
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereochemical matching determines both helix type and handedness in α/γ-peptides with a cyclic-constrained γ-amino acid†
Dayi
Liu
 a,
Ali T.
Mansour
ab,
Ogaritte
Yazbeck
b,
Daoud
Naoufal
b,
Sylvie
Robin
ac,
Eric
Gloaguen
d,
Valérie
Brenner
e,
Michel
Mons
*f and
David J.
Aitken
*a
a,
Ali T.
Mansour
ab,
Ogaritte
Yazbeck
b,
Daoud
Naoufal
b,
Sylvie
Robin
ac,
Eric
Gloaguen
d,
Valérie
Brenner
e,
Michel
Mons
*f and
David J.
Aitken
*a
aUniversité Paris-Saclay, CNRS, ICMMO, 91400 Orsay, France. E-mail: david.aitken@universite-paris-saclay.fr
bLebanese University, Faculty of Sciences I & PRASE-EDST, LCIO, Hadath, Lebanon
cUniversité Paris Cité, Faculté de Pharmacie, 75006 Paris, France
dUniversité Paris-Saclay, CNRS, ISMO, 91400 Orsay, France
eUniversité Paris-Saclay, CEA, DRF, 91191 Gif-sur-Yvette, France
fUniversité Paris-Saclay, CEA, LIDYL, 91191 Gif-sur-Yvette, France. E-mail: michel.mons@cea.fr
First published on 4th April 2025
Abstract
The folding preferences of α/γ-peptides containing a bespoke chiral cyclobutane-constrained γ-amino acid have been examined in a low-polarity solvent by quantum chemical calculations. With (S)-alanine, the preferred conformation is a right-handed 12/10 helix, whereas with (R)-alanine a left-handed 12 helical architecture is promoted. Experimental evidence for this dichotomy was obtained by detailed analysis of the IR amide I and II absorption bands and their assignments with assistance from theoretical simulations.
Foldamers are synthetic oligomers that adopt ordered conformations stabilized by intramolecular networks of non-covalent interactions.1–3 Peptide-based foldamers have been of particular interest, due to the modular nature of the requisite building blocks: these are not limited to α-amino acids (AAs), but include homologated (β-, γ-, and δ-) families too. Studies on homo-peptides and mixed peptides have revealed an impressive array of folded structures that are often reminiscent of the secondary structures found in Nature and are logically classified as helices, sheets and turns, although the topology of such architectures varies greatly as a function of the constitutive monomer units.4–7
Mixed α/γ-peptides alternate α- and γ-AAs in their primary sequence. Pioneering theoretical studies by Hofmann8 suggested that several types of stable helically-folded scaffolds might be adopted by α/γ-peptides. Subsequent experimental work, conducted by several groups,9–33 revealed examples of two main helical families in solution and in the solid state: the 12 helix and the 12/10 helix. There is a remarkable difference in the features that stabilize these two foldamer structures: the former relies on a uniformly oriented series of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(i)⋯H–N(i + 3) (12-ring) H-bonds whereas the latter features an alternation between CO(i)⋯H−N(i + 3) (12-ring) and CO(i)⋯H−N(i − 1) (10-ring) H-bonds that have opposite orientations (Fig. 1).
O(i)⋯H–N(i + 3) (12-ring) H-bonds whereas the latter features an alternation between CO(i)⋯H−N(i + 3) (12-ring) and CO(i)⋯H−N(i − 1) (10-ring) H-bonds that have opposite orientations (Fig. 1).
 | ||
| Fig. 1 Left: The H-bonding networks of 12 helix (top) and 12/10 helix (bottom) structures of α/γ-peptides. Right: Dihedral angles and numbering system in γ-AAs (top) and α-AAs (bottom). | ||
Efforts have been made to identify factors that may privilege one or other helical type (Fig. 2). For the most part, γ4-, γ4,4- and γ3,3-AAs (I–III) give rise to 12 helix structures when used in conjunction with an (S)-α-AA or the achiral Aib (α,α-dimethylglycine).16–21,25,28–31 The effects of backbone fluorinated γ-AAs on the 12 helix stability of oligomers of III were examined recently.33 Cyclic constraints have been employed with a view to imposing (or disfavoring) particular backbone torsion angles and thus dictating (or not) helical preferences.34 The γ2,3,4-AA (IV)14 in combination with an (R)-α-AA supports a 12 helix, whereas 12/10 helix formation is promoted by γ2,3-AA (V)32 and γ2,3,4-AA (VI)10 in alternation with an (S)-α-AA or by γ2,3,4-AA (VII)11 in alternation with an (R)-α-AA.
