
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBambusuril as an effective astatide sequestrating agent by hydrogen bonding†
Clémence
Maingueneau
 a,
Julie
Patissou
b,
Marine
Lafosse
b,
Elise
Cartier
b,
Jean-François
Gestin
a,
Grégory
Pieters
b,
Frédéric
Taran
b,
Jean-Pierre
Dognon
c,
François
Guérard
*a and
Marie-Pierre
Heck
*b
a,
Julie
Patissou
b,
Marine
Lafosse
b,
Elise
Cartier
b,
Jean-François
Gestin
a,
Grégory
Pieters
b,
Frédéric
Taran
b,
Jean-Pierre
Dognon
c,
François
Guérard
*a and
Marie-Pierre
Heck
*b
aNantes Université, Inserm, CNRS, Université d’Angers, CRCI2NA, Nantes, France. E-mail: françois.guerard@univ-nantes.fr
bUniversité Paris-Saclay, CEA, INRAE, Département Médicaments et Technologie pour la Santé (DMTS), SCBM, 91191 Gif-sur-Yvette, France. E-mail: marie-pierre.heck@cea.fr
cUniversité Paris-Saclay, CEA, CNRS, NIMBE, 91191 Gif-sur-Yvette, France
First published on 7th July 2025
Abstract
Herein, we report a molecular cage allowing strong chelation of the 211At radioanion. Propargylated bambus[6]uril shows good affinity towards iodide and astatide radiohalides, affording promising inclusion complexes that are stable in phosphate buffered saline and human serum. Density functional theory calculations support the presence of C–H⋯At non-covalent cooperative interactions governing the formation of astatinated cage complexes. To our knowledge, this work is the first to report 211At-labeling using encapsulation via hydrogen bonds, which opens new perspectives in the design of 211At−-based radiopharmaceuticals.
Radioisotopes of heavy halogens such as iodine I and astatine At are of significant interest in nuclear medicine for both imaging and therapeutic applications.1 Astatine, the heaviest halogen of group 17 periodic elements, exists as 32 unstable isotopes. Among them, 211At is considered as one of the most promising α-emitting radionuclides for targeted alpha therapy in cancer treatment. 211At exhibits several favorable properties for medical applications: a simple decay scheme leading in 100% of cases to the emission of one high energy (5.9–7.4 MeV), short track (50–90 μm) α-particle limiting irradiation of nearby healthy tissues, a short half-life (7.2 h) and a scalable production from a cheap 209Bi. Together, these features make 211At a radionuclide with high potential for effective targeted α-therapies.2,3 Consequently, several clinical trials of 211At-labeled drugs are currently underway.4 Among its identified oxidation states (−1, 0, 1, 5, and 7), astatide At− is the easiest species to obtain due to its stability in reducing media over a broad pH range.5 As astatine has no stable isotope, iodine, its closest halogen neighbour with similar physicochemical properties, is commonly used as a model of At. Therefore, iodine or general halogen chemistry is often applied to astatination although differences in chemical reactivity between the two elements have also been reported.6 Classical methods for At− labelling generally afford aryl astatine–carbon bonds either by nucleophilic or electrophilic reactions (see Fig. 1(A)). Electrophilic substitutions occur with At+ species including direct aromatic electrophilic substitution (SEAr, Fig. 1(a)), astatodestannylation (Fig. 1(b)), and astatodeboronation (Fig. 1(c)), while nucleophilic substitutions with At− species can be carried out through copper catalyzed astatodeboronation (Fig. 1(d)), halogen exchange (Fig. 1(e)), and SNAr reactions reported with aryliodonium salts (Fig. 1(f)) or spirocyclic aryliodonium ylides (Fig. 1(g)).7
 | ||
| Fig. 1 (A) Classical methods for astatination, and (B) our approach. | ||
Although several radiosynthesis routes can provide astato-aryl compounds, their potential for in vivo applications is questioned by the low C–At bond stability predicted by theoretical calculations8 and confirmed experimentally, leading to uncontrolled release of At in healthy organs.9 To limit de-astatination, other bonding modalities have been studied such as B–At bonds in boron clusters;10 metal–At bonds such as AtHg,11 Rh(III) or Ir(III)-(16aneS4-diol)-At;12 and Rh(I)13 or Au(I)-NHC-At14 complexes, which are, apart from metal toxicity, promising strategies for At-labeling.
