exception of tellurium, all chalcogens were able to insert into the Be–P bonds of 44Cl, which resulted in the formation of 66. While the oxygen and sulphur derivatives of 66 are stable, the weak Be–Se bonds destabilise the complex to an extent that transmetalation from boron onto beryllium occurs. These reactions lead to the formation of dinuclear beryllium complex 67 and phospha-selena-bora heterocycle 68. If an excess of selenium is present, ring strain in 67 is released through selenium insertion into the Be–C bond during the formation of five-membered phospha-selena-berylla cycle 69.104 | ||
| Scheme 14 Insertion of chalcogens into the Be–P bond of 44Cl and subsequent transmetalation and ring expansion in the case of selenium (En = O2, S8, and Se8).104 | ||
In contrast to the Be–S bonds in 66S,104 the respective bonds in thioether adducts to the beryllium halides (70 and 71, Scheme 15) are relatively weak.112 For this reason SMe2 is a versatile solubiliser for beryllium halides and their conversion into pseudo-halides or triflate.90,111 While in SMe2 solutions only diadducts 70 are present, careful removal of SMe2in vacuo or reaction of 70 with an additional equivalent of the respective beryllium halide gives dinuclear complexes 71.112 This behaviour is closely related to beryllium etherates.108 However, due to the weak Be–S bonds in 70, SMe2 partially dissociates during the formation of 71. In 71, the S–C bonds are highly polarised due to SMe2 coordination to the strongly Lewis acidic beryllium atoms. If non-coordinated SMe2 is present, it can nucleophilicly attack these carbon atoms, resulting in the formation of [SMe3]+ cations and thiolato beryllate 72, as depicted in Scheme 15.112 It is likely that similar processes are also accountable for the formation of triflato beryllate 59, as shown in Fig. 6.111
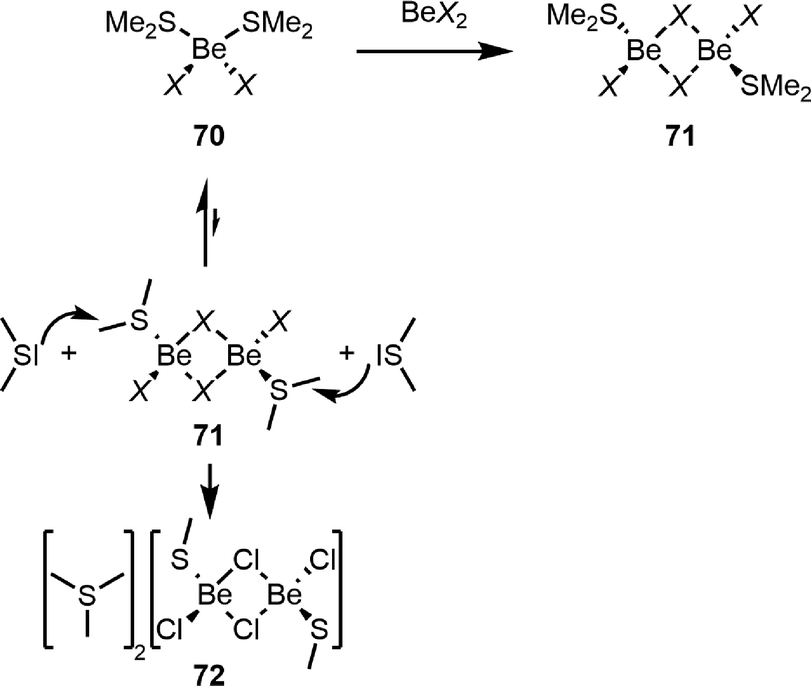 | ||
| Scheme 15 Nucleophilic attack of non-coordinated SMe2 at 71 during the formation of timethylsulfonium salt 72.112 | ||
During our research with the above-described ligand systems, we frequently encountered the formation of halido beryllate anions.31,99,112,117 These anions can be understood as molecular, anionic fragments from the solid state modifications of beryllium halides,119,120 as exemplified by the cholido beryllates 73–76, depicted in Fig. 7.99,117 Similar structural motifs have also been isolated by Dehnicke and colleagues.121–126 While it is plausible that the Be–halide bond strength and the μ-bridging properties decrease from fluoride via chloride and bromide to iodide, still no systematic investigations on the reactivity of the beryllates have been conducted.
 | ||
| Fig. 7 Structures of chlorido beryllates.99,117 | ||
As expected, donor ligands with softer donor sites coordinate relatively weakly to beryllium atoms. Therefore, complexes with ligands based on heavier elements are more promising considering their application in bond activation processes. However, this, of course, makes these compounds harder to isolate. At present, efforts are ongoing to prepare beryllium complexes with heavier p-block donor ligands and explore their bonding and reactivity.
