
Decarboxylative sulfinamidation of N-sulfinylamines with carboxylic acids via a photochemical iron-mediated ligand-to-metal charge transfer process†
Dan
Yang
 a,
Qiang
Xiao
*b,
Yan
Zhou
c,
Hui
Zhang
a,
Tingting
Chen
c and
Mingsong
Shi
*c
a,
Qiang
Xiao
*b,
Yan
Zhou
c,
Hui
Zhang
a,
Tingting
Chen
c and
Mingsong
Shi
*c
aPrecise Synthesis and Function Development Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637002, China
bInstitute of Ecology, China West Normal University, Nanchong 637002, China. E-mail: xiaoqiang1617@hotmail.com
cNHC Key Laboratory of Nuclear Technology Medical Transformation, Mianyang Central Hospital, School of Medicine, University of Electronic Science and Technology of China, Mianyang 621099, China. E-mail: shims90@sc-mch.cn
First published on 4th July 2025
Abstract
Sulfinamides are valuable four-valent sulfur compounds in medicinal chemistry, yet methods relying on simple and readily available feedstocks are scarce. Herein, a novel and efficient strategy was developed to facilitate the direct decarboxylative sulfinamidation of carboxylic acids via a photoinduced iron-mediated ligand-to-metal charge-transfer process. Notably, this protocol exemplifies an environmentally benign approach to the synthesis of a range of structurally diverse sulfinamides with an excellent functional group compatibility, thereby rendering it suitable for the late-stage modification of bioactive molecules.
The sulfinamide motif is widely recognized as one of the most important motifs in organic synthesis.1 The four-valent sulfinyl group has been demonstrated to facilitate the unification of a comprehensive organosulfur chemical library containing all of the predominant sulfur functional groups, including sulfonamides,2 sulfoximines,3 sulfonimidamide esters,4 and sulfonimidoyl halides.5 Moreover, the prevalence of this group is widely found in pharmaceutical compounds (and related compounds) that have been previously approved by the United States Food and Drug Association.6 In addition, enantiopure sulfonamides and their derivatives have been extensively used as chiral auxiliaries,7 ligands,8 and organocatalysts9 in asymmetric synthesis. Given their pivotal functions in organic synthesis, a considerable corpus of creative strategies has been dedicated to their preparation. Conventional access to sulfonamides primarily involves the two-electron nucleophilic substitution of amines with unstable sulfinyl chlorides, which in turn are derived from sulfinic acids and sulfinate salts in the presence of oxalyl chloride or thionyl chloride.10 Recent studies have demonstrated that the addition of organometallic reagents to N-sulfinylamines represents an efficient manner for the synthesis of sulfonamides and related organosulfur derivatives (Scheme 1a).11 Notwithstanding the ingenious methodologies that have been developed, the application of one-electron routes to sulfinamides remains in its infancy.
 | ||
| Scheme 1 Previous strategies for the synthesis of sulfinamides and our design. | ||
Photocatalysis has recently emerged as a reliable tool for the chemical community, facilitating the generation of carbon-centered intermediates, wherein coupling with N-sulfinylamines has been employed to prepare sulfinamides via one-electron processes under mild conditions. In the course of the synthesis of alkyl sulfonamides, 1,4-Dihydropyridines (DHPs)12 or potassium trifluoro(organo)borates13 (as nucleophilic alkyl radical precursors) were employed in combination with N-sulfinylamines (as electrophiles) in the presence of organic photosensitizers (Scheme 1b). Subsequently, Willis14 and Larionov15 independently described the use of decarboxylative sulfinamidation reactions to access sulfinamides using readily available carboxylic acids as alkyl radical sources (Scheme 1c). More recently, Hu et al. reported the direct C(sp3)–H bond sulfinamidation of simple hydrocarbons and N-sulfinylamine via a photochemical iron-mediated ligand-to-metal charge-transfer process (Scheme 1d).16 Despite these achievements, the development of green and efficient synthesis strategies still holds great opportunities.
