
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanical properties of graphene oxide from machine-learning-driven simulations
Zakariya
El-Machachi
 ,
Bowen
Cheng
and
Volker L.
Deringer
*
,
Bowen
Cheng
and
Volker L.
Deringer
*
Inorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, Oxford OX1 3QR, UK. E-mail: volker.deringer@chem.ox.ac.uk
First published on 16th June 2025
Abstract
Graphene oxide (GO) materials have complex chemical structures that are linked to their macroscopic properties. Here we show that first-principles simulations with a machine-learned interatomic potential can predict the mechanical properties of GO sheets in agreement with experiment and provide atomistic insights into the mechanisms of strain and fracture. Our work marks a step towards understanding and controlling mechanical properties of carbon-based materials with the help of atomistic machine learning.
The term ‘graphene oxide’ (GO) encompasses a class of carbonaceous 2D materials with applications in many fields.1 GO materials show various degrees of chemical functionalisation, introduced by oxidative and reductive processes, and it is important to understand how these structural aspects are connected to macroscopic properties. A key point here is the response of GO sheets to mechanical strain, and indeed it has been argued that the mechanical properties depend strongly on what functional groups (epoxide, hydroxyl, etc.) are present.2 GO and reduced GO (‘rGO’) have been used as sensors in biological applications,3 where small mechanically-induced structural changes from electromechanically linked cells alter the electronic properties.4
In 2007, there began renewed interest in multilayer GO and rGO, primarily focusing on mechanical, electrical, and catalytic properties.5 Atomic force microscopy indentation was used to study the mechanical properties of monolayer rGO.6 Independent, high-resolution TEM images of monolayer GO and rGO revealed the atomic structure,7,8 confirming a two-type domain structure of GO, where oxidised regions form a continuous network with smaller graphitic domains interspersed.9 Since then, various authors have investigated mechanical properties of (r)GO with varying degrees of functionalisation.10–13
In tandem, computational studies have been reported for GO including its mechanical properties,2,12–18 but have typically been limited by the length and time scales accessible to first-principles computations, or by the accuracy limits of empirical potentials.19 Machine learning (ML)-based interatomic potentials approximate the predictions of quantum-mechanical methods whilst adopting key aspects of empirical approaches, such as locality, to achieve first-principles accuracy with near-empirical speed.20 Such ML-driven simulations have been used to model defective,21 nanoporous,19 and amorphous graphene.22
Here, we study the links of atomic structure and mechanical properties in GO and rGO with ML-driven simulations, using the recently introduced GO-MACE-23 model.23 This model was trained using a domain-specific protocol that gradually explores relevant configurations, first through CASTEP + ML,24 then using the MACE architecture.25 We simulate mechanically straining a nano-scale rGO sheet and smaller-scale (r)GO structures across a range of parameters, viz. oxygen content and functionalisation. The predictions from our GO-MACE-23 model agree with experiments and with other computational simulations for GO. To our knowledge, limited work has been conducted on the mechanical properties of reduced GO, and so computations could provide new insight and lead to design rules based on the degree of functionalisation for next-generation (r)GO-based materials.