In this context, using host molecules, promoting noncovalent interactions with the anion guest, can be an interesting approach to capture At− through a host–guest complex (Fig. 1(B), this work). Nevertheless, encapsulation of anions with a large atomic radius, low charge density and low ability to engage in hydrogen bonds is an important challenge.15 In this regard, bambus[6]urils (BUs), synthetic neutral cavitands formed by six glycoluril units connected by six methylene bridges,16 are known as the best large anion chelators, and their complexation properties for iodide, phosphate and perchlorate anions have been described.17 Surprisingly, to the best of our knowledge, the use of BUs as radioactive anion-sequestering agents has not been reported so far. In this communication, we demonstrate the ability of R12BU[6] (R = propargyl) BU 1 to act as a powerful 125I− and 211At− receptor by sequestering these radioactive anions, thanks to twelve cooperative hydrogen-radioanion interactions. While our experimental data highlight the (radio)stability of the obtained radioactive complexes, DFT calculations were employed to gain deeper insight into the interaction energies between the host (BU) and the two radioactive guests, 125I and 211At.
As we previously reported, BU 1 (Scheme 1) exhibits high affinity for iodide, and crystals of I@propargyl12BU 2 confirmed the presence of the I− anion within BU's cavity.18 Accordingly, BU 1 was selected as a potential receptor for 211At−. First, we investigated the binding properties of Br− @BU 119 with I− by 1H NMR titrations (see the ESI† for experimental procedures and data processing details). In CD3CN, the stepwise addition of I− to BU 1 resulted in a slow exchange on the NMR timescale, indicating a strong affinity of BU 1 for I− (Ka = 4.4 × 103 M−1 in CD3CN, see Fig. S1 and S2 in the ESI†). This result is consistent with the well-established higher affinity of bambusurils for I− compared to Br− anions.17,20 The interaction between BU 1 and I− anions was also evaluated using isothermal titration calorimetry (ITC). In MeOH, BU 1 exhibited a strong affinity for iodide (Ka = 2.1 × 106 M−1, Fig. S3, ESI†). The ITC data clearly indicate that the formation of the I@BU 2 complex is enthalpy-driven (ΔH = −20.9 kJ mol−1) and follows a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry (Fig. S3, ESI†).20 The measured affinity for iodide is comparable to values previously reported for bambusurils.17
:1 binding stoichiometry (Fig. S3, ESI†).20 The measured affinity for iodide is comparable to values previously reported for bambusurils.17
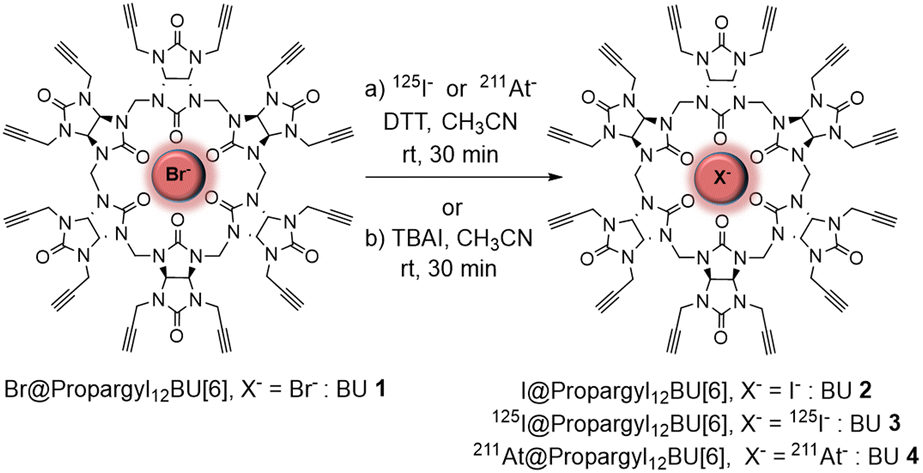 | ||
| Scheme 1 Synthesis of 125I@BU 3 and 211At@BU 4 radiocomplexes (conditions a) and I@BU 2 (conditions b). | ||
Based on these results, we investigated the complexation of 211At and 125I radioanions (used as a model of 211At) with BU 1 (Scheme 1). Preliminary experiments with sodium sulfite (Na2SO3), sodium metabisulfite (Na2S2O5), and dithiothreitol (DTT) were performed to identify a reductive agent capable of stabilizing 211At as the astatide species, amendable for the complexation reaction with BU 1 in CH3CN (see Table S1 and Fig. S4, ESI†). DTT appeared as the best reductive agent for astatine complexation with BU 1.