7 Conclusions
Beryllium element bonds are mostly covalent in nature but strongly polarised towards the ligand due to the higher electronegativity of the donating atom. Only in the case of very hard donor sites like in pseudo-halides, the bonding is mostly ionic. Electron deficiency at the beryllium atom is often reduced through hyperconjugation into empty atomic orbitals of beryllium, which results in the formation of (partial) beryllium–element multiple bonds. However, the electronic properties of the ligands have only a secondary effect on the structure of beryllium complexes, while the main factor is the steric demand. This is a direct result of the small size of the beryllium atom. Due to these strong steric influences, solid state parameters, like bond lengths, are not good descriptors for bond strengths. Therefore, these parameters can only be used cautiously when assessing charge distribution and reactivity of these complexes. A far superior probe for these properties is the NMR coupling constant obtained in solution. Unsurprisingly, beryllium forms the strongest bonds to other very hard elements, like oxygen, fluorine or nitrogen, which result in very stable compounds. However, the use of softer ligands like phosphines or thioethers results in well isolatable complexes, which show high reactivity in bond activation processes. Despite the advances in the understanding of beryllium–element bonds, still reactivity studies on fundamental compounds like beryllocenes are missing. Therefore, systematic investigations are required and ongoing. This is especially important for C-donor ligands, since these are the main substance class for the stabilisation of low valent beryllium complexes and also show unprecedented behaviour, as evident from the transmetalation from a more to a less electronegative element.Conflicts of interest
There are no conflicts to declare.Data availability
Since this is a review article, all the data are available in the study.Acknowledgements
The author expresses his gratitude to Dr. Chantsalmaa Berthold and Deniz F. Bekiş for discussions on the manuscript. Prof. Florian Kraus is thanked for moral and financial support as well as the provision of laboratory space. The DFG is gratefully acknowledged for financial support (BU2725/8-1).References
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Nat. Catal., 2018, 1, 40–47 CrossRef CAS.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef PubMed.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed.
- B. Rösch and S. Harder, Chem. Commun., 2021, 57, 9354–9365 RSC.
- C. Färber, P. Stegner, U. Zenneck, C. Knüpfer, G. Bendt, S. Schulz and S. Harder, Nat. Commun., 2022, 13, 3210 CrossRef PubMed.
- L. A. Freeman, J. E. Walley and R. J. Gilliard, Nat. Synth., 2022, 1, 439–448 CrossRef CAS.
- K. G. Pearce, H.-Y. Liu, S. E. Neale, H. M. Goff, M. F. Mahon, C. L. McMullin and M. S. Hill, Nat. Commun., 2023, 14, 8147 CrossRef CAS PubMed.
- I.-A. Bischoff, S. Danés, P. Thoni, B. Morgenstern, D. M. Andrada, C. Müller, J. Lambert, E. C. J. Gießelmann, M. Zimmer and A. Schäfer, Nat. Chem., 2024, 16, 1093–1100 CrossRef CAS PubMed.
- J. Mai, J. Maurer, J. Langer and S. Harder, Nat. Synth., 2024, 3, 368–377 CrossRef CAS.
- I.-A. Bischoff, B. Morgenstern, M. Zimmer and A. Schäfer, Angew. Chem., Int. Ed., 2024, 64, e202419688 CrossRef PubMed.
- M. R. Buchner, Chem. Commun., 2020, 56, 8895–8907 RSC.
- M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 405–412 CrossRef CAS.
- J. Elguero and I. Alkorta, Struct. Chem., 2023, 34, 391–398 CrossRef CAS.
- M. R. Buchner and M. Müller, ACS Chem. Health Saf., 2023, 30, 36–43 CrossRef CAS.