Carboxylic acids are structurally diverse compounds from nature, organisms and synthesis.17 Owing to their abundant, stable, and non-toxic properties, along with their widespread availability, carboxylic acids are regarded as optimal structural units for the assembly of intricate molecular frameworks through a one-electron process.18 Nevertheless, the direct conversion of carboxylic acids to other functional groups remains challenging due to the high oxidation potentials and reactivities of carboxylic acids, which prevent the decarboxylative radical generation protocol that is common in catalytic reactions. Iron-mediated ligand-to-metal charge transfer (LMCT), an electronic transition from the filled orbital of a ligand to the vacant orbit of a metal center, has recently emerged as a powerful strategy for the efficient generation of alkyl radicals from carboxylic acids. Notably, this approach has enabled the direct conversion of carboxylic acids into a variety of functionalities via an open–shell mechanism.19 In the context of our sustained research interest in the decarboxylative functionalization of carboxylic acids via one-electron processes,20 the work presented herein describes our most recent findings concerning the direct decarboxylative transformation of aliphatic carboxylic acids to generate sulfonamides via an iron-mediated LMCT process (Scheme 1e).
Initially, sulfinylamine 1a and pivalic acid 2a were employed as model substrates for the preparation of sulfinamide 3a (see ESI,† Tables S1–S5). To our delight, 3a was obtained in a 22% isolated yield in the presence of 10 mol% FeCl3 as the photocatalyst, along with K2CO3 as the base in acetonitrile (MeCN), under irradiation with a 40 W violet light-emitting diode (LED, 365–375 nm) at room temperature (Table S1, entry 1, ESI†). Subsequently, a range of iron salts were tested, and it was established that FeBr3 exhibited a considerable catalytic activity. Conversely, Fe2(SO4)3, Fe(OTf)3, and FeSO4 proved ineffective within the parameters of this protocol (Table S1, entries 2–7, ESI†). Switching the reaction solvent to CH2Cl2, tetrahydrofuran (THF), dimethylformamide (DMF), or dimethyl sulfoxide (DMSO) led to reduced product yields (Table S2, ESI†). Different bases were also investigated, including Na2CO3, Na3PO4, Et3N, and pyridine, with an enhanced yield of 69% being achieved using Et3N as the base (Table S3, entries 1–8, ESI†). Notably, the yield was further increased to 81% when the Et3N dosage was reduced to 0.5 equivalents (Table S3, entry 9, ESI†). In addition, light sources with different wavelengths were examined to replace the violet LED, with blue and white LEDs yielding the corresponding product 3a in yields of 18 and 74%, respectively (Table S4, ESI†). Additionally, control experiments revealed that FeCl3, Et3N, light irradiation, and an argon atmosphere were essential for the successful execution of this transformation (Table S5, ESI†).
With the optimized conditions in hand, various carboxylic acids 2 were treated with sulfinylamine 1a under the standard conditions to yield the corresponding sulfinamides 3, as summarized in Scheme 2. A survey of various tertiary carboxylic acids revealed the most suitable reaction coupling partners for this transformation, yielding the corresponding sulfinamides in 62–94% yields (3b–3e). This decarboxylative sulfinamidation reaction also proceeded well using an assortment of acyclic and cyclic secondary carboxylic acids, affording products 3f–3j in good yields (56–81%). Primary carboxylic acids were also suitable for application in this catalytic system, with the desired sulfinamides 3k–3q being produced in satisfactory yields. These results clearly demonstrate the potential application of the developed method for the subsequent modification of bioactive molecules. More specifically, ibuprofen and gemfibrozil were successfully converted into sulfonamides 3r and 3s in an equally straightforward manner under the optimized conditions.
 | ||
| Scheme 2 Substrate scope of carboxylic acids 2. Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), FeCl3 (10 mol%), Et3N (0.5 equiv.), MeCN (2 mL), 40 W violet LED (365–375 nm), Ar, rt, 24 h. Isolated yields. | ||
The scope of the N-sulfinylamine component was subsequently investigated using 1-adamantanecarboxylic acid 2d (Scheme 3). It was found that the electronic nature of the phenyl substituent had a negligible effect on reaction yield. However, the position of the substituent had a relatively significant impact on the yield of this transformation, wherein the substrates bearing an ortho-substituent gave slightly higher yields than those bearing para- and meta-substituents (3t–3aa). Of particular importance was the tolerance of the reaction toward chloride and bromide groups, which enabled modification of the halogenated position. Additionally, N-alkylsulfinylamines were efficiently converted to the corresponding sulfonamides 3ba and 3ca in yields of 51 and 41%, respectively. However, heteroary sulfinylamines 1da and 1ea proved incompatible with this catalytic system.