 | ||
| Fig. 2 Examples of previously-studied γ-AAs that promote helical structures in α/γ-peptides and the γ-AA considered in this work. | ||
In these studies, the focus has been on the nature of the γ-AA component and in general the configuration of the α-AA (in cases where this has been chiral) was selected in order to best accommodate the dihedral angles of the anticipated helical structure on the basis of Hofmann's theoretical work. A few clues have emerged suggesting that stereochemical matching between the α- and γ-AA components may be important: an α/γ-peptide comprising the enantiomer of one example of a γ4-AA (I) and (S)-alanine folded into a 12/10 helix rather than a 12 helix.15 However, no study has been conducted on the specific premise that a single chiral γ4-AA might participate in either 12 or 12/10 helical α/γ-peptide structures in which the configuration of the α-AA plays a determinant role.
To address this question, we perceived that the cyclobutane-constrained γ3,4-AA, (R,R)-3,4CB-GABA (Fig. 2),35 might serve as a useful probe in combination with (R)- or (S)-alanine as the α-AA component. To this end, we performed the detailed solution-state theoretical investigation described below and sought some experimental support for the findings. In the event, the limited solubility of the α/γ-peptide sequences led us to identify an unhabitual but crucial role for IR spectroscopy in the amide I and II regions in order to correlate experimental observations with theory. The α/γ-peptide sequences studied herein are presented in Fig. 3.
 | ||
| Fig. 3 α/γ-peptides considered in this work. | ||
Quantum chemistry calculations were carried out using DFT associated with a polarizable continuum model to assess the relative stabilities and the vibrational spectra of 12 and 12/10 helical structures formed by (R,R,S) amides 1A and 2A and (R,R,R) amides 3A and 4A in chloroform solution. The level of theory used for geometry optimization (RI-B97-D3-(BJ)-abc/def2-TZVPPD + COSMO model) was previously reported to reproduce successfully the structures and the energetic trends in the conformational landscape of short peptides.36–38 No significant variability of the helical architectures was observed along the series, with the exception of the C-terminus where the possibility of a C7 H-bond instead of a C10 H-bond arose, along with the benzyl group interacting with the neighboring cyclobutane ring. Vibrational data, obtained at a slightly more modest level of theory (RI-B97-D3-(BJ)/def2-TZVP + COSMO model), enabled us to provide relative Gibbs energies of the helical structures at 300 K (see ESI† Section S1.1). These high level energetic data for a chloroform solution (Fig. 4 and ESI† Table S1-1) were found to be strikingly dependent on the configuration of the Ala residue: in the (R,R,R) series, L-12 (Fig. 5) was the most stable helical form for 3A and 4A and the only one expected in solution, since its challengers, L-12/10 and R-12/10, were found to be much higher in energy (>25 kJ mol−1). In contrast, rather more competition was indicated in the (R,R,S) series, where the most stable R-12/10 structures (Fig. 5) for 1A and 2A were only 9 and 19 kJ mol−1 more stable, respectively, than the L-12 challengers. This suggested that, at least in the former case, both R-12/10 and L-12 helical forms might be expected in solution.
 | ||
| Fig. 4 Comparison of the relative energetics (ΔG at 300 K) of the four peptide amides 1A–4A in CHCl3 solution, as obtained by quantum chemistry calculations at the B97-D3 + COSMO model level of theory (details in ESI† Section S1.1). Peptides of the same length have the same energy reference. The precision, based on a previous estimate for capped single residue compounds (3 kJ mol−1),36–38 is considered accurate to within 10 kJ mol−1 for hexapeptides. Each level is labelled with the type of helix, its handedness (L or R; left or right) together with the Bn group orientation (g+ or g−). Additional labels in parentheses (CB and C7) indicate a Bn moiety interacting with a cyclobutane ring and the presence of a C7 H-bond instead of a C10 at the C-terminus, respectively. | ||
 | ||
| Fig. 5 Lowest energy helical conformations calculated for hexapeptide amides in CHCl3 solution: the R-12/10 helix of (R,R,S) 2A and the L-12 helix of (R,R,R) 4A. H-bonds are indicated with dotted lines and H-bonding distances are given in pm. | ||
These trends were rationalized from a further quantum chemistry study (see ESI† Section S1.2 for details) as being due to the combined effects of (i) configuration-dependent steric clashes that disfavor left-handed helices in the (R,R,S) series and (ii) solvation that facilitates 12 helix structures due to their larger macrodipole. The IR absorption spectra of the most stable helical forms of the four amides 1A–4A and of the two hexapeptide esters 2E and 4E were simulated using quantum chemistry at the RI-B97-D3-(BJ)/def2-TZVP + COSMO model level of theory.
The α/γ-peptides shown in Fig. 3 were synthesized using solution-state techniques (see ESI† Section S2) and turned out to be poorly soluble. Dilute chloroform solutions (1–3 mM) were examined to acquire experimental IR absorption data.
In the amide A region (see ESI† Section S3.1) all peptides showed free NH vibrations at 3440 cm−1. In the (R,R,R) series, tetrapeptide ester 3E showed minimal absorption below 3400 cm−1, suggesting little inclination towards H-bonded conformations. Peptides 3A, 4E and 4A each showed a strong low-frequency band, centered at 3332, 3325 and 3325 cm−1, respectively, that was compatible with a predominant helical conformation. In the (R,R,S) series, 1E showed a broadened band, centered at 3330 cm−1 with a shoulder at around 3300 cm−1. Tetrapeptide 1A showed a broadened band at 3300 cm−1 while hexapeptides 2E and 2A both showed a stronger band centered at 3295 cm−1. These data suggest that, with increasing peptide length, the (R,R,S) and (R,R,R) series adopt structures displaying amide A maxima at 3325 and 3295 cm−1, respectively, although the broadness of the absorption bands thwarts any conclusion that only one conformer type might be present in each case. Furthermore, ambiguity arose when comparison was made with the theoretical spectra (see ESI† Section S1.3.2), since these absorptions could be interpreted in terms of either of the two helical conformations, L-12 or R-12/10.
In order to better understand the solution-state peptide behavior patterns, we therefore focused attention on the amide I and II absorption bands. Indeed, the amide I region (CO stretch) is known to be strongly dependent on the relative disposition of the strongly coupled CO moieties and is therefore expected to be sensitive to the helical type. This facet was confirmed by an extensive theoretical study (see ESI† Section S1.3.1) and the good correspondence achieved between experimental and simulated spectra (Fig. 6) suggested it to be the method of choice for the diagnosis of the helical type.
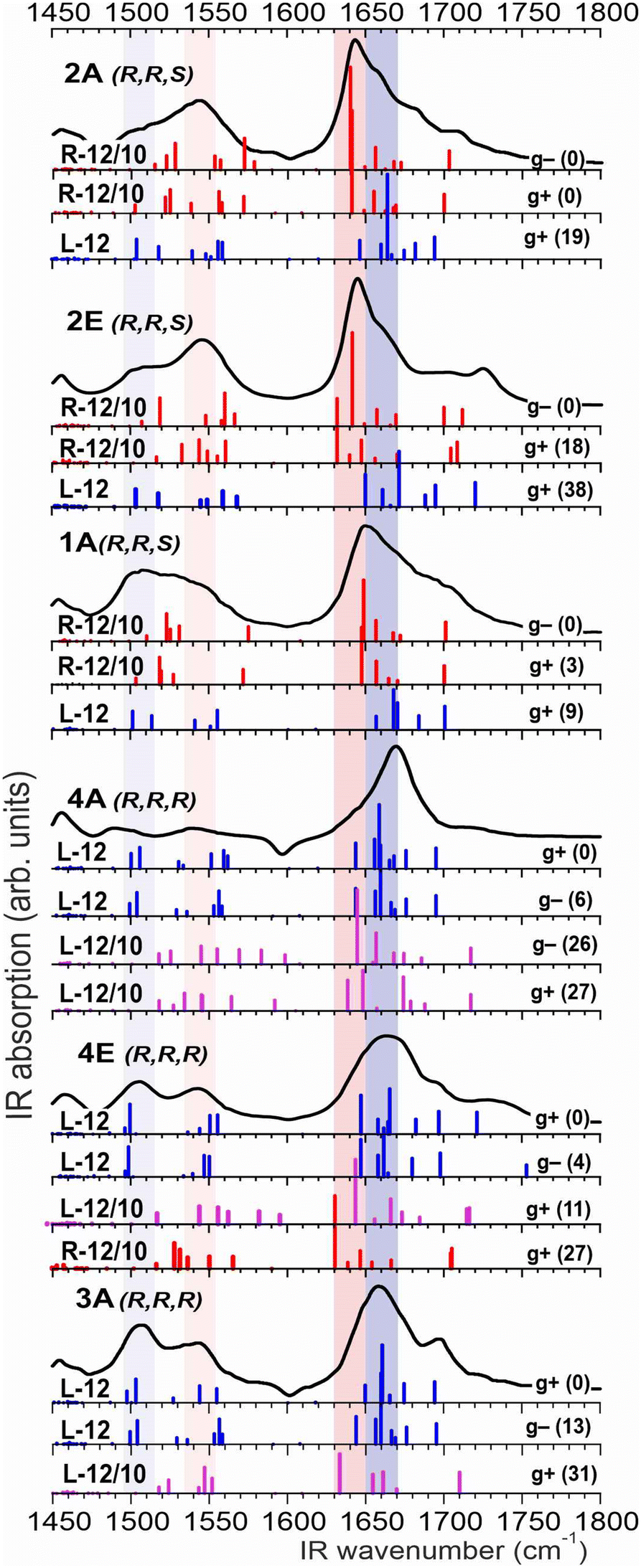 | ||
| Fig. 6 Experimental absorption spectra in the amide I and II regions (black traces) of 1A, 2E, 2A, 3A, 4E and 4A (CHCl3 solutions). For each peptide, the theoretical spectra (obtained at the RI-B97-D3-(BJ)/def2-TZVP + COSMO model level) of L-12, R-12/10 and L-12/10 forms (with kJ mol−1 relative energies in parentheses) are shown as colored sticks (blue, red and magenta, respectively). The colored zones (blue and pink) are diagnostic for the 12 and 12/10 backbones, respectively. | ||
In the (R,R,R) series, the experimental amide I and II absorption bands of 3A, 4E and 4A showed excellent agreement with the theoretical absorptions for L-12 structures, corroborating the predominance of this helical form in solution. Of note were the amide I feature at around 1660 cm−1 and the weaker amide II feature at 1505 cm−1 (Fig. 6, blue zones) which emerge as diagnostics for L-12. In the (R,R,S) series, the experimental absorptions are best interpreted in terms of a dominant R-12/10 form for 2E and 2A while contributions from both R-12/10 and L-12 forms prevail for 1A. The amide I feature at around 1640 cm−1 matches the main absorption bands calculated for the lower energy R-12/10 conformations (Fig. 6, pink zone). A detailed inspection, however, reveals that for the tetrapeptide 1A, this band is broad and straddles the diagnostic frequencies of R-12/10 and L-12 (Fig. 6, pink and blue zones), pointing to the presence of both of these conformers. This conclusion is supported by a significant amide II absorption around 1505 cm−1 (Fig. 6, blue zone), specific to the L-12 structure. In 2E and 2A, this absorption is depleted and replaced by another amide II band at 1540 cm−1, suggesting that the L-12 structure is diminished leaving the R-12/10 form as the major conformation. Reassuringly, these interpretations converge with the relative energetics (Fig. 4) and disambiguate the amide A data mentioned above. The IR spectra of representative peptides of each series, 1A and 3A, were unchanged after 5-fold dilution, indicating that only intramolecular interactions were implicated in solution-state behavior.
Solubility limitations largely precluded other spectroscopic studies, a fact that underlines the critical role of the solution-state IR studies. We were able to perform DMSO-d6 titration experiments of 1A and 3A in CDCl3 that confirmed the H-bonding status of all NH protons in complete agreement with 12/10 and 12 helix conformations, respectively. 1H NMR ROESY data was obtained for the (R,R,S) series peptides (1E, 1A, 2E, 2A) in the more polar solvent pyridine-d5; non-local correlations indicated the presence of 12/10 helical conformations (see ESI† Section S3.3). ECD spectra for these peptides in methanol showed Cotton effects that were in agreement with a right-handed 12/10 helix (see ESI† Section S3.4).39
Collectively, the conformational behaviour patterns are intriguing. The R-12/10 form of 2A (R,R,S series) shows an average γ-AA backbone dihedral θ = +14°, while the L-12 form of 4A (R,R,R series) shows an average value of θ = −28°. In the former case the puckered cyclobutane ring disposes the N atom in a pseudo-equatorial position and Cα in a pseudo-axial position, whereas in the latter case these positions are inversed, leading to a change of dihedral sign as required by a change of helix handedness. The φ/ψ dihedrals of the alanine components in 2A and 4A are very close to Hofmann's theoretical values for R-12/10 and L-12 helical forms, respectively (−64°/+142° and +65°/+35° respectively).