Our starting structure is taken from ref. 23 and is a partially disordered, fully sp2-bonded graphene sheet with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 368 atoms (17.7 × 15.3 nm2) generated using Monte–Carlo bond switching, following ref. 22. The structure was then functionalised with an O/C ratio of 40% and OH/O ratio of 0.5 decorated randomly, raising the atom count to 16645, then thermally reduced via MD over 1.9 ns at 1500 K, reducing the atom count to 11984 and yielding a simulated XPS spectrum in agreement with experimental spectra.23 To compute mechanical properties, we apply uniaxial tensile strain in the basal plane (Fig. 1a), then a fixed-volume geometry optimisation is then conducted, the uniaxial stress component recorded at that point, and the strain increased again in steps of 0.05%; the process repeats up to a strain of 0.50 (50%). A correction to the recorded stress value, σcell, is required to remove the dependence on the out-of-plane cell parameter (see, e.g., ref. 16): we report σ = (c/t) × σcell, where c is the cell parameter perpendicular to the basal plane and t is the thickness of the sheet (determined as the full width at half maximum for the z coordinates of all atoms in the sheet). We use 3.5 Å as a minimum thickness, since our small-scale monolayer structural models do not reach the experimentally-derived 7 Å for (r)GO,6,10 even at high O content: their effective thickness in our computations is therefore closer to graphene, as in ref. 14. (We note that the concept of a monolayer thickness must rely on an assumption,13 and that previous attempts of measurement for graphene have yielded various results.27)
368 atoms (17.7 × 15.3 nm2) generated using Monte–Carlo bond switching, following ref. 22. The structure was then functionalised with an O/C ratio of 40% and OH/O ratio of 0.5 decorated randomly, raising the atom count to 16645, then thermally reduced via MD over 1.9 ns at 1500 K, reducing the atom count to 11984 and yielding a simulated XPS spectrum in agreement with experimental spectra.23 To compute mechanical properties, we apply uniaxial tensile strain in the basal plane (Fig. 1a), then a fixed-volume geometry optimisation is then conducted, the uniaxial stress component recorded at that point, and the strain increased again in steps of 0.05%; the process repeats up to a strain of 0.50 (50%). A correction to the recorded stress value, σcell, is required to remove the dependence on the out-of-plane cell parameter (see, e.g., ref. 16): we report σ = (c/t) × σcell, where c is the cell parameter perpendicular to the basal plane and t is the thickness of the sheet (determined as the full width at half maximum for the z coordinates of all atoms in the sheet). We use 3.5 Å as a minimum thickness, since our small-scale monolayer structural models do not reach the experimentally-derived 7 Å for (r)GO,6,10 even at high O content: their effective thickness in our computations is therefore closer to graphene, as in ref. 14. (We note that the concept of a monolayer thickness must rely on an assumption,13 and that previous attempts of measurement for graphene have yielded various results.27)
 | ||
| Fig. 1 Mechanical properties of a reduced GO sheet from machine-learning-driven simulations. (a) Visualisation of the rGO sheet taken from ref. 23 now placed under progressive strain (arrows). Atoms and bonds are colour-coded by the non-affine displacement magnitude relative to the initial configuration using OVITO.26 Insets are shown below (C, grey; H, white; O, red). (b) Stress–strain curves for the rGO sheet axially strained in x or y direction. The fitted Young's moduli are Exx = 261 GPa, Eyy = 205 GPa; we include a value for rGO from ref. 6 (shaded area). (c) Change in aldehyde and ketone (purple) vs. ether count (red) relative to the unstrained structure. Solid (dashed) lines indicate strain in x (y) direction, respectively. | ||
We colour-code the rGO nano-structure in Fig. 1a by the non-affine displacement magnitude, which decouples displacement due to external strain and internal, non-affine deformations. This is done by mapping atomic positions onto the reference (initial) frame and computing displacement vectors using OVITO.26 In Fig. 1a, initially there is a uniform blue colour indicating large areas experiencing elastic deformation, with fewer pockets of red regions, which indicate local deformations due to non-affine displacements. These red regions involve plastic deformation in the sheet, arising due to rearrangements, defects, and bond cleavage. As the external strain is increased, there is an increase in local strain around holes and defects, such as O atoms in the carbon backbone. This can be seen in the first and second snapshot in Fig. 1a, where the sheet is still predominantly under elastic deformation—with the exception of the highlighted region which undergoes brittle fracture (dashed lines in the first inset), cleaving a large hole. In the second inset, arrows shows the motion of the 8 atoms (circled) previously involved in 4 C–C bonds which have been broken in a brittle manner. As the strain is increased further, we observe an increasing area of ‘red’ regions, indicating a change from elastic to plastic behaviour. In the third panel, we begin to see a few nanoribbons which join parts of the mostly fractured sheet. With increasing strain, the crack grows and the nanoribbon thins to just one ring thick: such structures, including their stress–strain curves, were recently studied with ML potentials.19 Here, there is a large degree of plastic deformation on the sheet (red regions in Fig. 1a).