- D. Naglav, B. Tobey, B. Lyhs, B. Römer, D. Bläser, C. Wölper, G. Jansen and S. Schulz, Angew. Chem., Int. Ed., 2017, 56, 8559–8563 CrossRef CAS PubMed.
- D. Naglav, D. Bläser, C. Wölper and S. Schulz, Inorg. Chem., 2014, 53, 1241–1249 CrossRef CAS PubMed.
- D. Naglav, B. Tobey, C. Wölper, D. Bläser, G. Jansen and S. Schulz, Eur. J. Inorg. Chem., 2016, 2424–2431 CrossRef CAS.
- D. Naglav, B. Tobey, K. Dzialkowski, D. Bäser, C. Wölper, G. Jansen and S. Schulz, Dalton Trans., 2018, 47, 12511–12515 RSC.
- M. R. Buchner and M. Müller, Z. Anorg. Allg. Chem., 2018, 644, 1186–1189 CrossRef CAS.
- M. R. Buchner, M. Müller, F. Dankert, K. Reuter and C. von Hänisch, Dalton Trans., 2018, 47, 16393–16397 RSC.
- M. Müller and M. R. Buchner, Angew. Chem., Int. Ed., 2018, 57, 9180–9184 CrossRef PubMed.
- B. Scheibe and M. R. Buchner, Eur. J. Inorg. Chem., 2018, 2300–2308 CrossRef CAS.
- O. Raymond, P. J. Brothers, M. R. Buchner, J. R. Lane, M. Müller, N. Spang, W. Henderson and P. G. Plieger, Inorg. Chem., 2019, 58, 6388–6398 CrossRef CAS PubMed.
- M. Müller and M. R. Buchner, Chem. – Eur. J., 2019, 25, 16257–16269 CrossRef PubMed.
- M. R. Buchner, M. Müller, O. Raymond, R. J. Severinsen, D. J. Nixon, W. Henderson, P. J. Brothers, G. J. Rowlands and P. G. Plieger, Eur. J. Inorg. Chem., 2019, 3863–3868 CrossRef CAS.
- M. Müller and M. R. Buchner, Inorg. Chem., 2019, 58, 13276–13284 CrossRef PubMed.
- M. Müller and M. R. Buchner, Chem. – Eur. J., 2019, 25, 11147–11156 CrossRef PubMed.
- M. R. Buchner, M. Müller and N. Spang, Dalton Trans., 2020, 49, 7708–7712 RSC.
- M. Müller, A. J. Karttunen and M. R. Buchner, Chem. Sci., 2020, 11, 5414–5422 RSC.
- M. Müller and M. R. Buchner, Chem. Commun., 2019, 55, 13649–13652 RSC.
- M. Müller and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 483–489 CrossRef.
- M. Müller and M. R. Buchner, Chem. – Eur. J., 2020, 26, 9915–9922 CrossRef PubMed.
- M. R. Buchner, M. Müller and S. S. Rudel, Angew. Chem., Int. Ed., 2017, 56, 1130–1134 CrossRef CAS PubMed.
- D. Naglav, B. Tobey, A. Neumann, D. Bläser, C. Wölper and S. Schulz, Organometallics, 2015, 34, 3072–3078 CrossRef CAS.
- M. Bayram, D. Naglav, C. Wölper and S. Schulz, Organometallics, 2016, 35, 2378–2383 CrossRef CAS.
- M. Bayram, D. Naglav, C. Wölper and S. Schulz, Organometallics, 2017, 36, 467–473 CrossRef CAS.
- J. E. Walley, G. Breiner, G. Wang, D. A. Dickie, A. Molino, J. L. Dutton, D. J. D. Wilson and R. J. Gilliard, Chem. Commun., 2019, 55, 1967–1970 RSC.
- J. E. Walley, Y.-O. Wong, L. A. Freeman, D. A. Dickie and R. J. Gilliard, Catalysts, 2019, 9 DOI:10.3390/catal9110934.
- J. E. Walley, A. D. Obi, G. Breiner, G. Wang, D. A. Dickie, A. Molino, J. L. Dutton, D. J. D. Wilson and R. J. J. Gilliard, Inorg. Chem., 2019, 58, 11118–11126 CrossRef CAS PubMed.
- J. E. Walley, D. A. Dickie and R. J. Gilliard, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 497–501 CrossRef CAS.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- G. Wang, J. E. Walley, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard, J. Am. Chem. Soc., 2020, 142, 4560–4564 CrossRef CAS PubMed.
- C. Czernetzki, M. Arrowsmith, F. Fantuzzi, A. Gärtner, T. Tröster, I. Krummenacher, F. Schorr and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 20776–20780 CrossRef CAS PubMed.
- M. Gimferrer, S. Danés, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2022, 13, 6583–6591 RSC.
- S. Pan and G. Frenking, Chem. Sci., 2023, 14, 379–383 RSC.
- M. Gimferrer, S. Danés, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2023, 14, 384–392 RSC.
- G. Wang, L. Freeman, D. Dickie, R. Mokrai, Z. Benk and R. J. Gilliard, Chem. – Eur. J., 2019, 25, 4335–4339 CrossRef CAS PubMed.
- A. Paparo, C. D. Smith and C. Jones, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed.
- J. T. Boronski, L. R. Thomas-Hargreaves, M. A. Ellwanger, A. E. Crumpton, J. Hicks, D. F. Bekiş, S. Aldridge and M. R. Buchner, J. Am. Chem. Soc., 2023, 145, 4408–4413 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Science, 2023, 380, 1147–1149 CrossRef CAS PubMed.
- J. T. Boronski, L. P. Griffin, C. Conder, A. E. Crumpton, L. L. Wales and S. Aldridge, Chem. Sci., 2024, 15, 15377–15384 RSC.
- C. Berthold, J. Maurer, L. Klerner, S. Harder and M. R. Buchner, Angew. Chem., Int. Ed., 2024, 63, e202408422 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton, A. F. Roper and S. Aldridge, Nat. Chem., 2024, 16, 1295–1300 CrossRef CAS PubMed.
- I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Science, 2004, 305, 1136–1138 CrossRef CAS PubMed.
- A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Río and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 693–703 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton and S. Aldridge, J. Am. Chem. Soc., 2024, 146, 35208–35215 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton, J. J. C. Struijs and S. Aldridge, J. Am. Chem. Soc., 2025, 147, 10073–10077 CrossRef CAS PubMed.
- S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973–2977 CrossRef CAS PubMed.
- M. del Mar Conejo, R. Fernández, E. Carmona, E. Gutiérrez-Puebla and A. Monge, Organometallics, 2001, 20, 2434–2436 CrossRef CAS.
- M. del Mar Conejo, R. Fernández, E. Carmona, R. A. Andersen, E. Gutiérrez-Puebla and M. A. Monge, Chem. – Eur. J., 2003, 9, 4462–4471 CrossRef CAS PubMed.
- M. del Mar Conejo, R. Fernández, E. Gutiérrez-Puebla, A. Monge, C. Ruiz and E. Carmona, Angew. Chem., Int. Ed., 2000, 39, 1949–1951 CrossRef CAS.
- M. del Mar Conejo, R. Fernández, D. del Río, E. Carmona, A. Monge and C. Ruiz, Chem. Commun., 2002, 2916–2917 RSC.
- R. Fernández and E. Carmona, Eur. J. Inorg. Chem., 2005, 3197–3206 CrossRef.
- M. R. Buchner, S. Pan, C. Poggel, N. Spang, M. Müller, G. Frenking and J. Sundermeyer, Organometallics, 2020, 39, 3224–3231 CrossRef CAS.
- M. R. Buchner, L. K. Kreuzer, L. R. Thomas-Hargreaves, M. Müller, S. I. Ivlev, G. Frenking and S. Pan, Chem. – Eur. J., 2024, 30, e202400966 CrossRef CAS PubMed.
- L. R. Thomas-Hargreaves, Y.-Q. Liu, Z.-H. Cui, S. Pan and M. R. Buchner, J. Comput. Chem., 2023, 44, 397–405 CrossRef CAS PubMed.
- H. Nöth and D. Schlosser, Chem. Ber., 1988, 121, 1711–1713 CrossRef.
- D. Naglav, A. Neumann, D. Bläser, C. Wölper, R. Haack, G. Jansen and S. Schulz, Chem. Commun., 2015, 51, 3889–3891 RSC.