In summary, the equivocal behavior of the (R,R)-3,4CB-GABA γ-AA allows it to support both R-12/10 and L-12 helical α/γ-peptide conformations in low-polarity solution, with the preferred structure being determined by the configuration of the α-AA. While it was known that the alteration of helix types could be achieved by changing configurations within γ-AA residues, the present discovery provides a significant alternative principle for helical foldamer control. These observations should be useful in furthering the development of design tools to control helix structure and handedness.40,41
D. L. was the recipient of a CSC Chinese PhD scholarship. A. T. M. was the recipient of a CIOES Lebanese PhD scholarship. Support from the French National Research Agency (Grant ANR-17-CE29-0008) is acknowledged.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC.
- Foldamers: Structure, Properties, and Applications, ed. S. Hecht and I. Huc, Wiley, Weinheim, Germany, 2007 Search PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS PubMed.
- T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687 RSC.
- D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366 CrossRef CAS PubMed.
- P. G. Vasudev, S. Chatterjee, N. Shamala and P. Balaram, Chem. Rev., 2011, 111, 657 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- C. Baldauf, R. Günther and H. J. Hofmann, J. Org. Chem., 2006, 71, 1200 CrossRef CAS PubMed.
- B. F. Fisher and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 10766 CrossRef CAS PubMed.
- B. F. Fisher, L. Guo, B. S. Dolinar, I. A. Guzei and S. H. Gellman, J. Am. Chem. Soc., 2015, 137, 6484 CrossRef CAS PubMed.
- M. W. Giuliano, S. J. Maynard, A. M. Almeida, L. Guo, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 15046 CrossRef CAS PubMed.
- M. W. Giuliano, S. J. Maynard, A. M. Almeida, A. G. Reidenbach, L. Guo, E. C. Ulrich, I. A. Guzei and S. H. Gellman, J. Org. Chem., 2013, 78, 12351 CrossRef CAS PubMed.
- L. Guo, W. C. Zhang, I. A. Guzei, L. C. Spencer and S. H. Gellman, Org. Lett., 2012, 14, 2582 CrossRef CAS PubMed.
- L. Guo, Y. G. Chi, A. M. Almeida, I. A. Guzei, B. K. Parker and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 16018 CrossRef CAS PubMed.
- G. V. M. Sharma, V. B. Jadhav, K. V. S. Ramakrishna, P. Jayaprakash, K. Narsimulu, V. Subash and A. C. Kunwar, J. Am. Chem. Soc., 2006, 128, 14657 CrossRef CAS PubMed.
- M. B. M. Reddy, K. Basuroy, S. Chandrappa, B. Dinesh, V. Basavalingappa, M. A. Venkatesha and P. Balaram, New J. Chem., 2015, 39, 3319 RSC.
- R. Sonti, B. Dinesh, K. Basuroy, S. Raghothama, N. Shamala and P. Balaram, Org. Lett., 2014, 16, 1656 CrossRef CAS PubMed.
- B. Dinesh, V. Vinaya, S. Raghothama and P. Balaram, Eur. J. Org. Chem., 2013, 3590 CrossRef CAS.
- K. Basuroy, B. Dinesh, N. Shamala and P. Balaram, Angew. Chem., Int. Ed., 2012, 51, 8736 CrossRef CAS PubMed.
- S. Chatterjee, P. G. Vasudev, S. Raghothama, C. Ramakrishnan, N. Shamala and P. Balaram, J. Am. Chem. Soc., 2009, 131, 5956 CrossRef CAS PubMed.
- K. Ananda, P. G. Vasudev, A. Sengupta, K. M. P. Raja, N. Shamala and P. Balaram, J. Am. Chem. Soc., 2005, 127, 16668 CrossRef CAS PubMed.
- R. Misra, G. George, A. Saseendran, S. Raghothama and H. N. Gopi, Chem. – Asian J., 2019, 14, 4408 CrossRef CAS PubMed.
- R. Misra, K. M. P. Raja, H. J. Hofmann and H. N. Gopi, Chem. – Eur. J., 2017, 23, 16644 CrossRef CAS PubMed.
- A. Malik, M. G. Kumar, A. Bandyopadhyay and H. N. Gopi, Pep. Sci., 2017, 108, e22978 CrossRef PubMed.
- R. Misra, A. Saseendran, G. George, K. Veeresh, K. M. P. Raja, S. Raghothama, H. J. Hofmann and H. N. Gopi, Chem. – Eur. J., 2017, 23, 3764 CrossRef CAS PubMed.
- M. G. Kumar, V. J. Thombare, M. M. Katariya, K. Veeresh, K. M. P. Raja and H. N. Gopi, Angew. Chem., Int. Ed., 2016, 55, 7847 CrossRef PubMed.
- A. Bandyopadhyay, A. Malik, M. G. Kumar and H. N. Gopi, Org. Lett., 2014, 16, 294 CrossRef CAS PubMed.
- S. V. Jadhav, R. Misra, S. K. Singh and H. N. Gopi, Chem. – Eur. J., 2013, 19, 16256 CrossRef CAS PubMed.
- S. V. Jadhav, A. Bandyopadhyay and H. N. Gopi, Org. Biomol. Chem., 2013, 11, 509 RSC.
- A. Bandyopadhyay, S. V. Jadhav and H. N. Gopi, Chem. Commun., 2012, 48, 7170 RSC.
- A. Bandyopadhyay and H. N. Gopi, Org. Lett., 2012, 14, 2770 CrossRef CAS PubMed.
- R. Fanelli, D. Berta, T. Földes, E. Rosta, R. A. Atkinson, H. J. Hofmann, K. Shankland and A. J. A. Cobb, J. Am. Chem. Soc., 2020, 142, 1382 CrossRef CAS PubMed.
- A. R. Patel, A. Lawer, M. Bhadbhade and L. Hunter, Org. Biomol. Chem., 2024, 22, 1608 RSC.
- Some cyclically-constrained γ-AAs have produced non-helical α/γ-peptide architectures: (a) C. Bonnel, B. Legrand, M. Simon, J. Martinez, J.-L. Bantignies, Y. K. Kang, E. Wenger, F. Hoh, N. Masurier and L. T. Maillard, Chem. – Eur. J., 2017, 23, 17584 CrossRef CAS PubMed; (b) F. Bouillère, D. Feytens, D. Gori, R. Guillot, C. Kouklovsky, E. Miclet and V. Alezra, Chem. Commun., 2012, 48, 1982 RSC; (c) D. Just, V. Palivec, K. Bártová, L. Bednárová, M. Pazderková, I. Cisarová, H. Martinez-Seara and U. Jahn, Commun. Chem., 2024, 7, 114 CrossRef CAS PubMed.
- H. Awada, S. Robin, R. Guillot, O. Yazbeck, D. Naoufal, N. Jaber, A. Hachem and D. J. Aitken, Eur. J. Org. Chem., 2014, 7148 CrossRef CAS.
- Z. Imani, V. R. Mundlapati, V. Brenner, E. Gloaguen, K. Le Barbu-Debus, A. Zehnacker-Rentien, S. Robin, D. J. Aitken and M. Mons, Chem. Commun., 2023, 59, 1161 RSC.
- D. Y. Liu, J. X. Bardaud, Z. Imani, S. Robin, E. Gloaguen, V. Brenner, D. J. Aitken and M. Mons, Molecules, 2023, 28, 5048 CrossRef CAS PubMed.
- D. Y. Liu, S. Robin, E. Gloaguen, V. Brenner, M. Mons and D. J. Aitken, Chem. Commun., 2024, 60, 2074 RSC.
- For the (R,R,R) series peptides, comparable ROESY experiments provided no exploitable data and ECD experiments revealed no regular folding pattern.
- I. M. Mándity, E. Wéber, T. A. Martinek, G. Olajos, G. K. Tóth, E. Vass and F. Fülöp, Angew. Chem., Int. Ed., 2009, 48, 2171 CrossRef PubMed.
- E. Rudzinska-Szostak and L. Berlicki, Chem. – Eur. J., 2017, 23, 14980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00746a |
| This journal is © The Royal Society of Chemistry 2025 |