The stress–strain curves, recorded separately for both directions, are shown in Fig. 1b. We note that in the ideal large-size limit, both directions would be equivalent. However, as the sheet has been thermally reduced for 2 ns,23 the introduction of holes and defects in the sheet appears to have introduced a (slight) stochastic anisotropy, which is revealed under uniaxial straining. The curves in Fig. 1b qualitatively have the same profile as other stress–strain curves for GO: (i) an initial elastic regime showing a linear response; (ii) a brief plastic region where the curve plateaus; and (iii) a sharp, brittle failure followed by plastic deformation.6,10,13 There is a notable difference between the two curves: one (σxx) indicates higher ultimate tensile strength and a single catastrophic brittle-failure event, where the sheet fractures completely; for the other (σyy), we find that the sheet has lower ultimate tensile strength in this direction and undergoes staggered brittle-fracture events leading to a wider plastic deformation phase. Our estimation of the Young's modulus for the rGO sheet agrees with ref. 6 (Fig. 1b).
Given that our GO-MACE-23-driven simulations agree well with experiments, we are now able to extract and analyse structural information from the simulation trajectory. One of the key structural features in (r)GO is the oxygen functionalisation. In our previous work, we highlighted the sp3-to-sp2 transition at high temperatures resulting in the epoxide and hydroxyl groups transforming into Csp2–O groups, such as ether.23 These groups become part of the carbon backbone in the material, and thus, we show here that these groups play a key role in the response to mechanical strain. In Fig. 1c, we show the evolution of the Csp2–O and Csp2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O count, indicating a change from O being present within the carbon backbone to cleaving off to form a carbonyl group such as a ketone. This rapid decrease of Csp2–O groups coinciding with the brittle failure of the material (under straining in x; solid lines in Fig. 1c) suggests that these groups contribute substantially to the failure mode of (r)GO. In the y direction (dashed lines in Fig. 1c), the stress–strain profile differs slightly and shows an extended period of plastic deformation. Here, we see a more gradual decrease in Csp2–O groups as the latter slowly transform into carbonyl groups.
O count, indicating a change from O being present within the carbon backbone to cleaving off to form a carbonyl group such as a ketone. This rapid decrease of Csp2–O groups coinciding with the brittle failure of the material (under straining in x; solid lines in Fig. 1c) suggests that these groups contribute substantially to the failure mode of (r)GO. In the y direction (dashed lines in Fig. 1c), the stress–strain profile differs slightly and shows an extended period of plastic deformation. Here, we see a more gradual decrease in Csp2–O groups as the latter slowly transform into carbonyl groups.
We then tested whether the properties of GO can be controlled by varying functional groups. Simulations were run for an ensemble of graphene decorated randomly with oxygen groups above and below the basal plane. We varied the O/C ratio from 10% to 50%, and the initial OH/O ratio (cf. ref. 23), in line with experimental reports.1 We observe that generally, with increasing O/C ratio, there is a lower ultimate tensile strength in the material, and the gradient in the elastic region decreases with increasing O content, corresponding to a lower Young's modulus. Another important feature is the type of functional group. We observe that sheets with 100% epoxide and 0% hydroxyl (Fig. 2a) display plastic behaviour, being able to withstand higher strain at higher O content. At equal ratio (Fig. 2b), we find a reduced plastic response, with failure at lower strain. Finally, for fully hydroxyl-decorated sheets (Fig. 2c), the behaviour shifts towards an elastic response and failure at even lower strain. This difference is due to the epoxide groups flattening under strain and incorporating into the sheet, whereas hydroxyl groups cannot be incorporated into the sheet in the same manner, resulting in brittle failure due to the cleavage of C–C bonds.