Although commercially available 125I is provided as sodium iodide in basic solution (NaOH), DTT was added to the 125I[NaI] solution to study the complexation under the same experimental conditions as those used for 211At. The non-radioactive iodinated complex I@propargyl12BU 218 was first prepared as an analytical reference of 125I@BU 3 and 211At@BU 4 radiocomplexes. BU 2 was synthesized from Br−@BU 1 and TBAI (tetrabutylammonium iodide) in CH3CN (90% yield), as reported in Scheme 1, conditions (b). Then, radiolabelled complexes 125I@BU 3 and 211At@BU 4 were prepared from BU 1 (1.2 mM in CH3CN) by adding a solution of 125I or 211At in the presence of DTT (see ESI† and Fig. S5 for details). After 30 min at room temperature, the complexes 125I@BU 3 and 211At@BU 4 were obtained in very good radiochemical yields (RCYs), 99% and 90%, respectively. These results showed that the radiocomplexation reactions of 125I and 211At are rapid and that the radiolabelled complexes BU 3–4 are stable and detectable at trace radionuclide concentrations, demonstrating the capacity of BU 1 to efficiently encapsulate radioactive iodide and astatide anions. Complexes BU 3–4 were identified by radio-HPLC analysis. As expected, nearly identical retention times were obtained for 211At@BU 4, 125I@BU 3 and non-radioactive iodinated reference I@BU 2 (see Fig. S6, ESI†).
Subsequently, various BU concentrations and solvents were studied as they can influence radioanion complexation (see Fig. S7–S10, ESI†). 125I and 211At radioanions reacted at room temperature for 30 min with varying concentrations of BU 1 in CH3CN, CH2Cl2 or CHCl3 solutions (see Fig. 2(A) and (B)). Starting from BU 1 (a concentration of 293.9 μM), the corresponding complexes 211At@BU 4 (Fig. 2(A) and Fig. S7, ESI†) and 125I @BU 3 (Fig. 2(B) and Fig. S8 and S9, ESI†) were obtained in high RCYs (90%, 88%, and 91%) for 211At@BU 4 and (99%, 99%, and 99%) for 125I @BU 3 in CH3CN, CH2Cl2, and CHCl3, respectively. The efficient complexation of 211At @BU 4 in CHCl3 is very promising as purified 211At is frequently delivered in CHCl3. Given the limited radiolabelling reactions described in CHCl3, complexation can be performed immediately after astatide purification, avoiding the additional concentration evaporation step usually required. We observed that 125I@BU 3 and 211At@BU 4 complexes are formed within 10 min of reaction, indicating rapid anion capture kinetics (Fig. S9 and S10, ESI†). The radiolabelled 125I@BU 3 and 211At@BU 4 complexes were subsequently purified on a silica cartridge before further evaluation (see the ESI† for details).
