
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly active sulfur based pincer ruthenium catalyst for CO2 hydrogenation†
Alexander
Mondragón-Díaz
 a,
Steven P.
Kelley
a,
Nilay
Hazari
b and
Wesley H.
Bernskoetter
*a
a,
Steven P.
Kelley
a,
Nilay
Hazari
b and
Wesley H.
Bernskoetter
*a
aDepartment of Chemistry, University of Missouri, Columbia, MO, USA. E-mail: bernskoetterwh@missouri.edu
bDepartment of Chemistry, Yale University, New Haven, CT, USA
First published on 15th April 2025
Abstract
The synthesis of a new air-stable SPS pincer ligand that supports a Ru catalyst for CO2 hydrogenation to formate is described. This rare S-donor based pincer system gives higher activity compared to related PNP supported Ru catalysts and is less dependent on Lewis acidic Li co-catalysts for achieving high turnover numbers. The SPS ligated Ru catalyst is also active for N-formylation of amines with CO2.
Pincer ligands are commonly used to support homogeneous transition metal catalysts because of their electronic and structural versatility.1 In particular, PNP type pincer ligands can support active catalysts for a plethora of transformations using metals from across the transition series.2,3 For example, our own laboratories and others have employed group VIII complexes ligated with PNP pincer ligands as catalysts for CO2 hydrogenation and related processes.4–6 While the steric and electronic properties of PNP ligands are frequently modified by varying the phosphine substituents,7–9 this is often synthetically challenging. Further, PNP ligands can be air-sensitive and typically their syntheses require expensive precursors. In principle, replacing the phosphine donors in PNP ligands with weaker σ-donating thioethers provides advantages in terms of ligand cost and improved air stability.10,11 Nevertheless, examples of pincer ligands with sulfur donors are limited. Recent studies of S-donor analogues of widely utilized Ru-MACHO PNP and Ru-pyridyl PNP catalysts report inferior catalytic performance compared to PNP ligated complexes, likely because the Ru centers are less electron rich and have significantly higher reduction potentials.10a,12 To address this issue, in our current study we have a prepared a new air-stable P- and S-donor hybrid pincer ligand, CH3P(C6H4StBu)2 (tBuSPMeS), and used it to develop an SPS Ru dihydride catalyst, (tBuSPMeS)Ru(PPh3)H2, for CO2 hydrogenation and amine formylation. This catalyst displays superior CO2 hydrogenation activity compared to related PNP Ru systems. Preliminary mechanistic studies identified an SPS Ru formate hydride complex as the likely resting state in catalytic CO2 reduction.
We hypothesized that a SPS pincer ligand would be more electron rich than a SNS pincer ligand and potentially give comparable reactivity to PNP ligated complexes. Therefore, we targeted tBuSPMeS, which contains a strongly donating central P-donor, alongside two S-donors, with sterically bulky tBu-substituents to prevent dimerization. tBuSPMeS was prepared in three high yielding steps from inexpensive and commercially available starting materials (Scheme 1).13 Initially 2-bromothiophenol was converted into (2-bromophenyl)(tert-butyl)sulfane, which was treated with nBuLi and PCl3 to form tBuSPClS. Reaction of tBuSPClS with methyl Grignard generated the tBuSPMeS ligand. Notably, benzene solutions of tBuSPMeS are stable to air at ambient temperature for more than one week.
 | ||
| Scheme 1 Synthesis of the tBuSPMeS ligand. | ||
Starting from commercially available [Ru(PPh3)3]Cl2, the Ru dichloride complex (tBuSPMeS)Ru(PPh3)Cl2 (1-Cl2) was prepared in nearly quantitative yield by ligand substitution over 1 h at ambient temperature (Scheme 2). The 1H NMR spectrum of the isolated yellow solid displays one doublet at 0.82 ppm for the P–(CH3) group, and two equal intensity resonances for the S–tBu groups at 1.75 and 0.90 ppm, consistent with two thioethers groups occupying distinct chemical environments. These are consistent with X-ray diffraction studies of 1-Cl2 that indicate the tBuSPMeS ligand is bound in a fac mode with thioether groups positioned trans to the Cl and PPh3 ligands (Fig. 1, left). The PPh3 ligand in 1-Cl2 is oriented cis to the P-donor of the tBuSPMeS, in agreement with the two doublets at δ 86.72 and δ 28.55 ppm observed in the 31P{1H} NMR spectrum. The fac-ligation of the pincer ligand in 1-Cl2 contrasts with most reported PNP and SNS Ru complexes, which bind in a mer orientation.2e,10b,12b
 | ||
| Scheme 2 Synthesis of the [(tBuSPMeS)Ru] complexes 1-Cl2, 1-BH4, 1H2, 1-CO2H and 2. | ||
 | ||
| Fig. 1 Solid State structure of 1-Cl2 (left), 1-H2 (center) and 2 (right). Thermal ellipsoids are shown at 50% probability. Hydrogen atoms, solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (deg) for 1-Cl2: Ru(1)–P(1) 2.213(2), Ru(1)–P(2) 2.366(2), Ru(1)–S(1) 2.430(2), Ru(1)–S(2) 2.344(2), Ru(1)–Cl(1) 2.505(2), Ru(1)–Cl(2) 2.453(2); P(1)–Ru(1)–P(2) 100.24(6), S(1)–Ru(1)–S(2) 96.46(7), Cl(1)–Ru(1)–Cl(2) 95.25(6), P(1)–Ru(1)–Cl(1) 170.29(7), 1-H2: Ru(1)–P(1) 2.262(6), Ru(1)–P(2) 2.263(6), Ru(1)–S(1) 2.373(7), Ru(1)–S(2) 2.399(7), Ru(1)–H(1) 1.500(5), Ru(1)–H(2) 1.650(5); P(1)–Ru(1)–P(2) 100.17(3), S(1)–Ru(1)–S(2) 101.95(2), H–Ru(1)–H(A) 87.00(3), P(1)–Ru(1)–H(A) 168.29(2), S(1)–Ru(1)–P(2) 160.40(3), S(2)–Ru(1)–H 172.00(2), P(1)–Ru(1)–H(A) 168.00(2).for 2: Ru(1)–P(1) 2.366(1), Ru(1)–P(2) 2.302(1), Ru(1)–S(2) 2.391(1), Ru(1)–S(3) 2.458(1), Ru(1)–S(4) 2.395(1), Ru(1)–C(35) 1.858(5); P(1)–Ru(1)–P(2) 167.98(4), S(3)–Ru(1)–S(4) 90.95(4), S(4)–Ru(1)–S(2) 171.05(4), C(36)–Ru(1)–S(3) 175.20(2). | ||
Treatment of 1-Cl2 with NaBH4 in EtOH initially afforded a species tentatively identified as (tBuSPMeS)Ru(PPh3)(H)(BH4) (1-BH4) which exhibited a 1H NMR spectrum containing a Ru–H resonance at δ −13.05 ppm (dd, 2JP–H = 29, 23 Hz) integrating to one proton, as well as a broad resonance centered at δ −0.80 ppm, integrating to four protons (Scheme 2). This signal is similar to those observed for other rapidly exchanging M–BH4 complexes.3d,4a,6a,14a Exposure of 1-BH4 to low pressures or standing under N2 at ambient temperature for 1 h resulted in partial conversion to the corresponding Ru dihydride complex, (tBuSPMeS)Ru(PPh3)H2 (1-H2), obviating isolation of pure samples of 1-BH4. 1-H2 was obtained in 90% yield by treatment of toluene solutions of 1-BH4 with triethylamine over 4 h (Scheme 2). The 1H NMR spectrum of 1-H2 in C6D6 displays two sets of 8-line resonances consistent with Ru–H ligands bound cis (−12.04 ppm) and trans (−5.05 ppm) to the P-atom in tBuSPMeS.14 The molecular structure of 1-H2 was confirmed by X-ray diffraction (Fig. 1, center). The tBuSPMeS ligand is again bound in a fac geometry with two Ru–H moieties located in a mutually cis arrangement, consistent with the solution NMR data. The Ru–H bond lengths differ significantly, with Ru(1)–H(1) approximately 0.16 Å longer than Ru(1)–H(2) owing to the strongly σ-donating properties of the phosphorus donor compared with the thioether donor. Significantly, these differences in Ru–H bond length impact the relative nucleophilic activity of these hydride ligands and lead to a marked difference in their tendency to undergo insertion reactions (vide infra).15
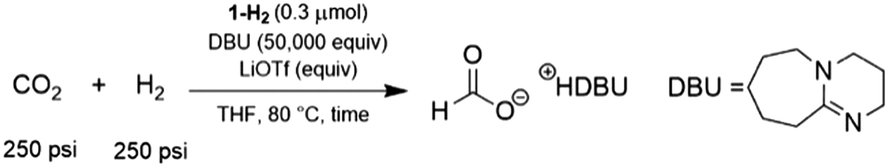