 | ||
| Fig. 2 Mechanical behaviour of GO and its dependence on the oxygen content. We show stress–strain curves for 20 GO sheets of ∼200 atoms each, at varying O/C ratio (indicated by colour-coding). The shaded areas indicate one standard deviation. We characterise sheets that have been functionalised: (a) fully with epoxide groups; (b) with half epoxide, half hydroxyl groups; and (c) fully with hydroxyl groups. | ||
We note briefly that this dependence of mechanical properties on functionalisation type (Fig. 2) was not observed in earlier simulations using an empirically fitted ReaxFF potential, originally developed for hydrocarbon combustion:2a in the latter case, the weakening with higher O content was independent of the functional group type (hydroxyl or epoxide), and the material was predicted to exclusively show plastic behaviour.2a We speculate that the (more accurate, but more costly) ML potential can uncover additional nuance here which is difficult or impossible to describe with empirically fitted interatomic potentials.
Beyond the overall oxygen content (which can be tuned by gradual thermal reduction), the properties of the sheets could be controlled more finely if one found ways to modify the chemical groups present. We explore this idea in Fig. 3a: we plot averaged Young's moduli for independent structures which start from a pristine graphene backbone and are subsequently decorated with different O/C and OH/O ratios, with 10 strained in x (filled) and 10 strained in y (Fig. 3a). Fig. 3b shows the same, but for structures which have been thermally reduced for 10 ps at 1500 K using GO-MACE-23, resulting in gas evolution. There is a clear relationship between increasing hydroxyl-to-epoxide ratio and Young's modulus, with a more profound effect at higher O content. Literature values reported for GO are 290–430 GPa with O/C ratio from 0.1–0.7;10,12,13,16 for rGO, 250 ± 150 GPa has been reported in ref. 6.
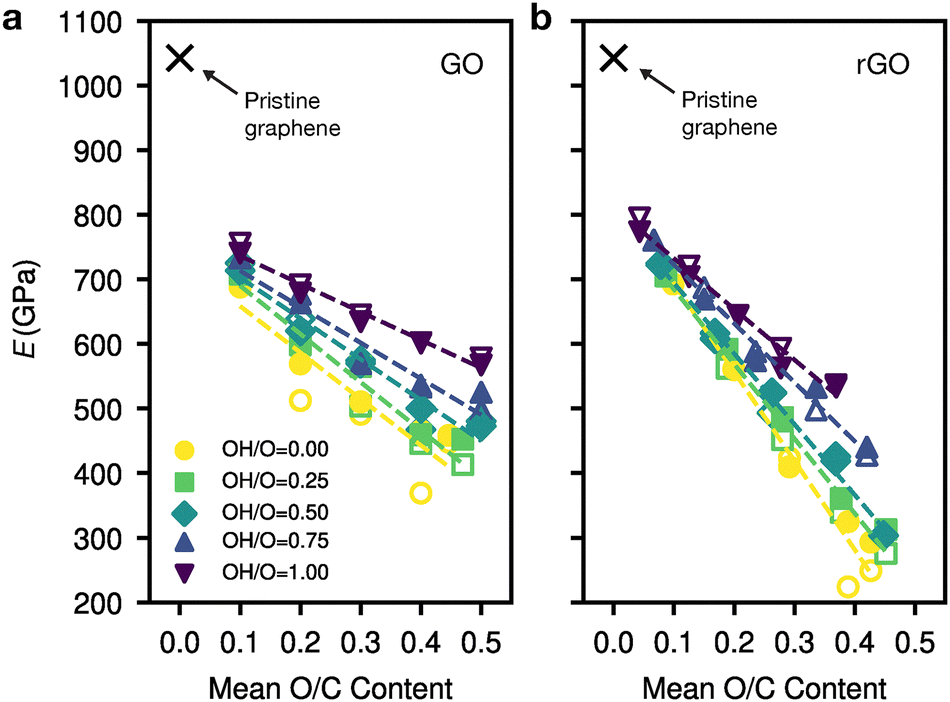 | ||
| Fig. 3 Controlling the Young's modulus, E, of GO by tuning the hydroxyl content. Results are shown for varying initial OH/O ratios. (a) E values for GO: averaged for 10 structures of ∼200 atoms each, strained uniaxially in x (filled markers), and 10 strained uniaxially in y (unfilled), respectively. The cross represents pristine graphene. (b) As for panel (a), except that the structures have been thermally reduced for 10 ps. | ||
Once our GO structures have been heated for a relatively short time, we observe a significant change in the Young's modulus. These heated structures, which we call rGO, have a lower Young's modulus at higher O content compared to that of GO. This is because oxygen leaves the sheet upon heating, mainly as H2O and CO2.23 It is the latter which causes a rearrangement of the graphene backbone, introducing defects into the sheet in the form of point defects or topological defects. There is a drastic shift in the Young's modulus for structures with more epoxide groups once heated. These defects reduce the homogeneity of the sheet, resulting in ‘weak points’ in the sheet, thus lowering the Young's modulus. At the other end, at low initial O/C ratio (0.1) there is minimal disruption to the sheet as gas evolves, and we observe an increase in the Young's modulus compared with GO, due to lower O content as the system is heated.