 | ||
| Fig. 2 (A) Influence of the BU 1 concentration on radiolabelling of 211At in DCM (●), CHCl3 (■), and CH3CN (▲); (B) influence of the BU 1 concentration on radiolabelling of 125I in DCM (●), CHCl3 (■), and CH3CN (▲), standard conditions: 0.5–1.5 MBq, DTT (0.16 μmol), at rt for 30 min, with RCY determined by TLC; (C) stability study of 211At@BU 4 in PBS (●) and in HS (■) at 37 °C; (D) stability study of 125I @ BU 3 in PBS (●) and in HS (■) at 37 °C with RCY (%) as a function of time (hours); (E) bonding energy analysis using ZORA-DFT with the revDOD-PBE-D4 functional, focusing on interactions between At−/I− and BU 1 fragments (see the ESI† for details); (F) isosurface map to visualize C–H⋯At non-covalent interactions in BU 4; for ease of reading, only H atom interactions with At are shown. The molecular structure is color-coded according to the contribution of various atoms to the interfragment interaction, using a blue-green-red color scale. Atoms appearing in red indicate a greater involvement in the At-BU cage interaction, highlighting regions of stronger interactions. | ||
Then, the stabilities of radiocomplexes 125I@BU 3 and 211At@BU 4 were evaluated in human blood serum (HS) at 37 °C and phosphate buffered saline (PBS) at room temperature (see Fig. 2(C), (D) and Fig. S11–S14, ESI†). In fact, as bambusurils have a strong affinity for anions, PBS is an interesting medium to study as it contains chlorides, at the same concentration and same pH (7.4) as in the blood, which are potential competitors of At−. Both 125I@BU 3 and 211At@BU 4 showed some dehalogenation in PBS but remained mostly intact after 6 h of incubation (60% and 55%, respectively, see Fig. 2(C), (D) and Fig. S11, S12, ESI†). In HS, the stability of the complexes was higher than in PBS, again with a slight superiority of 125I@BU 3 over 211At@BU 4 (80% and 70% of intact complex after 6 h, respectively, see Fig. 2(C) and (D) and Fig. S13 and S14, ESI†). Overall, observing the retention of 125I and 211At in the BU's cage over several hours validates our complexation concept and the strong encapsulation of these halides.
To gain deeper insights into the stability of the 125I@BU 3 and 211At@BU 4 complexes, density functional theory (DFT) calculations were performed. The factors affecting the stability of the BU 1 complexes with I− and At− anions were analyzed using an energy decomposition approach (see Fig. 2(E) and Fig. S15, ESI,† for details). For both complexes 211At@BU 4 and 125I@BU 3, similar trends were observed in the noncovalent interactions involved. The dominant contribution to the interaction energy arises from the electrostatic interaction, accounting for approximately 64% of the total attractive interactions in both complexes. This is followed by orbital interactions, contributing around 22.5% for 211At@BU 4 and 24.5% for 125I@BU 3. To a lesser extent, the MP2 interaction energies associated with the noncovalent C–H⋯X interaction are also notable, with values of approximately 10.9 kcal mol−1 for 211At@BU 4 compared to 8.6 kcal mol−1 for 125I@BU 3. Finally, dispersion interactions further contribute to the overall stability of the complexes. The total bonding energy for I− and At− exhibits a difference of 3 kcal mol−1, with greater stabilization observed in the 125I@BU 3 complex (−68.8 versus −65.8 kcal mol−1 for I and At, respectively). This indicates that BU 1 has a slightly higher affinity for iodide than for astatide. This trend has been confirmed experimentally since the BU 1 concentration can be decreased by 8-fold (34 μmol L−1) for complexation with 211At and by 32-fold (8.6 μmol L−1) for 125I@BU 3 formation, without affecting the RCY (see Fig. S7–S10, ESI†). Here, the difference in the stability of the two complexes (211At@BU 4 and 125I@BU 3) could be explained by either this slightly lower affinity of At−versus I− or the tendency of At− to be oxidized in the At+ cation exhibiting no affinity for the BU 1 cage.5
Quantum chemical calculations were also employed to determine the volume of the central bambusuril cavity, for both the empty BU cage and the anions encapsulated (see Fig. S16 and S17, ESI†). The volume of the central cavity in the empty BU's cage BU 1 was found to be 32.6 Å. In the presence of astatide and iodide anions, the cavity volume decreased to 32.0 Å and 31.5 Å, respectively. The smaller cavity size observed with iodide compared to astatine suggests a stronger interaction between the iodide anion and the cage, consistent with the interaction energy analysis. Furthermore, the non-covalent C–H⋯I and C–H⋯At interactions in I@BU 3 and At@BU 4 were analyzed using Multiwfn software21 (Fig. 2(F) and Fig. S14, ESI†). The computed isosurfaces (green regions) clearly highlight the presence of C–H⋯I interactions (Fig. S16(a), ESI†) and C–H⋯At interactions (Fig. 2(F) and Fig. S16(b), ESI†).
In summary, we report the first radiolabelling of a BU[6] cage with astatine-211 and iodine-125, yielding stable 211At@BU 4 and 125I@BU 3 complexes in both organic and biologically relevant media. These experimental and theoretical results constitute the first evidence of astatide sequestration within a host molecule through hydrogen bond interactions, demonstrated here using a bambusuril cage. To our knowledge, radiopharmaceuticals based on anion chelation remain unexplored, in contrast to the widespread use of cation-complexing agents with radiometals in nuclear medicine.22 This preliminary study thus paves the way for new opportunities in the field of radiopharmaceuticals.
This work was financially supported by the “Ministère de l'Education nationale de l'Enseignement supérieur et de la Recherche”, the French National Agency for Research (ANR-24-CE07-5913 and ANR-11-LABX-18-01 (Labex IRON)), and the INCa-DGOS-INSERM-ITMO Cancer_18011. The Radioactivity Technical Platform (SFR Santé François Bonamy) is thanked for the technical support, and the Arronax GIP is acknowledged for providing 211At. The “Service de Chimie Bioorganique et de Marquage” (SCBM, CEA) is a partner of NOMATEN, a Centre of Excellence in Multifunctional Materials for Industrial and Medical Applications (EU H2020 Teaming #857470).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- R. Eychenne, C. Alliot, J.-F. Gestin and F. Guérard, Nuclear Medicine and Molecular Imaging, Elsevier, Oxford, 2022, pp. 121–132 Search PubMed.
- S. Lindegren, P. Albertsson, T. Bäck, H. Jensen, S. Palm and E. Aneheim, Cancer Biother. Radiopharm., 2020, 35, 425 CrossRef PubMed.
- (a) R. Eychenne, M. Chérel, F. Haddad, F. Guérard and J.-F. Gestin, Pharmaceutics, 2021, 13, 906 CrossRef CAS PubMed; (b) R. Zimmermann, J. Nucl. Med., 2025, 66, 681 CrossRef PubMed.
- P. Albertsson, T. Bäck, K. Bergmark, A. Hallqvist, M. Johansson, E. Aneheim, S. Lindegren, C. Timperanza, K. Smerud and S. Palm, Front. Med., 2022, 9, 1076210 CrossRef PubMed.
- (a) L. Liu, R. Maurice, N. Galland, P. Moisy, J. Champion and G. Montavon, Inorg. Chem., 2022, 34, 13462 CrossRef PubMed; (b) A. N. Espino-Vásquez, F. C. Rojas-Castro and L. Mitzuko Fajardo-Yamamoto, Future Pharmacol., 2022, 4, 377 CrossRef.
- F. Guérard, C. Maingueneau, L. Liu, R. Eychenne, J.-F. Gestin, G. Montavon and N. Galland, Acc. Chem. Res., 2021, 54, 3264 CrossRef PubMed.
- (a) M. Vanermen, M. Ligeour, M.-C. Oliveira, J.-F. Gestin, F. Elvas, L. Navarro and F. Guérard, EJNMMI Radiopharmacy Chem., 2024, 9, 69 CrossRef PubMed; (b) T. Dong, Z. Zhang, W. Li, W. Zhuo, T. Cui and Z. Li, J. Org. Chem., 2024, 89, 11837 CrossRef CAS PubMed.
- M. Amaouch, G. Montavon, N. Galland and J. Pilmé, Mol. Phys., 2016, 114, 1326 CrossRef CAS.
- (a) D. S. Wilbur, Curr. Radiopharm., 2008, 1, 144 CrossRef CAS; (b) D. Teze, D.-C. Sergentu, V. Kalichuk, J. Barbet, D. Deniaud, N. Galland, R. Maurice and G. Montavon, Sci. Rep., 2017, 7, 2579 CrossRef PubMed.
- D. S. Wilbur, M.-K. Chyan, D. K. Hamlin, R. L. Vessella, T. J. Wedge and M. F. Hawthorne, Bioconjugate Chem., 2007, 18, 1226 CrossRef CAS PubMed.
- M. Pruszinski, A. Bilewicz, B. Was and B. Petelenz, J. Radioanal. Nucl. Chem., 2006, 268, 91 CrossRef.
- M. Pruszyński, A. Bilewicz and M. R. Zalutsky, Bioconjugate Chem., 2008, 19, 958 CrossRef PubMed.
- H. Rajerison, F. Guérard, M. Mougin-Degraef, M. Bourgeois, I. Da Silva, M. Chérel, J. Barbet, A. Faivre-Chauvet and J.-F. Gestin, Nucl. Med. Biol., 2014, 41, e23 CrossRef CAS PubMed.
- M. Ligeour, S. G. Guillet, C. Maingueneau, M. Croyal, A. Planchat, J.-F. Gestin, N. Galland and F. Guérard, Chem. – Eur. J., 2025, e202500826 CrossRef CAS PubMed.
- (a) M. J. Langton, Angew. Chem., Int. Ed., 2016, 55, 1974 CrossRef CAS PubMed; (b) F. Biedermann, W. M. Nau and H.-J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158 CrossRef CAS PubMed; (c) S. Kubik, Chem. Soc. Rev., 2010, 39, 3648 RSC; (d) Q.-X. Liu, J.-R. Chen, X.-F. Sun, X.-J. Zhao and K.-Q. Cai, RSC Adv., 2016, 6, 12256 RSC; (e) H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924 CrossRef CAS PubMed.
- (a) J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378–2381 CrossRef CAS PubMed; (b) T. Fiala, K. Sleziakova, K. Marsalek, K. Salvadori and V. Sindelar, J. Org. Chem., 2018, 83, 1903 CrossRef CAS PubMed; (c) J. Rivollier, P. Thuéry and M.-P. Heck, Org. Lett., 2013, 15, 480 CrossRef CAS PubMed; (d) D. Azazna, M. Lafosse, J. Rivollier, J. Wang, I. Ben Cheikh, M. Meyer, P. Thuéry, J.-P. Dognon, G. Huber and M.-P. Heck, Chem. – Eur. J., 2018, 24, 10793 CrossRef CAS PubMed; (e) M. Singh, E. Solel, E. Keinan and O. Reany, Chem. – Eur. J., 2016, 22, 8848 CrossRef CAS PubMed; (f) P. Mondal, E. Solel, N. Fridman, E. Keinan and O. Reany, Chem. – Eur. J., 2019, 25, 13336 CrossRef CAS PubMed.
- (a) V. Havel and V. Sindelar, ChemPlusChem, 2015, 80, 1601 CrossRef CAS PubMed; (b) T. Lizal and V. Sindelar, Isr. J. Chem., 2018, 58, 326 CrossRef CAS; (c) M. Chvojka, D. Madea, H. Valkenier and V. Šindelář, Angew. Chem., Int. Ed., 2024, 63, e202318261 CrossRef CAS PubMed; (d) M. Lafosse, Y. Liang, J. P. Schneider, E. Cartier, A. Bodlenner, P. Compain and M.-P. Heck, Molecules, 2022, 27, 4772 CrossRef CAS PubMed.
- M. Lafosse, E. Cartier, K. Solmont, J. Rivollier, D. Azazna, P. Thuéry, Y. Boulard, A. Gontier, J.-B. Charbonnier, B. Kuhnast and M.-P. Heck, Org. Lett., 2020, 22, 3099 CrossRef CAS PubMed.
- Br−@BU 1 was chosen due to its good solubility in CH3CN compared to insoluble anion-free BU 1.
- (a) R. Ghai, R. J. Falconer and B. M. Collins, J. Mol. Recognit., 2012, 25, 32 CrossRef CAS PubMed; (b) T. Wiseman, S. Williston, J.-F. Brandts and L. Lin, Anal. Biochem., 1989, 179, 131 CrossRef CAS PubMed.
- T. Lu, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS PubMed.
- Q. Peña, A. Wang, O. Zaremba, Y. Shi, H. W. Scheeren, J. M. Metselaar, F. Kiessling, R. M. Pallares, S. Wuttke and T. Lammers, Chem. Soc. Rev., 2022, 51, 2544 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: https://doi.org/10.1039/d5cc02762d |
| This journal is © The Royal Society of Chemistry 2025 |