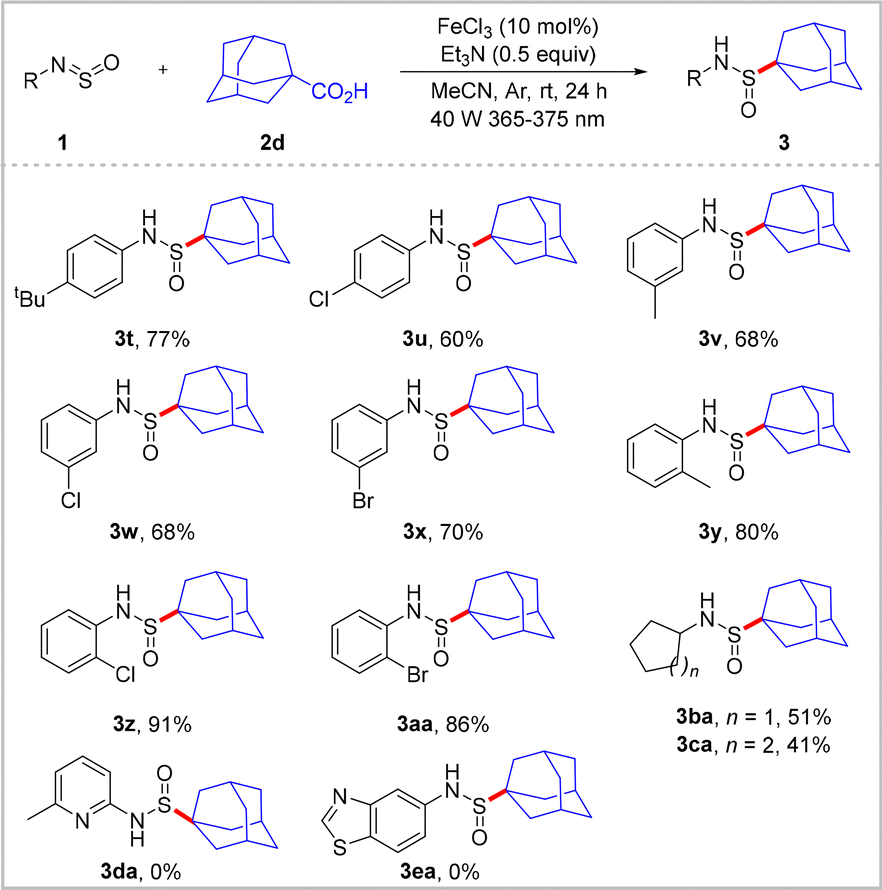 | ||
| Scheme 3 Substrate scope of N-sulfinylamine 1. 1 (0.2 mmol), 2d (0.3 mmol), FeCl3 (10 mol%), Et3N (0.5 equiv), MeCN (2 mL), 40 W violet LED (365–375 nm), Ar, rt, 24 h. Isolated yields. | ||
To demonstrate the practicality of the developed method, a 5 mmol scale-up experiment was carried out under standard conditions, giving target compound 3a in 73% yield (Scheme 4a). Subsequently, the obtained sulfinamides were easily converted into valuable S(VI) congeners. More specifically, sulfonimidate ester 4 was conveniently constructed in 78% yield via an iodosylbenzene-mediated oxidation/nucleophilic addition cascade using methanol as the nucleophile (Scheme 4b, right).21 In addition, sulfinamidation product 3i was efficiently oxidized to sulfonamide 5 in 52% yield using meta-chloroperbenzoic acid (mCPBA) as the oxidant (Scheme 4b, left). A control experiment was also performed to elucidate the reaction mechanism. More specifically, when 4 equiv. of the radical scavenger 2,2,6,6-tetramethylpiperidine-1-oxy (TEMPO) was added to the reaction system under standard conditions, the reaction was completely inhibited, and the corresponding TEMPO-adduct 6 was detected by high-resolution mass spectrometry (HRMS) (Scheme 4c). A radical clock experiment involving 1a with 2t yielded in a mixture of non-ring-opened product 3t and the ring-opened product 3t’ in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 ratio (Scheme 4d). Both the radical trapping experiment and the radical clock experiment confirmed that the reaction proceeds via a decarboxylation-induced alkyl radical pathway. Cyclic sulfinamide 7 was observed when 1a reacted with 4-bromobutyric acid 2u, supporting the generation of a sulfinamide anion intermediate in this reaction (Scheme 4e).
:1.5 ratio (Scheme 4d). Both the radical trapping experiment and the radical clock experiment confirmed that the reaction proceeds via a decarboxylation-induced alkyl radical pathway. Cyclic sulfinamide 7 was observed when 1a reacted with 4-bromobutyric acid 2u, supporting the generation of a sulfinamide anion intermediate in this reaction (Scheme 4e).
 | ||
| Scheme 4 Synthetic applications and control experiment. | ||
Based on the control experiments and previous reports,22 a possible mechanism for this transformation was proposed, as shown in Scheme 5. Initially, the in situ-generated carboxylate anions coordinate with Fe(III) cations to form intermediate A. This intermediate undergoes intramolecular carboxylate-to-Fe(III) charge transfer process to produce the Fe(II) species and the corresponding carboxylate radical. The reaction is initiated by the release of CO2 from the carboxylate radical, which subsequently generates a key alkyl radical. Then addition of alkyl radical to Fe(III) coordinated N-arylsulfinylamine 1 to form aminosulfinyl radical B, where O-binding is proposed in analogy to the prior report with MgX2.23 Density functional theory (DFT) computations have also elucidated that FeCl3 acts as a Lewis acid in facilitating nucleophilic attack processes (see ESI,† Fig. S1). Subsequently, the single-electron reduction of aminosulfinyl radical B by leads to the generation of the corresponding Fe(III) species and anion C. Finally, the protonation of anion C provides the desired sulfonamide product 3.
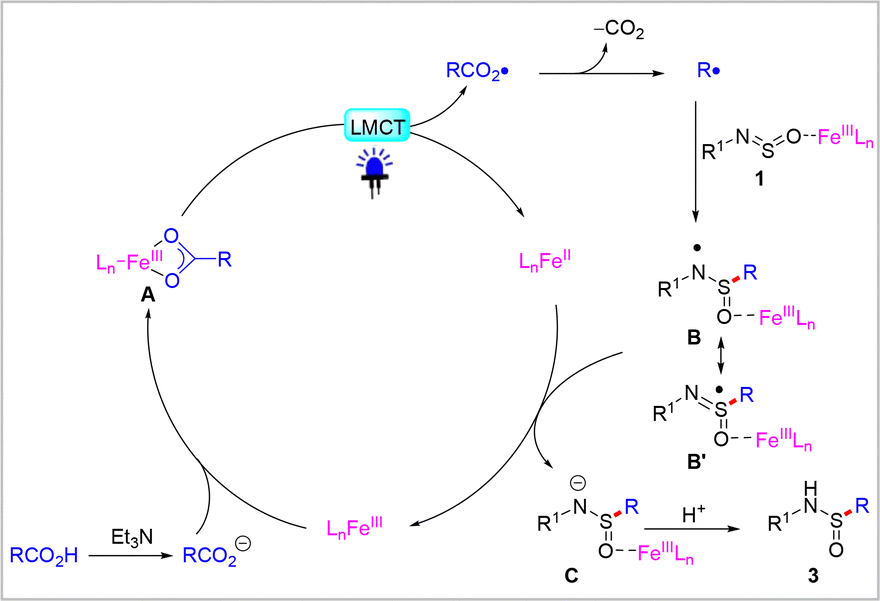 | ||
| Scheme 5 Proposed mechanism. | ||
In conclusion, a novel and direct protocol for the decarboxylative sulfinamidation of carboxylic acids was developed via a photoinduced iron-mediated LMCT process. This approach features mild conditions and is a straightforward procedure for the preparation of a range of structurally diverse sulfonamides, demonstrating an excellent functional group compatibility. Furthermore, the obtained sulfonamides were smoothly transformed into valuable sulfur(VI) derivatives including sulfonamides and sulfonimidate esters. It is expected that the versatility of this method will result in its widespread application in the preparation of sulfur-containing derivatives.
