
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical S-vinylation of sulfinamides with β-bromostyrenes†
Yaseen
Hussain
a,
Ivan
Sliusarevskyi
a,
Claire
Empel
ac,
Magnus
Rueping
 *b and
Rene M.
Koenigs
*ac
*b and
Rene M.
Koenigs
*ac
aInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, D-52074 Aachen, Germany
bKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
cDepartment of Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: rene.koenigs@uni-bayreuth.de
First published on 6th June 2025
Abstract
We present an electrochemical method for sulfoximine synthesis via radical cross-coupling of sulfonimidoyl and styryl radicals, generated from sulfinamides and bromostyrenes. This approach enables the efficient synthesis of vinyl sulfoximines, including bioactive-tethered derivatives, in moderate to good yields.
Sulfoximine-containing molecules have gained significant attention in medicinal chemistry due to their unique structural features, including a stereogenic center at the sulfur atom and a small, hydrophilic core.1 These compounds exhibit diverse biological properties; for example as a proline-rich tyrosine kinase inhibitor (A),2 cyclin-dependent kinases (CDK) inhibitors (B),3 or CYP24 hydroxylase inhibitor (C).4 Beyond their pharmaceutical applications, sulfoximines have also been employed as pesticides in crop protection.5 Given their widespread importance in drug discovery and agrochemicals, the development of efficient and sustainable synthetic methodologies to access sulfoximines remains a crucial research goal. Several strategies have been explored for sulfoximine synthesis. One of the most common approaches involves the amination of sulfoxides using aminating reagents.6–9 However, such approach is often limited by the need for stoichiometric oxidants and low substrate tolerance, restricting its broader applicability.7 More recently, radical methodologies have emerged as promising alternatives for sulfoximine synthesis.10–13 Bolm11 and Gau12 reported methods using sulfonimidoyl radicals, generated from sulfonimidoyl chloride or fluoride.
Electrochemical synthesis—the use of electric current to mediate redox transformations in organic synthesis—has witnessed a renaissance in recent years and is developing as an inherently green and sustainable method to access radical intermediates under precise reaction conditions.13,14 In this context, Ling and co-workers developed an electrochemical approach that generates sulfonimidoyl radicals in situ via paired electrolysis, enabling vinyl sulfoximine synthesis.15 Against the background of recent developments in sulfoximine synthesis and our interest in this research area,16 we herein present a modified electrochemical approach that circumvents the need for DBU and triethylamine salts. Our protocol employs carbonate salts as the sole additive, enabling the efficient synthesis of vinyl sulfoximines under ambient conditions (Scheme 1).
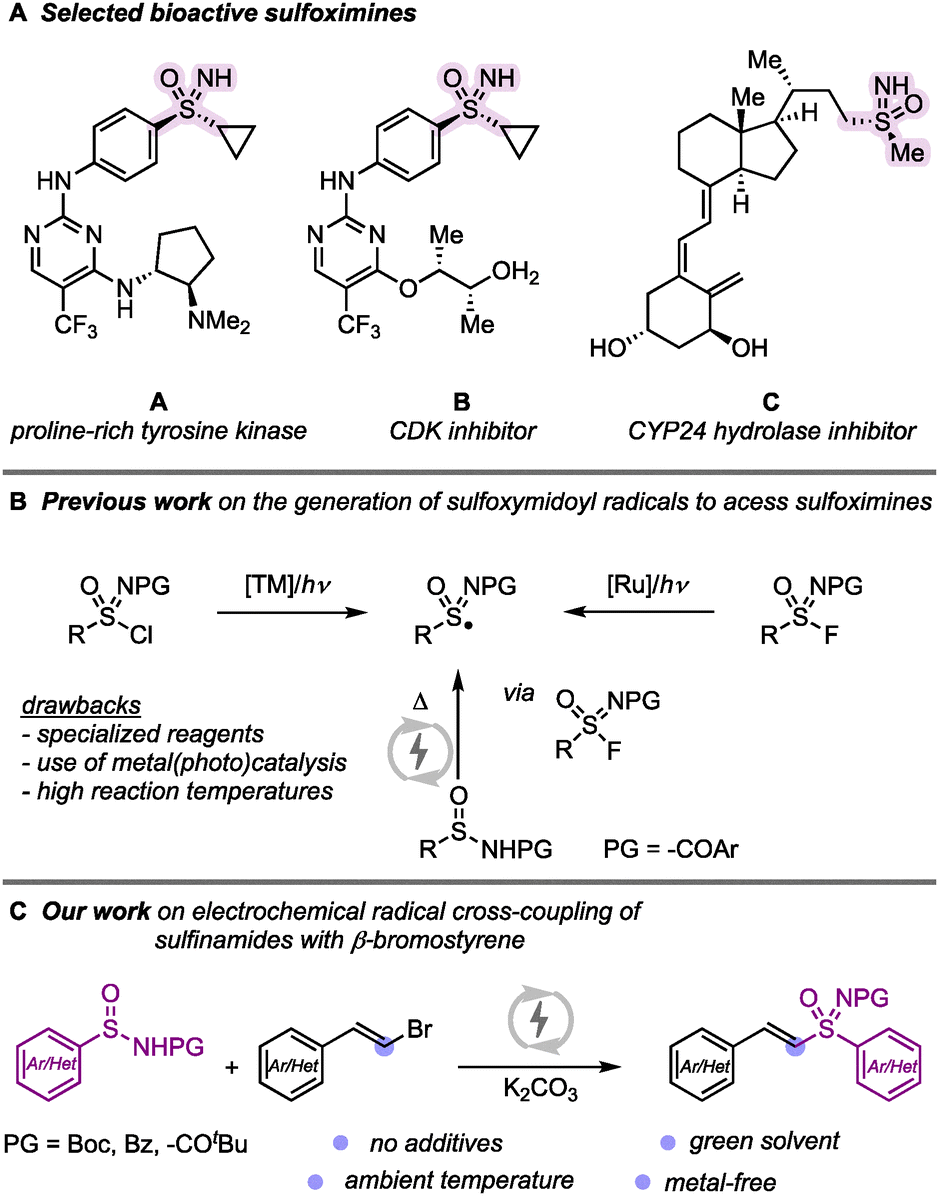 | ||
| Scheme 1 Biologically relevance sulfoximines bearing molecules and strategies for the generation of sulfonimidoyl radicals. | ||
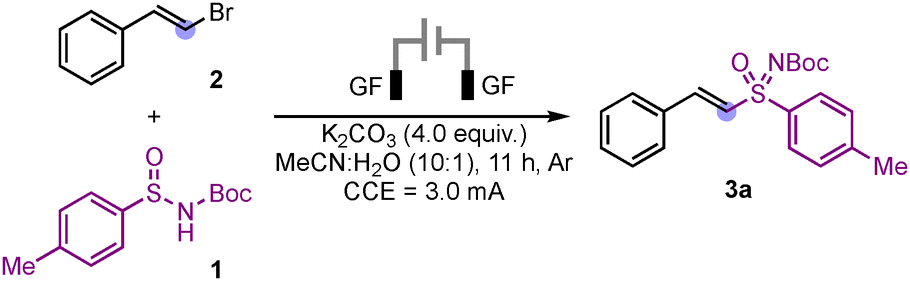
Initially, we investigated the reaction of sulfinamide 1a with β-bromostyrene (2a) under electrochemical conditions, using a graphite electrode and K2CO3 as the base. To our delight, the desired product 3a was formed in 41% yield (Table 1, entry 1). With this positive result we then proceeded to screen other solvents, yet both THF or methanol gave inferior results. On increasing the ratio of water, a sharp decrease in the yield of 3a was observed (Table 1, entry 4). We next went for evaluation of organic and inorganic bases; using pyridine and KOH as base the product 3a was formed only in traces while Na2CO3 or K3PO4 gave 3a in 39% and 33%, respectively (Table 1, entries 5–8). When the reaction was performed in presence of electrolytes in acetonitrile, the product 3a was not detected (Table 1, entry 9); similarly, a significant decrease in the yield of 3a was observed under aerobic conditions (Table 1, entry 10). Changing the cathode to Ni plate resulted in a significant increase in the yield of the desired product 3a, which could be isolated in 77% yield (Table 1, entry 11). The reaction failed to deliver the product in absence of electric current indicating the necessity of electricity. Further, on increasing or decreasing the reaction current does not provide a better than at 3 mA (Table 1, entry 12).
|
|
||
|---|---|---|
| Entry | Variation from condition | Yield [%] of 3a |
a Reaction conditions: 1a (0.1 mmol), 2a (3.0 equiv.) and K2CO3 in MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (10:1, 2.0 mL) under argon. 1H NMR yields were calculated using mesitylene as internal standard.
b Isolated yield.
c Nickel as cathode. :H2O (10:1, 2.0 mL) under argon. 1H NMR yields were calculated using mesitylene as internal standard.
b Isolated yield.
c Nickel as cathode.
|
||
| 1 | None | 41 |
| 2 | THF:H2O |
39 |
| 3 | MeOH:H2O |
Traces |
| 4 | MeCN:H2O (1:5) |
13 |
| 5 | Pyridine instead of K2CO3 | Traces |
| 6 | KOH instead of K2CO3 | Traces |
| 7 | Na2CO3 instead of K2CO3 | 39 |
| 8 | K3PO4 instead of K2CO3 | 33 |
| 9 | TBAI, TBClO4, TBABF4 in MeCN | — |
| 10 | Under air | 14 |
| 11 | Ni as cathode | 84 (77)b |
| 12 | No current | NR |
| 13c | At 5 mA/1 mA current | 75/63 |
We first investigated the substrate scope (Scheme 2) by varying the sulfinamide (1). Both pivolyl and benzoyl protecting groups were well tolerated in this transformation; however, the benzoyl-protected sulfinamide yielded product 3c in only 22%, whereas the Boc- and pivolyl-protected variants afforded products 3a and 3b in significantly higher yields of 77% and 67%, respectively.
 | ||
| Scheme 2 Substrates scope; 1 (0.2 mmol), 2 (3.0 equiv.) and K2CO3 (4.0 equiv.) in MeCN:H2O (10:1, 2.0 mL) were electrolyzed at 3 mA CCE using GF (anode) and Ni (cathode) under argon. FE = faradaic efficiency. | ||
Next, we explored the effect of the aryl ring substitutions on the sulfinamide. A wide range of substituents, including electron-donating, electron-withdrawing groups, and halogens, were well-tolerated. Phenylsulfinamide yielded product 3d in 70%. Halogenated sulfinamides produced 3e and 3f gave slightly lower yields, whereas the methoxy-substituted sulfinamide gave product 3g in 56%. Notably, mono-CF3-substituted arylsulfinamide provided product 3h in only 22% yield, while 3,5-CF3 and CN-substituted sulfinamides failed to generate the corresponding sulfoximines (see ESI†). The presence of two CF3 groups (see, ESI†) at the meta positions of the sulfinamide failed to provide the desired product, possibly due to their strong electron-withdrawing nature. Similarly, substitutions at the ortho position e.g. Cl or Me, (see ESI†) did not yield the corresponding sulfoximines, which may be attributed to steric hindrance. The thiophene-derived sulfinamide afforded product 3i in a good yield of 75%, whereas the naphthalene analogue provided 3j in 43%. We then assessed the influence of the substituents on the aryl ring of bromo styrene 2. The reaction tolerated diverse substitution patterns, including electron-donating, electron-withdrawing groups, and halogens, affording the respective products in moderate to good yields. Electron-donating groups positively influenced the yields (3k, 3l), while halogenated substrates led to moderate yields (3m, 46%; 3n, 55%). Strongly electron-withdrawing substituents such as CN and CO2Me gave 3o and 3p in lower yields (30% and 32%, respectively), whereas CF3-substituted styrene gave 3q in an excellent 73% yield. meta- and ortho-substituted bromostyrenes reacted efficiently, delivering products 3r (50%) and 3s (56%). The tri-methoxy-substituted bromostyrene provided 3t in 75% yield. Additionally, heterocyclic bromostyrenes were well tolerated; pyridine- and furan-containing derivatives gave products 3w and 3v in lower yields, while the thiophene analogue afforded 3u in 73%. Finally, bioactive tethered bromostyrene derivatives were also compatible, yielding products 3x–3z in moderate yields. The observed faradaic efficiencies (6.9–30.9%) are relatively low, which may be due to competitive hydrogen evolution resulting from water present in the reaction medium.
To demonstrate the applicability of the developed electrochemical protocol, a scale up reaction at 2 mmol scale were performed under the standard reaction condition to provide the desired product in 72% yield (Scheme 3A). Additionally, Michael addition reaction of 3a with thiol and pyrrolidine have been performed and the products 4 and 5 were isolated in 83% and 57% yield, respectively (Scheme 3B). Furthermore, the Boc-protecting group could be easily removed under acidic condition to deliver the free sulfoximine 6 (Scheme 3B).
 | ||
| Scheme 3 Scale up reaction and post-synthetic transformations. | ||
We then performed several control experiments to get insight into the reaction mechanism (Scheme 4A). When the reaction was performed using TEMPO as radical quencher the reaction failed to provide the product 3a suggesting the radical nature of the reaction. Further, using 1,1-diphenyl ethene as radical trapping agent, we observed the formation of the adduct 7. Based on these control experiments and previous literature reports,17 we postulated that the sulfinamide anion 8 undergoes anodic oxidation to generate the N-centred radical intermediate which could undergo electronic rearrangement to generate the sulfonimidoyl radical 9 (Scheme 4B). At the cathode, the bromostyrene undergoes one-electron reduction to generate the styryl radical 10, which finally undergoes radical cross coupling with 9 to provide the sulfoximine 3a (Scheme 4B).
 | ||
| Scheme 4 Control experiment and possible reaction mechanism. | ||
In conclusion, we have demonstrated the application of electrochemistry to access vinyl sulfoximines through a radical cross-coupling of the sulfonimidoyl and styryl radicals under metal- and additive-free conditions. The developed protocol successfully delivered a variety of sulfoximine derivatives in moderate to good yields. This strategy was further applied to synthesize bioactive tethered vinyl sulfoximines in moderate yields. Additionally, the synthetic applicability of the protocol was demonstrated through a scale-up reaction and subsequent post-synthetic modifications.
This publication is based upon work partially supported by King Abdullah University of Science and Technology (KAUST) under Award No. ORFS-CRG12-2024-6438.
Data availability
All experimental data, and detailed experimental procedures are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Reggelin and C. Zur, Synthesis, 2000, 1–64 CrossRef CAS; (b) U. Lücking, Angew. Chem., Int. Ed., 2013, 52, 9399–9408 CrossRef PubMed; (c) J. A. Sirvent and U. Lücking, ChemMedChem, 2017, 12, 487–501 CrossRef CAS PubMed; (d) M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225–245 CrossRef CAS PubMed; (e) P. Mader and L. Kattner, J. Med. Chem., 2020, 63, 14243–14275 CrossRef PubMed; (f) Y. Han, K. Xing, J. Zhang, T. Tong, Y. Shi, H. Cao, H. Yu, Y. Zhang, D. Liu and L. Zhao, Eur. J. Med. Chem., 2021, 209, 112885–112907 CrossRef CAS PubMed; (g) U. Lücking, Chem. – Eur. J., 2022, 28, e202201993 CrossRef PubMed; (h) H. J. Gais, Eur. J. Org. Chem., 2024, e202301143 CrossRef CAS.
- D. P. Walker, M. P. Zawistoski, M. A. McGlynn, J.-C. Li, D. W. Kung, P. C. Bonnette, A. Baumann, L. Buckbinder, J. A. Houser, J. Boer, A. Mistry, S. Han, L. Xing and A. Guzman-Perez, Bioorg. Med. Chem. Lett., 2009, 19, 3253 CrossRef CAS PubMed.
- G. Siemeister, U. Lücking, A. M. Wengner, P. Lienau, W. Steinke, C. Schatz, D. Mimberg and K. Ziegelbauer, Mol. Cancer Ther., 2012, 11, 2265 CrossRef CAS PubMed.
- M. Kahraman, S. Sinishtaj, P. M. Dolan, T. W. Kensler, S. Peleg, U. Saha, S. S. Chuang, G. Bernstein, B. Korczak and G. H. Posner, J. Med. Chem., 2004, 47, 6854 CrossRef CAS PubMed.
- Y. Zhu, M. R. Loso, B. M. Nugent, J. X. Huang and R. B. Rogers, Dow AgroSciences LLC, Indianapolis, USA, WO/2008/057129 A1, 2008 Search PubMed.
- (a) P. Ghosh, B. Ganguly and S. Das, Asian J. Org. Chem., 2020, 9, 2035–2082 CrossRef CAS; (b) M. Andresini, A. Tota, L. Degennaro, J. A. Bull and R. Luisi, Chem. Eur. J., 2021, 27, 17293–17321 CrossRef CAS PubMed.
- W. Zheng, X. Chen, F. Chen, Z. He and Q. Zeng, Chem. Rec., 2021, 21, 396–416 CrossRef CAS PubMed.
- (a) Y. Aota, T. Kano and K. Maruoka, Angew. Chem., Int. Ed., 2019, 58, 17661–17665 CrossRef CAS PubMed; (b) Y. Maeda, S. Hamada, Y. Aota, K. Otsubo, T. Kano and K. Maruoka, J. Org. Chem., 2022, 87, 3652–3660 CrossRef CAS PubMed; (c) X. Zou, H. Wang and B. Gao, Org. Lett., 2023, 25, 7656–7660 CrossRef CAS PubMed; (d) G. Jersovs, D. Melgalvis, A. Kinens, P. A. Donetsa and E. Suna, Org. Chem. Front., 2025, 12, 14–23 RSC.
- X. Zou, B. Shen, G.-L. Li, Q. Liang, Y. Ouyang, B. Yang, P. Yu and B. Gao, Sci. China Chem., 2024, 67, 928–935 CrossRef CAS.
- For selected review articles, see; (a) J. Zhu, W.-C. Yang, X.-D. Wang and L. Wu, Adv. Synth. Catal., 2018, 360, 386–400 CrossRef CAS; (b) G. M. Martins, A. G. Meirinho, N. Ahmed, A. L. Braga and R. S. Mendes, ChemElectroChem, 2019, 6, 5928–5940 CrossRef CAS; (c) V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, RSC Adv., 2020, 10, 20046–20056 RSC; (d) B.-C. Qian, C.-Z. Zhu and G.-B. Shen, ACS Omega, 2022, 7, 39531–39561 CrossRef CAS PubMed; (e) J. Liu, J.-P. Wan and Y. Liu, Org. Chem. Front., 2024, 11, 597–630 RSC; (f) Z. Ye, X. Zhang, W. Ma and F. Zhang, Green Chem., 2023, 25, 2524–2540 RSC ; for selected recent research articles on sulfur radical, see ; (g) Y. Li, H. Wang, Z. Wang, H. Alhumade, Z. Huang and A. Lei, Chem. Sci., 2023, 14, 372–378 RSC; (h) G. Liu, S. Xu, Y. Yue, C. Sua and W. Song, Chem. Commun., 2024, 60, 6154–6157 RSC; (i) J. Kumar, D. Sharma, Y. Hussain Solaim, J. Sinhmar Muskan, A. Changotra and P. Chauhan, Org. Lett., 2025, 27, 1608–1613 CrossRef CAS PubMed; (j) Y. Hussain, C. Empel, R. M. Koenigs and P. Chauhan, Angew. Chem., Int. Ed., 2023, 62, e202309184 CrossRef CAS PubMed; (k) R. I. Patel, B. Saxena and A. Sharma, Green Chem., 2024, 26, 10265–10274 RSC; (l) D. Sharma, Y. Hussain, M. Sharma and P. Chauhan, Green Chem., 2022, 24, 4783–4788 RSC.
- (a) P. Shi, Y. Tu, D. Zhang, C. Wang, K.-N. Truong, K. Rissanen and C. Bolm, Adv. Synth. Catal., 2021, 363, 2552–2556 CrossRef CAS; (b) P. Shi, Y. Tu, C. Wang, D. Ma and C. Bolm, J. Org. Chem., 2022, 87, 3817–3824 CrossRef CAS PubMed; (c) D. Kong, M. M. Amer and C. Bolm, Green Chem., 2022, 24, 3125–3129 RSC.
- X. Wu, W. Zhang, G. Sun, X. Zou, X. Sang, Y. He and B. Gao, Nat. Commun., 2023, 14, 5168–5178 CrossRef CAS PubMed.
- (a) S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122–7123 CrossRef CAS PubMed; (b) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (c) N. Li, R. Sitdikov, A. P. Kale, J. Steverlynck, B. Li and M. Rueping, Beilstein J. Org. Chem., 2024, 20, 2500–2566 CrossRef CAS PubMed; (d) B. Huang, Z. Sun and G. Sun, eScience, 2024, 2, 243–277 CrossRef.
- (a) A. Wiebe, T. Gieshoff, S. Mçhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (b) K. S. McClymont, F.-Y. Wang, A. Minakar and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 8608–8613 CrossRef CAS PubMed; (c) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (d) N. Kaeffer and W. Leitner, JACS Au, 2022, 2, 1266–1289 CrossRef CAS PubMed ; For selected recent research articles on electrocatalysis, see ; (e) H. Chen, C. Zhai, C. Zhu and M. Rueping, Chem. Catal., 2025, 5, 101257 CrossRef CAS; (f) S. Panja, A. Pan, S. Biswas, C. Das, A. Guha, R. Y. Nimje, T. G. M. Dhar, A. Gupta, A. Mathur, A. Dutta, L. Roy and D. Maiti, Angew. Chem., Int. Ed., 2025, 64, e202422876 CrossRef CAS PubMed; (g) T. von Münchow, N. K. Pandit, S. Dana, P. Boos, S. E. Peters, J. Boucat, Y.-R. Liu, A. Scheremetjew and L. Ackermann, Nat. Catal., 2025, 8, 257–269 CrossRef; (h) F. Bu, Y. Deng, L. Lu, Y. Li, W. Song, Z. Yang, X. Luo, X. Dong, R. Yi, D. Yang, S. Wang, A. Lei and W. Li, J. Am. Chem. Soc., 2025, 147, 5785–5795 CrossRef CAS PubMed; (i) M. Jafarzadeh, M. Nassir, L. Gherardi, N. Raheja, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2025, 64, e202421163 CrossRef CAS PubMed.
- T. Liu, Y. Tang, J. Guo, Y. Hang, K. Zhang, C. Zheng, W. Zhong, D. Song and F. Ling, Org. Lett., 2024, 26, 8463–8467 CrossRef CAS PubMed.
- Selected references: (a) C. Empel and R. M. Koenigs, Chem. Catal., 2022, 2, 2506–2514 CrossRef CAS; (b) F. Li, W. F. Zhou, C. Empel, O. Datsenko, A. Kumar, Y. Xu, J. H. M. Ehrler, I. Atodiresei, S. Knapp, P. M. Mykhailiuk, E. Proschak and R. M. Koenigs, Science, 2024, 383, 498–503 CrossRef CAS PubMed; (c) X. Zhao, Z. Tang, L. Shi, Y. Guo, R. M. Koenigs and X. Hao, Green Synth. Catal., 2024 DOI:10.1016/j.gresc.2024.05.001; (d) Y. Guo, C. Pei, C. Empel, S. Jana and R. M. Koenigs, ChemPhotoChem, 2022, 6, e202100293 CrossRef CAS; (e) C. Zhu, H. Chen, H. Yue and M. Rueping, Nat. Synth., 2023, 2, 1068–1081 CrossRef CAS; (f) G. S. Kumar, C. Zhu, R. Kancherla, P. S. Shinde and M. Rueping, ACS Catal., 2023, 13, 8813–8820 CrossRef CAS; (g) C. Zhu, A. P. Kale, H. Yue and M. Rueping, JACS Au, 2021, 1, 1057–1065 CrossRef CAS PubMed.
- (a) E. A. Noten, C. H. Ng, R. M. Wolesensky and C. R. J. Stephenson, Nat. Chem., 2024, 16, 599–606 CrossRef CAS PubMed; (b) H. F. Piedra, V. Gebler, C. Valdés and M. Plaza, Chem. Sci., 2023, 14, 12767–12773 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02045j |
| This journal is © The Royal Society of Chemistry 2025 |