Pincer ligated Ru hydride complexes are leading catalysts for carbonyl reduction,4b,c,e,5,9 which motivated our evaluation of 1-H2 for catalytic CO2 hydrogenation. Using conditions previously optimized for (iPrPNRP)RuHCl(CO) (iPrPNRP = [RN(CH2CH2PiPr2)2] R = H, Me or Ph),5a 0.3 μmol of 1-H2 under 500 psi H2/CO2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of excess 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) resulted in the hydrogenation of CO2 to formate (Table 1). After 1 h, 1-H2 produced formate with a turnover number (TON) of 5600 (entry 1), demonstrating an activity roughly 10 times higher than that of (iPrPNMeP)Ru(H)2(CO) and 6 times greater than (iPrPNHP)Ru(H)2(CO) under comparable conditions.5a The TON at this short interval is determined largely by the initial rate of catalysis and serves as an approximation of the turnover frequency (TOF) for 1-H2. Our results indicate that air stable S-donor pincer ligands can provide superior catalytic performance compared to state-of-the-art PNP ligands.5a Excellent catalytic activity was also observed for 1-H2 in the presence of co-catalytic amounts of lithium triflate (LiOTf) (1.5 mmol) and a TON of 15700 was observed after 1 h (entry 2). The beneficial effects of Lewis acids (LAs), such as LiOTf, in CO2 hydrogenation reactions has been observed in many related pincer-supported systems for CO2 hydrogenation.4e,5a,16 Extending reaction times to 6 h narrows the gap in catalytic productivity between trials with and without LA (entries 3 & 4). After 24 h, the enhancing effects of LiOTf are minor (entries 5 & 6), with experiments using only 1-H2 achieving a TON of over 30000. In fact, there is essentially no increase in TON between 6 h and 24 h in the presence of LiOTf, suggesting the catalyst is largely deactivated after 6 h. In the absence of LiOTf, the catalyst is active up to 24 h, with no increase between 24 h and 48 h (entry 7). These trends suggest that while LiOTf, a common co-catalyst for these reactions, enhances the already leading activity of 1-H2, additives are not required to achieve high productivity and that LAs may accelerate catalyst deactivation in addition to improving the rate of formate production. This hypothesis is further supported by experiments with lower catalyst loadings (0.03 μmol) which achieve a maximum TON of 160000 without LiOTf (entry 8). This corresponds to an average TOF of 6600 h−1 over the first 24 h of reaction time. In the presence of LiOTf these values decrease to a TON of 105000 and an average TOF of 4400 h−1 (entry 9). Comparing TOF values between catalysts is often complicated by variation in reaction conditions and the length of time reactions are performed. Nevertheless, 1-H2 delivers activity which is clearly higher than the most comparable Ru-MACHO PNP frameworks,5a though initial TOF values in excess of 1 × 106 h−1 have been reported for Ru-pyridyl PNP catalysts in CO2 hydrogenation to formate.4c
:1) in the presence of excess 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) resulted in the hydrogenation of CO2 to formate (Table 1). After 1 h, 1-H2 produced formate with a turnover number (TON) of 5600 (entry 1), demonstrating an activity roughly 10 times higher than that of (iPrPNMeP)Ru(H)2(CO) and 6 times greater than (iPrPNHP)Ru(H)2(CO) under comparable conditions.5a The TON at this short interval is determined largely by the initial rate of catalysis and serves as an approximation of the turnover frequency (TOF) for 1-H2. Our results indicate that air stable S-donor pincer ligands can provide superior catalytic performance compared to state-of-the-art PNP ligands.5a Excellent catalytic activity was also observed for 1-H2 in the presence of co-catalytic amounts of lithium triflate (LiOTf) (1.5 mmol) and a TON of 15700 was observed after 1 h (entry 2). The beneficial effects of Lewis acids (LAs), such as LiOTf, in CO2 hydrogenation reactions has been observed in many related pincer-supported systems for CO2 hydrogenation.4e,5a,16 Extending reaction times to 6 h narrows the gap in catalytic productivity between trials with and without LA (entries 3 & 4). After 24 h, the enhancing effects of LiOTf are minor (entries 5 & 6), with experiments using only 1-H2 achieving a TON of over 30000. In fact, there is essentially no increase in TON between 6 h and 24 h in the presence of LiOTf, suggesting the catalyst is largely deactivated after 6 h. In the absence of LiOTf, the catalyst is active up to 24 h, with no increase between 24 h and 48 h (entry 7). These trends suggest that while LiOTf, a common co-catalyst for these reactions, enhances the already leading activity of 1-H2, additives are not required to achieve high productivity and that LAs may accelerate catalyst deactivation in addition to improving the rate of formate production. This hypothesis is further supported by experiments with lower catalyst loadings (0.03 μmol) which achieve a maximum TON of 160000 without LiOTf (entry 8). This corresponds to an average TOF of 6600 h−1 over the first 24 h of reaction time. In the presence of LiOTf these values decrease to a TON of 105000 and an average TOF of 4400 h−1 (entry 9). Comparing TOF values between catalysts is often complicated by variation in reaction conditions and the length of time reactions are performed. Nevertheless, 1-H2 delivers activity which is clearly higher than the most comparable Ru-MACHO PNP frameworks,5a though initial TOF values in excess of 1 × 106 h−1 have been reported for Ru-pyridyl PNP catalysts in CO2 hydrogenation to formate.4c
|
|
|||||
|---|---|---|---|---|---|
| Entry | LiOTf (mmol) | T (°C) | Time (h) | TONb | Yieldc (%) |
| a Reaction conditions: 250 psi of CO2/250 psi of H2, 1-H2 (0.3 μmol), DBU (2.34 g, 15.0 mmol), THF (10 mL), 80 °C. b TONs were quantified using 1H NMR spectroscopy with DMF as an internal standard; reported values are the average of three trials with the standard deviation in parentheses. c The yield is based on DBU. d 1-H2 = 0.03 μmol. | |||||
| 1 | 0 | 80 | 1 | 5600 (600) | 10 |
| 2 | 1.5 | 80 | 1 | 15700 (1000) |
32 |
| 3 | 0 | 80 | 6 | 22000 (600) |
44 |
| 4 | 1.5 | 80 | 6 | 34500 (200) |
69 |
| 5 | 0 | 80 | 24 | 30200 (200) |
60 |
| 6 | 1.5 | 80 | 24 | 34000 (1000) |
68 |
| 7 | 0 | 80 | 48 | 32800 (1300) |
66 |
| 8d | 0 | 80 | 24 | 160000 (10000) |
32 |
| 9d | 1.5 | 80 | 24 | 105000 (5000) |
21 |
To study the pathway of formate production, a sample of 1-H2 was exposed to 13CO2 and the reaction monitored by NMR spectroscopy. Spectra collected immediately following gas addition indicated complete conversion to a Ru formate hydride complex, [(tBuSPMeS)RuH(PPh3)(HCO2)] (1-HCO2) (Scheme 2). The 13CO2 derived formate ligand appears as a doublet at 8.92 ppm in the 1H NMR with a corresponding resonance at 169.0 ppm in the 13C NMR spectrum. Multinuclear 1D and 2D NMR studies indicate that the formate group is coordinated trans to the P atom of the BuSPMeS ligand, consistent with insertion occurring at the Ru–H opposite the more trans influencing phosphine site. Analysis after several days under 13CO2 or at elevated temperatures up to 80 °C gave no indication of insertion into the remaining Ru–H of 1-HCO2. Unfortunately, isolation of 1-HCO2 in pure form was not possible as exposure to vacuum caused immediate reversion to 1-H2, similar to reports of related Fe and Ru formate complexes.5a Previously, our laboratories and others have identified the insertion of CO2 into Ru–H or Fe–H bonds as a key step in the catalytic hydrogenation of CO2 to formate.4a,5a,15,17In situ NMR experiments starting from 1-H2 under modified catalytic conditions (1:3:40 of 1-H2:LiBF4:DBU in THF-d8 under 2 atm of CO2/H2 at 23 °C) indicate 1-HCO2 is the primary Ru complex present during formate production (Fig. S26, ESI†), consistent with 1-HCO2 acting as the catalytic resting state. This suggests that loss of the formate ligand is the turnover limiting step in catalysis.
Many leading pincer supported Ru and Fe catalysts for carbonyl hydrogenation reactions contain CO ancillary ligands.3d–5 The presence of these π-acids have been variously postulated to influence the fac/mer coordination mode of the pincer ligand, improve the stability of the catalysts and attenuate the steric and electronic properties of the metal.18 Seeking to develop a CO coordinated analog of 1-H2, we attempted ligand substitution of PPh3. However, exposure of 1-H2 to CO gave no reaction at ambient temperature and led to unselective reactivity at elevated temperatures. Alternatively, treating [RuH2(PPh3)3(CO)] with tBuSPMeS in toluene at 100 °C resulted in the formation of a single SPS pincer containing product, [(κ3-SPMeS)Ru(κ2-tBuSPMeS)(CO)] (2) (Scheme 2), in moderate yield based on the 2:1 SPS to Ru stoichiometry. Complex 2 is the result of two C–S bond activations of the ligand S–tBu groups and κ2 coordination of an additional equivalent of tBuSPMeS. The identity of the S–tBu derived organic products has remained elusive, but 2 was characterized by NMR and XRD analysis (Fig. 1, right). Catalytic trials using 2 indicate it is largely inactive for CO2 hydrogenation (Table S1, ESI†), suggesting that activation of S-donor substituents in the presence of Ru–H groups may present a path toward catalyst deactivation. Analogous ligand activation reactions are rare for PNP complexes and should be carefully considered in the design of S-donor pincer ligands.
Metal-catalyzed formylation of amines with CO2 and H2 is another application of CO2 hydrogenation which provides valuable chemicals.19,20 A preliminary investigation into N-formylation using pyrrolidine was conducted using 0.2 mol% of 1-H2 under 500 psi of H2/CO2 (1:1) at 120 °C for 24 h. A 96% yield of 1-formylpyrrolidine corresponding to a TON of 480 was observed (Fig. 2). A preliminary screening of other secondary and primary amine substrates revealed only modest activity and scope for this reaction (Table S3, ESI†). Reducing the loading of 1-H2 to 0.002 mol% increased TON significantly to 4080, albeit with a yield decreased to 8%. Still, the activity of 1-H2 towards CO2 based N-formylation indicates that SPS or other S-donor pincer ligands are viable scaffolds for the development of new catalysts for other carbonyl reduction processes.
 | ||
| Fig. 2 N-Formylation of pyrrolidine with H2 and CO2 catalyzed by 1-H2. | ||
In summary, this work identifies a new, air stable SPS pincer ligand that can support a highly active and productive Ru catalyst for CO2 hydrogenation to formate. The SPS-ligated Ru catalyst surpasses the activity of related PNP ligated Ru catalysts for CO2 reduction and is less dependent on Lewis acidic LiOTf co-catalyst for achieving maximum productivity. Preliminary mechanistic studies suggest that CO2 insertion into the strongly trans influenced Ru–H of 1-H2 produces a formate complex 1-HCO2 that likely serves as the catalyst resting state. Attempts to incorporate a CO ancillary ligand into the coordination sphere of a SPS-ligated Ru complex resulted in deleterious C–S activation and formation of a poorly active Ru thiolate complex. This work provides rare examples of S-donor congeners of widely used PNP-ligated Ru hydrogenation catalysts and demonstrates the potential for enhancing catalytic performance with S-donor pincer ligands. Our laboratories are currently seeking to elucidate the origins for the improved catalyst performance, characterize deactivation pathways, and design superior air stable S-donor pincer ligand platforms.
Data availability
The experimental details supporting this article have been included as part of the ESI.† CCDC numbers 2427581–2427583 contain supplementary crystallographic data for 1-Cl2, 1-H2, and 2.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) L. González-Sebastián, A. Reyes-Sanchez and D. Morales-Morales, Organometallics, 2023, 42, 2426–2446 CrossRef; (b) M. Esfandiari, G. Mohammadnezhad, O. Akintola, F. Otto, T. Fritz and W. Plass, Dalton Trans., 2023, 52, 11875–11885 RSC; (c) S. Martínez-Vivas, D. G. Gusev, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2023, 62, e2023138 Search PubMed.
- (a) T. Mitsumoto, Y. Ashida, K. Arashiba, S. Kuriyama, A. Egi, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2023, 62, e2023066 Search PubMed; (b) B. Wang, C. Rong, M. Lei, S. Liu and F. De Proft, Inorg. Chem., 2023, 62(19), 7366–7375 CrossRef CAS PubMed; (c) Y. Liu, X. Yue, L. Li, Z. Li, L. Zhang, M. Pu, Z. Yang, C. Wang, J. Xiao and M. Lei, Inorg. Chem., 2020, 59(12), 8404–8411 CrossRef CAS PubMed; (d) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (e) A. Friedrich, M. Drees, M. Käss, E. Herdtweck and S. Schneider, Inorg. Chem., 2010, 49, 5482–5494 CrossRef CAS PubMed.
- (a) P. Hermosilla, P. López, P. García-Orduña, F. J. Lahoz, V. Polo and M. A. Casado, Organometallics, 2018, 37(15), 2618–2629 CrossRef CAS; (b) S. Kar and D. Milstein, Chem. Commun., 2022, 58, 3731–3746 RSC; (c) A. V. Zabula, Y. Qiao, A. J. Kosanovich, T. Cheisson, B. C. Manor, P. J. Carroll, O. V. Ozerov and E. J. Schelter, Chem. – Eur. J., 2017, 23, 17923–17934 CrossRef CAS PubMed; (d) R. Langer, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 7196–7210 CrossRef CAS PubMed.
- (a) Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC; (b) R. Sen, A. Goeppert and G. K. S. Prakash, Angew. Chem., Int. Ed., 2022, 61, e202207278 CrossRef CAS PubMed; (c) G. A. Filonenko, R. V. Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS; (d) J. B. Curley, N. E. Smith, W. H. Bernskoetter, N. Hazari and B. Q. Mercado, Organometallics, 2018, 37, 3846–3853 CrossRef CAS; (e) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- (a) J. B. Curley, C. Hert, W. H. Bernskoetter, N. Hazari and B. Q. Mercado, Inorg. Chem., 2022, 61, 643–656 CrossRef CAS PubMed; (b) A. Agapova, E. Alberico, A. Kammer, H. Junge and M. Beller, Chem. Cat. Chem., 2019, 11, 1910–1914 CrossRef CAS.
- (a) E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed; (b) X.-F. Liu, X.-Y. Li and L.-N. He, Eur. J. Org. Chem., 2019, 2437–2447 CrossRef CAS; (c) Z. Chen, S. Du, J. Zhanga and X.-F. Wu, Green Chem., 2020, 22, 8169–8182 RSC.
- (a) S. Lapointe, E. Khaskin, R. R. Fayzullin and J. R. Khusnutdinova, Organometallics, 2019, 38(7), 1581–1594 CrossRef CAS; (b) D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41(2), 201–213 CrossRef CAS PubMed; (c) S. Gu, R. J. Nielsen, K. H. Taylor, G. C. Fortman, J. Chen, D. A. Dickie, W. A. Goddard III and T. B. Gunnoe, Organometallics, 2020, 39(10), 1917–1933 CrossRef CAS.
- D. Benito-Garagorri, E. Becker, J. Wiedermann, W. Lackner, M. Pollak, K. Mereiter, J. Kisala and K. Kirchner, Organometallics, 2006, 25(8), 1900–1913 CrossRef CAS.
- S. Kar, R. Sen, J. Kothandaraman, A. Goeppert, R. Chowdhury, S. B. Munoz, R. Haiges and G. K. S. Prakash, J. Am. Chem. Soc., 2019, 141, 3160–3170 CrossRef CAS PubMed.
- (a) D. N. Chirdon, S. P. Kelley, N. Hazari and W. H. Bernskoetter, Organometallics, 2021, 40, 4066–4076 CrossRef CAS; (b) C. Vinas, P. Angles, G. Sanchez, N. Lucena, F. Teixidor, L. Escriche, J. Casabo, J. F. Piniella, A. Alvarez-Larena, R. Kivekäs and R. Sillanpää, Inorg. Chem., 1998, 37, 701–707 CrossRef CAS.
- M. R. Elsby and R. T. Baker, Acc. Chem. Res., 2023, 56, 798–809 CrossRef CAS PubMed.
- (a) D. S. McGuinness, P. Wasserscheid, W. Keim, D. Morgan, J. T. Dixon, A. Bollmann, H. Maumela, F. Hess and U. Englert, J. Am. Chem. Soc., 2003, 125, 5272–5273 CrossRef CAS PubMed; (b) D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., 2013, 125, 2598–2602 CrossRef; (c) J. Schörgenhumer, A. Zimmermann and M. Waser, Org. Process Res. Dev., 2018, 22, 862–870 CrossRef.
- (a) S. B. Harkins and J. C. Peters, J. Am. Chem. Soc., 2004, 126, 2885–2893 CrossRef CAS PubMed; (b) Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458–11461 RSC.
- (a) C. Bianchini, P. J. Perez, M. Peruzzini, F. Zanobini and A. Vacca, Inorg. Chem., 1991, 30, 279–287 CrossRef CAS; (b) M. Hirano, S. Togashi, M. Ito, Y. Sakaguchi, N. Komine and S. Komiya, Organometallics, 2010, 29, 3146–3159 CrossRef CAS; (c) M. M. Bhadbhade, L. D. Field, R. Gilbert-Wilson, R. W. Guest and P. Jensen, Inorg. Chem., 2011, 50, 6220–6228 CrossRef CAS PubMed; (d) P. Lorusso, G. R. Easthamb and D. J. Cole-Hamilton, Dalton Trans., 2018, 47, 9411–9417 RSC.
- (a) Y.-Y. Ohnishi, T. Matsunaga, Y. Nakao, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2005, 127, 4021–4032 CrossRef CAS PubMed; (b) T. Liu, Z. Liu, L. Tang, J. Li and Z. Yang, Catalysts, 2021, 11(1356), 1–10 Search PubMed.
- (a) L. Pavlovic and K. H. Hopmann, Organometallics, 2023, 42, 3025–3035 CrossRef CAS; (b) A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed.
- (a) G. Liang and Min Zhang, Chem. – Eur. J., 2024, 30, e202402114 CrossRef CAS PubMed; (b) S. Ramakrishnan, K. M. Waldie, I. Warnke, A. G. De Crisci, V. S. Batista, R. M. Waymouth and C. E. D. Chidsey, Inorg. Chem., 2016, 55, 1623–1632 CrossRef CAS PubMed; (c) P. Zhang, S.-F. Ni and L. Dang, Chem. – Asian J., 2016, 11, 2528–2536 CrossRef CAS PubMed.
- (a) D. Benito-Garagorri, M. Puchberger, K. Mereiter and K. Kirchner, Angew. Chem., Int. Ed., 2008, 47, 9142–9145 CrossRef CAS; (b) I. Koehne, T. J. Schmeier, E. A. Bielinski, C. J. Pan, P. O. Lagaditis, W. H. Bernskoetter, M. K. Takase, C. Würtele, N. Hazari and S. Schneider, Inorg. Chem., 2014, 53, 2133–2143 CrossRef CAS PubMed; (c) N. E. Smith, W. H. Bernskoetter, N. Hazari and B. Q. Mercado, Organometallics, 2017, 36, 3995–4004 CrossRef CAS; (d) M. Halder, D. Castillo Cardenas, A. M. Chartouni and D. B. Culver, Dalton Trans., 2025, 54, 2851–2859 RSC.
- (a) L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS PubMed; (b) U. Jayarathne, N. Hazari and W. H. Bernskoetter, ACS Catal., 2018, 8, 1338–1345 CrossRef CAS; (c) M. Hulla, G. Laurenczy and P. J. Dyson, ACS Catal., 2018, 8, 10619–10630 CrossRef CAS; (d) M. Rahman, I. Dutta, S. S. Gholap, G. N. Ngô, Y. Rachuri, L. Alrais and K.-W. Huang, ChemCatChem, 2024, 16, e202401202 CrossRef CAS.
- (a) K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Translated by C. R. Lindley, Wiley-VCH, Weinheim, 3rd edn, 1997 CrossRef; (b) H. Bipp and H. Kieczka, Formamides. Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2003, vol. 15, pp. 36–47 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2427581–2427583. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01194a |
| This journal is © The Royal Society of Chemistry 2025 |