Looking forward, our results support the idea that the mechanical properties of GO might be amenable to computationally-guided ‘design’2 (see ref. 28 for a general perspective). This goal could be achieved if the chemical structure can be fine-tuned, in this case between hydroxyl (more elastic behaviour) and epoxide groups (more plastic behaviour), or through nitrogen doping which increases the Young's modulus.29 Applications such as wearable electronics30 may benefit from a flexible GO structure with more epoxide groups. Conversely, high OH content may be useful for reinforcing composites: e.g., adding 1 vol% of GO to an Al–Mg alloy enhanced the bending strength substantially.31
We thank F. C. Mocanu for helpful comments and technical advice. This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/L015722/1, EP/V049178/1, EP/W524311/1] and UK Research and Innovation [grant number EP/X016188/1]. We acknowledge the use of the University of Oxford Advanced Research Computing facility (https://dx.doi.org/10.5281/zenodo.22558).
Conflicts of interest
There are no conflicts to declare.Data availability
Data and code supporting this work are openly available at https://github.com/zakmachachi/GO-mechanical-properties.References
- (a) D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC; (b) Graphene Oxide: Fundamentals and Applications, ed. A. M. Dimiev and S. Eigler, John Wiley & Sons, Chichester, 2017 Search PubMed; (c) S. Guo, S. Garaj, A. Bianco and C. Ménard-Moyon, Nat. Rev. Phys., 2022, 4, 247 CrossRef CAS; (d) J. Wu, H. Lin, D. J. Moss, K. P. Loh and B. Jia, Nat. Rev. Chem., 2023, 7, 162 CrossRef PubMed.
- (a) Y. Sun, X. Tang, H. Bao, Z. Yang and F. Ma, RSC Adv., 2020, 10, 29610 RSC; (b) M. Tavakol, A. Montazeri, S. H. Aboutalebi and R. Asgari, Appl. Surf. Sci., 2020, 525, 146554 CrossRef CAS.
- T. Laurila, S. Sainio and M. A. Caro, Prog. Mater. Sci., 2017, 88, 499 CrossRef CAS.
- R. Kempaiah, A. Chung and V. Maheshwari, ACS Nano, 2011, 5, 6025 CrossRef CAS PubMed.
- (a) D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457 CrossRef CAS PubMed; (b) S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- C. Gómez-Navarro, M. Burghard and K. Kern, Nano Lett., 2008, 8, 2045 CrossRef PubMed.
- C. Gómez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144 CrossRef PubMed.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467 CrossRef CAS PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477 CrossRef CAS.
- J. W. Suk, R. D. Piner, J. An and R. S. Ruoff, ACS Nano, 2010, 4, 6557 CrossRef CAS PubMed.
- T. Sakuma, R. Sato, A. Yamaguchi, H. Imai, N. Arai and Y. Oaki, J. Am. Chem. Soc., 2025, 147, 11564 CrossRef CAS PubMed.