- G. Wang, J. E. Walley, D. A. Dickie, A. Molino, D. J. Wilson and R. J. Gilliard, Angew. Chem., Int. Ed., 2021, 60, 9407–9411 CrossRef CAS PubMed.
- D. F. Bekiş, L. R. Thomas-Hargreaves, S. I. Ivlev and M. R. Buchner, Dalton Trans., 2024, 53, 15551–15564 RSC.
- C. Helling, D. J. D. Wilson and C. Jones, J. Am. Chem. Soc., 2025, 147, 16620–16629 CrossRef CAS PubMed.
- W. Petz, K. Dehnicke, N. Holzmann, G. Frenking and B. Neumüller, Z. Anorg. Allg. Chem., 2011, 637, 1702–1710 CrossRef CAS.
- A. Paparo, A. J. R. Matthews, C. D. Smith, A. J. Edwards, K. Yuvaraj and C. Jones, Dalton Trans., 2021, 50, 7604–7609 RSC.
- L. R. Thomas-Hargreaves, C. Berthold, W. Augustinov, M. Müller, S. I. Ivlev and M. R. Buchner, Chem. – Eur. J., 2022, 28, e202200851 CrossRef CAS PubMed.
- L. R. Thomas-Hargreaves, S. Pan, S. I. Ivlev, G. Frenking and M. R. Buchner, Inorg. Chem., 2022, 61, 700–705 CrossRef CAS PubMed.
- J. Gottfriedsen and S. Blaurock, Organometallics, 2006, 25, 3784–3786 CrossRef CAS.
- R. J. Gilliard, M. Y. Abraham, Y. Wang, P. Wei, Y. Xie, B. Quillian, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2012, 134, 9953–9955 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2015, 34, 653–662 CrossRef CAS.
- M. R. Buchner and D. F. Bekiş, Dalton Trans., 2023, 52, 13864–13867 RSC.
- L. R. Thomas-Hargreaves, M. Müller, N. Spang, S. I. Ivlev and M. R. Buchner, Organometallics, 2021, 40, 3797–3807 CrossRef CAS.
- A. Paparo, S. P. Best, K. Yuvaraj and C. Jones, Organometallics, 2020, 39, 4208–4213 CrossRef CAS.
- M. R. Buchner, L. R. Thomas-Hargreaves, C. Berthold, D. F. Bekiş and S. I. Ivlev, Chem. – Eur. J., 2023, 29, e202302495 CrossRef CAS PubMed.
- M. Niemeyer and P. P. Power, Inorg. Chem., 1997, 36, 4688–4696 CrossRef CAS PubMed.
- A. Paparo and C. Jones, Chem. – Asian J., 2019, 14, 486–490 CrossRef CAS PubMed.
- C. Helling and C. Jones, Chem. – Eur. J., 2023, 29, e202302222 CrossRef CAS PubMed.
- C. Berthold, G. Stebens, B. Butschke, I.-A. Bischoff, A. Schäfer, C. Ding, S. Pan and M. R. Buchner, Inorg. Chem. Front., 2025, 12, 2844–2855 RSC.
- C. Berthold, M. Müller, S. I. Ivlev, D. M. Andrada and M. R. Buchner, Dalton Trans., 2023, 52, 13547–13554 RSC.
- N. Spang, M. Müller, W. Augustinov and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 939–949 CrossRef CAS.
- M. R. Buchner and M. Müller, Dalton Trans., 2021, 50, 7246–7255 RSC.
- F. Kraus, S. A. Baer, M. R. Buchner and A. J. Karttunen, Chem. – Eur. J., 2012, 18, 2131–2142 CrossRef CAS PubMed.
- J. K. Buchanan, R. J. Severinsen, M. R. Buchner, L. R. Thomas-Hargreaves, N. Spang, K. D. John and P. G. Plieger, Dalton Trans., 2021, 50, 16950–16953 RSC.
- D. Naglav-Hansen, K. Dzialkowski, B. Tobey, C. Wölper, G. Jansen and S. Schulz, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 503–508 CrossRef CAS.
- M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon and I. Mallov, Inorg. Chem., 2012, 51, 13408–13418 CrossRef CAS PubMed.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill and G. Kociok-Köhn, Dalton Trans., 2013, 42, 9720–9726 RSC.
- T. J. Hadlington, Dalton Trans., 2024, 53, 882–886 RSC.
- M. R. Buchner, N. Spang, M. Müller and S. S. Rudel, Inorg. Chem., 2018, 57, 11314–11317 CrossRef CAS PubMed.
- T. J. Hadlington and T. Szilvási, Nat. Commun., 2022, 13, 461 CrossRef CAS PubMed.
- M. R. Buchner, D. Ćoćić, S. I. Ivlev, N. Spang, M. Müller and R. Puchta, Dalton Trans., 2023, 52, 5287–5296 RSC.
- M. R. Buchner and S. I. Ivlev, Eur. J. Inorg. Chem., 2023, e202300199 CrossRef CAS.
- C. Berthold, G. Hoß, M. H. Lochte and M. R. Buchner, Inorg. Chem., 2024, 63, 24392–24399 CrossRef CAS PubMed.
- C. Berthold, M. H. Lochte and M. R. Buchner, Chem. – Eur. J., 2025, 31, e202500673 CrossRef CAS PubMed.
- C. Berthold, D. F. Bekiş, J. Moritz, G. Stebens, B. Butschke and M. R. Buchner, ChemistryEurope, 2025, 3, e202500142 Search PubMed.
- C. Jones and A. Stasch, Anal. Sci.: X-Ray Struct. Anal. Online, 2007, 23, 115–116 CrossRef PubMed.
- K. Ruhlandt-Senge, R. A. Bartlett, M. M. Olmstead and P. P. Power, Inorg. Chem., 1993, 32, 1724–1728 CrossRef CAS.
- D. F. Bekiş, L. R. Thomas-Hargreaves, C. Berthold, S. I. Ivlev and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2023, 78, 165–173 CrossRef.
- A. Paparo, C. N. de Bruin-Dickason and C. Jones, Aust. J. Chem., 2020, 73, 1144–1148 CrossRef CAS.
- M. R. Buchner, F. Dankert, C. Berthold, M. Müller and C. von Hänisch, Chem. – Eur. J., 2023, e202302652 CrossRef CAS PubMed.
- W. Augustinov, M. Müller, L. R. Thomas-Hargreaves, S. I. Ivlev and M. R. Buchner, Inorg. Chem., 2024, 63, 5208–5219 CrossRef CAS PubMed.
- M. R. Buchner, L. R. Thomas-Hargreaves, L. K. Kreuzer, N. Spang and S. I. Ivlev, Eur. J. Inorg. Chem., 2021, 4990–4997 CrossRef CAS.
- M. R. Buchner and M. Müller, Inorg. Chem., 2021, 60, 17379–17387 CrossRef CAS PubMed.
- M. R. Buchner, M. Müller and S. I. Ivlev, Inorg. Chem., 2024, 63, 17901–17906 CrossRef CAS PubMed.
- R. J. Berger, P. Håkansson and R. Mera-Adasme, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 413–419 CrossRef CAS.
- M. R. Buchner and M. Müller, Z. Anorg. Allg. Chem., 2023, 649, e202200347 CrossRef CAS.
- M. R. Buchner, N. Spang and S. I. Ivlev, Z. Naturforsch., B: J. Chem. Sci., 2022, 77, 381–390 CrossRef CAS.
- C. Berthold, L. R. Thomas-Hargreaves, S. I. Ivlev and M. R. Buchner, Z. Naturforsch., B: J. Chem. Sci., 2021, 76, 651–658 CrossRef CAS.
- M. Müller, F. Pielnhofer and M. R. Buchner, Dalton Trans., 2018, 47, 12506–12510 RSC.
- M. R. Buchner, F. Dankert, N. Spang, F. Pielnhofer and C. von Hänisch, Inorg. Chem., 2020, 59, 16783–16788 CrossRef CAS PubMed.
- B. Neumüller, F. Weller and K. Dehnicke, Z. Anorg. Allg. Chem., 2003, 629, 2195–2199 CrossRef.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2004, 630, 1374–1376 CrossRef.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2004, 630, 347–349 CrossRef.
- B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2005, 631, 2535–2537 CrossRef.
- R. Tonner, G. Frenking, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2007, 633, 1183–1188 CrossRef CAS.
- K. Dehnicke and B. Neumüller, Z. Anorg. Allg. Chem., 2008, 634, 2703–2728 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |