
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free alkene hydroboration with pinacolborane employing C6F5BH2·SMe2 as a precatalyst†
Nikita
Slesarchuk
a,
George
Doerksen
 ab,
Petra
Vasko
*a and
Timo
Repo
*a
ab,
Petra
Vasko
*a and
Timo
Repo
*a
aDepartment of Chemistry, Laboratory of Inorganic Chemistry, University of Helsinki, P.O. Box 55, FIN-00014, Finland. E-mail: petra.vasko@helsinki.fi; timo.repo@helsinki.fi
bDepartment of Chemistry, University of Calgary, 2500 University Drive, Calgary, T2N 1N4, Canada
First published on 27th March 2025
Abstract
We have developed C6F5BH2·SMe2 as a unique, metal-free precatalyst for alkene hydroboration. It combines high reactivity and excellent regio- and chemoselectivity. Mechanistic studies reveal that the catalyst's structure is nearly ideal: the transborylation step occurs via an [sp3-C–B/B–H] transition state and the hydroborylation step goes through a low barrier (ΔG‡ = 15.2 kcal mol−1) with cyclohexene as a substrate.
Boronic esters are highly valuable compounds in both the chemical industry and scientific research. Unlike alkylboranes, they are air-stable, isolable and relatively inexpensive.1 Boronic esters are used in a wide range of transformations2 including Suzuki–Miyaura coupling,3 Chan–Evans–Lam coupling,4 Zweifel olefination5 and Matteson homologation,6 providing access to multiple structurally complex compounds.7 Accordingly, boronic esters are one of the most important building blocks in the pharmaceutical industry, leading to significant and increasing demand for their synthesis.8 There are many outstanding methods for the synthesis of boronic esters using Li- or Mg-containing catalysts9 and transition-metal catalysis.10 Despite its undoubted advantages, transition-metal catalysis has also shortcomings. Scarcity and high cost of metals along with the energetically expensive procedure for removing traces of metals from target molecules are relevant concerns for the modern pharmaceutical industry.11 Metal-free catalysis is void of these disadvantages, making it a substantial augmentation of current synthetic methods.12
Hydroboration reaction is a crucial method for synthesizing boronic esters.1 In general, pinacol-substituted boronic esters are highly stable allowing their easy isolation, purification, and storage,1 as well as transformations targeted at other functional groups in their presence.5,6,7a Metal-free approaches for the production of pinacol boronic esters can be divided into two categories. The classical method involves an attack by a boron electrophile followed by esterification of the resulting compound with a 1,2-diol.13 In the second method a boron electrophile reacts first and subsequent transborylation takes place with pinacolborane (HBpin) to yield the desired product (Scheme 1A).14 The latter option is more appealing for future development as it can be integrated into a catalytic cycle using a B–H electrophile: during the final transborylation step, the B–H electrophile is regenerated and, thus, could be available for the next cycle. Various methods of metal-free catalytic hydroboration range from alcohols, ketones, imines, and C–C triple bonds to yield boronic esters in combination with HBpin.12a,13d,15 However, there are only few reported studies on metal-free catalytic hydroboration of C–C double bonds using pinacolborane as a boron source.14b,c,16 Particularly, the leading examples are B(3,5-(CF3)2C6H3)3 (BArF3)14b and BH3.14c The first example, BArF3, requires an additional transborylation step to generate in situ the catalytically active B–H species. It shows notable reactivity towards substituted styrene derivatives and terminal alkenes, achieving an average of 95% anti-Markovnikov regioselectivity (Scheme 1B). Although BH3 represents the simplest possible catalyst candidate for hydroboration, it exhibits similar catalytic reactivity towards styrenes and terminal alkenes (Scheme 1B). Herein, we report a powerful catalytic method for hydroboration of terminal and internal olefins using C6F5BH2·SMe2 as a catalyst precursor (Scheme 1C). This compound combines a boron centre with high electrophilicity and steric accessibility. Those features ensure high regioselectivity and reactivity. Notably, 1 can successfully hydroborate R-(+)-limonene, β-(−)-pinene, α-(+)-pinene, and a TBS-protected pregnenolone derivative, demonstrating its potential in natural product synthesis.
 | ||
| Scheme 1 Metal-free synthetic routes (A–C) to pinacol boronic esters (RBpin). | ||
The borane C6F5BH2·SMe2 (1) has traditionally been synthesized from (C6F5)3B·OEt2,17 but we pursued its preparation from more affordable reagents. A recent report introduced C6F5BH2 as a fleeting intermediate in a one-pot reaction starting from C6F5Br.18 We slightly modified this procedure and were able to isolate C6F5BH2·SMe2 with an acceptable 72% yield (Scheme 2). Selective Li–Br exchange of pentafluorobromobenzene at −78 °C produces C6F5Li, which is capable of attacking the BH3·SMe2 adduct to form its lithium salt (C6F5BH3Li). Following hydride abstraction with TMSBr and recrystallization from hexane gave colourless needle-shaped crystals of C6F5BH2·SMe2. Surprisingly, the dimethyl sulfide cannot be removed by applying vacuum overnight and it appears to be crucial in stabilizing the highly electrophilic boron centre.
 | ||
| Scheme 2 Synthetic procedure for C6F5BH2·SMe2 (1). | ||
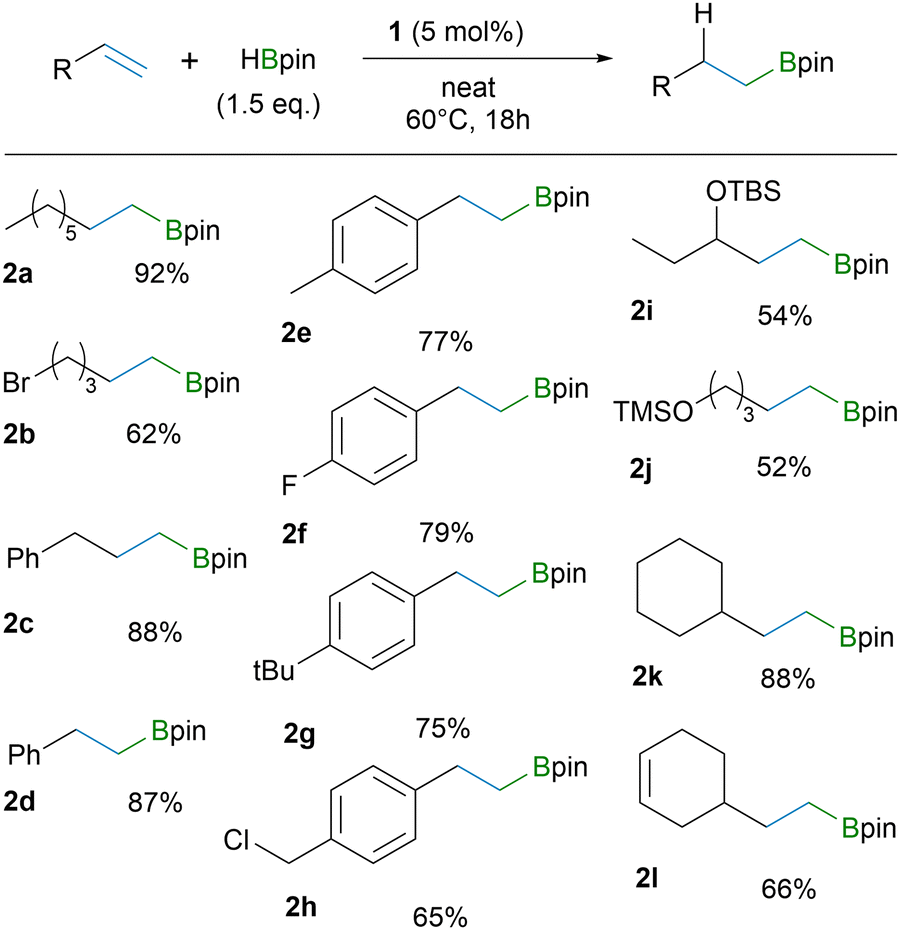
Initial reactivity studies and catalytic performance of 1 were probed using 1-octene and cyclohexene as substrates (see ESI† for further information). For 1-octene, hydroboration in neat conditions with 5 mol% catalyst loading and 60 °C temperature gave the highest yield of the product. Furthermore, by using 3 equivalents of HBpin we were able to obtain quantitative isolated yield of the product, while using 1.5 eq. gave 92% yield. For cyclohexene, a higher temperature (80 °C) and catalyst loading of 10% with extended reaction time gave the optimal result (isolated yield of 96% for 2-cyclohexyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, CyBpin).
The optimized reaction conditions were then applied to a variety of terminal alkenes (Tables 1 and 2). The results clearly demonstrate functional group tolerance towards Br (2b), Cl (2h), F (2f), OTBS (2i) and OTMS (2j) and that the reaction is effective for styrene derivatives (2d–h). The hydroboration maintains exclusive regioselectivity forming only the anti-Markovnikov products (>99% based on GC-MS; see ESI†). The only outlier in the substrate series is C6F5-substituted styrene 3e by giving two different regioisomers in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (Table 2). Presumably, the strong electron withdrawing effect of perfluoroaromatic unit makes also the Markovnikov addition feasible.
:2 ratio (Table 2). Presumably, the strong electron withdrawing effect of perfluoroaromatic unit makes also the Markovnikov addition feasible.
| a Reported yields are isolated. |
|---|
|

The catalyst system demonstrates noteworthy chemoselectivity towards a terminal C–C double bond instead of an internal one (Table 1,2l). However, when treating vinylcyclohexene with a larger excess HBpin (4 eq.), the double hydroboration product 3g was isolated in 97% yield. Applying the optimized cyclohexene conditions to various other alkenes, it is evident that 1,1-disubstituted C–C double bonds react more compliantly compared to 1,2-disubstituted C–C double bonds (Table 2). Notwithstanding, the hydroboration occurs also for the 1,1,2-trisubstituted alkene substrate (3f), which is a highly challenging substrate even for transition-metal catalysts.2b Here, four diastereomers are seen for 3f. It is also worth to note that chloride in an allyl position remains unaffected to yield borylated methallyl chloride (3c), which was isolated in an almost quantitative yield. These results above demonstrate the high reactivity of 1 towards terminal and internal olefins and its complimentary character for metal catalysed hydroboration reactions. However, the catalyst system 1 has also its inherent limitations. Substrates containing ketone or unprotected alcohol groups prohibit the catalytic reactivity.19 And, unexpectedly, neither trans- nor cis-stilbene yielded any product, even though B(3,5-(CF3)2C6H3)3 was amenable in hydroboration with cis-stilbene.14b We assume that breaking the conjugation between the two aromatic cores has a moderately high energy barrier that cannot be overcome under these reaction conditions.
To explore the potential of the hydroboration further, we focused on different enantiomerically pure terpenes to investigate whether stereoselectivity is attained. R-(+)-limonene (3h) and β-(−)-pinene (3j) gave 84% and 90% yields, respectively (Table 2). NMR analysis revealed the presence of two diastereomers for the product of R-(+)-limonene, whereas β-(−)-pinene product was found as one main diastereomer (dr = 95:5) in both NMR and GC-MS (see ESI†). Applicability of the catalytic method was further evaluated by hydroboration of a natural product derivative, pregnenolone (Scheme 3). Despite the presence of an OTBS-substituent and an internal C–C π-bond, the product 5 was isolated with a 71% yield, in line with the catalyst's high functional group tolerance and chemoselectivity.
 | ||
| Scheme 3 Hydroboration of a pregnenolone derivative with HBpin. | ||
The mechanism of the hydroboration catalysis is likely to involve two stages: addition of the C6F5BH2·SMe2 to the C–C double bond and subsequent transborylation reaction with HBpin. This would be in agreeance with previously reported mechanistic studies.14b,20 An alternative mechanism, including an in situ generation of BH3 through nucleophilic attack on HBpin, has been proposed.21 We conducted mechanistic investigations which ruled out the involvement of BH3 (see ESI†). However, these studies revealed minor decomposition of HBpin at elevated temperatures, therefore its slight excess is preferable to ensure high yields.
To gain a more detailed understanding of the hydroboration mechanism, we performed quantum chemical calculations using density functional theory (DFT) at the PBE0-GD3BJ/Def2-TZVP level. We excluded the effect of SMe2 in our calculations as it is not present in any of the product NMR spectra and used the hydroboration of cyclohexene as our model reaction (Fig. 1). The first step in the calculated mechanism includes the addition of C6F5BH2 to cyclohexene, which proceeds in an almost barrierless fashion (TS1, ΔG‡ = 2.1 kcal mol−1) to produce an intermediate C6F5BHCy (Int1). In the next step, the transborylation between Int1 and HBpin can happen directly via a barrier of 17.7 kcal mol−1 (TS2a), but interestingly, the addition of a second cyclohexene to Int1 is more facile and requires only 11 kcal mol−1 of energy (TS2b). This difference in barrier heights can be related to the degree of polarization of the B–H bonds in TS2a and TS2b. The higher polarization of the bond in TS2b (the difference of calculated Mulliken charges is 0.54 in TS2b and 0.40 in TS2a) is in agreement with a lower transition state energy compared to TS2a. Subsequently, the activation barrier for the transborylation of C6F5BCy2 was calculated to be 15.2 kcal mol−1 (TS3), which is 2.5 kcal mol−1 lower than the transborylation of C6F5BHCy (TS2a). The overall reaction is exergonic in nature (ΔG = −37.3 kcal mol−1), and furthermore, the relatively modest energy barrier for TS3 indicates that the sp3-C–B fragment is feasible to undergo transborylation in the presence of a C6F5–B bond.
 | ||
| Fig. 1 Gibbs free energy diagram of the calculated reaction mechanism based on cyclohexene. The energies are given in kcal mol−1 in the gas phase at 298 K. The effect of SMe2 was omitted. | ||
In conclusion, we have developed a highly efficient metal-free hydroboration reaction for olefinic C–C double bonds using easy-to-synthesize C6F5BH2·SMe2 as a catalyst precursor. Noteworthy are high regioselectivity of the reaction with terminal alkenes and catalyst's good tolerance toward halides and silicon-protected alcohols. Our approach is also effective not only with terminal alkenes but also with internal ones. Computational analysis together with experimental data unfolds that B–C6F5 motif of the catalyst is averse to undesired decomposition, providing a pathway for selective transborylation of an alkyl sp3-C–B bond. This method proves to be effective with a model steroid and terpenes, providing stereoselectivity with the latter. We believe that this approach can serve as a straightforward and inexpensive way to convert alkenes to pinacol boronic esters, which are essential for various synthetic applications.
This work has been supported by CHEMS – The Doctoral Programme in Chemistry and Molecular Sciences at University of Helsinki. This project has received funding from the European Union's Horizon Europe research and innovation programme under grant agreement No. 101057816 – TRANSPHARM. P. V. wishes to thank the Research Council of Finland (grant no. 338271 and 346565) for funding. P. V. and G. D. wish to acknowledge Prof. Roland Roesler for facilitating the student exchange and The Mathematics of Information Technology and Complex Systems (Mitacs) for Globalink Research Award Ref. IT41864 to G. D. The authors wish to acknowledge CSC – IT Center for Science, Finland for computational resources. The authors also thank S. Heikkinen for help with NMR measurements, S. Seefried for providing tert-butyldimethyl(pent-1-en-3-yloxy)silane and A. Nudler for corrections during the preparation of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 Search PubMed.
- (a) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 Search PubMed; (b) S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877–8922 Search PubMed; (c) S. Nandy, S. Paul, K. K. Das, P. Kumar, D. Ghorai and S. Panda, Org. Biomol. Chem., 2021, 19, 7276–7297 Search PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 Search PubMed; (b) J. Xu, O. P. Bercher and M. P. Watson, J. Am. Chem. Soc., 2021, 143, 8608–8613 Search PubMed; (c) J. Li, X. Zhang, Y. Yao, Y. Gao, W. Yang and W. Zhao, J. Org. Chem., 2022, 87, 6951–6959 CrossRef; (d) M. Zhang, P. S. Lee, C. Allais, R. A. Singer and J. P. Morken, J. Am. Chem. Soc., 2023, 145, 8308–8313 Search PubMed.
- (a) J. C. Vantourout, H. N. Miras, A. Isidro-Llobet, S. Sproules and A. J. B. Watson, J. Am. Chem. Soc., 2017, 139, 4769–4779 CrossRef PubMed; (b) J. D. Grayson, F. M. Dennis, C. C. Robertson and B. M. Partridge, J. Org. Chem., 2021, 86, 9883–9897 Search PubMed.
- (a) G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 Search PubMed; (b) R. Armstrong and V. Aggarwal, Synthesis, 2017, 3323–3336 Search PubMed.
- (a) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588–7590 Search PubMed; (b) J. L. Stymiest, G. Dutheuil, A. Mahmood and V. K. Aggarwal, Angew. Chem., Int. Ed., 2007, 46, 7491–7494 CrossRef.
- (a) R. E. Shade, A. M. Hyde, J.-C. Olsen and C. A. Merlic, J. Am. Chem. Soc., 2010, 132, 1202–1203 CrossRef PubMed; (b) T. Bootwicha, J. M. Feilner, E. L. Myers and V. K. Aggarwal, Nat. Chem., 2017, 9, 896–902 Search PubMed; (c) A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117–122 CrossRef PubMed.
- Y. Wang, I. Haight, R. Gupta and A. Vasudevan, J. Med. Chem., 2021, 64, 17115–17122 CrossRef.
- (a) M. K. Bisai, S. Yadav, T. Das, K. Vanka and S. S. Sen, Chem. Commun., 2019, 55, 11711–11714 Search PubMed; (b) R. Kumar, S. Dutta, V. Sharma, P. P. Singh, R. G. Gonnade, D. Koley and S. S. Sen, Chem. – Eur. J., 2022, 28, e202201896 Search PubMed.
- (a) S. A. Westcott, H. P. Blom, T. B. Marder and R. T. Baker, J. Am. Chem. Soc., 1992, 114, 8863–8869 CrossRef; (b) D. A. Evans, G. C. Fu and A. H. Hoveyda, J. Am. Chem. Soc., 1992, 114, 6671–6679 Search PubMed; (c) Y. Yamamoto, R. Fujikawa, T. Umemoto and N. Miyaura, Tetrahedron, 2004, 60, 10695–10700 CrossRef; (d) L. Zhang, Z. Zuo, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 2696–2700 Search PubMed; (e) W. N. Palmer, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2015, 5, 622–626 Search PubMed; (f) Y. Xi and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 6703–6706 Search PubMed; (g) S. Kisan, V. Krishnakumar and C. Gunanathan, ACS Catal., 2017, 7, 5950–5954 CrossRef; (h) W. Zhao, K.-Z. Chen, A.-Z. Li and B.-J. Li, J. Am. Chem. Soc., 2022, 144, 13071–13078 CrossRef PubMed.
- (a) A. J. Hunt, T. J. Farmer and J. H. Clark, Elemental Sustainability and the Importance of Scarce Element Recovery, The Royal Society of Chemistry, 2013, p. 1 RSC; (b) J. D. Hayler, D. K. Leahy and E. M. Simmons, Organometallics, 2019, 38, 36–46 CrossRef.
- (a) M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef PubMed; (b) Z.-H. Shang, J. Pan, Z. Wang, Z.-X. Zhang and J. Wu, Eur. J. Org. Chem., 2023, e202201379 Search PubMed; (c) C. Guo, P. Li, S. Wang, N. Liu, Q. Bu, Y. Wang and Y. Qiu, J. Org. Chem., 2023, 88, 4569–4580 CrossRef CAS PubMed; (d) N. Slesarchuk, E. Ma, J. Miranda-Pizarro, S. Heikkinen, D. Schollmeyer, M. Nieger, P. Vasko and T. Repo, Dalton Trans., 2024, 53, 9590–9595 RSC.
- (a) A. G. Karatjas and E. Vedejs, J. Org. Chem., 2008, 73, 9508–9510 CrossRef CAS PubMed; (b) S. Li, C. Hu, X. Cui, J. Zhang, L. L. Liu and L. Wu, Angew. Chem., Int. Ed., 2021, 60, 26238 Search PubMed; (c) C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714–719 Search PubMed; (d) S. Rej and N. Chatani, J. Am. Chem. Soc., 2021, 143, 2920–2929 CrossRef CAS PubMed; (e) M. Huang, J. Hu, S. Shi, A. Friedrich, J. Krebs, S. A. Westcott, U. Radius and T. B. Marder, Chem. – Eur. J., 2022, 28, e202200480 Search PubMed; (f) X. Tan, X. Wang, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2022, 144, 23286–23291 CAS.
- (a) A. Arase, M. Hoshi, A. Mijin and K. Nishi, Synth. Commun., 1995, 25, 1957–1962 CAS; (b) Q. Yin, S. Kemper, H. F. T. Klare and M. Oestreich, Chem. – Eur. J., 2016, 22, 13840 CAS; (c) N. Ang, C. Buettner, S. Docherty, A. Bismuto, J. Carney, J. Docherty, M. Cowley and S. Thomas, Synthesis, 2018, 803–808 CAS; (d) A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CAS; (e) A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, Synthesis, 2023, 62–74 CAS; (f) F. Wech, N. Koch, T. Müller and U. Gellrich, Org. Chem. Front., 2024, 11, 5921–5927 CAS; (g) J. H. Docherty, K. Nicholson, A. P. Dominey and S. P. Thomas, ACS Catal., 2020, 10, 4686–4691 Search PubMed.
- A. Das and T. K. Panda, ChemCatChem, 2023, 15, e202201011 CAS.
- (a) C. E. Tucker, J. Davidson and P. Knochel, J. Org. Chem., 1992, 57, 3482–3485 CrossRef CAS; (b) N. N. H. Ton, B. K. Mai and T. V. Nguyen, J. Org. Chem., 2021, 86, 9117–9133 CrossRef CAS; (c) P. Huninik, J. Szyling, A. Czapik and J. Walkowiak, Green Chem., 2023, 25, 3715–3722 RSC.
- A.-M. Fuller, D. L. Hughes, S. J. Lancaster and C. M. White, Organometallics, 2010, 29, 2194–2197 CrossRef CAS.
- R. J. Blagg and G. G. Wildgoose, RSC Adv., 2016, 6, 42421–42427 RSC.
- K. Nicholson, J. Dunne, P. DaBell, A. B. Garcia, A. D. Bage, J. H. Docherty, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2021, 11, 2034–2040 CrossRef CAS.
- (a) R. Köster, Ann. N. Y. Acad. Sci., 1969, 159, 73–88 CrossRef; (b) P. Vasko, I. A. Zulkifly, M. Á. Fuentes, Z. Mo, J. Hicks, P. C. J. Kamer and S. Aldridge, Chem. – Eur. J., 2018, 24, 10531 CrossRef CAS PubMed; (c) E. Nieto-Sepulveda, A. D. Bage, L. A. Evans, T. A. Hunt, A. G. Leach, S. P. Thomas and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2019, 141, 18600–18611 CrossRef CAS PubMed; (d) A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, Synthesis, 2023, 62–74 Search PubMed.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization, computational analysis and additional information. See DOI: https://doi.org/10.1039/d5cc01110h |
| This journal is © The Royal Society of Chemistry 2025 |