We thank the National Natural Science Foundation of China (No. 22203056, 42171045), the Mianyang Science and Technology Bureau (Mianyang Science and Technology Program) (No. 2023ZYDF073), the NHC Key Laboratory of Nuclear Technology Medical Transformation (Mianyang Central Hospital) (No. 2024HYX014) and the Opening Project of Key Laboratory of Green Catalysis of Higher Education Institutes of Sichuan (No. LZJ2402) for financial support.
Conflicts of interest
There are no conflicts to declare.Data availability
All experimental procedures, characterization data and NMR spectra for all new compounds can be found in the ESI.†Notes and references
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS.
- H. X. Dai, A. F. Stepan, M. S. Plummer, Y. H. Zhang and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef CAS.
- Y. Aota, T. Kano and K. Maruoka, J. Am. Chem. Soc., 2019, 141, 19263–19268 CrossRef CAS.
- P. M. Matos and R. A. Stockman, Org. Biomol. Chem., 2020, 18, 6429–6442 RSC.
- S. Greed, E. L. Briggs, F. I. M. Idiris, A. J. P. White, U. Lücking and J. A. Bull, Chem. – Eur. J., 2020, 26, 12533–12538 CrossRef CAS.
- S. A. Shah, G. Rivera and M. Ashfaq, Mini-Rev. Med. Chem., 2013, 13, 70–86 CrossRef CAS.
- J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS.
- (a) W. Li and J. Zhang, Acc. Chem. Res., 2024, 57, 489–513 CAS; (b) X. Yu, S. Liu, L. Wang, Y. Li, M. Huang, Z. Y. Yan, H. C. Li, Q. Zhang, X. Zhang and B. F. Shi, ACS Catal., 2025, 15, 2540–2549 CrossRef CAS.
- E. Wojaczyńska and J. Wojaczyńska, Chem. Rev., 2020, 120, 4578–4611 CrossRef.
- J. A. Vogel, R. Hammami, A. Ko, H. Datta, Y. N. Eiben, K. J. Labenne, E. C. McCarver, E. Z. Yilmaz and P. R. Melvin, Org. Lett., 2022, 24, 5962–5966 CrossRef CAS.
- (a) T. Q. Davies, A. Hall and M. C. Willis, Angew. Chem., Int. Ed., 2017, 56, 14937–14941 CrossRef CAS; (b) L. Bayeh, P. Q. Le and U. K. Tambar, Nature, 2017, 547, 196–200 CrossRef CAS; (c) M. Bremerich, C. M. Conrads, T. Langletz and C. Bolm, Angew. Chem., Int. Ed., 2019, 58, 19014–19020 CrossRef CAS; (d) Z. X. Zhang, T. Q. Davies and M. C. Willis, J. Am. Chem. Soc., 2019, 141, 13022–13027 CrossRef CAS; (e) T. Q. Davies, M. J. Tilby, D. Skolc, A. Hall and M. C. Willis, Org. Lett., 2020, 22, 9495–9499 CrossRef CAS; (f) T. Q. Davies, M. J. Tilby, J. Ren, N. A. Parker, D. Skolc, A. Hall, F. Duarte and M. C. Willis, J. Am. Chem. Soc., 2020, 142, 15445–15453 CrossRef CAS; (g) T. Q. Davies and M. C. Willis, Chem. – Eur. J., 2021, 27, 8918–8927 CrossRef CAS; (h) S. Tummanapalli, K. C. Gulipalli, S. Bodige, D. Vemula, S. Endoori, A. K. Pommidi and S. K. Punna, Tetrahedron Lett., 2021, 73, 153118 CrossRef CAS; (i) P. K. T. Lo and M. C. Willis, J. Am. Chem. Soc., 2021, 143, 15576–15581 CrossRef CAS; (j) M. Ding, Z. X. Zhang, T. Q. Davies and M. C. Willis, Org. Lett., 2022, 24, 1711–1715 CrossRef CAS; (k) L. Xi, X. Fang, M. Wang and Z. Shi, J. Am. Chem. Soc., 2024, 146, 17587–17594 CrossRef CAS; (l) M. K. Wei, D. F. Moseley, R. M. Bär, Y. Sempere and M. C. Willis, J. Am. Chem. Soc., 2024, 146, 19690–19695 CrossRef CAS; (m) Y. Yuan, X. Tian, H. Zheng, Y. Li, J. Zhang and J. Yang, Angew. Chem., Int. Ed., 2025, e202506243 CAS; (n) M. Malik, R. Senatore, D. Castiglione, A. Roller-Prado and V. Pace, Chem. Commun., 2023, 59, 11065–11608 RSC.
- L. Li, S. q Zhang, Y. Chen, X. Cui, G. Zhao, Z. Tang and G. x Li, ACS Catal., 2022, 12, 15334–15340 CrossRef CAS.
- (a) M. Yan, S. f Wang, Y. p Zhang, J. z Zhao, Z. Tang and G. x Li, Org. Biomol. Chem., 2024, 22, 348–352 RSC; (b) S. Das, P. P. Mondal, A. Dhibar, A. Ruth and B. Sahoo, Org. Lett., 2024, 26, 3679–3684 CrossRef CAS.
- J. A. Andrews, J. Kalepu, C. F. Palmer, D. L. Poole, K. E. Christensen and M. C. Willis, J. Am. Chem. Soc., 2023, 145, 21623–21629 CrossRef CAS.
- H. T. Dang, A. Porey, S. Nand, R. Trevino, P. Manning-Lorino, W. B. Hughes, S. O. Fremin, W. T. Thompson, S. K. Dhakal, H. D. Arman and O. V. Larionov, Chem. Sci., 2023, 14, 13384–13391 RSC.
- G. D. Xia, R. Li, L. Zhang, Y. Wei and X. Q. Hu, Org. Lett., 2024, 26, 3703–3708 CrossRef CAS.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS.
- C. Q. Deng and J. Deng, Green Chem., 2025, 27, 275–292 RSC.
- (a) J. Stahl and B. König, Green Chem., 2024, 26, 3058–3071 RSC; (b) J. Qin, H. Lei, C. Gao, Y. Zheng, Y. Zhao and W. Xia, Org. Biomol. Chem., 2024, 22, 6034–6044 RSC; (c) X. Y. Yuan, C. C. Wang and B. Yu, Chin. Chem. Lett., 2024, 35, 109517 CrossRef CAS.
- D. Yang, Y. T. Mei, Z. Y. Guo, Q. Y. Hou, H. Zhang, Y. X. Zheng, L. H. Jing, D. J. Cheng and M. S. Shi, J. Org. Chem., 2025, 90, 3665–3672 CrossRef CAS.
- P. M. Matos, W. Lewis, J. Moore and R. A. Stockman, Org. Lett., 2018, 20, 3674–3677 CrossRef CAS.
- (a) Y. Zhang, J. Qian, M. Wang, Y. Huang and P. Hu, Org. Lett., 2022, 24, 5972–5976 CrossRef CAS; (b) X. K. Qi, L. J. Yao, M. J. Zheng, L. Zhao, C. Yang, L. Guo and W. Xia, ACS Catal., 2024, 14, 1300–1310 CrossRef CAS; (c) H. Gao, L. Guo, Y. Zhu, C. Yang and W. Xia, Chem. Commun., 2023, 59, 2771–2774 RSC; (d) Q. Huang, C. Lou, L. Lv and Z. Li, Chem. Commun., 2024, 60, 12389–12392 RSC; (e) Y. Yang, X. Huang and Y. Jin, Chem. Commun., 2025, 61, 1944–1961 RSC.
- H. Gilman and H. L. Morris, J. Am. Chem. Soc., 1926, 48, 2399–2404 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02696b |
| This journal is © The Royal Society of Chemistry 2025 |