- X. Wei, L. Mao, R. A. Soler-Crespo, J. T. Paci, J. Huang, S. T. Nguyen and H. D. Espinosa, Nat. Commun., 2015, 6, 8029 CrossRef CAS PubMed.
- C. Cao, M. Daly, C. V. Singh, Y. Sun and T. Filleter, Carbon, 2015, 81, 497 CrossRef CAS.
- J. T. Paci, T. Belytschko and G. C. Schatz, J. Phys. Chem. C, 2007, 111, 18099 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581 CrossRef CAS.
- L. Liu, J. Zhang, J. Zhao and F. Liu, Nanoscale, 2012, 4, 5910 RSC.
- B. D. Jensen, K. E. Wise and G. M. Odegard, J. Phys. Chem. A, 2015, 119, 9710 CrossRef CAS PubMed.
- C. D. Williams, P. Carbone and F. R. Siperstein, Nanoscale, 2018, 10, 1946 RSC.
- A. Kabylda, B. Mortazavi, X. Zhuang and A. Tkatchenko, Adv. Funct. Mater., 2024, 35, 2417891 CrossRef.
- (a) J. Behler, Angew. Chem., Int. Ed., 2017, 56, 12828 CrossRef CAS PubMed; (b) V. L. Deringer, M. A. Caro and G. Csányi, Adv. Mater., 2019, 31, 1902765 CrossRef CAS PubMed; (c) P. Friederich, F. Häse, J. Proppe and A. Aspuru-Guzik, Nat. Mater., 2021, 20, 750 CrossRef CAS PubMed.
- F. L. Thiemann, P. Rowe, A. Zen, E. A. Müller and A. Michaelides, Nano Lett., 2021, 21, 8143 CrossRef CAS PubMed.
- Z. El-Machachi, M. Wilson and V. L. Deringer, Chem. Sci., 2022, 13, 13720 RSC.
- Z. El-Machachi, D. Frantzov, A. Nijamudheen, T. Zarrouk, M. A. Caro and V. L. Deringer, Angew. Chem., Int. Ed., 2024, 63, e202410088 CrossRef CAS PubMed.
- T. K. Stenczel, Z. El-Machachi, G. Liepuoniute, J. D. Morrow, A. P. Bartók, M. I. J. Probert, G. Csányi and V. L. Deringer, J. Chem. Phys., 2023, 159, 044803 CrossRef CAS PubMed.
- (a) I. Batatia, D. P. Kovács, G. N. C. Simm, C. Ortner and G. Csányi, Adv. Neural Inf. Process Syst., 2022, 11423–11436 Search PubMed; (b) I. Batatia, S. Batzner, D. P. Kovács, A. Musaelian, G. N. C. Simm, R. Drautz, C. Ortner, B. Kozinsky and G. Csányi, Nat. Mach. Intell., 2025, 7, 56 CrossRef PubMed.
- A. Stukowski, Model. Simul. Mater. Sci. Eng., 2009, 18, 015012 CrossRef.
- C. J. Shearer, A. D. Slattery, A. J. Stapleton, J. G. Shapter and C. T. Gibson, Nanotechnology, 2016, 27, 125704 CrossRef PubMed.
- Y. Liu, A. Madanchi, A. S. Anker, L. Simine and V. L. Deringer, Nat. Rev. Mater., 2025, 10, 228 CrossRef.
- K. Tschoppe, F. Beckert, M. Beckert and R. Mülhaupt, Macromol. Mater. Eng., 2015, 300, 140 CrossRef CAS.
- H. Jang, Y. J. Park, X. Chen, T. Das, M.-S. Kim and J.-H. Ahn, Adv. Mater., 2016, 28, 4184 CrossRef CAS PubMed.
- H. Kwon, J. Mondal, K. A. AlOgab, V. Sammelselg, M. Takamichi, A. Kawaski and M. Leparoux, J. Alloys Compd., 2017, 698, 807 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |